
Towards Engineering Novel PE-Based Immunotoxins by Targeting Them to the Nucleus

Abstract
:1. Introduction
2. Results and Discussion
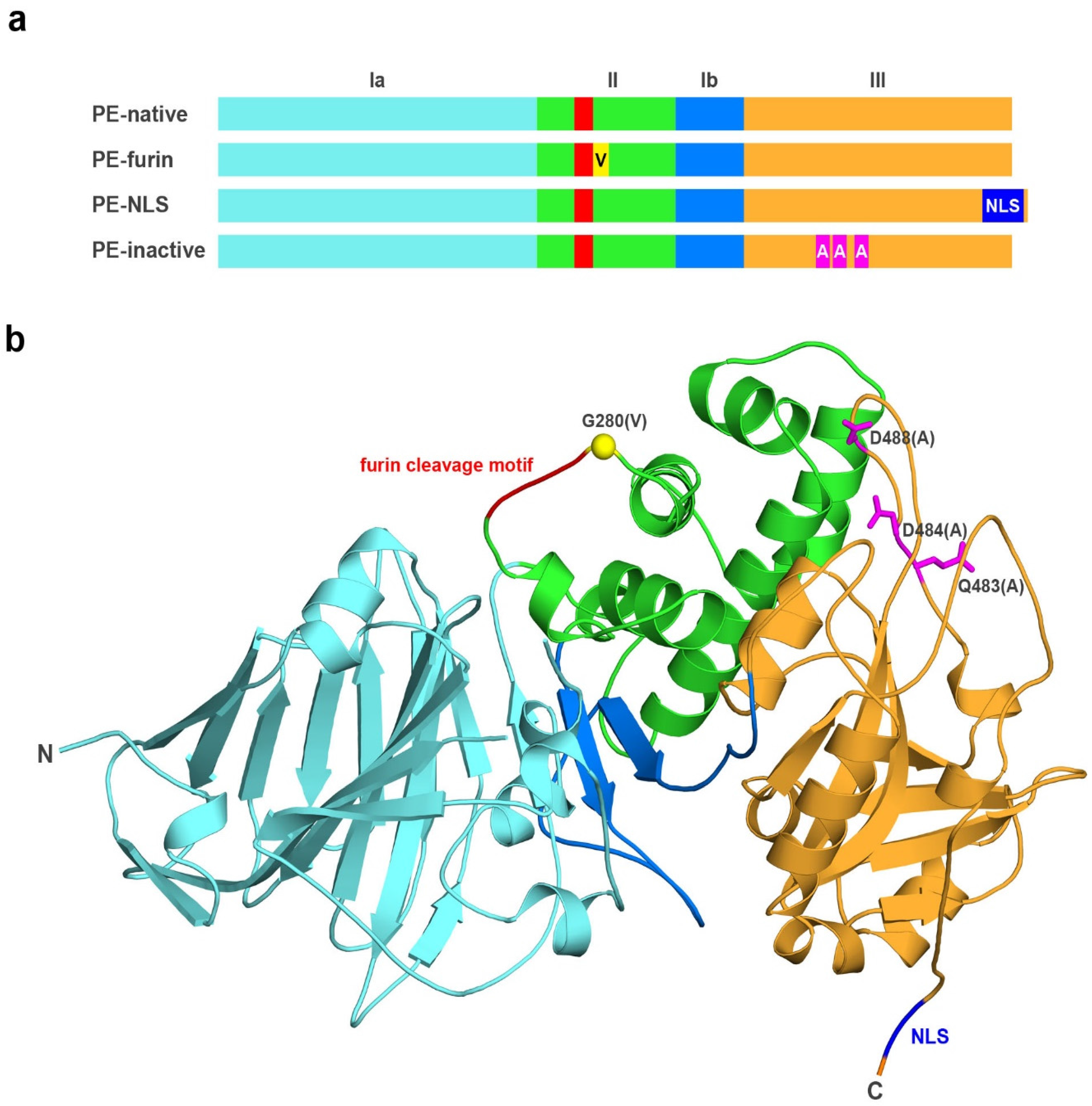
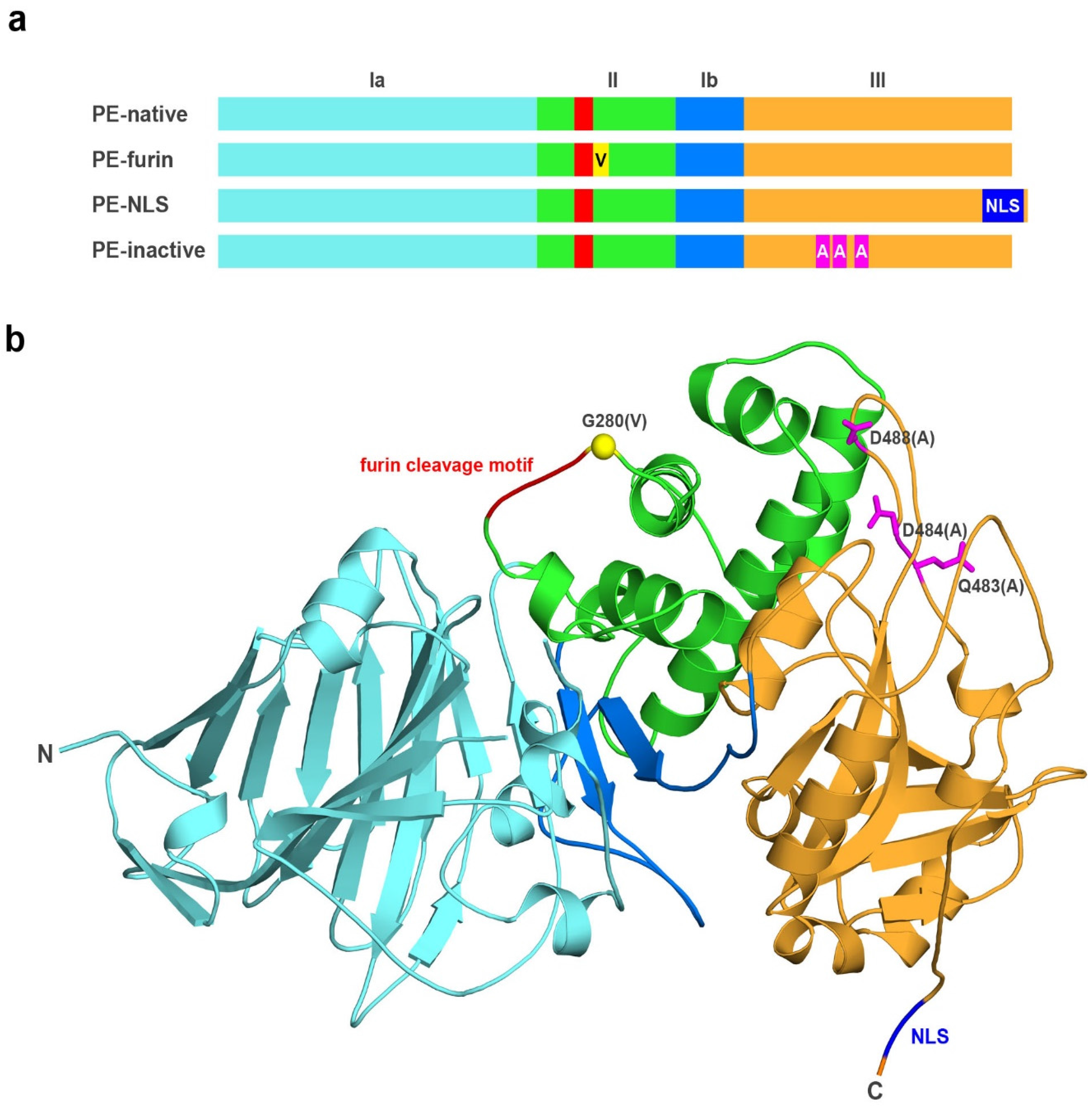
2.1. Analyzed Muteins
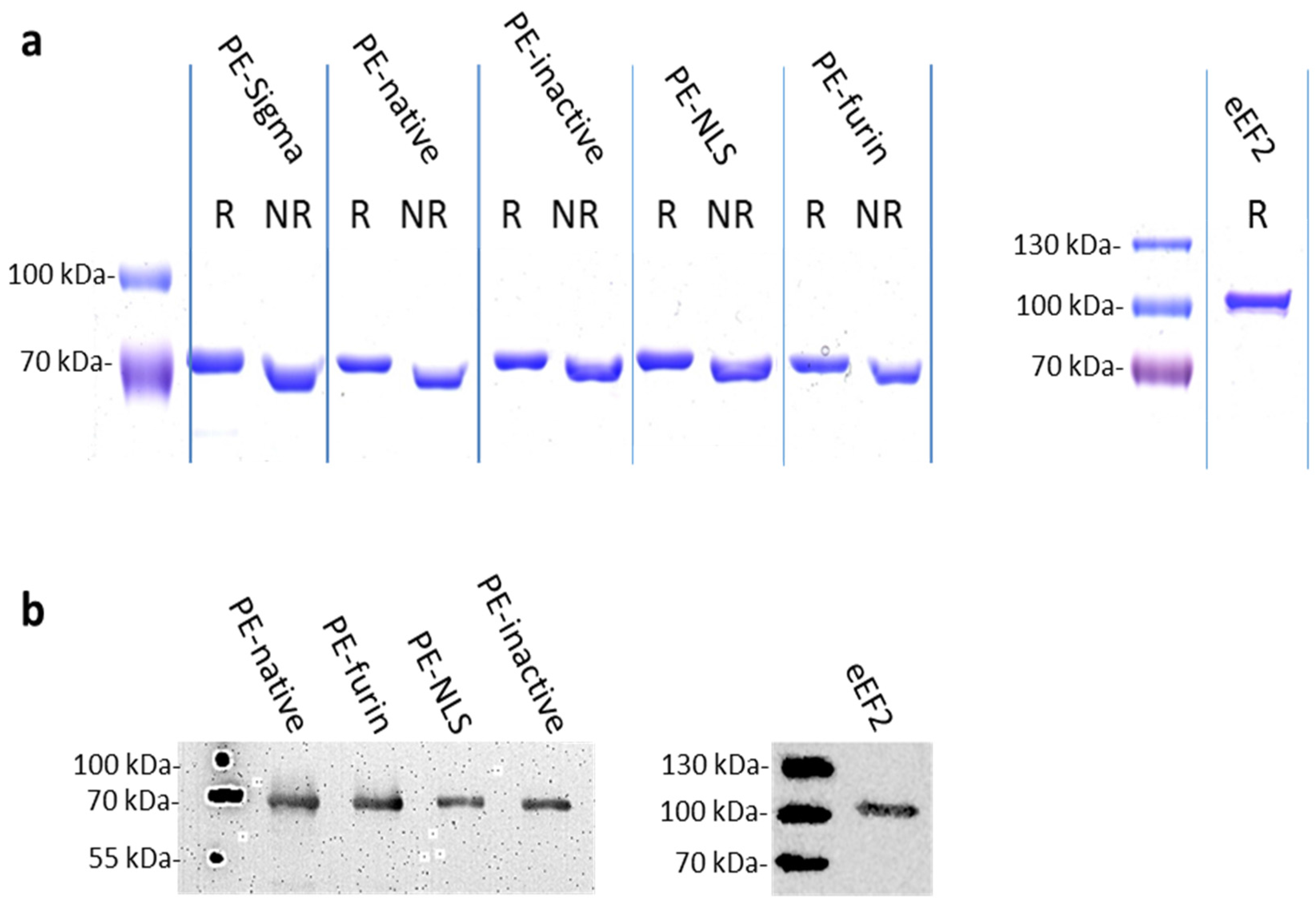
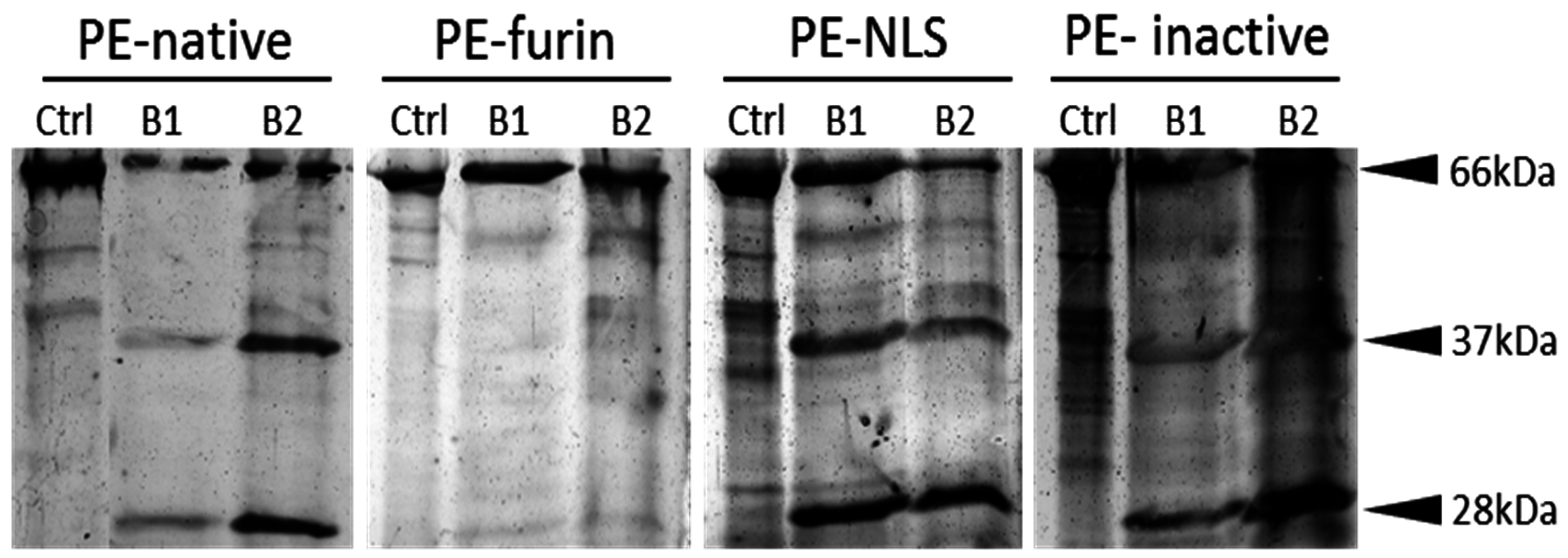
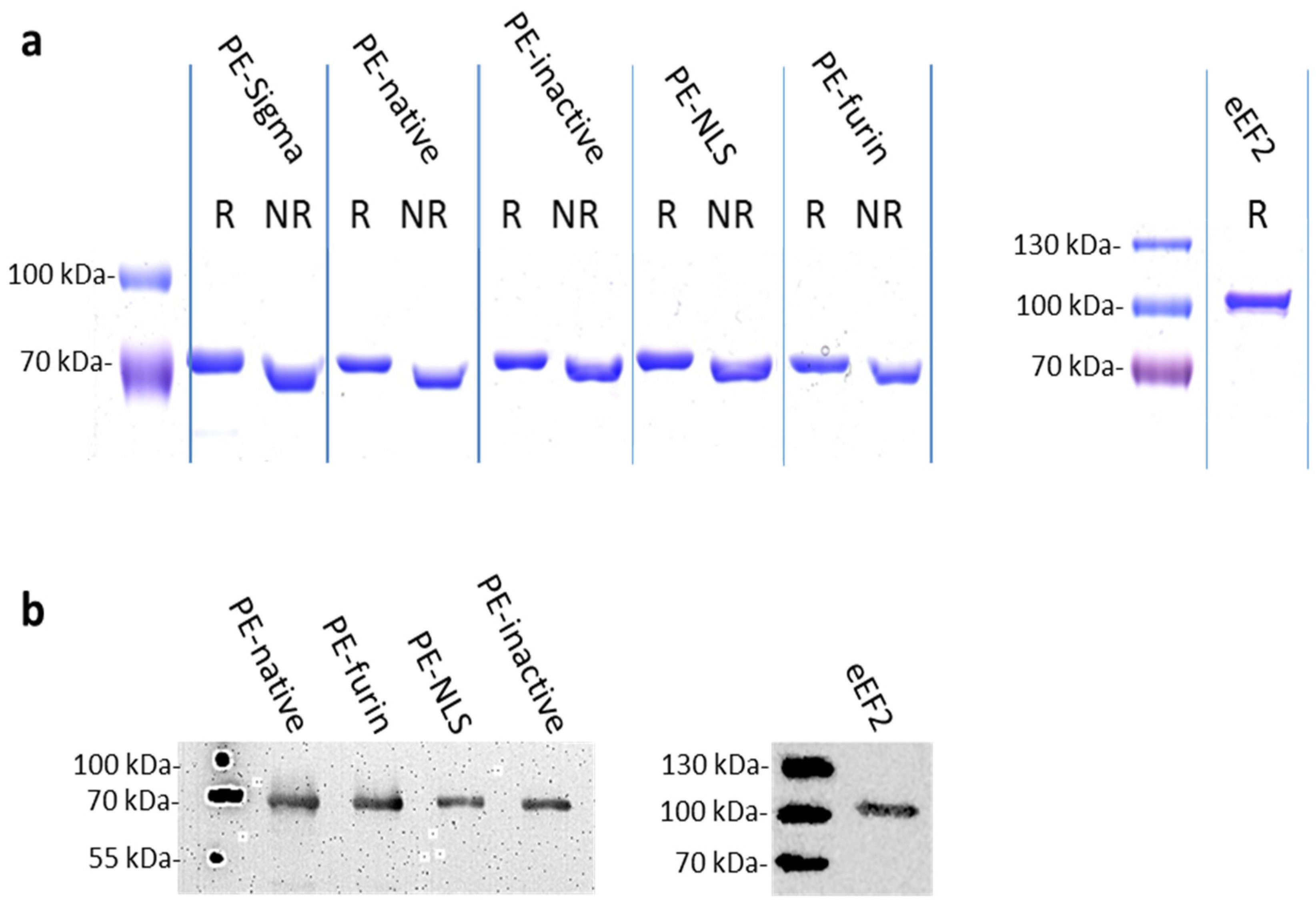
2.2. Protein Purification
2.3. ADP-Ribosylation Activity of Analyzed Toxins
2.4. Cytotoxicity of Analyzed Toxins
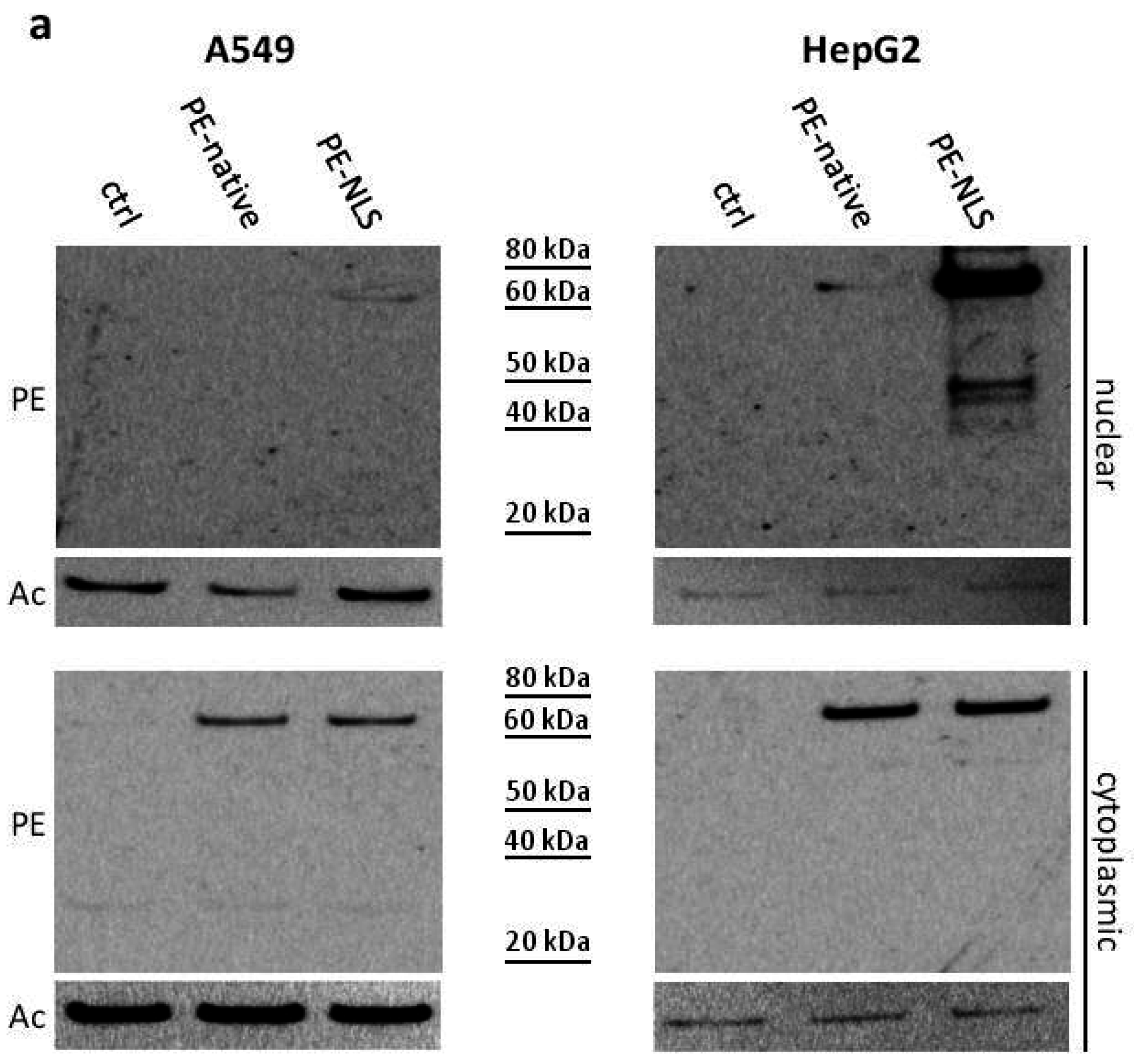
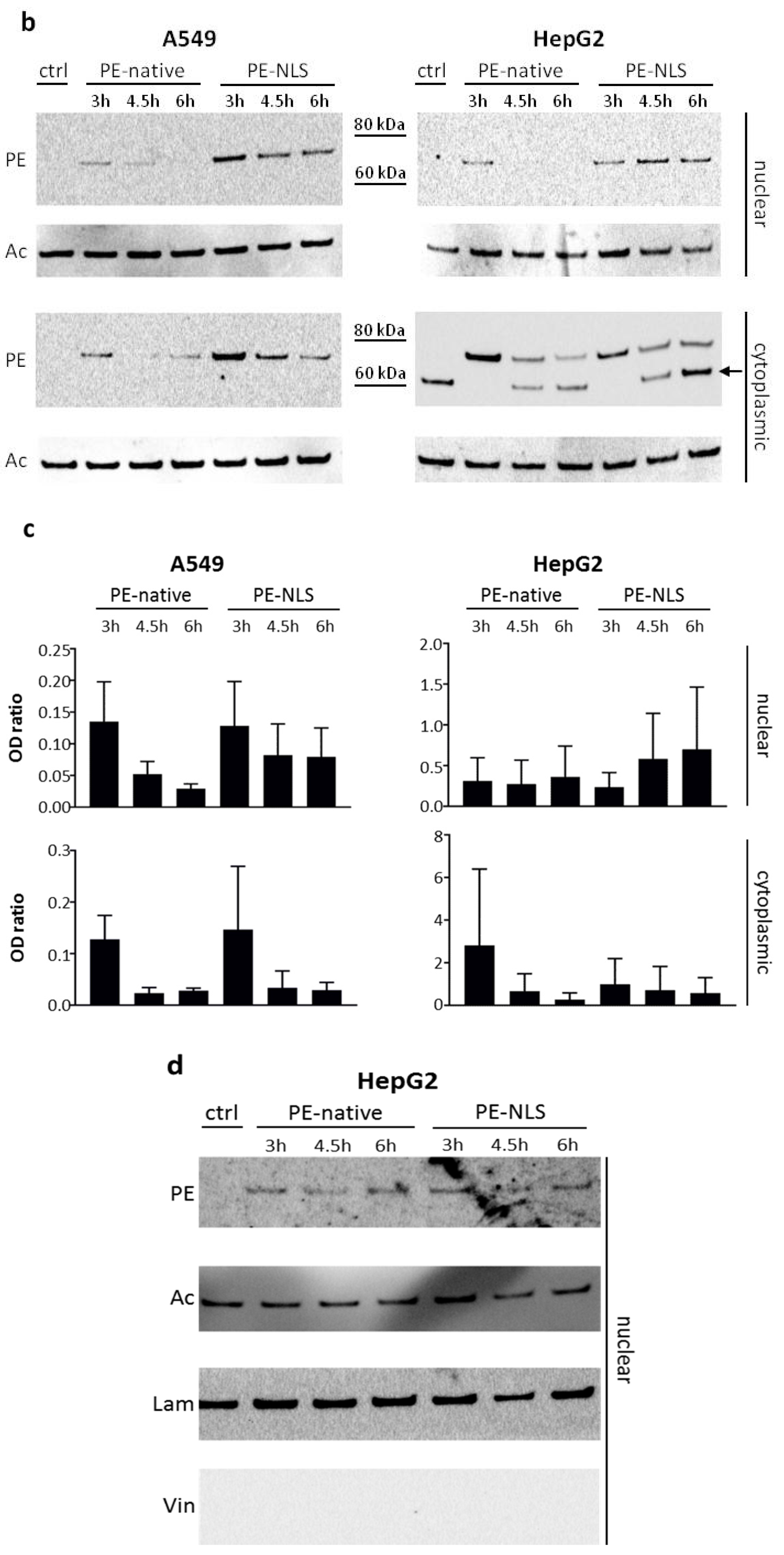
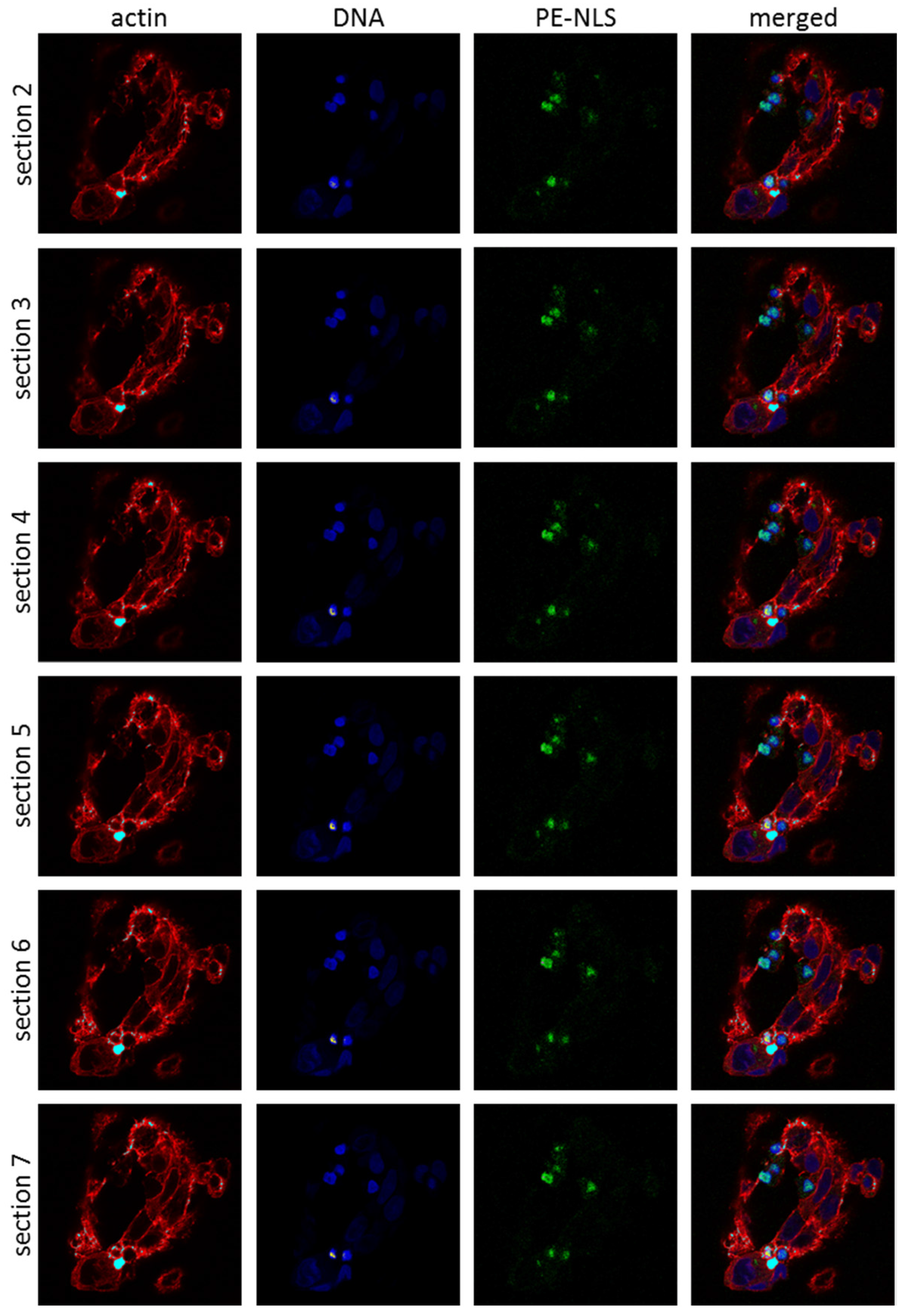
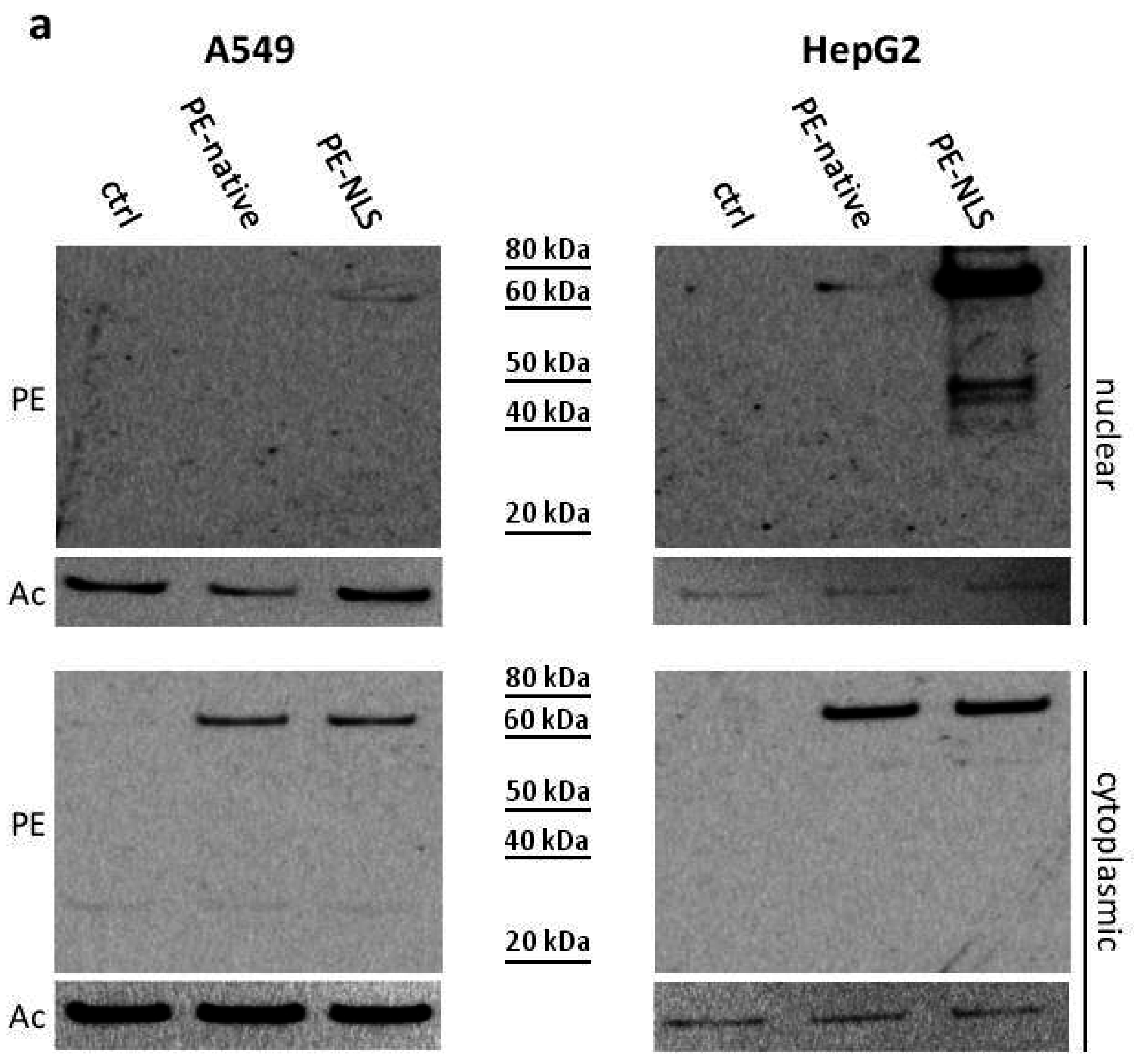
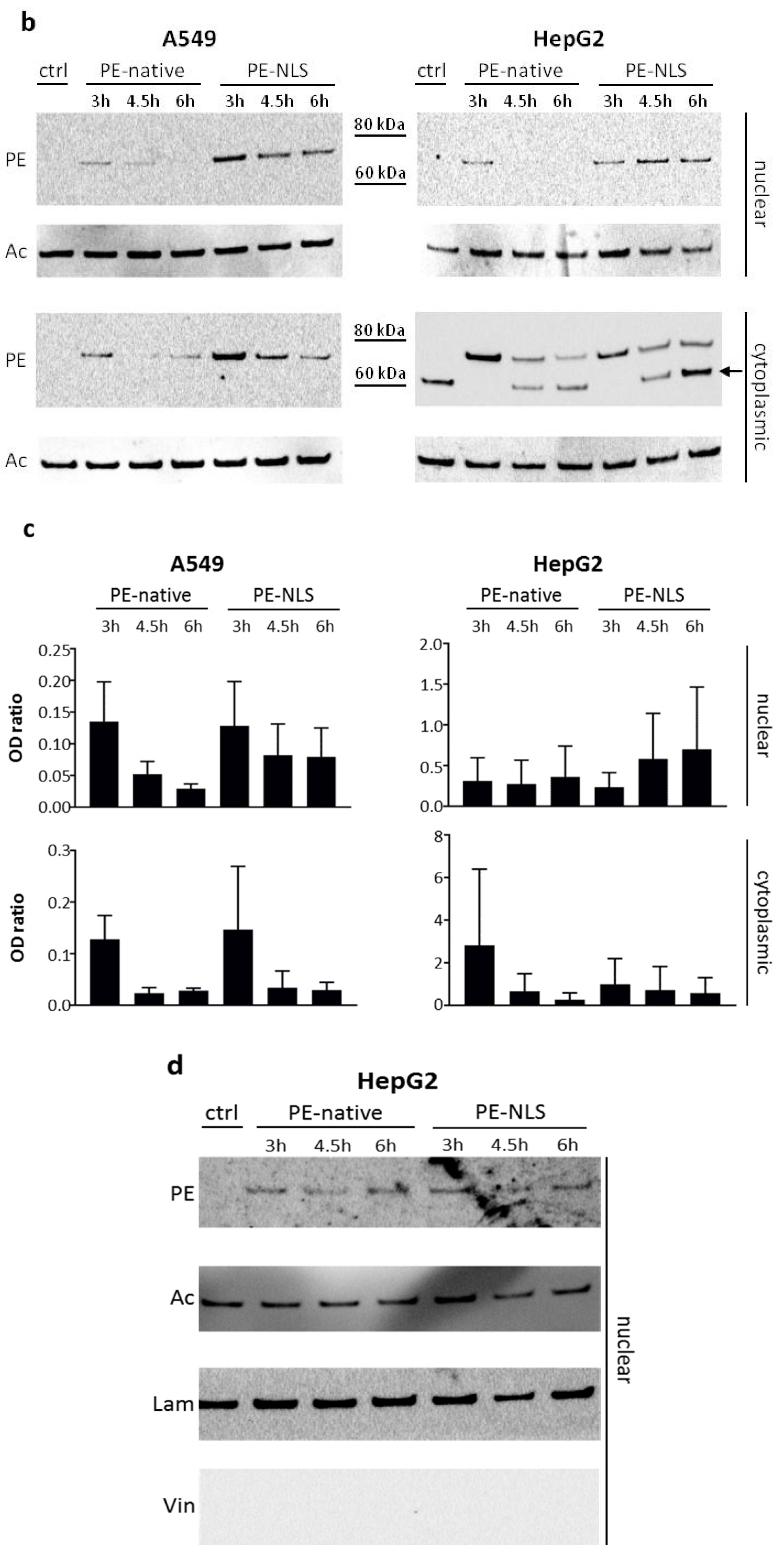
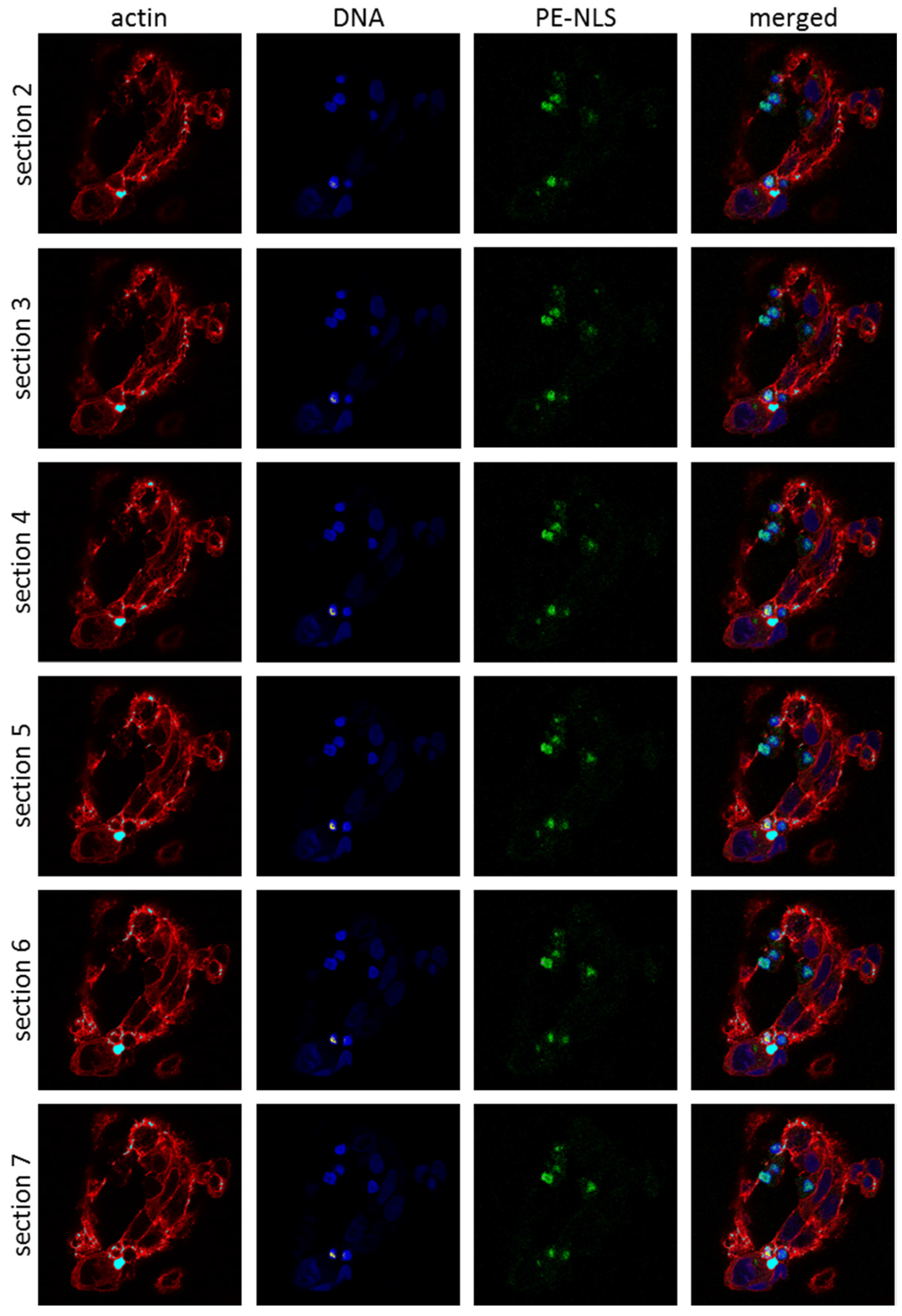
2.5. Toxins Accumulation inside the Cell
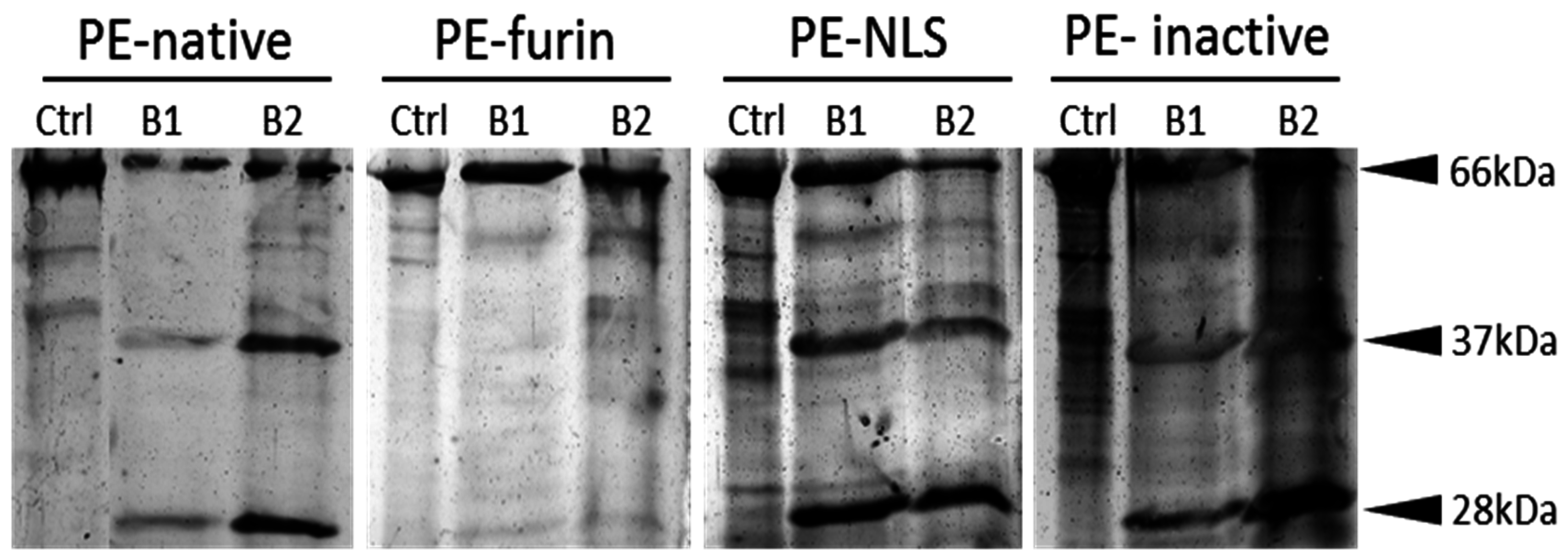
2.6. Furin Cleavage
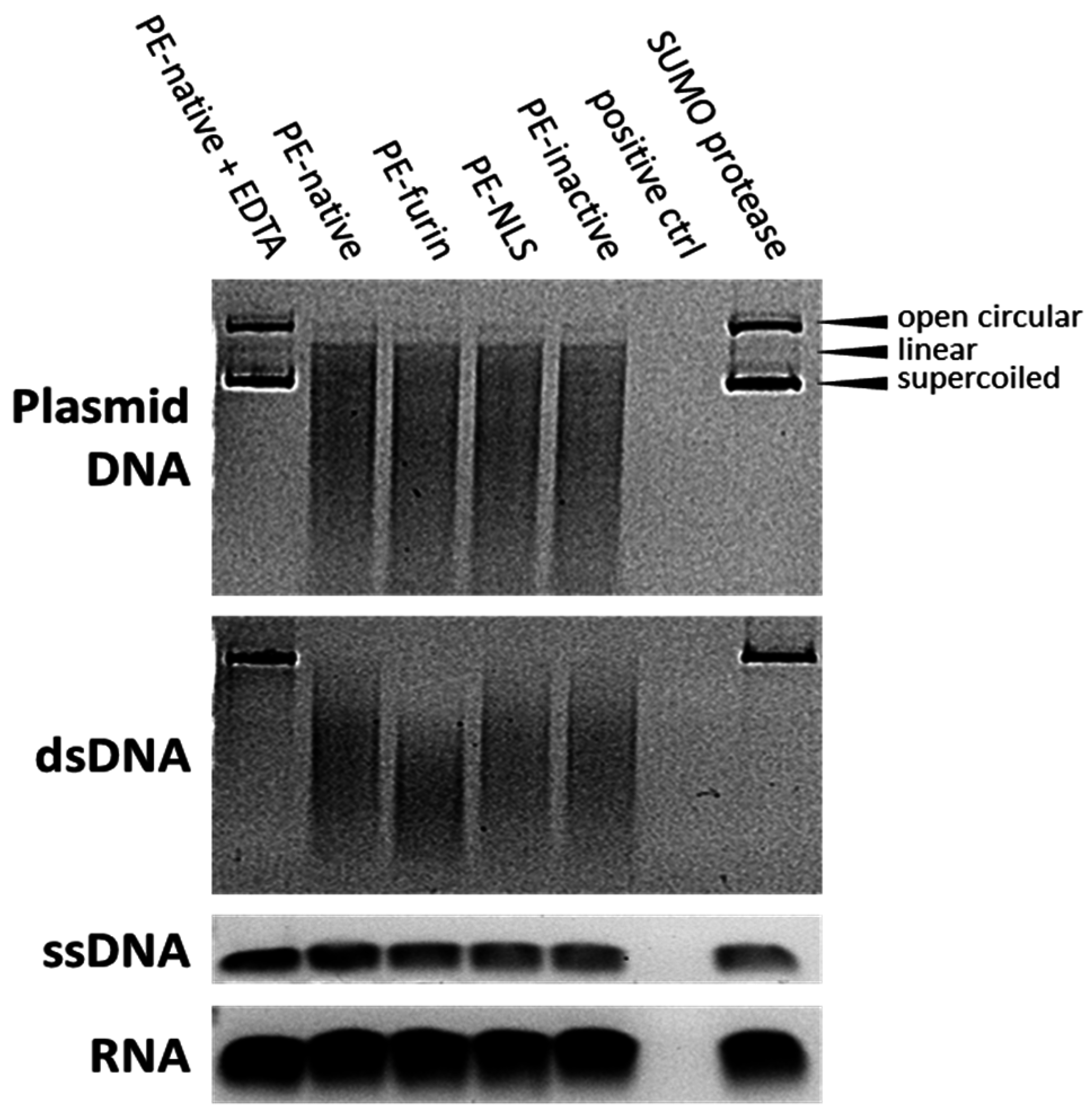
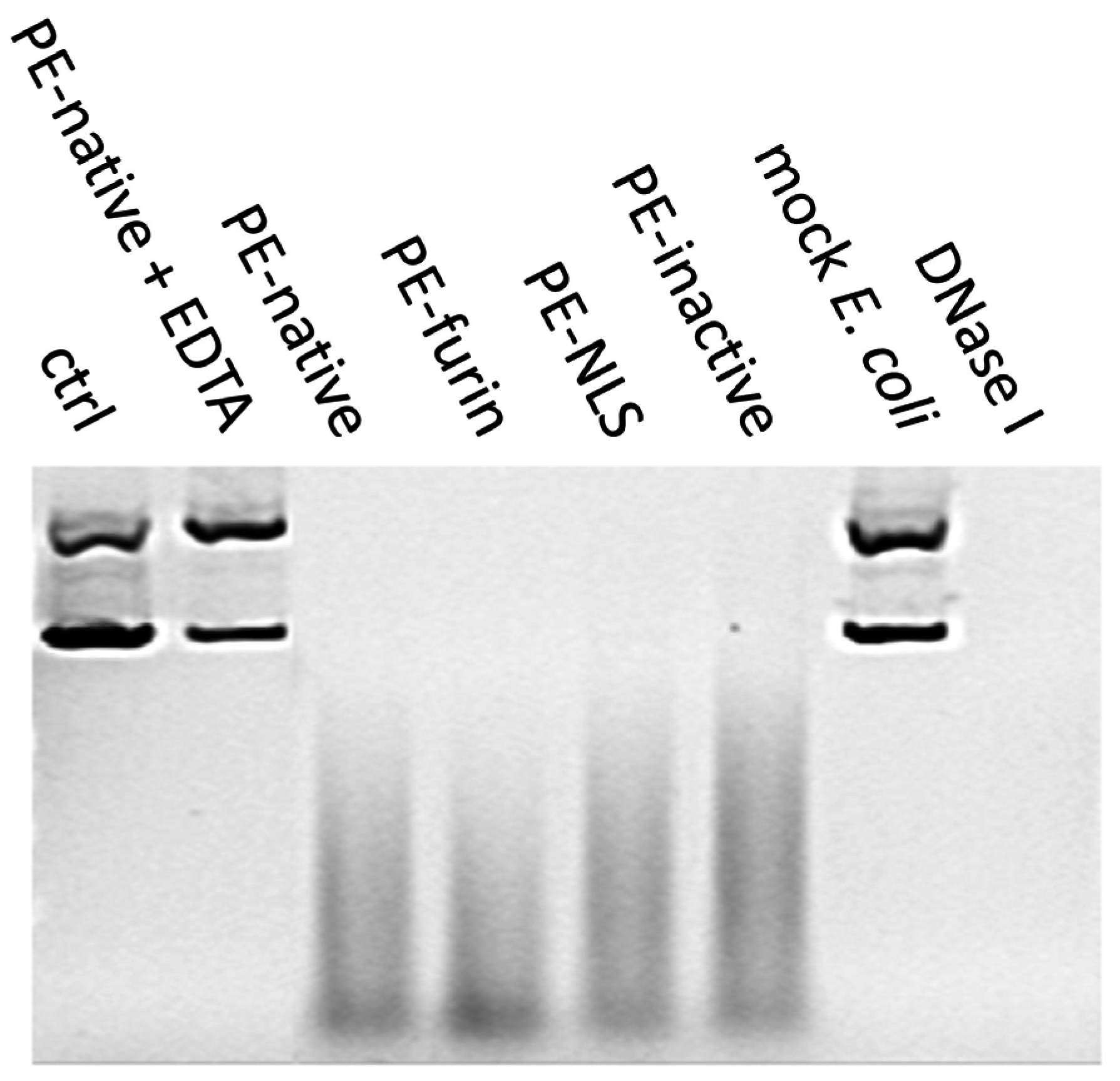
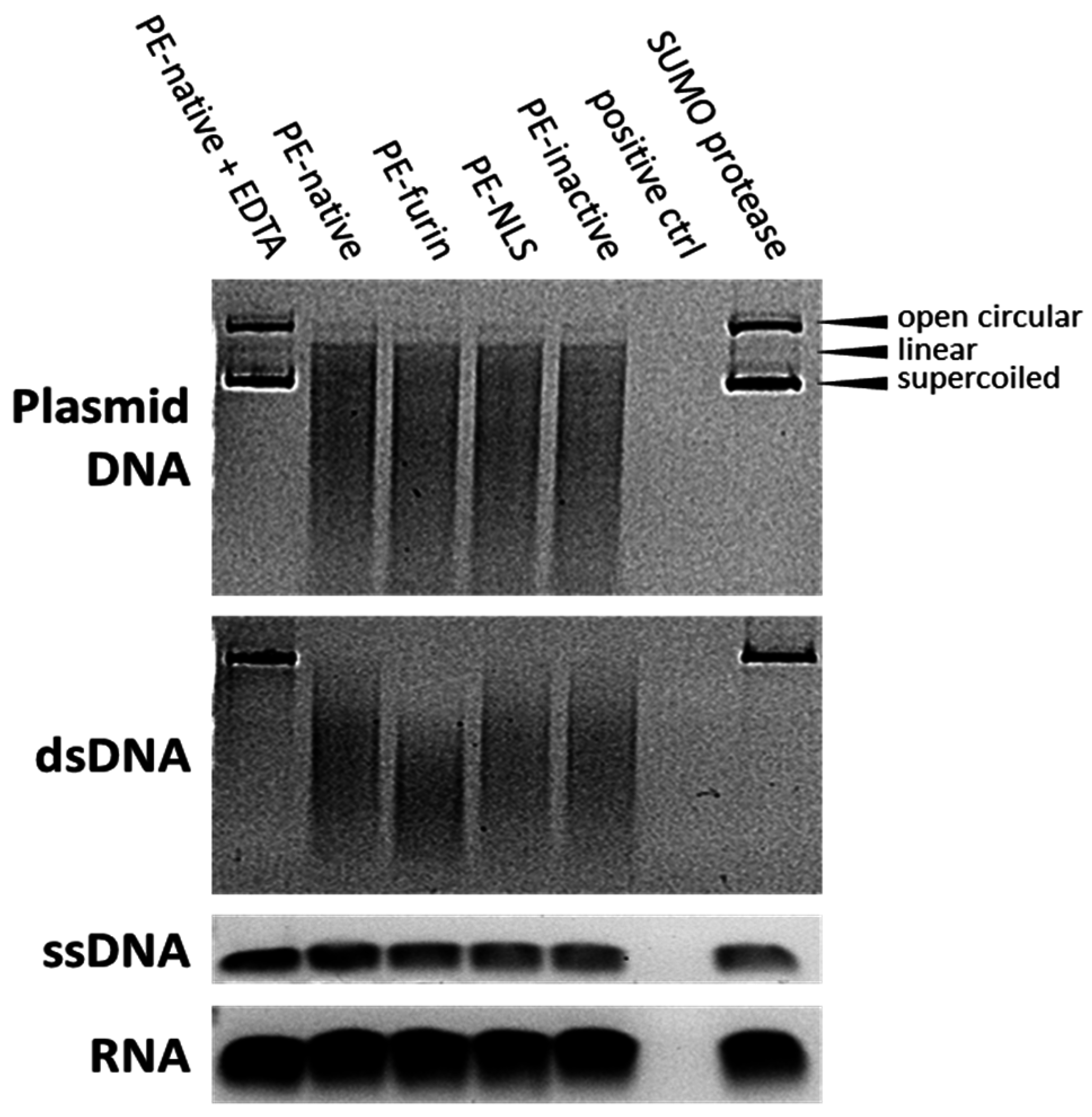
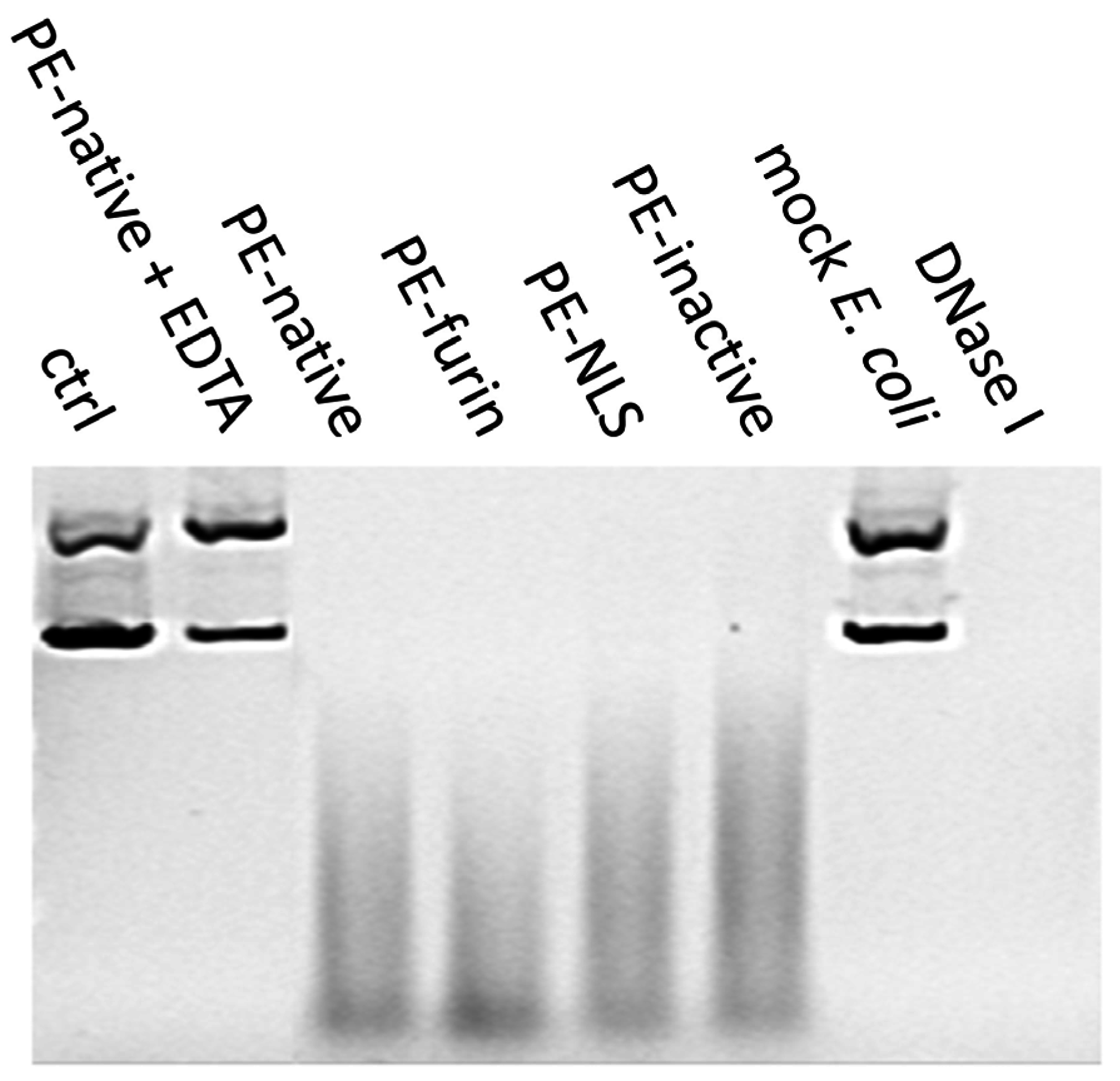
2.7. Deoxyribonclease Activity of PE
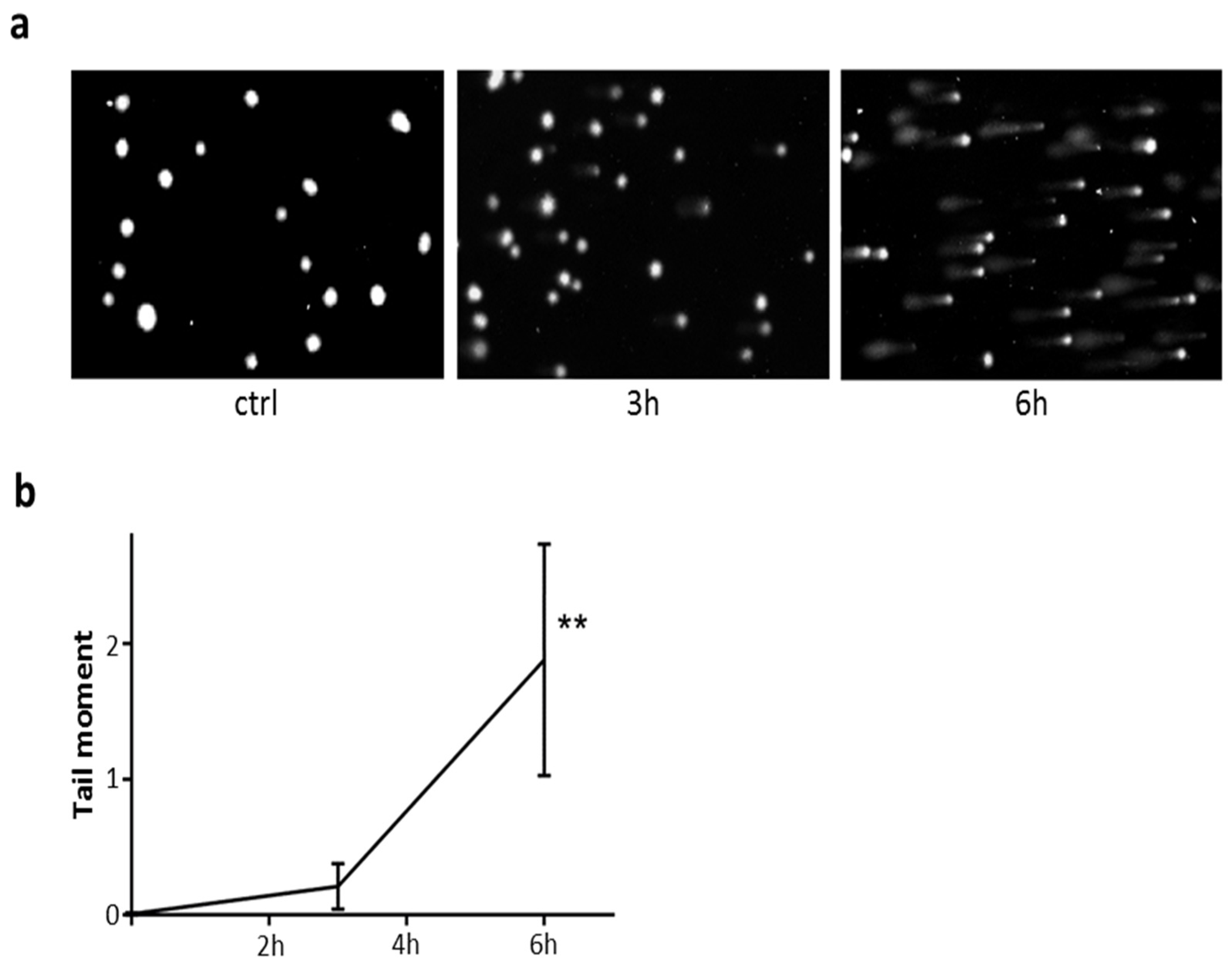
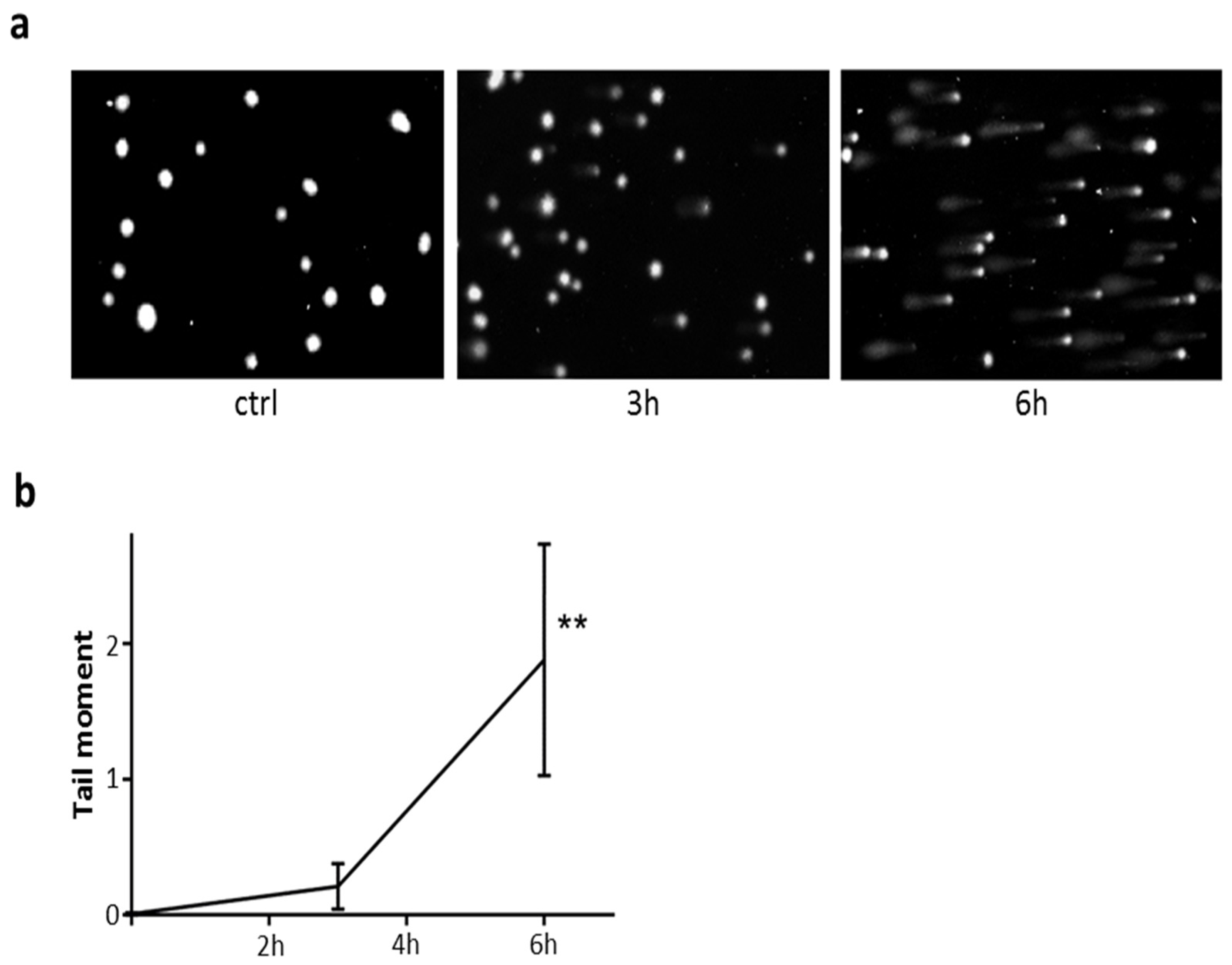
2.8. DNA Damage in Cells Exposed to PE-NLS
3. Conclusions
4. Materials and Methods
4.1. Proteins Expression and Purification
4.2. ADP-Ribosylation Assays
4.2.1. Western Blotting with Anti-Biotin
4.2.2. Solid-Phase Assay
4.3. Cytotoxicity Measurements
4.4. Viability Measurements
4.5. Intracellular Localization Studies
4.5.1. Western Blotting with Anti-Exotoxin A
4.5.2. Confocal Microscopy
4.6. Furin Digestion Assay
4.7. Agarose Gel Nuclease Assays
4.7.1. DNA/RNA Degradation
4.7.2. Electroelution
4.8. Alkaline Comet Assay
4.9. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Weldon, J.E.; Pastan, I. A guide to taming a toxin-recombinant immunotoxins constructed from Pseudomonas Exotoxin A for the treatment of cancer. FEBS J. 2011, 278, 4683–4700. [Google Scholar] [CrossRef]
- Wilson, B.A.; Collier, R.J. Diphtheria toxin and Pseudomonas aeruginosa Exotoxin A: Active-site structure and enzymic mechanism. Curr. Top. Microbiol. Immunol. 1992, 175, 27–41. [Google Scholar]
- Falnes, P.O.; Sandvig, K. Penetration of protein toxins into cells. Curr. Opin. Cell Biol. 2000, 12, 407–413. [Google Scholar] [CrossRef]
- Lord, J.M.; Smith, D.C.; Roberts, L.M. Toxin entry: How bacterial proteins get into mammalian cells. Cell. Microbiol. 1999, 1, 85–91. [Google Scholar] [CrossRef]
- Brinkmann, U.; Buchner, J.; Pastan, I. Independent domain folding of Pseudomonas Exotoxin and single-chain immunotoxins—Influence of interdomain connections. Proc. Natl. Acad. Sci. USA 1992, 89, 3075–3079. [Google Scholar] [CrossRef]
- Wedekind, J.E.; Trame, C.B.; Dorywalska, M.; Koehl, P.; Raschke, T.M.; McKee, M.; FitzGerald, D.; Collier, R.J.; McKay, D.B. Refined crystallographic structure of Pseudomonas aeruginosa Exotoxin A and its implications for the molecular mechanism of toxicity. J. Mol. Biol. 2001, 314, 823–837. [Google Scholar] [CrossRef]
- Jackson, M.E.; Simpson, J.C.; Girod, A.; Pepperkok, R.; Roberts, L.M.; Lord, J.M. The kdel retrieval system is exploited by Pseudomonas Exotoxin A, but not by shiga-like toxin-1, during retrograde transport from the golgi complex to the endoplasmic reticulum. J. Cell Sci. 1999, 112, 467–475. [Google Scholar]
- Chiron, M.F.; Fryling, C.M.; FitzGerald, D.J. Cleavage of Pseudomonas Exotoxin and diphtheria toxin by a furin-like enzyme prepared from beef liver. J. Biol. Chem. 1994, 269, 18167–18176. [Google Scholar]
- Wolf, P.; Elsasser-Beile, U. Pseudomonas Exotoxin A: From virulence factor to anti-cancer agent. Int. J. Med. Microbiol. 2009, 299, 161–176. [Google Scholar] [CrossRef]
- Michalska, M.; Wolf, P. Pseudomonas Exotoxin A: Optimized by evolution for effective killing. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef]
- Antignani, A.; Fitzgerald, D. Immunotoxins: The role of the toxin. Toxins 2013, 5, 1486–1502. [Google Scholar] [CrossRef]
- Brinkmann, U.; Pastan, I. Immunotoxins against cancer. Biochim. Biophys. Acta 1994, 1198, 27–45. [Google Scholar] [CrossRef]
- Alewine, C.; Hassan, R.; Pastan, I. Advances in anticancer immunotoxin therapy. Oncologist 2015, 20, 176–185. [Google Scholar] [CrossRef]
- FitzGerald, D.J.; Kreitman, R.; Wilson, W.; Squires, D.; Pastan, I. Recombinant immunotoxins for treating cancer. Int. J. Med. Microbiol. 2004, 293, 577–582. [Google Scholar] [CrossRef]
- Aruna, G. Immunotoxins: A review of their use in cancer treatment. J. Stem. Cells Regen. Med. 2006, 1, 31–36. [Google Scholar]
- Pennell, C.A.; Erickson, H.A. Designing immunotoxins for cancer therapy. Immunol. Res. 2002, 25, 177–191. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Ragin, A.D.; Morgan, R.A.; Chmielewski, J. Cellular import mediated by nuclear localization signal peptide sequences. Chem. Biol. 2002, 9, 943–948. [Google Scholar] [CrossRef]
- Pandya, H.; Gibo, D.M.; Debinski, W. Molecular targeting of intracellular compartments specifically in cancer cells. Genes Cancer 2010, 1, 421–433. [Google Scholar] [CrossRef]
- Gerber, D.E. Targeted therapies: A new generation of cancer treatments. Am. Fam. Phys. 2008, 77, 311–319. [Google Scholar]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Liu, B.; Ezeogu, L.; Zellmer, L.; Yu, B.; Xu, N.; Joshua Liao, D. Protecting the normal in order to better kill the cancer. Cancer Med. 2015, 4, 1394–1403. [Google Scholar] [CrossRef]
- Ojima, I.; Zuniga, E.S.; Berger, W.T.; Seitz, J.D. Tumor-targeting drug delivery of new-generation taxoids. Future Med. Chem. 2012, 4, 33–50. [Google Scholar] [CrossRef]
- Lange, A.; Mills, R.E.; Lange, C.J.; Stewart, M.; Devine, S.E.; Corbett, A.H. Classical nuclear localization signals: Definition, function, and interaction with importin alpha. J. Biol. Chem. 2007, 282, 5101–5105. [Google Scholar] [CrossRef]
- Watanabe, T.; Nakagawa, T.; Ikemizu, J.; Nagahama, M.; Murakami, K.; Nakayama, K. Sequence requirements for precursor cleavage within the constitutive secretory pathway. J. Biol. Chem. 1992, 267, 8270–8274. [Google Scholar]
- Duckert, P.; Brunak, S.; Blom, N. Prediction of proprotein convertase cleavage sites. Protein Eng. Des. Sel. 2004, 17, 107–112. [Google Scholar] [CrossRef]
- Yates, S.P.; Merrill, A.R. A catalytic loop within Pseudomonas aeruginosa Exotoxin A modulates its transferase activity. J. Biol. Chem. 2001, 276, 35029–35036. [Google Scholar] [CrossRef]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant protein expression in Escherichia coli: Advances and challenges. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef]
- Singh, S.M.; Panda, A.K. Solubilization and refolding of bacterial inclusion body proteins. J. Biosci. Bioeng. 2005, 99, 303–310. [Google Scholar] [CrossRef]
- Beattie, B.K.; Merrill, A.R. In vitro enzyme activation and folded stability of Pseudomonas aeruginosa Exotoxin A and its C-terminal peptide. Biochemistry 1996, 35, 9042–9051. [Google Scholar] [CrossRef]
- Bolanos-Garcia, V.M.; Davies, O.R. Structural analysis and classification of native proteins from E. coli commonly co-purified by immobilised metal affinity chromatography. Biochim. Biophys. Acta 2006, 1760, 1304–1313. [Google Scholar] [CrossRef]
- Bachran, C.; Sutherland, M.; Bachran, D.; Fuchs, H. Quantification of diphtheria toxin mediated adp-ribosylation in a solid-phase assay. Clin. Chem. 2007, 53, 1676–1683. [Google Scholar] [CrossRef]
- Chaumet, A.; Wright, G.D.; Seet, S.H.; Tham, K.M.; Gounko, N.V.; Bard, F. Nuclear envelope-associated endosomes deliver surface proteins to the nucleus. Nat. Commun. 2015, 6, 8218. [Google Scholar] [CrossRef]
- Sekimoto, T.; Miyamoto, Y.; Arai, S.; Yoneda, Y. Importin alpha protein acts as a negative regulator for snail protein nuclear import. J. Biol. Chem. 2011, 286, 15126–15131. [Google Scholar] [CrossRef]
- Nguyen, T.; Sherratt, P.J.; Huang, H.C.; Yang, C.S.; Pickett, C.B. Increased protein stability as a mechanism that enhances NRF2-mediated transcriptional activation of the antioxidant response element. Degradation of NRF2 by the 26 S proteasome. J. Biol. Chem. 2003, 278, 4536–4541. [Google Scholar] [CrossRef]
- Risberg, K.; Fodstad, O.; Andersson, Y. The melanoma specific 9.2.27pe immunotoxin efficiently kills melanoma cells in vitro. Int. J. Cancer 2009, 125, 23–33. [Google Scholar] [CrossRef]
- Morlon-Guyot, J.; Mere, J.; Bonhoure, A.; Beaumelle, B. Processing of Pseudomonas aeruginosa Exotoxin A is dispensable for cell intoxication. Infect. Immun. 2009, 77, 3090–3099. [Google Scholar] [CrossRef]
- Falnes, P.O.; Olsnes, S. Modulation of the intracellular stability and toxicity of diphtheria toxin through degradation by the N-end rule pathway. EMBO J. 1998, 17, 615–625. [Google Scholar] [CrossRef]
- Soderholm, J.F.; Bird, S.L.; Kalab, P.; Sampathkumar, Y.; Hasegawa, K.; Uehara-Bingen, M.; Weis, K.; Heald, R. Importazole, a small molecule inhibitor of the transport receptor importin-beta. ACS Chem. Biol. 2011, 6, 700–708. [Google Scholar] [CrossRef]
- Melen, K.; Kinnunen, L.; Fagerlund, R.; Ikonen, N.; Twu, K.Y.; Krug, R.M.; Julkunen, I. Nuclear and nucleolar targeting of influenza a virus NS1 protein: Striking differences between different virus subtypes. J. Virol. 2007, 81, 5995–6006. [Google Scholar] [CrossRef]
- Liou, J.S.; Liu, B.R.; Martin, A.L.; Huang, Y.W.; Chiang, H.J.; Lee, H.J. Protein transduction in human cells is enhanced by cell-penetrating peptides fused with an endosomolytic HA2 sequence. Peptides 2012, 37, 273–284. [Google Scholar] [CrossRef]
- Kuo, H.Y.; Chen, Y.C.; Chang, H.Y.; Jeng, J.C.; Lin, E.H.; Pan, C.M.; Chang, Y.W.; Wang, M.L.; Chou, Y.T.; Shih, H.M.; et al. The PML isoform IV is a negative regulator of nuclear EGFR’s transcriptional activity in lung cancer. Carcinogenesis 2013, 34, 1708–1716. [Google Scholar] [CrossRef]
- Hsu, S.C.; Hung, M.C. Characterization of a novel tripartite nuclear localization sequence in the EGFR family. J. Biol. Chem. 2007, 282, 10432–10440. [Google Scholar] [CrossRef]
- McKee, M.L.; FitzGerald, D.J. Reduction of furin-nicked Pseudomonas Exotoxin A: An unfolding story. Biochemistry 1999, 38, 16507–16513. [Google Scholar] [CrossRef]
- Gu, M.; Gordon, V.M.; Fitzgerald, D.J.; Leppla, S.H. Furin regulates both the activation of Pseudomonas Exotoxin A and the quantity of the toxin receptor expressed on target cells. Infect. Immun. 1996, 64, 524–527. [Google Scholar]
- Mere, J.; Morlon-Guyot, J.; Bonhoure, A.; Chiche, L.; Beaumelle, B. Acid-triggered membrane insertion of Pseudomonas Exotoxin A involves an original mechanism based on ph-regulated tryptophan exposure. J. Biol. Chem. 2005, 280, 21194–21201. [Google Scholar] [CrossRef]
- Zdanovsky, A.G.; Chiron, M.; Pastan, I.; FitzGerald, D.J. Mechanism of action of Pseudomonas Exotoxin. Identification of a rate-limiting step. J. Biol. Chem. 1993, 268, 21791–21799. [Google Scholar]
- Lee, J.W.; Nakamura, L.T.; Chang, M.P.; Wisnieski, B.J. Mechanistic aspects of the deoxyribonuclease activity of diphtheria toxin. Biochim. Biophys. Acta 2005, 1747, 121–131. [Google Scholar] [CrossRef]
- Nakamura, L.T.; Wisnieski, B.J. Characterization of the deoxyribonuclease activity of diphtheria toxin. J. Biol. Chem. 1990, 265, 5237–5241. [Google Scholar]
- Bruce, C.; Baldwin, R.L.; Lessnick, S.L.; Wisnieski, B.J. Diphtheria toxin and its adp-ribosyltransferase-defective homologue CRM197 possess deoxyribonuclease activity. Proc. Natl. Acad. Sci. USA 1990, 87, 2995–2998. [Google Scholar] [CrossRef]
- Cooper, G.M. The nuclear envelope and traffic between the nucleus and cytoplasm. In The Cell: A Molecular Approach, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2000. [Google Scholar]
- Chang, M.P.; Baldwin, R.L.; Bruce, C.; Wisnieski, B.J. Second cytotoxic pathway of diphtheria toxin suggested by nuclease activity. Science 1989, 246, 1165–1168. [Google Scholar] [CrossRef]
- Miller, R.; Wiedmann, M. Dynamic duo-the salmonella cytolethal distending toxin combines adp-ribosyltransferase and nuclease activities in a novel form of the cytolethal distending toxin. Toxins 2016, 8. [Google Scholar] [CrossRef]
- Chiron, M.F.; Ogata, M.; FitzGerald, D.J. Pseudomonas Exotoxin exhibits increased sensitivity to furin when sequences at the cleavage site are mutated to resemble the arginine-rich loop of diphtheria toxin. Mol. Microbiol. 1996, 22, 769–778. [Google Scholar] [CrossRef]
- Robichon, C.; Luo, J.; Causey, T.B.; Benner, J.S.; Samuelson, J.C. Engineering escherichia coli BL21(DE3) derivative strains to minimize E. coli protein contamination after purification by immobilized metal affinity chromatography. Appl. Environ. Microbiol. 2011, 77, 4634–4646. [Google Scholar] [CrossRef]
- Repetto, G.; del Peso, A.; Zurita, J.L. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat. Protoc. 2008, 3, 1125–1131. [Google Scholar] [CrossRef]
- Smith, J.A. Electroelution of proteins from stained gels. Curr. Protoc. Immunol. 2002. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Toxin | EC50 ± SD (μg/mL) |
|---|---|
| PE-native | 0.54 ± 0.01 |
| PE-inactive | - |
| PE-NLS | 0.57 ± 0.19 |
| PE-furin | 0.40 ± 0.08 |
| PE-Sigma | 3.00 ± 0.40 |
| Toxin | IC50 ± SD (ng/mL) | |
|---|---|---|
| A549 | HepG2 | |
| PE-native | 12 ± 4 | 22 ± 4 * |
| PE-inactive | - | - |
| PE-NLS | 17 ± 6 | 13 ± 4 |
| PE-furin | 10 ± 1 | 13 ± 2 |
| PE-Sigma | 425 ± 65 | 3197 ± 1030 * |
| Toxin | Viability (%) | |||||
|---|---|---|---|---|---|---|
| A549 | A549 + MG132 | A549 + Importazole | HepG2 | HepG2 + Lactacystin | HepG2 + Importazole | |
| PE-native | 49.1 ± 2.8 | 24.7 ± 2.7 * | 53.3 ± 6.2 | 49.7 ± 2.0 | 27.9 ± 2.3 * | 54.3 ± 1.0 |
| PE-NLS | 52.2 ± 2.7 | 32.6 ± 4.9 * | 56.2 ± 3.8 | 52.9 ± 3.6 | 42.5 ± 3.1 * | 68.7 ± 1.0 ** |
| Ctrl | - | 82.6 ± 0.5 | 122.5 ± 3.3 | - | 100.3 ± 0.8 | 100.1 ± 4.3 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borowiec, M.; Gorzkiewicz, M.; Grzesik, J.; Walczak-Drzewiecka, A.; Salkowska, A.; Rodakowska, E.; Steczkiewicz, K.; Rychlewski, L.; Dastych, J.; Ginalski, K. Towards Engineering Novel PE-Based Immunotoxins by Targeting Them to the Nucleus. Toxins 2016, 8, 321. https://doi.org/10.3390/toxins8110321
Borowiec M, Gorzkiewicz M, Grzesik J, Walczak-Drzewiecka A, Salkowska A, Rodakowska E, Steczkiewicz K, Rychlewski L, Dastych J, Ginalski K. Towards Engineering Novel PE-Based Immunotoxins by Targeting Them to the Nucleus. Toxins. 2016; 8(11):321. https://doi.org/10.3390/toxins8110321
Chicago/Turabian StyleBorowiec, Marta, Michal Gorzkiewicz, Joanna Grzesik, Aurelia Walczak-Drzewiecka, Anna Salkowska, Ewelina Rodakowska, Kamil Steczkiewicz, Leszek Rychlewski, Jaroslaw Dastych, and Krzysztof Ginalski. 2016. "Towards Engineering Novel PE-Based Immunotoxins by Targeting Them to the Nucleus" Toxins 8, no. 11: 321. https://doi.org/10.3390/toxins8110321
APA StyleBorowiec, M., Gorzkiewicz, M., Grzesik, J., Walczak-Drzewiecka, A., Salkowska, A., Rodakowska, E., Steczkiewicz, K., Rychlewski, L., Dastych, J., & Ginalski, K. (2016). Towards Engineering Novel PE-Based Immunotoxins by Targeting Them to the Nucleus. Toxins, 8(11), 321. https://doi.org/10.3390/toxins8110321