
Structure, Biology, and Therapeutic Application of Toxin–Antitoxin Systems in Pathogenic Bacteria

Abstract
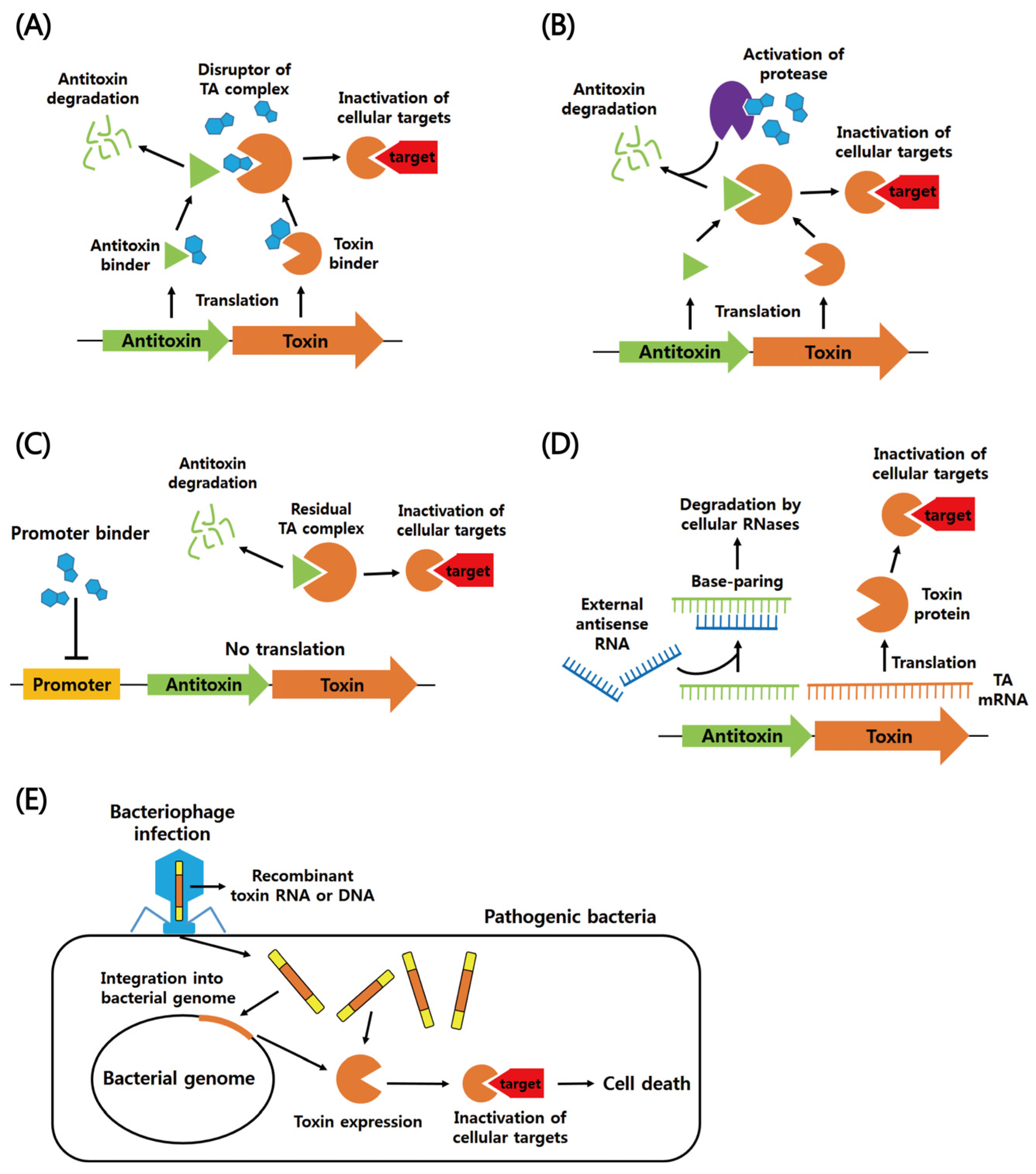
:1. Biological Roles of TA Systems
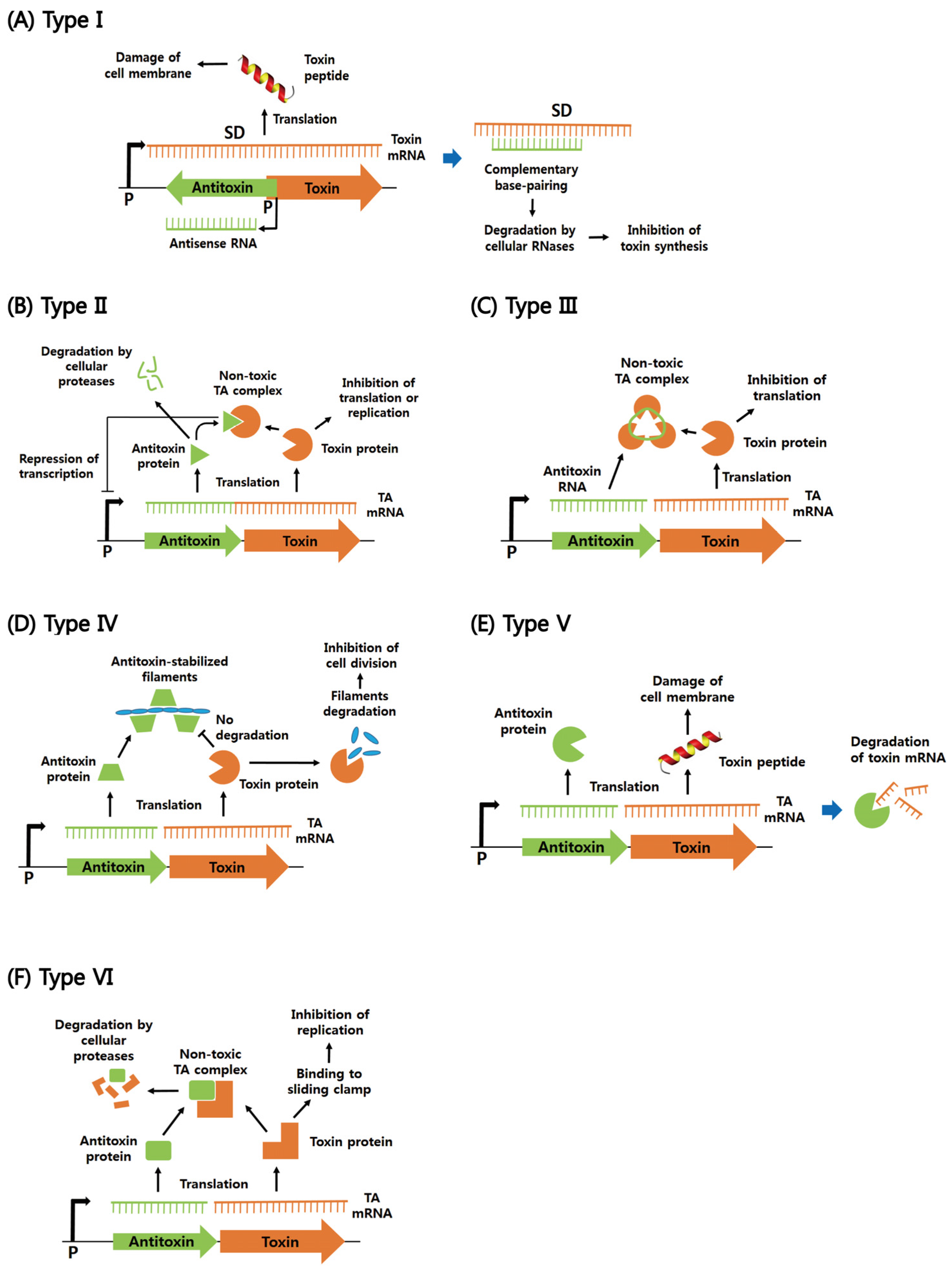
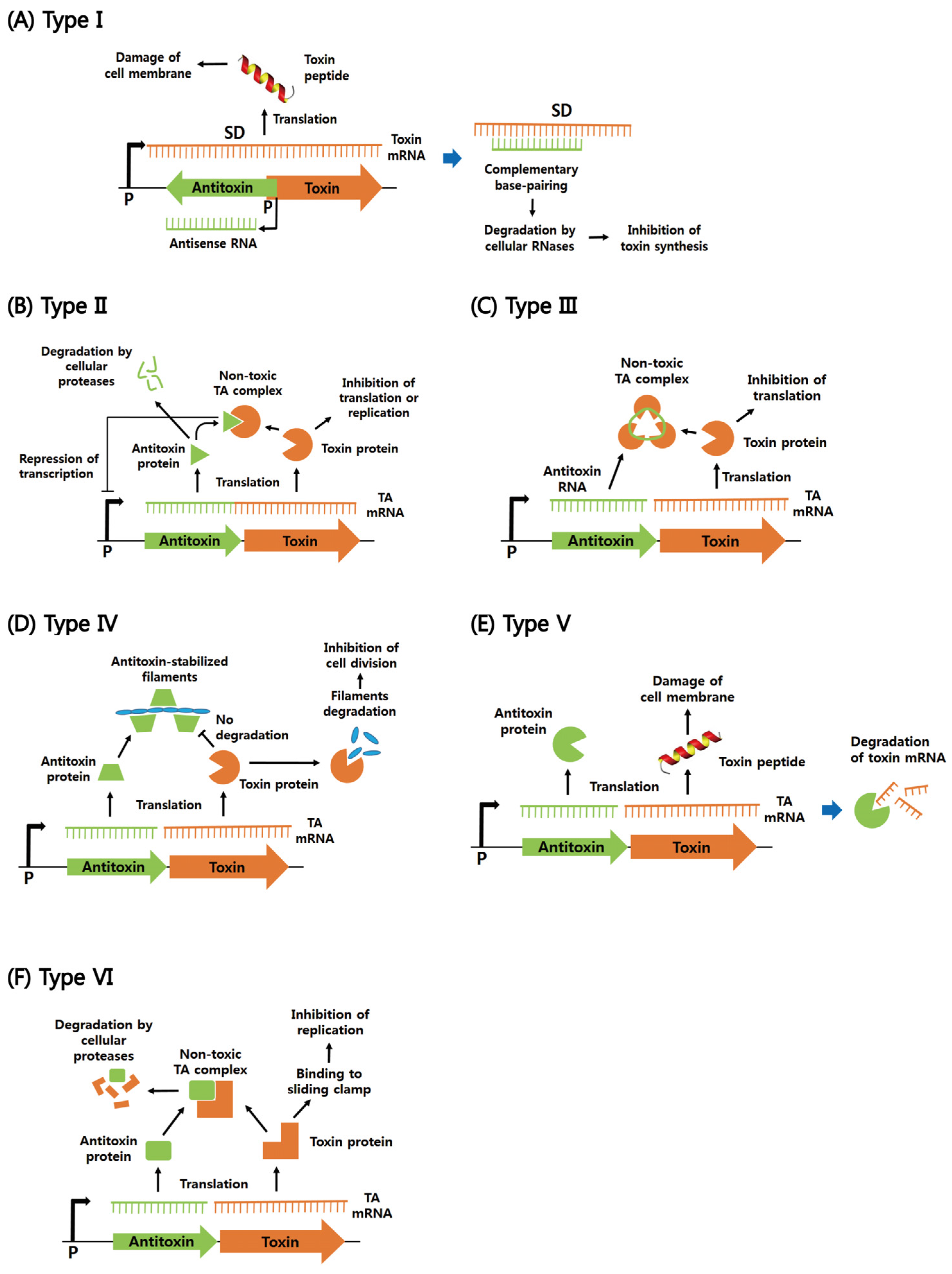
2. Functions of Six Different Types of Bacterial TA Systems
2.1. Type I
2.2. Type II
2.3. Types III–VI
3. TA Modules from Pathogenic Bacteria
3.1. Gram-Positive Bacteria
3.2. Gram-Negative Bacteria
3.3. Mycobacterium Tuberculosis
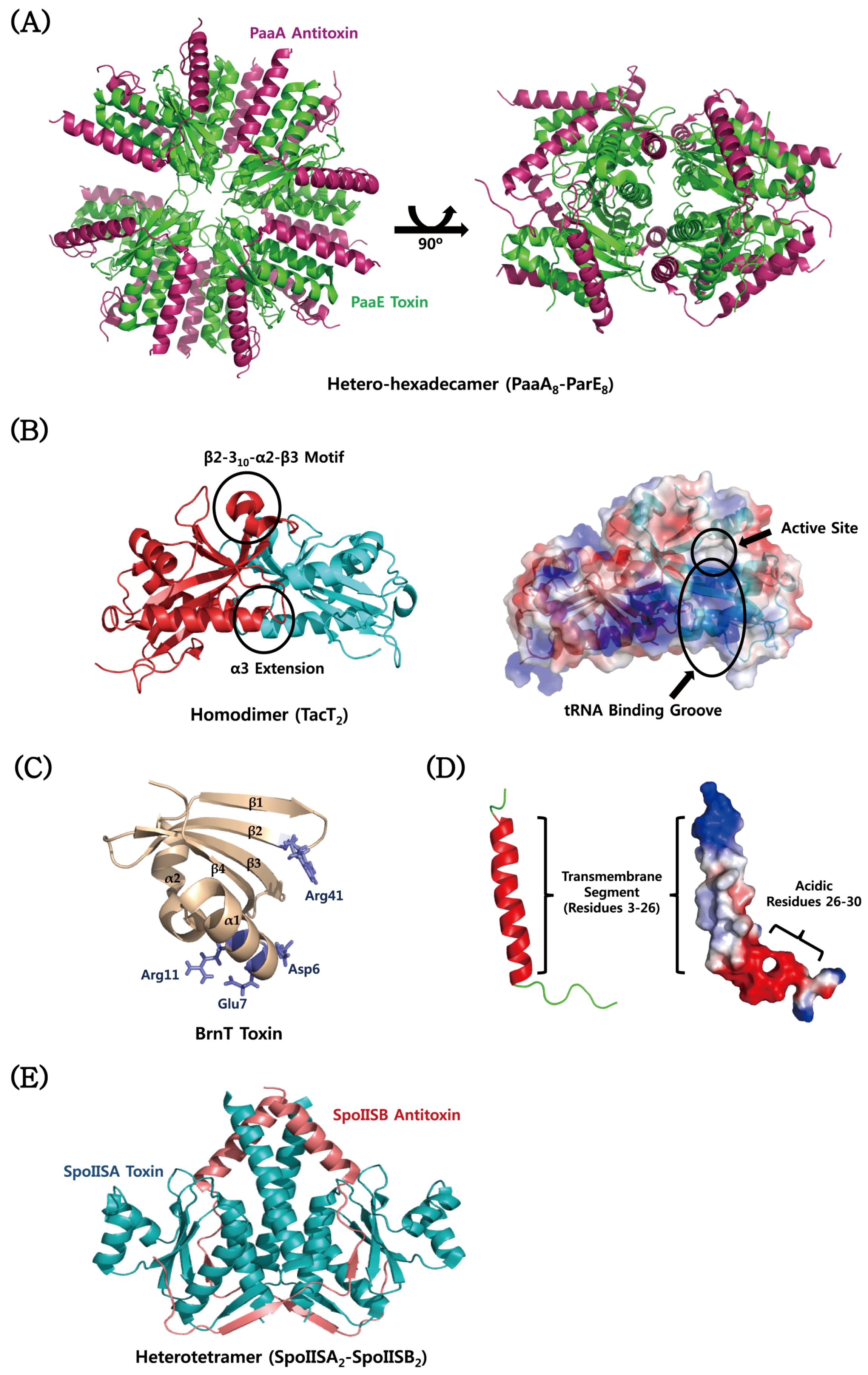
4. Structural Characteristics of TA Proteins from Pathogenic Bacteria
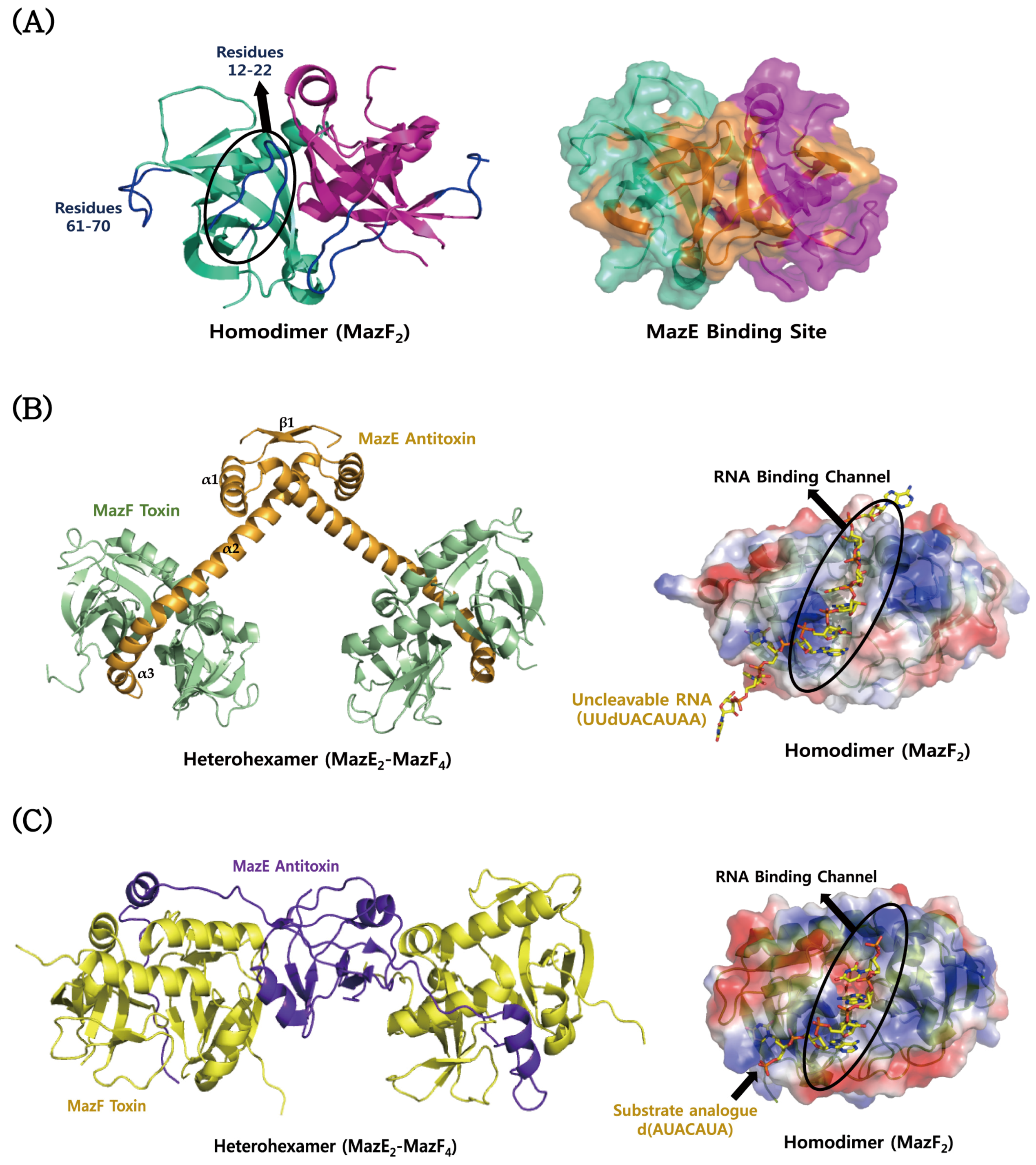
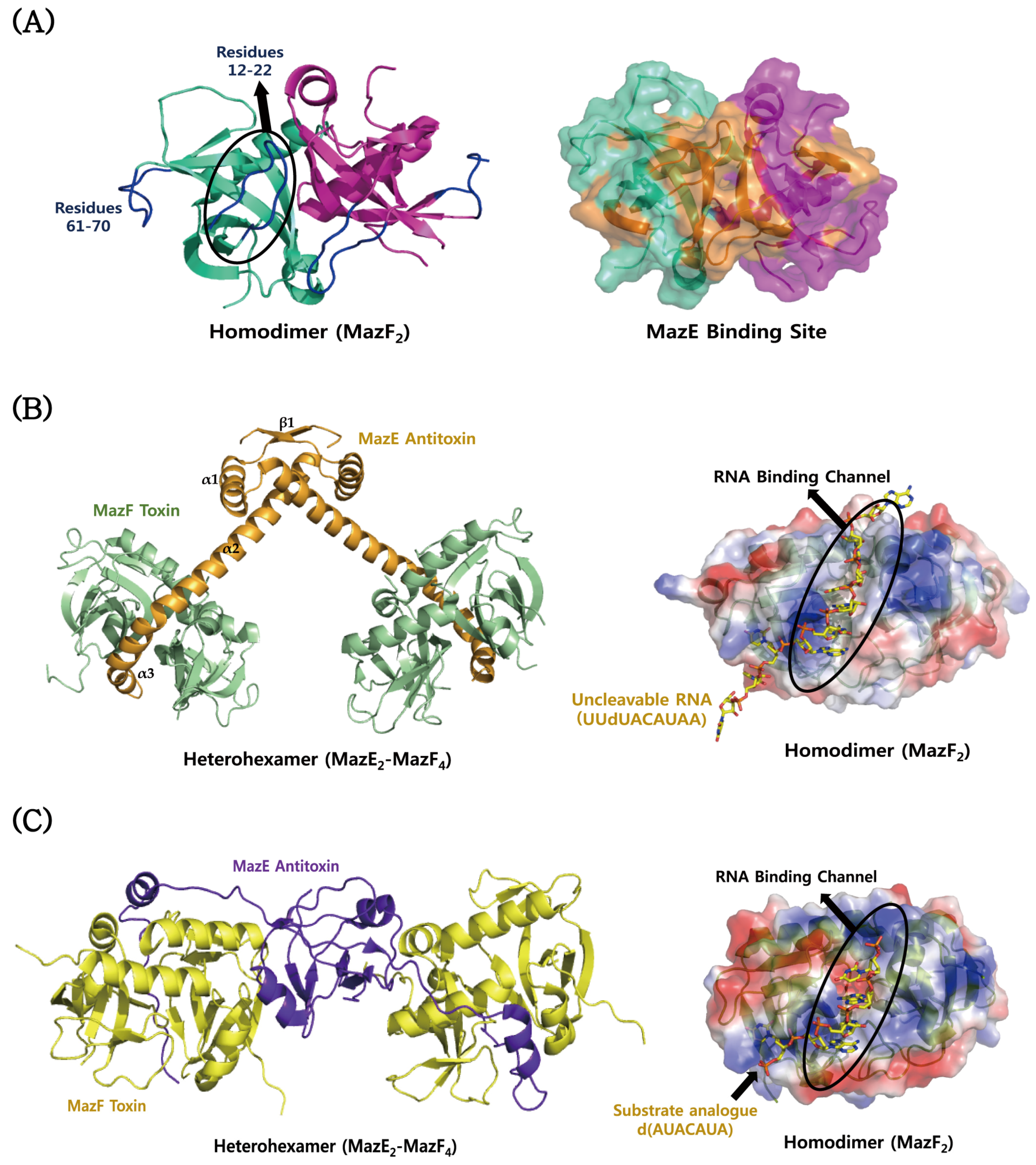
4.1. MazEF
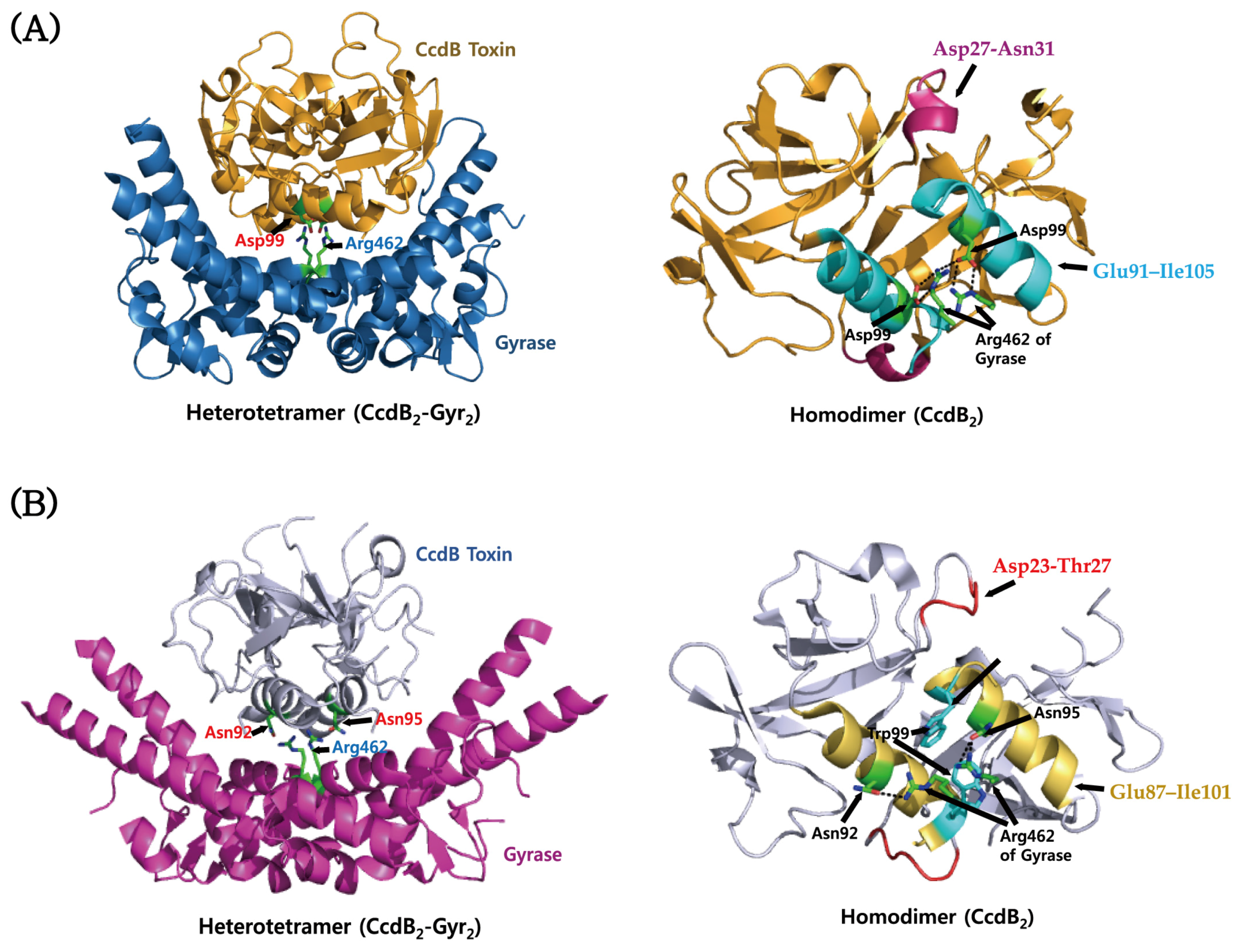
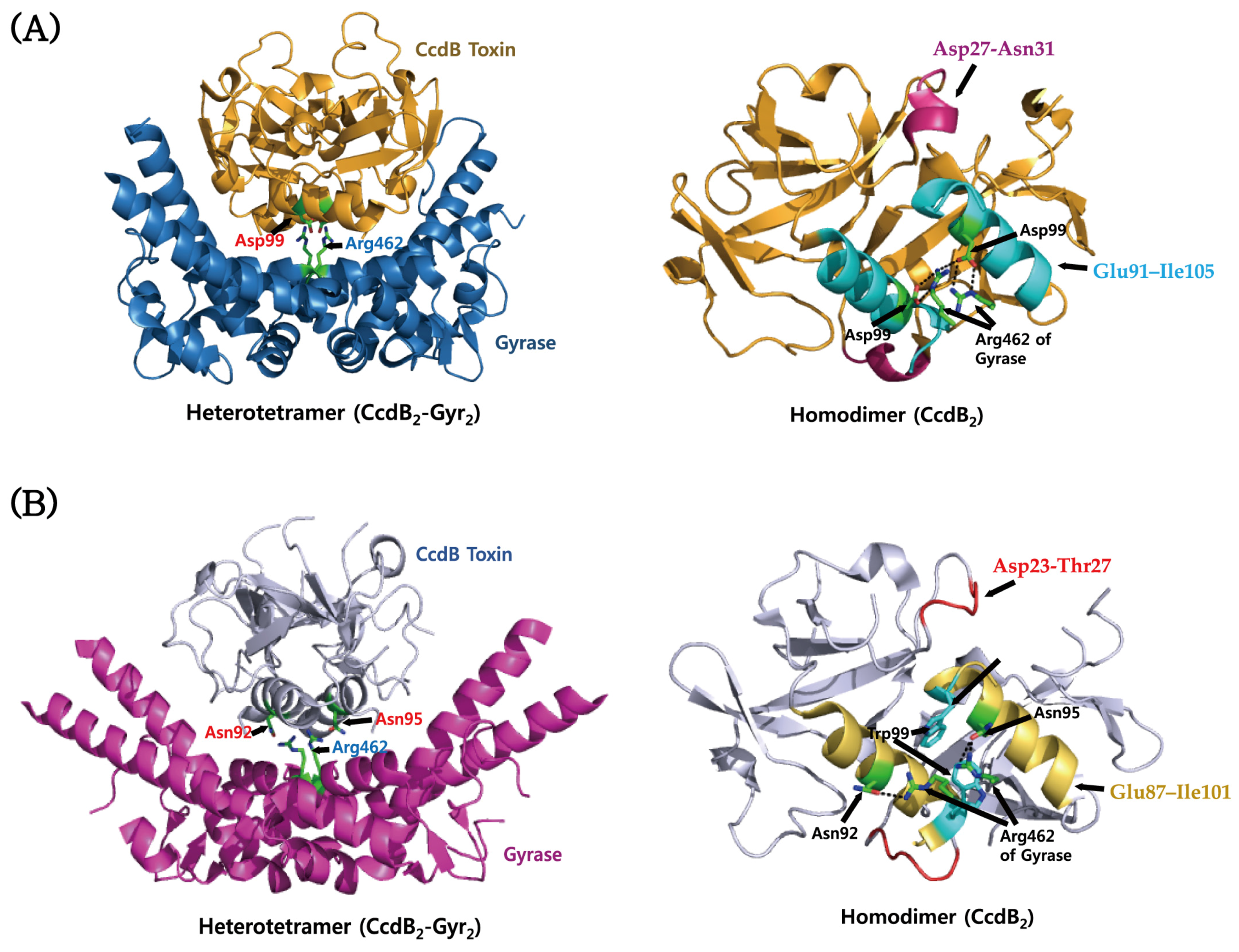
4.2. CcdAB
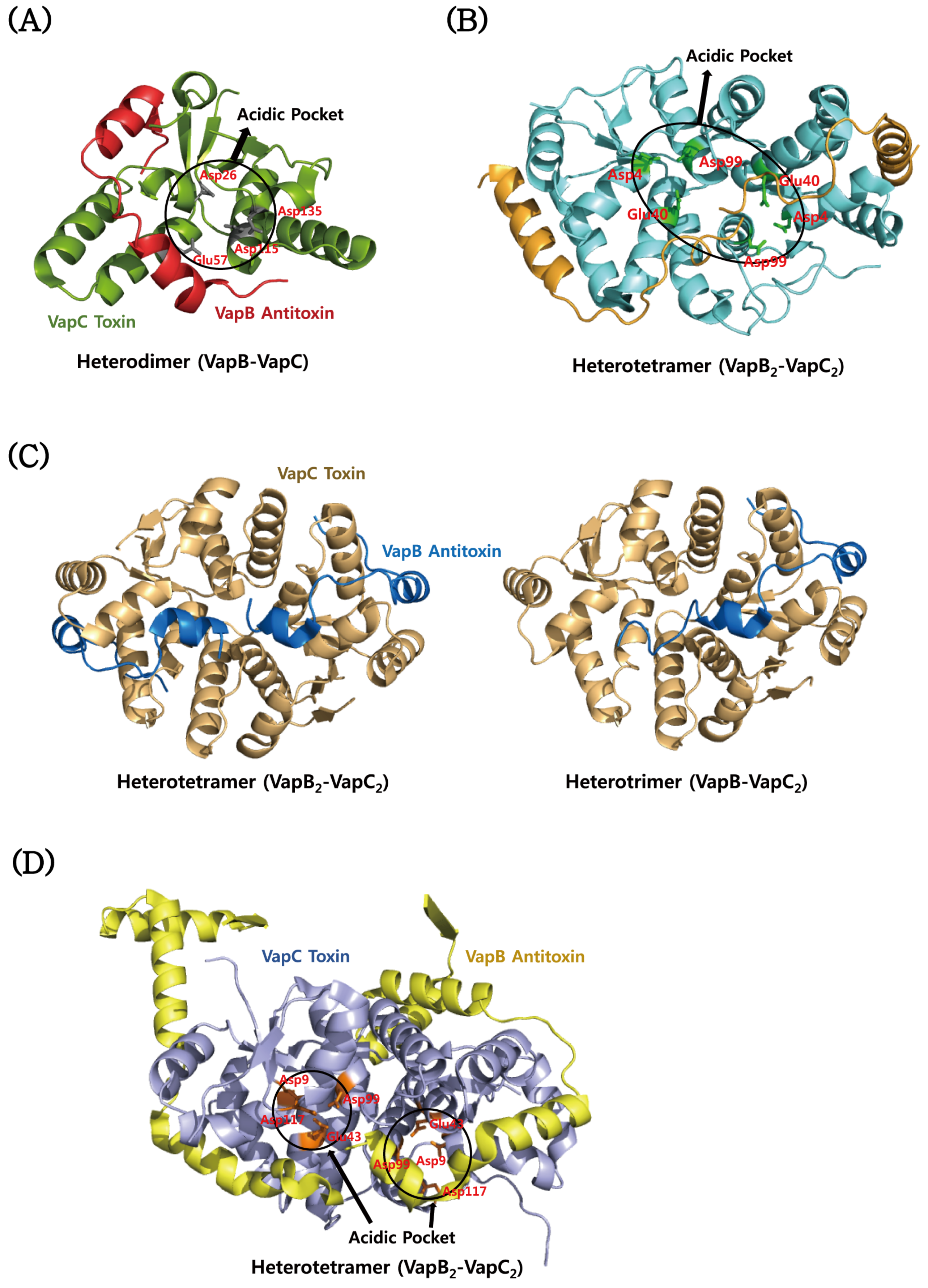
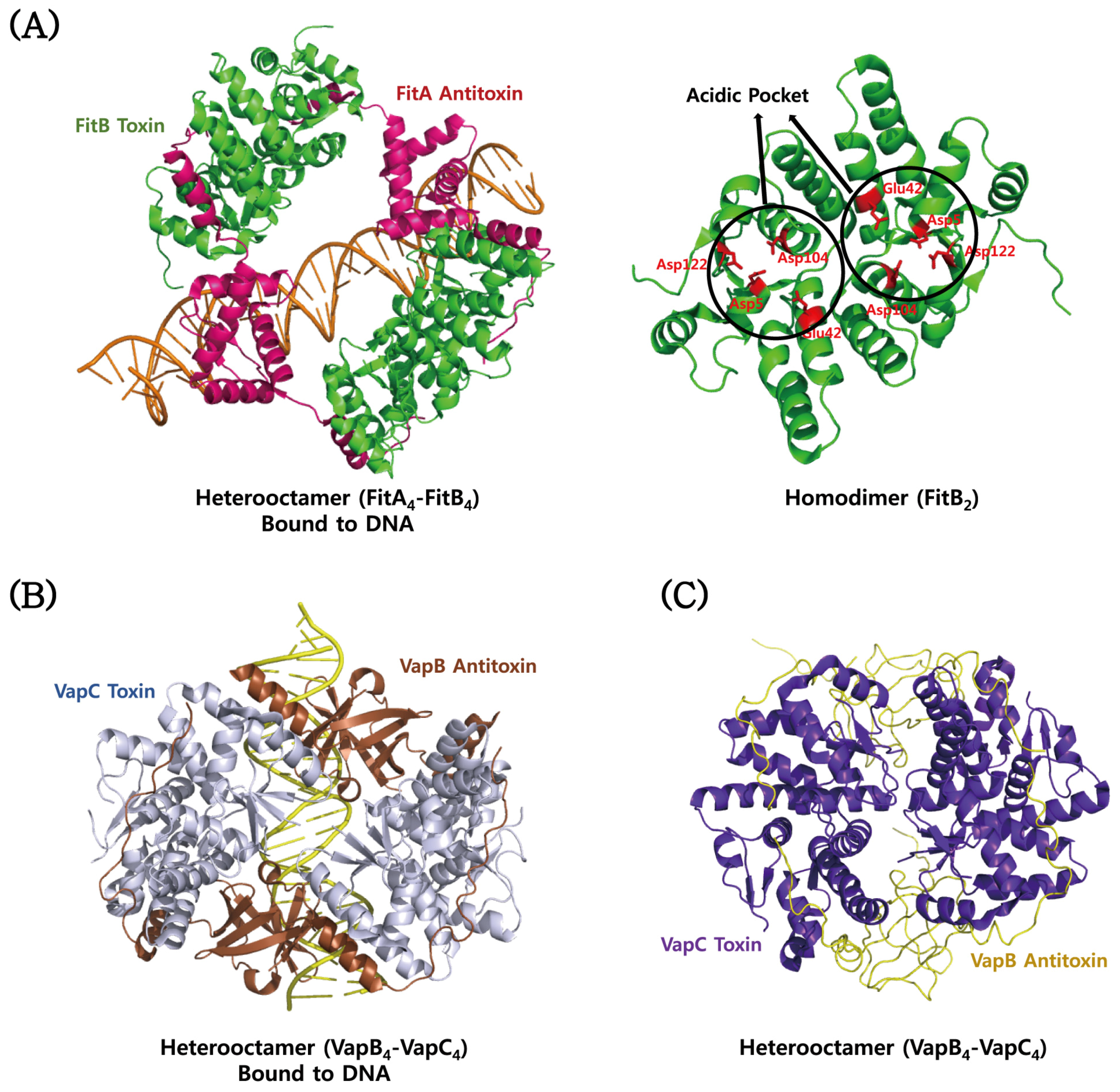
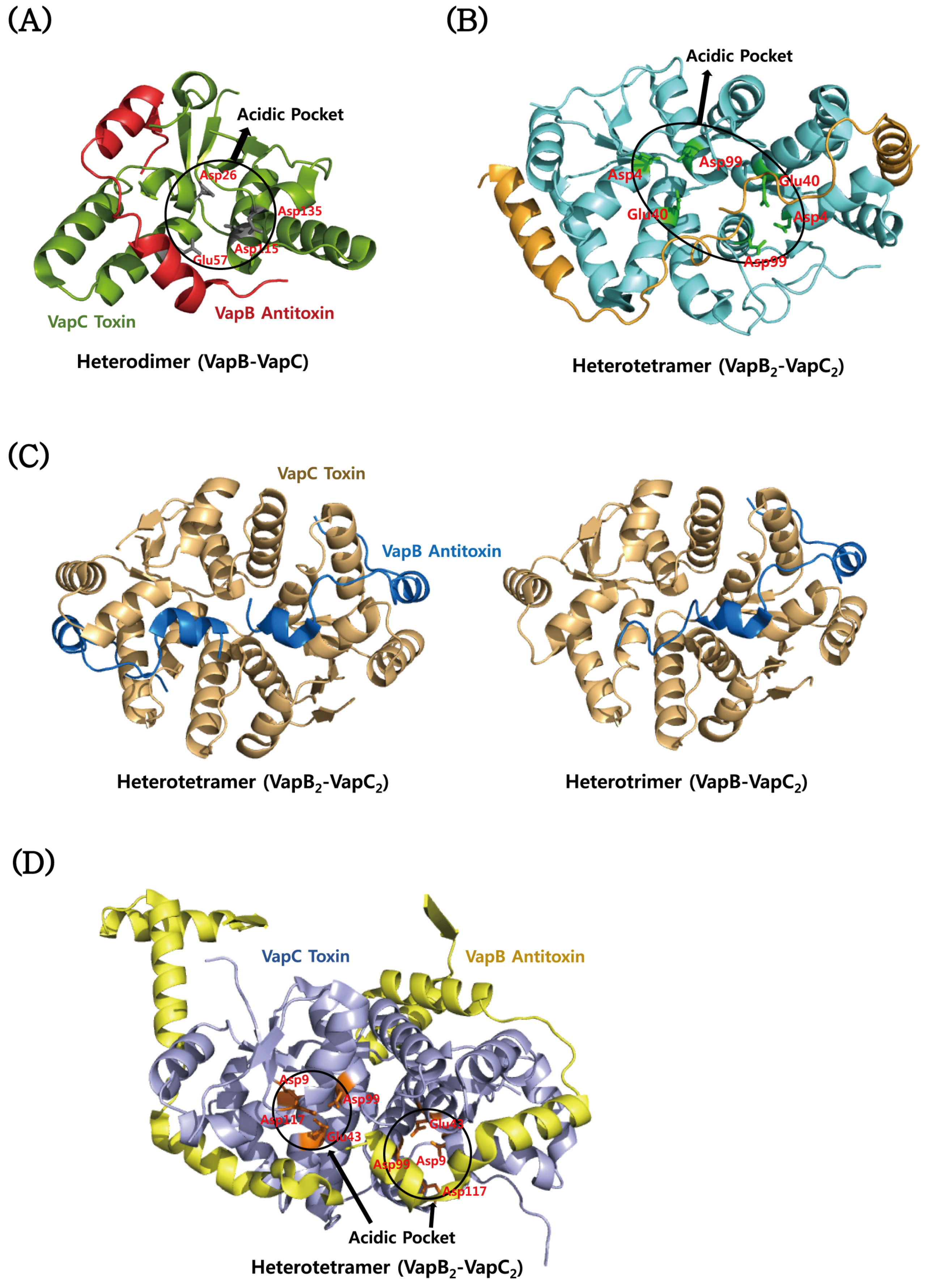
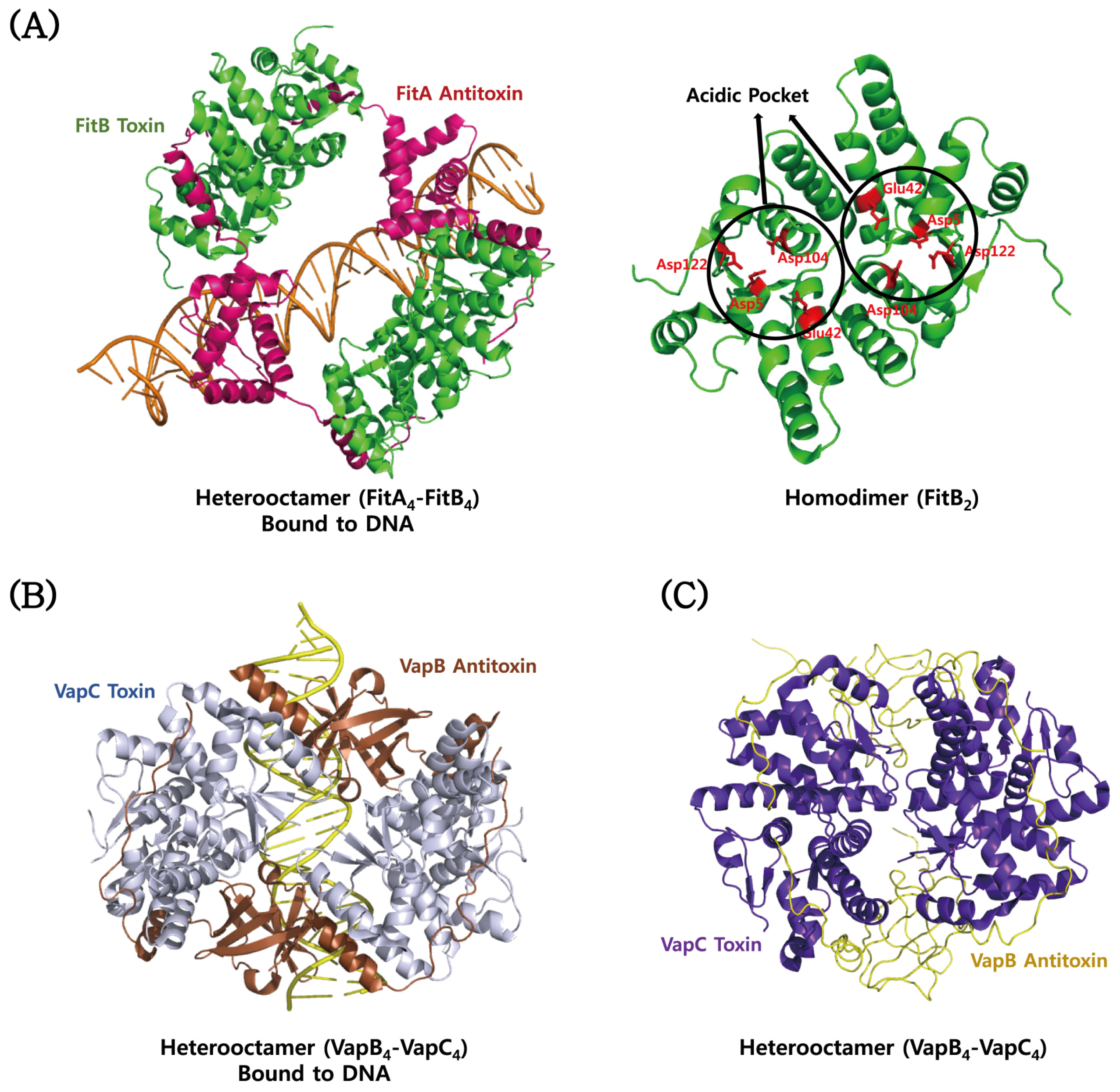
4.3. VapBC
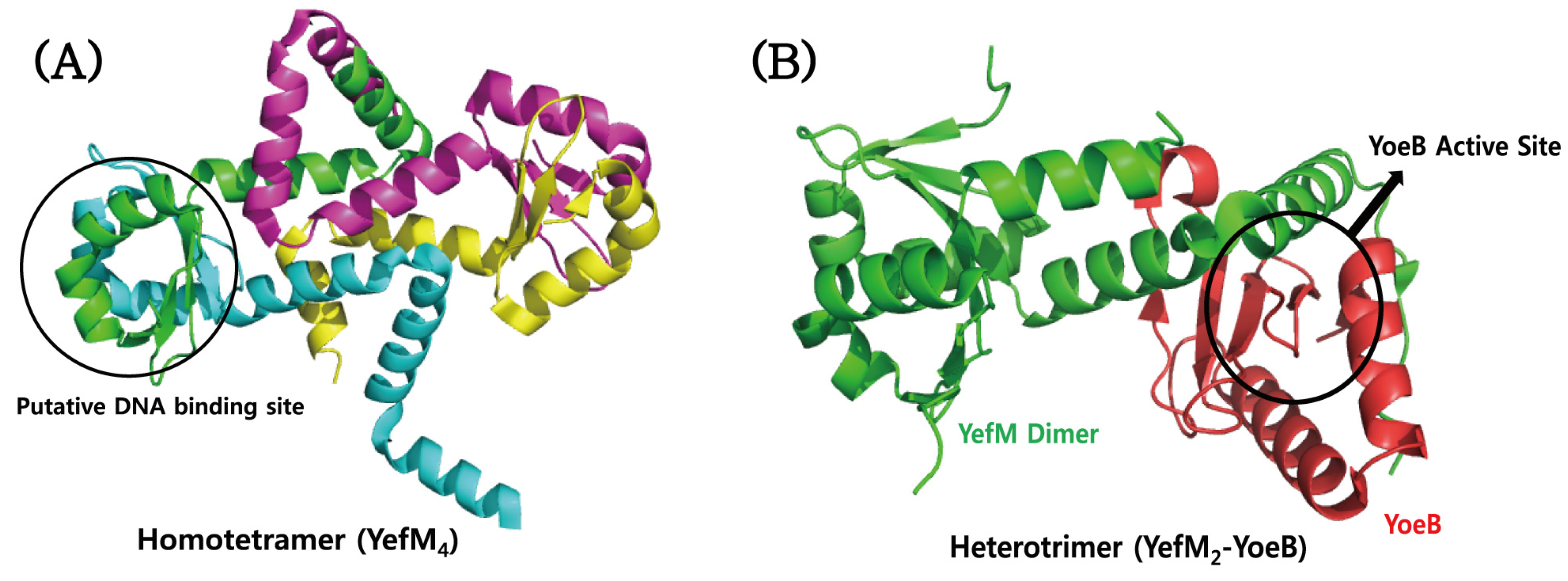
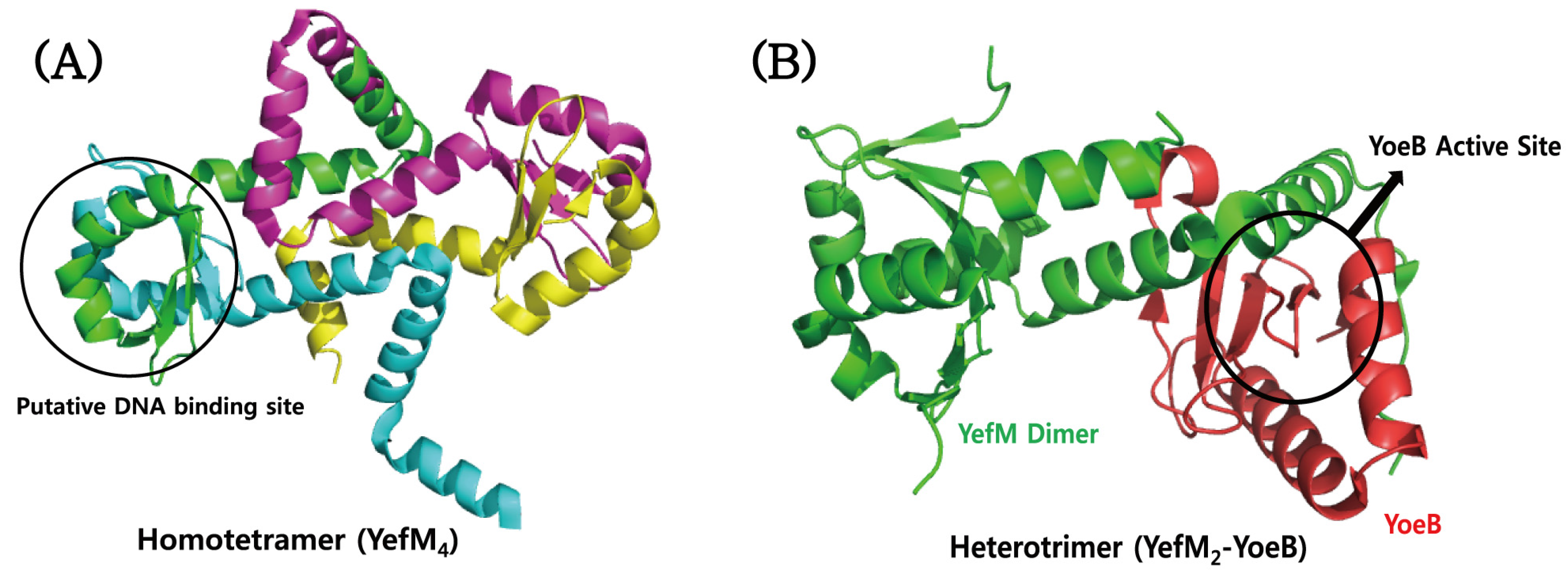
4.4. YefM–YoeB
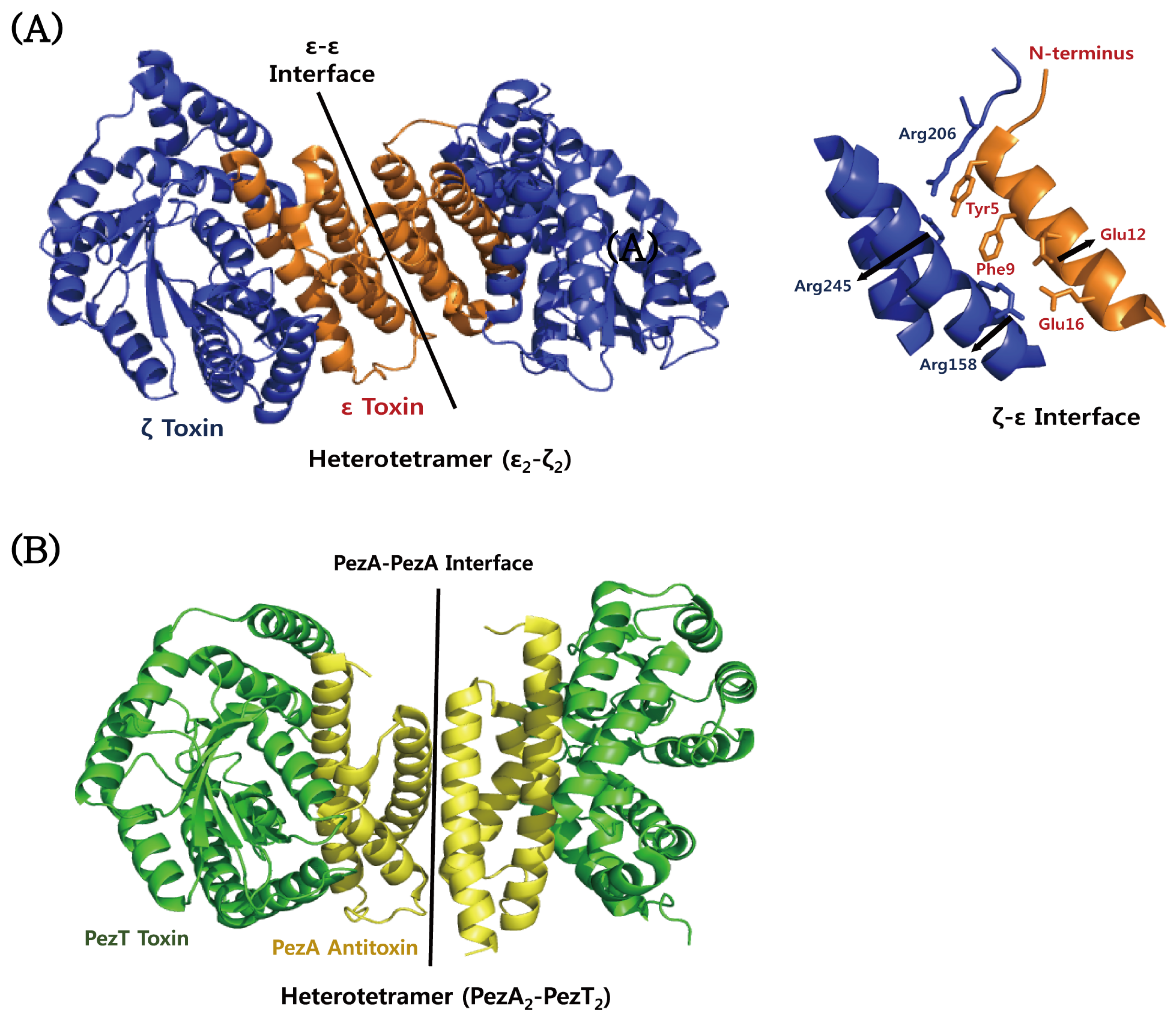
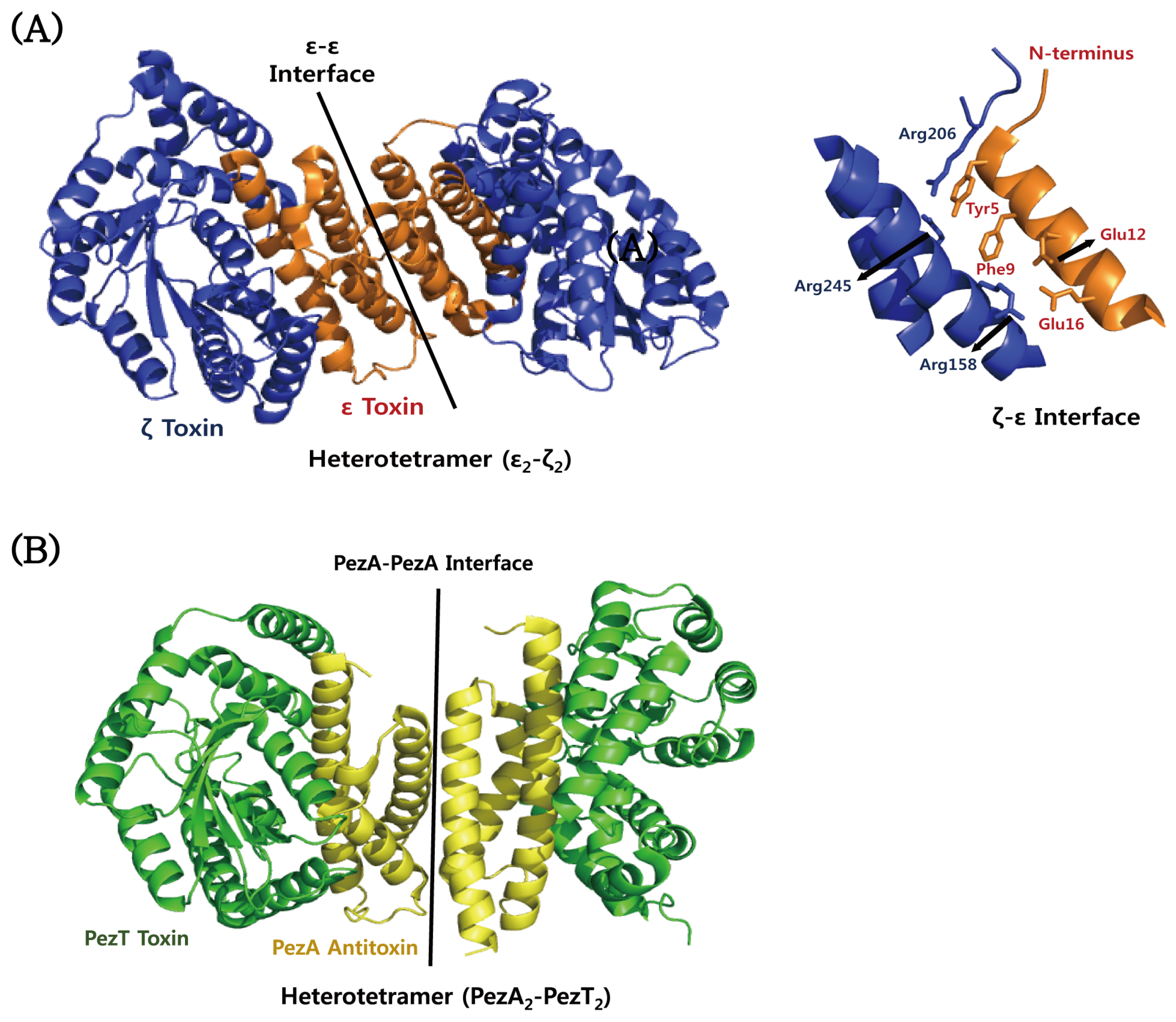
4.5. ε–ζ
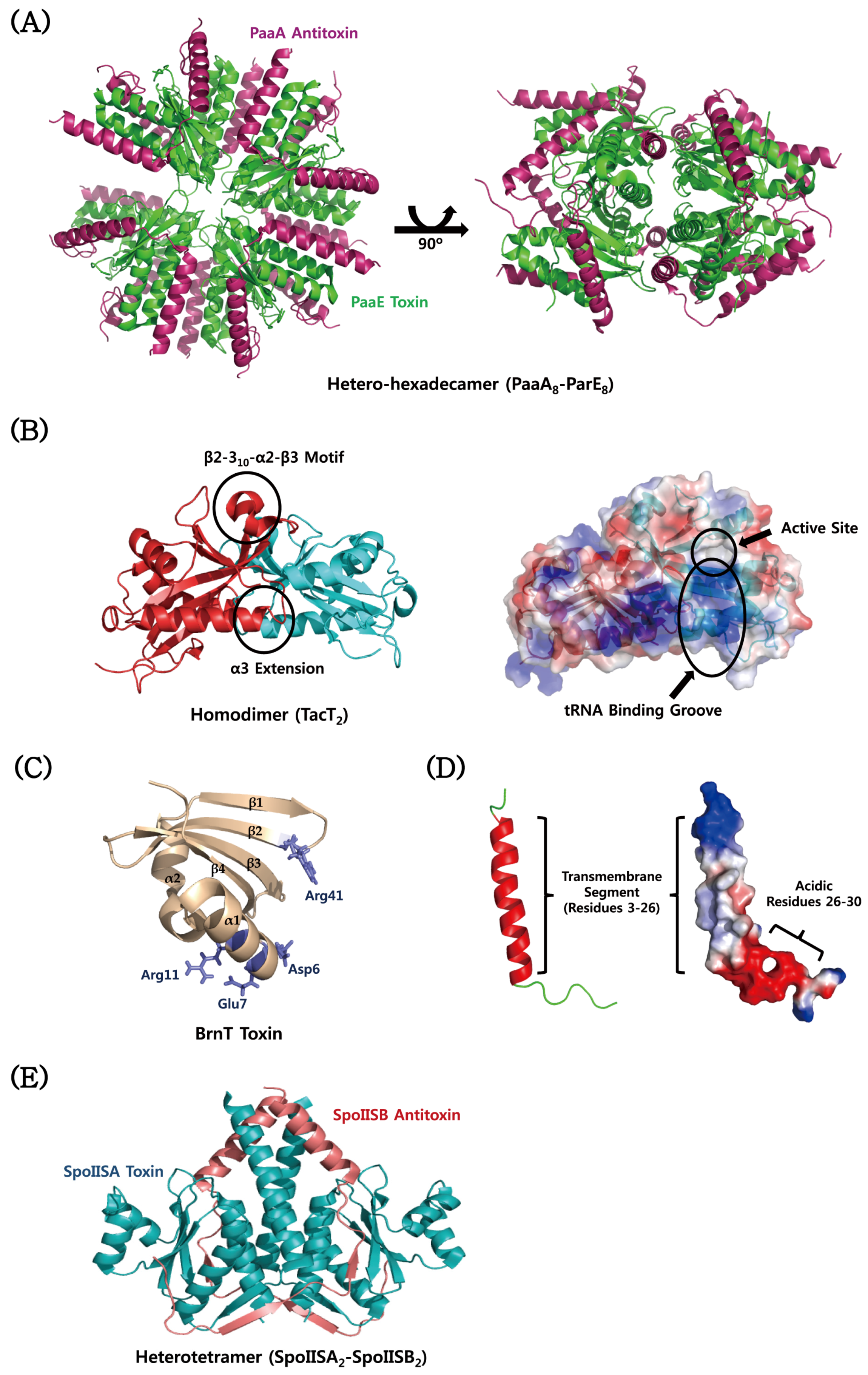
4.6. Atypical TA Modules
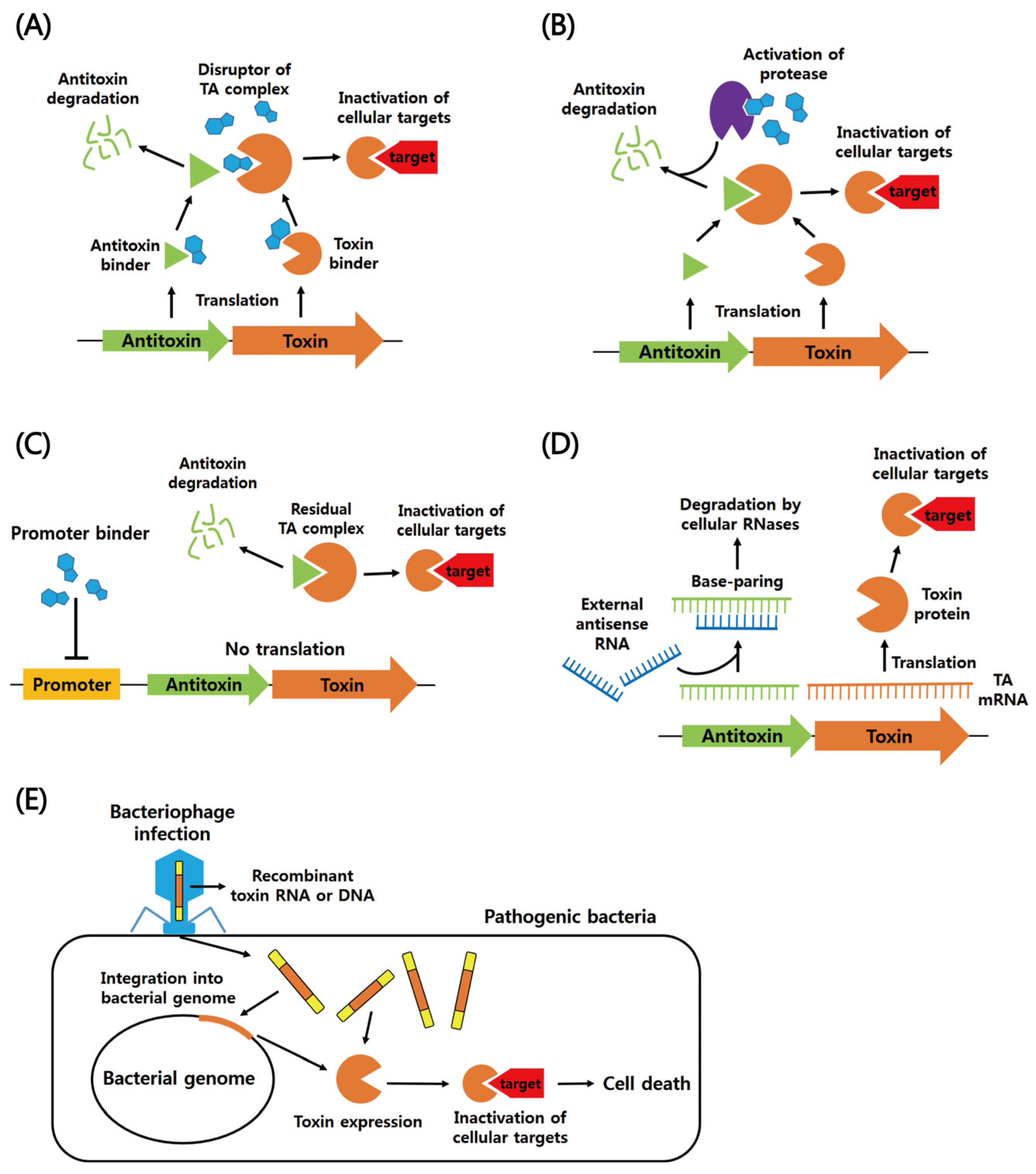
5. Exploitation of TA Systems for the Development of Novel Antibiotic Drugs
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Shao, Y.; Harrison, E.M.; Bi, D.; Tai, C.; He, X.; Ou, H.Y.; Rajakumar, K.; Deng, Z. TADB: A web-based resource for Type 2 toxin–antitoxin loci in bacteria and archaea. Nucleic Acids Res. 2011, 39, D606–D611. [Google Scholar] [CrossRef] [PubMed]
- Leplae, R.; Geeraerts, D.; Hallez, R.; Guglielmini, J.; Dreze, P.; van Melderen, L. Diversity of bacterial type II toxin–antitoxin systems: A comprehensive search and functional analysis of novel families. Nucleic Acids Res. 2011, 39, 5513–5525. [Google Scholar] [CrossRef] [PubMed]
- Sala, A.; Bordes, P.; Genevaux, P. Multiple toxin–antitoxin systems in Mycobacterium tuberculosis. Toxins 2014, 6, 1002–1020. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.T.; Moreno-Cordoba, I.; Yeo, C.C.; Espinosa, M. Toxin–antitoxin genes of the Gram-positive pathogen Streptococcus pneumoniae: So few and yet so many. Microbiol. Mol. Biol. Rev. 2012, 76, 773–791. [Google Scholar] [CrossRef] [PubMed]
- Schuster, C.F.; Bertram, R. Toxin-Antitoxin Systems of Staphylococcus aureus. Toxins 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Page, R.; Peti, W. Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat. Chem. Biol. 2016, 12, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Son, W.S.; Lee, B.J. Structural overview of toxin-antitoxin systems in infectious bacteria: A target for developing antimicrobial agents. Biochim. Biophys. Acta 2013, 1834, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Kedzierska, B.; Hayes, F. Emerging Roles of Toxin–Antitoxin Modules in Bacterial Pathogenesis. Molecules 2016, 21. [Google Scholar] [CrossRef] [PubMed]
- Ogura, T.; Hiraga, S. Mini-F plasmid genes that couple host cell division to plasmid proliferation. Proc. Natl. Acad. Sci. USA 1983, 80, 4784–4788. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, K.; Rasmussen, P.B.; Molin, S. Unique type of plasmid maintenance function: Postsegregational killing of plasmid-free cells. Proc. Natl. Acad. Sci. USA 1986, 83, 3116–3120. [Google Scholar] [CrossRef] [PubMed]
- Van Melderen, L.; Saavedra de Bast, M. Bacterial toxin–antitoxin systems: More than selfish entities? PLoS Genet. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Coussens, N.P.; Daines, D.A. Wake me when it’s over—Bacterial toxin-antitoxin proteins and induced dormancy. Exp. Biol. Med. 2016, 241, 1332–1342. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, K.; Christensen, S.K.; Lobner-Olesen, A. Prokaryotic toxin-antitoxin stress response loci. Nat. Rev. Microbiol. 2005, 3, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Van Melderen, L. Toxin–antitoxin systems: Why so many, what for? Curr. Opin. Microbiol. 2010, 13, 781–785. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Behiels, E.; Devreese, B. Toxin–Antitoxin systems: Their role in persistence, biofilm formation, and pathogenicity. Pathog. Dis. 2014, 70, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Pandey, D.P.; Gerdes, K. Toxin–antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 2005, 33, 966–976. [Google Scholar] [CrossRef] [PubMed]
- Arcus, V.L.; Rainey, P.B.; Turner, S.J. The PIN-domain toxin–antitoxin array in mycobacteria. Trends Microbiol. 2005, 13, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Ramage, H.R.; Connolly, L.E.; Cox, J.S. Comprehensive functional analysis of Mycobacterium tuberculosis toxin-antitoxin systems: Implications for pathogenesis, stress responses, and evolution. PLoS Genet. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Robson, J.; McKenzie, J.L.; Cursons, R.; Cook, G.M.; Arcus, V.L. The vapBC operon from Mycobacterium smegmatis is an autoregulated toxin–antitoxin module that controls growth via inhibition of translation. J. Mol. Biol. 2009, 390, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Moyed, H.S.; Bertrand, K.P. hipA, a newly recognized gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J. Bacteriol. 1983, 155, 768–775. [Google Scholar] [PubMed]
- Korch, S.B.; Henderson, T.A.; Hill, T.M. Characterization of the hipA7 allele of Escherichia coli and evidence that high persistence is governed by (p)ppGpp synthesis. Mol. Microbiol. 2003, 50, 1199–1213. [Google Scholar] [CrossRef] [PubMed]
- Maisonneuve, E.; Castro-Camargo, M.; Gerdes, K. (p)ppGpp controls bacterial persistence by stochastic induction of toxin-antitoxin activity. Cell 2013, 154, 1140–1150. [Google Scholar] [CrossRef] [PubMed]
- Maisonneuve, E.; Gerdes, K. Molecular mechanisms underlying bacterial persisters. Cell 2014, 157, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Verstraeten, N.; Knapen, W.J.; Kint, C.I.; Liebens, V.; van den Bergh, B.; Dewachter, L.; Michiels, J.E.; Fu, Q.; David, C.C.; Fierro, A.C.; et al. Obg and Membrane Depolarization Are Part of a Microbial Bet-Hedging Strategy that Leads to Antibiotic Tolerance. Mol. Cell 2015, 59, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, K.; Maisonneuve, E. Remarkable Functional Convergence: Alarmone ppGpp Mediates Persistence by Activating Type I and II Toxin–Antitoxins. Mol. Cell 2015, 59, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Dorr, T.; Vulic, M.; Lewis, K. Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol. 2010, 8. [Google Scholar] [CrossRef] [PubMed]
- Maisonneuve, E.; Shakespeare, L.J.; Jorgensen, M.G.; Gerdes, K. Bacterial persistence by RNA endonucleases. Proc. Natl. Acad. Sci. USA 2011, 108, 13206–13211. [Google Scholar] [CrossRef] [PubMed]
- Helaine, S.; Cheverton, A.M.; Watson, K.G.; Faure, L.M.; Matthews, S.A.; Holden, D.W. Internalization of Salmonella by macrophages induces formation of nonreplicating persisters. Science 2014, 343, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Lobato-Marquez, D.; Moreno-Cordoba, I.; Figueroa, V.; Diaz-Orejas, R.; Garcia-del Portillo, F. Distinct type I and type II toxin–antitoxin modules control Salmonella lifestyle inside eukaryotic cells. Sci. Rep. 2015, 5, 9374. [Google Scholar] [CrossRef] [PubMed]
- Cos, P.; Tote, K.; Horemans, T.; Maes, L. Biofilms: An extra hurdle for effective antimicrobial therapy. Curr. Pharm. Des. 2010, 16, 2279–2295. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wood, T.K. Toxin–antitoxin systems influence biofilm and persister cell formation and the general stress response. Appl. Environ. Microbiol. 2011, 77, 5577–5583. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Bedzyk, L.A.; Thomas, S.M.; Ye, R.W.; Wood, T.K. Gene expression in Escherichia coli biofilms. Appl. Microbiol. Biotechnol. 2004, 64, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Van Houdt, R.; Michiels, C.W. Role of bacterial cell surface structures in Escherichia coli biofilm formation. Res. Microbiol. 2005, 156, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kim, Y.; Hong, S.H.; Ma, Q.; Brown, B.L.; Pu, M.; Tarone, A.M.; Benedik, M.J.; Peti, W.; Page, R.; et al. Antitoxin MqsA helps mediate the bacterial general stress response. Nat. Chem. Biol. 2011, 7, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Soo, V.W.; Wood, T.K. Antitoxin MqsA represses curli formation through the master biofilm regulator CsgD. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Barrios, A.F.; Zuo, R.; Hashimoto, Y.; Yang, L.; Bentley, W.E.; Wood, T.K. Autoinducer 2 controls biofilm formation in Escherichia coli through a novel motility quorum-sensing regulator (MqsR, B3022). J. Bacteriol. 2006, 188, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.S.; Garcia-Contreras, R.; Wood, T.K. Escherichia coli transcription factor YncC (McbR) regulates colanic acid and biofilm formation by repressing expression of periplasmic protein YbiM (McbA). ISME J. 2008, 2, 615–631. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.J.; Wade, W.D.; Akierman, S.; Vacchi-Suzzi, C.; Stremick, C.A.; Turner, R.J.; Ceri, H. The chromosomal toxin gene yafQ is a determinant of multidrug tolerance for Escherichia coli growing in a biofilm. Antimicrob. Agents Chemother. 2009, 53, 2253–2258. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, Q.; Li, M.; Heijstra, B.D.; Wang, S.; Liang, Q.; Qi, Q. Escherichia coli toxin gene hipA affects biofilm formation and DNA release. Microbiology 2013, 159, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Wang, X.; Ma, Q.; Zhang, X.S.; Wood, T.K. Toxin-antitoxin systems in Escherichia coli influence biofilm formation through YjgK (TabA) and fimbriae. J. Bacteriol. 2009, 191, 1258–1267. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.; Argaman, L.; Wagner, E.G.; Altuvia, S. The small RNA IstR inhibits synthesis of an SOS-induced toxic peptide. Curr. Biol. 2004, 14, 2271–2276. [Google Scholar] [CrossRef] [PubMed]
- Unoson, C.; Wagner, E.G. A small SOS-induced toxin is targeted against the inner membrane in Escherichia coli. Mol. Microbiol. 2008, 70, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, K.; Thisted, T.; Martinussen, J. Mechanism of post-segregational killing by the hok/sok system of plasmid R1: Sok antisense RNA regulates formation of a hok mRNA species correlated with killing of plasmid-free cells. Mol. Microbiol. 1990, 4, 1807–1818. [Google Scholar] [CrossRef] [PubMed]
- Pecota, D.C.; Osapay, G.; Selsted, M.E.; Wood, T.K. Antimicrobial properties of the Escherichia coli R1 plasmid host killing peptide. J. Biotechnol. 2003, 100, 1–12. [Google Scholar] [CrossRef]
- Mok, W.W.; Patel, N.H.; Li, Y. Decoding toxicity: Deducing the sequence requirements of IbsC, a type I toxin in Escherichia coli. J. Biol. Chem. 2010, 285, 41627–41636. [Google Scholar] [CrossRef] [PubMed]
- Fozo, E.M.; Kawano, M.; Fontaine, F.; Kaya, Y.; Mendieta, K.S.; Jones, K.L.; Ocampo, A.; Rudd, K.E.; Storz, G. Repression of small toxic protein synthesis by the Sib and OhsC small RNAs. Mol. Microbiol. 2008, 70, 1076–1093. [Google Scholar] [CrossRef] [PubMed]
- Silvaggi, J.M.; Perkins, J.B.; Losick, R. Genes for small, noncoding RNAs under sporulation control in Bacillus subtilis. J. Bacteriol. 2006, 188, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Silvaggi, J.M.; Perkins, J.B.; Losick, R. Small untranslated RNA antitoxin in Bacillus subtilis. J. Bacteriol. 2005, 187, 6641–6650. [Google Scholar] [CrossRef] [PubMed]
- Weaver, K.E.; Weaver, D.M.; Wells, C.L.; Waters, C.M.; Gardner, M.E.; Ehli, E.A. Enterococcus faecalis plasmid pAD1-encoded Fst toxin affects membrane permeability and alters cellular responses to lantibiotics. J. Bacteriol. 2003, 185, 2169–2177. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Weaver, K.E. Addiction toxin Fst has unique effects on chromosome segregation and cell division in Enterococcus faecalis and Bacillus subtilis. J. Bacteriol. 2006, 188, 5374–5384. [Google Scholar] [CrossRef] [PubMed]
- Kawano, M.; Aravind, L.; Storz, G. An antisense RNA controls synthesis of an SOS-induced toxin evolved from an antitoxin. Mol. Microbiol. 2007, 64, 738–754. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, J.; Hoeflich, K.P.; Ikura, M.; Qing, G.; Inouye, M. MazF cleaves cellular mRNAs specifically at ACA to block protein synthesis in Escherichia coli. Mol. Cell 2003, 12, 913–923. [Google Scholar] [CrossRef]
- Vesper, O.; Amitai, S.; Belitsky, M.; Byrgazov, K.; Kaberdina, A.C.; Engelberg-Kulka, H.; Moll, I. Selective translation of leaderless mRNAs by specialized ribosomes generated by MazF in Escherichia coli. Cell 2011, 147, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Gomez, A.J.; Lemonnier, M.; Santos-Sierra, S.; Berzal-Herranz, A.; Diaz-Orejas, R. RNase/anti-RNase activities of the bacterial parD toxin-antitoxin system. J. Bacteriol. 2005, 187, 3151–3157. [Google Scholar] [CrossRef] [PubMed]
- Budde, P.P.; Davis, B.M.; Yuan, J.; Waldor, M.K. Characterization of a higBA toxin–antitoxin locus in Vibrio cholerae. J. Bacteriol. 2007, 189, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.M.; Woychik, N.A. Bacterial toxin HigB associates with ribosomes and mediates translation-dependent mRNA cleavage at A-rich sites. J. Biol. Chem. 2009, 284, 18605–18613. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.K.; Gerdes, K. RelE toxins from bacteria and Archaea cleave mRNAs on translating ribosomes, which are rescued by tmRNA. Mol. Microbiol. 2003, 48, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Winther, K.S.; Gerdes, K. Enteric virulence associated protein VapC inhibits translation by cleavage of initiator tRNA. Proc. Natl. Acad. Sci. USA 2011, 108, 7403–7407. [Google Scholar] [CrossRef] [PubMed]
- Winther, K.S.; Brodersen, D.E.; Brown, A.K.; Gerdes, K. VapC20 of Mycobacterium tuberculosis cleaves the sarcin-ricin loop of 23S rRNA. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Daines, D.A.; Wu, M.H.; Yuan, S.Y. VapC-1 of nontypeable Haemophilus influenzae is a ribonuclease. J. Bacteriol. 2007, 189, 5041–5048. [Google Scholar] [CrossRef] [PubMed]
- Castro-Roa, D.; Garcia-Pino, A.; De Gieter, S.; van Nuland, N.A.; Loris, R.; Zenkin, N. The Fic protein Doc uses an inverted substrate to phosphorylate and inactivate EF-Tu. Nat. Chem. Biol. 2013, 9, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Cruz, J.W.; Rothenbacher, F.P.; Maehigashi, T.; Lane, W.S.; Dunham, C.M.; Woychik, N.A. Doc toxin is a kinase that inactivates elongation factor Tu. J. Biol. Chem. 2014, 289, 7788–7798. [Google Scholar] [CrossRef] [PubMed]
- Germain, E.; Castro-Roa, D.; Zenkin, N.; Gerdes, K. Molecular mechanism of bacterial persistence by HipA. Mol. Cell 2013, 52, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Kaspy, I.; Rotem, E.; Weiss, N.; Ronin, I.; Balaban, N.Q.; Glaser, G. HipA-mediated antibiotic persistence via phosphorylation of the glutamyl-tRNA-synthetase. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Simic, M.; De Jonge, N.; Loris, R.; Vesnaver, G.; Lah, J. Driving forces of gyrase recognition by the addiction toxin CcdB. J. Biol. Chem. 2009, 284, 20002–20010. [Google Scholar] [CrossRef] [PubMed]
- Wilbaux, M.; Mine, N.; Guerout, A.M.; Mazel, D.; van Melderen, L. Functional interactions between coexisting toxin–antitoxin systems of the ccd family in Escherichia coli O157:H7. J. Bacteriol. 2007, 189, 2712–2719. [Google Scholar] [CrossRef] [PubMed]
- Mutschler, H.; Gebhardt, M.; Shoeman, R.L.; Meinhart, A. A novel mechanism of programmed cell death in bacteria by toxin–antitoxin systems corrupts peptidoglycan synthesis. PLoS Biol. 2011, 9. [Google Scholar] [CrossRef] [PubMed]
- Meinhart, A.; Alonso, J.C.; Strater, N.; Saenger, W. Crystal structure of the plasmid maintenance system epsilon/zeta: Functional mechanism of toxin zeta and inactivation by epsilon 2 zeta 2 complex formation. Proc. Natl. Acad. Sci. USA 2003, 100, 1661–1666. [Google Scholar] [CrossRef] [PubMed]
- Fineran, P.C.; Blower, T.R.; Foulds, I.J.; Humphreys, D.P.; Lilley, K.S.; Salmond, G.P. The phage abortive infection system, ToxIN, functions as a protein-RNA toxin–antitoxin pair. Proc. Natl. Acad. Sci. USA 2009, 106, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Masuda, H.; Tan, Q.; Awano, N.; Wu, K.P.; Inouye, M. YeeU enhances the bundling of cytoskeletal polymers of MreB and FtsZ, antagonizing the CbtA (YeeV) toxicity in Escherichia coli. Mol. Microbiol. 2012, 84, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Masuda, H.; Tan, Q.; Awano, N.; Yamaguchi, Y.; Inouye, M. A novel membrane-bound toxin for cell division, CptA (YgfX), inhibits polymerization of cytoskeleton proteins, FtsZ and MreB, in Escherichia coli. FEMS Microbiol. Lett. 2012, 328, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lord, D.M.; Cheng, H.Y.; Osbourne, D.O.; Hong, S.H.; Sanchez-Torres, V.; Quiroga, C.; Zheng, K.; Herrmann, T.; Peti, W.; et al. A new type V toxin–antitoxin system where mRNA for toxin GhoT is cleaved by antitoxin GhoS. Nat. Chem. Biol. 2012, 8, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Aakre, C.D.; Phung, T.N.; Huang, D.; Laub, M.T. A bacterial toxin inhibits DNA replication elongation through a direct interaction with the beta sliding clamp. Mol. Cell 2013, 52, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Brielle, R.; Pinel-Marie, M.L.; Felden, B. Linking bacterial type I toxins with their actions. Curr. Opin. Microbiol. 2016, 30, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Brantl, S.; Jahn, N. sRNAs in bacterial type I and type III toxin–antitoxin systems. FEMS Microbiol. Rev. 2015, 39, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, K.; Wagner, E.G. RNA antitoxins. Curr. Opin. Microbiol. 2007, 10, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Wessner, F.; Lacoux, C.; Goeders, N.; Fouquier d’Herouel, A.; Matos, R.; Serror, P.; Van Melderen, L.; Repoila, F. Regulatory crosstalk between type I and type II toxin-antitoxin systems in the human pathogen Enterococcus faecalis. RNA Biol. 2015, 12, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Dufourc, E.J.; Buchoux, S.; Toupe, J.; Sani, M.A.; Jean-Francois, F.; Khemtemourian, L.; Grelard, A.; Loudet-Courreges, C.; Laguerre, M.; Elezgaray, J.; et al. Membrane interacting peptides: From killers to helpers. Curr. Protein Pept. Sci. 2012, 13, 620–631. [Google Scholar] [CrossRef] [PubMed]
- Weaver, K.E.; Tritle, D.J. Identification and characterization of an Enterococcus faecalis plasmid pAD1-encoded stability determinant which produces two small RNA molecules necessary for its function. Plasmid 1994, 32, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Weaver, K.E.; Reddy, S.G.; Brinkman, C.L.; Patel, S.; Bayles, K.W.; Endres, J.L. Identification and characterization of a family of toxin–antitoxin systems related to the Enterococcus faecalis plasmid pAD1 par addiction module. Microbiology 2009, 155, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, T.J.; Weaver, K.E. Antisense RNA regulation of the pAD1 par post-segregational killing system requires interaction at the 5′ and 3′ ends of the RNAs. Mol. Microbiol. 2000, 37, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Chukwudi, C.U.; Good, L. The role of the hok/sok locus in bacterial response to stressful growth conditions. Microb. Pathog. 2015, 79, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Kawano, M.; Reynolds, A.A.; Miranda-Rios, J.; Storz, G. Detection of 5′- and 3′-UTR-derived small RNAs and cis-encoded antisense RNAs in Escherichia coli. Nucleic Acids Res. 2005, 33, 1040–1050. [Google Scholar] [CrossRef] [PubMed]
- Kawano, M.; Oshima, T.; Kasai, H.; Mori, H. Molecular characterization of long direct repeat (LDR) sequences expressing a stable mRNA encoding for a 35-amino-acid cell-killing peptide and a cis-encoded small antisense RNA in Escherichia coli. Mol. Microbiol. 2002, 45, 333–349. [Google Scholar] [CrossRef] [PubMed]
- Weel-Sneve, R.; Kristiansen, K.I.; Odsbu, I.; Dalhus, B.; Booth, J.; Rognes, T.; Skarstad, K.; Bjoras, M. Single transmembrane peptide DinQ modulates membrane-dependent activities. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Fozo, E.M. New type I toxin–antitoxin families from “wild” and laboratory strains of E. coli: Ibs-Sib, ShoB-OhsC and Zor-Orz. RNA Biol. 2012, 9, 1504–1512. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Quiroga, C.; Chen, Q.; McAnulty, M.J.; Benedik, M.J.; Wood, T.K.; Wang, X. RalR (a DNase) and RalA (a small RNA) form a type I toxin–antitoxin system in Escherichia coli. Nucleic Acids Res. 2014, 42, 6448–6462. [Google Scholar] [CrossRef] [PubMed]
- Durand, S.; Jahn, N.; Condon, C.; Brantl, S. Type I toxin-antitoxin systems in Bacillus subtilis. RNA Biol. 2012, 9, 1491–1497. [Google Scholar] [CrossRef] [PubMed]
- Pinel-Marie, M.L.; Brielle, R.; Felden, B. Dual toxic-peptide-coding Staphylococcus aureus RNA under antisense regulation targets host cells and bacterial rivals unequally. Cell Rep. 2014, 7, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Unterholzner, S.J.; Poppenberger, B.; Rozhon, W. Toxin–antitoxin systems: Biology, identification, and application. Mob. Genet. Elements 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.T.; Espinosa, M.; Yeo, C.C. Keeping the Wolves at Bay: Antitoxins of Prokaryotic Type II Toxin-Antitoxin Systems. Front. Mol. Biosci. 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Mruk, I.; Kobayashi, I. To be or not to be: Regulation of restriction-modification systems and other toxin-antitoxin systems. Nucleic Acids Res. 2014, 42, 70–86. [Google Scholar] [CrossRef] [PubMed]
- Loris, R.; Garcia-Pino, A. Disorder- and dynamics-based regulatory mechanisms in toxin-antitoxin modules. Chem. Rev. 2014, 114, 6933–6947. [Google Scholar] [CrossRef] [PubMed]
- Gazit, E.; Sauer, R.T. The Doc toxin and Phd antidote proteins of the bacteriophage P1 plasmid addiction system form a heterotrimeric complex. J. Biol. Chem. 1999, 274, 16813–16818. [Google Scholar] [CrossRef] [PubMed]
- Korch, S.B.; Hill, T.M. Ectopic overexpression of wild-type and mutant hipA genes in Escherichia coli: Effects on macromolecular synthesis and persister formation. J. Bacteriol. 2006, 188, 3826–3836. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhang, Y.; Teh, J.S.; Zhang, J.; Connell, N.; Rubin, H.; Inouye, M. Characterization of mRNA interferases from Mycobacterium tuberculosis. J. Biol. Chem. 2006, 281, 18638–18643. [Google Scholar] [CrossRef] [PubMed]
- Overgaard, M.; Borch, J.; Jorgensen, M.G.; Gerdes, K. Messenger RNA interferase RelE controls relBE transcription by conditional cooperativity. Mol. Microbiol. 2008, 69, 841–857. [Google Scholar] [CrossRef] [PubMed]
- Christensen-Dalsgaard, M.; Gerdes, K. Translation affects YoeB and MazF messenger RNA interferase activities by different mechanisms. Nucleic Acids Res. 2008, 36, 6472–6481. [Google Scholar] [CrossRef] [PubMed]
- Christensen-Dalsgaard, M.; Jorgensen, M.G.; Gerdes, K. Three new RelE-homologous mRNA interferases of Escherichia coli differentially induced by environmental stresses. Mol. Microbiol. 2010, 75, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Inouye, M. mRNA interferases, sequence-specific endoribonucleases from the toxin-antitoxin systems. Prog. Mol. Biol. Transl. Sci. 2009, 85, 467–500. [Google Scholar] [PubMed]
- Takagi, H.; Kakuta, Y.; Okada, T.; Yao, M.; Tanaka, I.; Kimura, M. Crystal structure of archaeal toxin–antitoxin RelE-RelB complex with implications for toxin activity and antitoxin effects. Nat. Struct. Mol. Biol. 2005, 12, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.; Zavialov, A.V.; Pavlov, M.Y.; Elf, J.; Gerdes, K.; Ehrenberg, M. The bacterial toxin RelE displays codon-specific cleavage of mRNAs in the ribosomal A site. Cell 2003, 112, 131–140. [Google Scholar] [CrossRef]
- Bravo, A.; de Torrontegui, G.; Diaz, R. Identification of components of a new stability system of plasmid R1, ParD, that is close to the origin of replication of this plasmid. Mol. Gen. Genet. 1987, 210, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Tsuchimoto, S.; Ohtsubo, H.; Ohtsubo, E. Two genes, pemK and pemI, responsible for stable maintenance of resistance plasmid R100. J. Bacteriol. 1988, 170, 1461–1466. [Google Scholar] [PubMed]
- Schifano, J.M.; Edifor, R.; Sharp, J.D.; Ouyang, M.; Konkimalla, A.; Husson, R.N.; Woychik, N.A. Mycobacterial toxin MazF-mt6 inhibits translation through cleavage of 23S rRNA at the ribosomal A site. Proc. Natl. Acad. Sci. USA 2013, 110, 8501–8506. [Google Scholar] [CrossRef] [PubMed]
- Schifano, J.M.; Vvedenskaya, I.O.; Knoblauch, J.G.; Ouyang, M.; Nickels, B.E.; Woychik, N.A. An RNA-seq method for defining endoribonuclease cleavage specificity identifies dual rRNA substrates for toxin MazF-mt3. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Schifano, J.M.; Cruz, J.W.; Vvedenskaya, I.O.; Edifor, R.; Ouyang, M.; Husson, R.N.; Nickels, B.E.; Woychik, N.A. tRNA is a new target for cleavage by a MazF toxin. Nucleic Acids Res. 2016, 44, 1256–1270. [Google Scholar] [CrossRef] [PubMed]
- Dao-Thi, M.H.; Charlier, D.; Loris, R.; Maes, D.; Messens, J.; Wyns, L.; Backmann, J. Intricate interactions within the ccd plasmid addiction system. J. Biol. Chem. 2002, 277, 3733–3742. [Google Scholar] [CrossRef] [PubMed]
- Van Melderen, L.; Bernard, P.; Couturier, M. Lon-dependent proteolysis of CcdA is the key control for activation of CcdB in plasmid-free segregant bacteria. Mol. Microbiol. 1994, 11, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Critchlow, S.E.; O’Dea, M.H.; Howells, A.J.; Couturier, M.; Gellert, M.; Maxwell, A. The interaction of the F plasmid killer protein, CcdB, with DNA gyrase: Induction of DNA cleavage and blocking of transcription. J. Mol. Biol. 1997, 273, 826–839. [Google Scholar] [CrossRef] [PubMed]
- Maki, S.; Takiguchi, S.; Miki, T.; Horiuchi, T. Modulation of DNA supercoiling activity of Escherichia coli DNA gyrase by F plasmid proteins. Antagonistic actions of LetA (CcdA) and LetD (CcdB) proteins. J. Biol. Chem. 1992, 267, 12244–12251. [Google Scholar] [PubMed]
- Maki, S.; Takiguchi, S.; Horiuchi, T.; Sekimizu, K.; Miki, T. Partner switching mechanisms in inactivation and rejuvenation of Escherichia coli DNA gyrase by F plasmid proteins LetD (CcdB) and LetA (CcdA). J. Mol. Biol. 1996, 256, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Pogliano, J.; Helinski, D.R.; Konieczny, I. ParE toxin encoded by the broad-host-range plasmid RK2 is an inhibitor of Escherichia coli gyrase. Mol. Microbiol. 2002, 44, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Nakayasu, E.S.; Zhang, L.Q.; Luo, Z.Q. Identification of Fic-1 as an enzyme that inhibits bacterial DNA replication by AMPylating GyrB, promoting filament formation. Sci. Signal. 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Harms, A.; Stanger, F.V.; Scheu, P.D.; de Jong, I.G.; Goepfert, A.; Glatter, T.; Gerdes, K.; Schirmer, T.; Dehio, C. Adenylylation of Gyrase and Topo IV by FicT Toxins Disrupts Bacterial DNA Topology. Cell Rep. 2015, 12, 1497–1507. [Google Scholar] [CrossRef] [PubMed]
- Justice, S.S.; Hunstad, D.A.; Cegelski, L.; Hultgren, S.J. Morphological plasticity as a bacterial survival strategy. Nat. Rev. Microbiol. 2008, 6, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Inouye, M. RatA (YfjG), an Escherichia coli toxin, inhibits 70S ribosome association to block translation initiation. Mol. Microbiol. 2011, 79, 1418–1429. [Google Scholar] [CrossRef] [PubMed]
- Ainelo, A.; Tamman, H.; Leppik, M.; Remme, J.; Horak, R. The toxin GraT inhibits ribosome biogenesis. Mol. Microbiol. 2016, 100, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Al Refaii, A.; Alix, J.H. Ribosome biogenesis is temperature-dependent and delayed in Escherichia coli lacking the chaperones DnaK or DnaJ. Mol. Microbiol. 2009, 71, 748–762. [Google Scholar] [CrossRef] [PubMed]
- Li, G.Y.; Zhang, Y.; Inouye, M.; Ikura, M. Inhibitory mechanism of Escherichia coli RelE-RelB toxin–antitoxin module involves a helix displacement near an mRNA interferase active site. J. Biol. Chem. 2009, 284, 14628–14636. [Google Scholar] [CrossRef] [PubMed]
- Christensen-Dalsgaard, M.; Gerdes, K. Two higBA loci in the Vibrio cholerae superintegron encode mRNA cleaving enzymes and can stabilize plasmids. Mol. Microbiol. 2006, 62, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Kamada, K.; Hanaoka, F.; Burley, S.K. Crystal structure of the MazE/MazF complex: Molecular bases of antidote-toxin recognition. Mol. Cell 2003, 11, 875–884. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Zhu, L.; Suzuki, M.; Inouye, M. Interference of mRNA function by sequence-specific endoribonuclease PemK. J. Biol. Chem. 2004, 279, 20678–20684. [Google Scholar] [CrossRef] [PubMed]
- Kamada, K.; Hanaoka, F. Conformational change in the catalytic site of the ribonuclease YoeB toxin by YefM antitoxin. Mol. Cell 2005, 19, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Arbing, M.A.; Handelman, S.K.; Kuzin, A.P.; Verdon, G.; Wang, C.; Su, M.; Rothenbacher, F.P.; Abashidze, M.; Liu, M.; Hurley, J.M.; et al. Crystal structures of Phd-Doc, HigA, and YeeU establish multiple evolutionary links between microbial growth-regulating toxin-antitoxin systems. Structure 2010, 18, 996–1010. [Google Scholar] [CrossRef] [PubMed]
- Short, F.L.; Pei, X.Y.; Blower, T.R.; Ong, S.L.; Fineran, P.C.; Luisi, B.F.; Salmond, G.P. Selectivity and self-assembly in the control of a bacterial toxin by an antitoxic noncoding RNA pseudoknot. Proc. Natl. Acad. Sci. USA 2013, 110. [Google Scholar] [CrossRef] [PubMed]
- Blower, T.R.; Pei, X.Y.; Short, F.L.; Fineran, P.C.; Humphreys, D.P.; Luisi, B.F.; Salmond, G.P. A processed noncoding RNA regulates an altruistic bacterial antiviral system. Nat. Struct. Mol. Biol. 2011, 18, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Samson, J.E.; Spinelli, S.; Cambillau, C.; Moineau, S. Structure and activity of AbiQ, a lactococcal endoribonuclease belonging to the type III toxin–antitoxin system. Mol. Microbiol. 2013, 87, 756–768. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lord, D.M.; Hong, S.H.; Peti, W.; Benedik, M.J.; Page, R.; Wood, T.K. Type II toxin/antitoxin MqsR/MqsA controls type V toxin/antitoxin GhoT/GhoS. Environ. Microbiol. 2013, 15, 1734–1744. [Google Scholar] [CrossRef] [PubMed]
- Ceglowski, P.; Boitsov, A.; Chai, S.; Alonso, J.C. Analysis of the stabilization system of pSM19035-derived plasmid pBT233 in Bacillus subtilis. Gene 1993, 136, 1–12. [Google Scholar] [CrossRef]
- Camacho, A.G.; Misselwitz, R.; Behlke, J.; Ayora, S.; Welfle, K.; Meinhart, A.; Lara, B.; Saenger, W.; Welfle, H.; Alonso, J.C. In vitro and in vivo stability of the epsilon2zeta2 protein complex of the broad host-range Streptococcus pyogenes pSM19035 addiction system. Biol. Chem. 2002, 383, 1701–1713. [Google Scholar] [CrossRef] [PubMed]
- Ceglowski, P.; Boitsov, A.; Karamyan, N.; Chai, S.; Alonso, J.C. Characterization of the effectors required for stable inheritance of Streptococcus pyogenes pSM19035-derived plasmids in Bacillus subtilis. Mol. Gen. Genet. 1993, 241, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Lioy, V.S.; Martin, M.T.; Camacho, A.G.; Lurz, R.; Antelmann, H.; Hecker, M.; Hitchin, E.; Ridge, Y.; Wells, J.M.; Alonso, J.C. pSM19035-encoded zeta toxin induces stasis followed by death in a subpopulation of cells. Microbiology 2006, 152, 2365–2379. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.J.; Ray, C.G.; Sherris, J.C. Sherris Medical Microbiology: An Introduction to Infectious Diseases, 4th ed.; McGraw-Hill: New York, NY, USA, 2004; pp. 375–380. [Google Scholar]
- Chan, W.T.; Yeo, C.C.; Sadowy, E.; Espinosa, M. Functional validation of putative toxin-antitoxin genes from the Gram-positive pathogen Streptococcus pneumoniae: Phd-doc is the fourth bona-fide operon. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Nieto, C.; Pellicer, T.; Balsa, D.; Christensen, S.K.; Gerdes, K.; Espinosa, M. The chromosomal relBE2 toxin-antitoxin locus of Streptococcus pneumoniae: Characterization and use of a bioluminescence resonance energy transfer assay to detect toxin–antitoxin interaction. Mol. Microbiol. 2006, 59, 1280–1296. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Cordoba, I.; Diago-Navarro, E.; Barendregt, A.; Heck, A.J.; Alfonso, C.; Diaz-Orejas, R.; Nieto, C.; Espinosa, M. The toxin–antitoxin proteins relBE2Spn of Streptococcus pneumoniae: Characterization and association to their DNA target. Proteins 2012, 80, 1834–1846. [Google Scholar] [CrossRef] [PubMed]
- Li, G.Y.; Zhang, Y.; Inouye, M.; Ikura, M. Structural mechanism of transcriptional autorepression of the Escherichia coli RelB/RelE antitoxin/toxin module. J. Mol. Biol. 2008, 380, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Nieto, C.; Cherny, I.; Khoo, S.K.; de Lacoba, M.G.; Chan, W.T.; Yeo, C.C.; Gazit, E.; Espinosa, M. The yefM–yoeB toxin–antitoxin systems of Escherichia coli and Streptococcus pneumoniae: Functional and structural correlation. J. Bacteriol. 2007, 189, 1266–1278. [Google Scholar] [CrossRef] [PubMed]
- Khoo, S.K.; Loll, B.; Chan, W.T.; Shoeman, R.L.; Ngoo, L.; Yeo, C.C.; Meinhart, A. Molecular and structural characterization of the PezAT chromosomal toxin-antitoxin system of the human pathogen Streptococcus pneumoniae. J. Biol. Chem. 2007, 282, 19606–19618. [Google Scholar] [CrossRef] [PubMed]
- Zielenkiewicz, U.; Ceglowski, P. The toxin-antitoxin system of the streptococcal plasmid pSM19035. J. Bacteriol. 2005, 187, 6094–6105. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; Gilliland, S.M.; Holden, D.W. A Streptococcus pneumoniae pathogenicity island encoding an ABC transporter involved in iron uptake and virulence. Mol. Microbiol. 2001, 40, 572–585. [Google Scholar] [CrossRef] [PubMed]
- Croucher, N.J.; Walker, D.; Romero, P.; Lennard, N.; Paterson, G.K.; Bason, N.C.; Mitchell, A.M.; Quail, M.A.; Andrew, P.W.; Parkhill, J.; et al. Role of conjugative elements in the evolution of the multidrug-resistant pandemic clone Streptococcus pneumonia Spain23F ST81. J. Bacteriol. 2009, 191, 1480–1489. [Google Scholar] [CrossRef] [PubMed]
- Murray, B.E. The life and times of the Enterococcus. Clin. Microbiol. Rev. 1990, 3, 46–65. [Google Scholar] [CrossRef] [PubMed]
- Gobl, C.; Kosol, S.; Stockner, T.; Ruckert, H.M.; Zangger, K. Solution structure and membrane binding of the toxin Fst of the par addiction module. Biochemistry 2010, 49, 6567–6575. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.Y.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G., Jr. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, R.V.; Fowler, V.G., Jr.; Skov, R.; Bruun, N.E. Future challenges and treatment of Staphylococcus aureus bacteremia with emphasis on MRSA. Future Microbiol. 2011, 6, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Adler, E.; Barak, I.; Stragier, P. Bacillus subtilis locus encoding a killer protein and its antidote. J. Bacteriol. 2001, 183, 3574–3581. [Google Scholar] [CrossRef] [PubMed]
- Hopper, S.; Wilbur, J.S.; Vasquez, B.L.; Larson, J.; Clary, S.; Mehr, I.J.; Seifert, H.S.; So, M. Isolation of Neisseria gonorrhoeae mutants that show enhanced trafficking across polarized T84 epithelial monolayers. Infect. Immun. 2000, 68, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Merz, A.J.; So, M. Interactions of pathogenic Neisseriae with epithelial cell membranes. Annu. Rev. Cell Dev. Biol. 2000, 16, 423–457. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.L.; Apicella, M.A. The molecular mechanisms used by Neisseria gonorrhoeae to initiate infection differ between men and women. Clin. Microbiol. Rev. 2004, 17, 965–981. [Google Scholar] [CrossRef] [PubMed]
- Raoult, D.; La Scola, B.; Enea, M.; Fournier, P.E.; Roux, V.; Fenollar, F.; Galvao, M.A.; de Lamballerie, X. A flea-associated Rickettsia pathogenic for humans. Emerg. Infect. Dis. 2001, 7, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Renesto, P.; Audic, S.; Robert, C.; Blanc, G.; Fournier, P.E.; Parinello, H.; Claverie, J.M.; Raoult, D. The genome sequence of Rickettsia felis identifies the first putative conjugative plasmid in an obligate intracellular parasite. PLoS Biol. 2005, 3. [Google Scholar] [CrossRef] [PubMed]
- Audoly, G.; Vincentelli, R.; Edouard, S.; Georgiades, K.; Mediannikov, O.; Gimenez, G.; Socolovschi, C.; Mege, J.L.; Cambillau, C.; Raoult, D. Effect of rickettsial toxin VapC on its eukaryotic host. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Tamparo, C.D.; Lewis, M.A. Diseases of the Human Body, 5th ed.; F.A. Davis: Philadelphia, PA, USA, 2011; p. 591. [Google Scholar]
- Karch, H.; Tarr, P.I.; Bielaszewska, M. Enterohaemorrhagic Escherichia coli in human medicine. Int. J. Med. Microbiol. 2005, 295, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Hallez, R.; Geeraerts, D.; Sterckx, Y.; Mine, N.; Loris, R.; Van Melderen, L. New toxins homologous to ParE belonging to three-component toxin-antitoxin systems in Escherichia coli O157:H7. Mol. Microbiol. 2010, 76, 719–732. [Google Scholar] [CrossRef] [PubMed]
- De la Hoz, A.B.; Ayora, S.; Sitkiewicz, I.; Fernandez, S.; Pankiewicz, R.; Alonso, J.C.; Ceglowski, P. Plasmid copy-number control and better-than-random segregation genes of pSM19035 share a common regulator. Proc. Natl. Acad. Sci. USA 2000, 97, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Cheverton, A.M.; Gollan, B.; Przydacz, M.; Wong, C.T.; Mylona, A.; Hare, S.A.; Helaine, S. A Salmonella Toxin Promotes Persister Formation through Acetylation of tRNA. Mol. Cell 2016, 63, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Dworkin, M.; Falkow, S. The Prokaryotes: A Handbook on the Biology of Bacteria, 3rd ed.; Springer: New York, NY, USA; London, UK, 2006; p. 315. [Google Scholar]
- Zorzini, V.; Buts, L.; Sleutel, M.; Garcia-Pino, A.; Talavera, A.; Haesaerts, S.; De Greve, H.; Cheung, A.; van Nuland, N.A.; Loris, R. Structural and biophysical characterization of Staphylococcus aureus SaMazF shows conservation of functional dynamics. Nucleic Acids Res. 2014, 42, 6709–6725. [Google Scholar] [CrossRef] [PubMed]
- Mattison, K.; Wilbur, J.S.; So, M.; Brennan, R.G. Structure of FitAB from Neisseria gonorrhoeae bound to DNA reveals a tetramer of toxin-antitoxin heterodimers containing pin domains and ribbon-helix-helix motifs. J. Biol. Chem. 2006, 281, 37942–37951. [Google Scholar] [CrossRef] [PubMed]
- Mate, M.J.; Vincentelli, R.; Foos, N.; Raoult, D.; Cambillau, C.; Ortiz-Lombardia, M. Crystal structure of the DNA-bound VapBC2 antitoxin/toxin pair from Rickettsia felis. Nucleic Acids Res. 2012, 40, 3245–3258. [Google Scholar] [CrossRef] [PubMed]
- Dienemann, C.; Boggild, A.; Winther, K.S.; Gerdes, K.; Brodersen, D.E. Crystal structure of the VapBC toxin–antitoxin complex from Shigella flexneri reveals a hetero-octameric DNA-binding assembly. J. Mol. Biol. 2011, 414, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Sterckx, Y.G.; Volkov, A.N.; Vranken, W.F.; Kragelj, J.; Jensen, M.R.; Buts, L.; Garcia-Pino, A.; Jove, T.; van Melderen, L.; Blackledge, M.; et al. Small-angle X-ray scattering- and nuclear magnetic resonance-derived conformational ensemble of the highly flexible antitoxin PaaA2. Structure 2014, 22, 854–865. [Google Scholar] [CrossRef] [PubMed]
- Sterckx, Y.G.; Jove, T.; Shkumatov, A.V.; Garcia-Pino, A.; Geerts, L.; De Kerpel, M.; Lah, J.; De Greve, H.; Van Melderen, L.; Loris, R. A unique hetero-hexadecameric architecture displayed by the Escherichia coli O157 PaaA2-ParE2 antitoxin-toxin complex. J. Mol. Biol. 2016, 428, 1589–1603. [Google Scholar] [CrossRef] [PubMed]
- Zorzini, V.; Mernik, A.; Lah, J.; Sterckx, Y.G.; de Jonge, N.; Garcia-Pino, A.; De Greve, H.; Versees, W.; Loris, R. Substrate Recognition and Activity Regulation of the Escherichia coli mRNA Endonuclease MazF. J. Biol. Chem. 2016, 291, 10950–10960. [Google Scholar] [CrossRef] [PubMed]
- Loris, R.; Dao-Thi, M.H.; Bahassi, E.M.; Van Melderen, L.; Poortmans, F.; Liddington, R.; Couturier, M.; Wyns, L. Crystal structure of CcdB, a topoisomerase poison from E. coli. J. Mol. Biol. 1999, 285, 1667–1677. [Google Scholar] [CrossRef] [PubMed]
- Dao-Thi, M.H.; Van Melderen, L.; de Genst, E.; Afif, H.; Buts, L.; Wyns, L.; Loris, R. Molecular basis of gyrase poisoning by the addiction toxin CcdB. J. Mol. Biol. 2005, 348, 1091–1102. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, N.; Hohlweg, W.; Garcia-Pino, A.; Respondek, M.; Buts, L.; Haesaerts, S.; Lah, J.; Zangger, K.; Loris, R. Structural and thermodynamic characterization of Vibrio fischeri CcdB. J. Biol. Chem. 2010, 285, 5606–5613. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, N.; Simic, M.; Buts, L.; Haesaerts, S.; Roelants, K.; Garcia-Pino, A.; Sterckx, Y.; De Greve, H.; Lah, J.; Loris, R. Alternative interactions define gyrase specificity in the CcdB family. Mol. Microbiol. 2012, 84, 965–978. [Google Scholar] [CrossRef] [PubMed]
- Heaton, B.E.; Herrou, J.; Blackwell, A.E.; Wysocki, V.H.; Crosson, S. Molecular structure and function of the novel BrnT/BrnA toxin–antitoxin system of Brucella abortus. J. Biol. Chem. 2012, 287, 12098–12110. [Google Scholar] [CrossRef] [PubMed]
- Miallau, L.; Faller, M.; Chiang, J.; Arbing, M.; Guo, F.; Cascio, D.; Eisenberg, D. Structure and proposed activity of a member of the VapBC family of toxin–antitoxin systems. VapBC-5 from Mycobacterium tuberculosis. J. Biol. Chem. 2009, 284, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Min, A.B.; Miallau, L.; Sawaya, M.R.; Habel, J.; Cascio, D.; Eisenberg, D. The crystal structure of the Rv0301-Rv0300 VapBC-3 toxin–antitoxin complex from M. tuberculosis reveals a Mg(2)(+) ion in the active site and a putative RNA-binding site. Protein Sci. 2012, 21, 1754–1767. [Google Scholar] [CrossRef] [PubMed]
- Das, U.; Pogenberg, V.; Subhramanyam, U.K.; Wilmanns, M.; Gourinath, S.; Srinivasan, A. Crystal structure of the VapBC-15 complex from Mycobacterium tuberculosis reveals a two-metal ion dependent PIN-domain ribonuclease and a variable mode of toxin–antitoxin assembly. J. Struct. Biol. 2014, 188, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.G.; Lee, S.J.; Chae, S.; Lee, K.Y.; Kim, J.H.; Lee, B.J. Structural and functional studies of the Mycobacterium tuberculosis VapBC30 toxin-antitoxin system: Implications for the design of novel antimicrobial peptides. Nucleic Acids Res. 2015, 43, 7624–7637. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Issac, B.; Dodson, E.J.; Turkenburg, J.P.; Mande, S.C. Crystal structure of Mycobacterium tuberculosis YefM antitoxin reveals that it is not an intrinsically unstructured protein. J. Mol. Biol. 2008, 383, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Florek, P.; Levdikov, V.M.; Blagova, E.; Lebedev, A.A.; Skrabana, R.; Resetarova, S.; Pavelcikova, P.; Barak, I.; Wilkinson, A.J. The structure and interactions of SpoIISA and SpoIISB, a toxin-antitoxin system in Bacillus subtilis. J. Biol. Chem. 2011, 286, 6808–6819. [Google Scholar] [CrossRef] [PubMed]
- Simanshu, D.K.; Yamaguchi, Y.; Park, J.H.; Inouye, M.; Patel, D.J. Structural basis of mRNA recognition and cleavage by toxin MazF and its regulation by antitoxin MazE in Bacillus subtilis. Mol. Cell 2013, 52, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Yamaguchi, Y.; Inouye, M. Bacillus subtilis MazF-bs (EndoA) is a UACAU-specific mRNA interferase. FEBS Lett. 2011, 585, 2526–2532. [Google Scholar] [CrossRef] [PubMed]
- Arcus, V.L.; Backbro, K.; Roos, A.; Daniel, E.L.; Baker, E.N. Distant structural homology leads to the functional characterization of an archaeal PIN domain as an exonuclease. J. Biol. Chem. 2004, 279, 16471–16478. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.T.; Balsa, D.; Espinosa, M. One cannot rule them all: Are bacterial toxins–antitoxins druggable? FEMS Microbiol. Rev. 2015, 39, 522–540. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.J.; Hergenrother, P.J. Artificial activation of toxin-antitoxin systems as an antibacterial strategy. Trends Microbiol. 2012, 20, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Mutschler, H.; Meinhart, A. Epsilon/zeta systems: Their role in resistance, virulence, and their potential for antibiotic development. J. Mol. Med. 2011, 89, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, S. Speculative strategies for new antibacterials: All roads should not lead to Rome. J. Antibiot. 2013, 66, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Bienstock, R.J. Computational drug design targeting protein-protein interactions. Curr. Pharm. Des. 2012, 18, 1240–1254. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.K.; Maenhaut-Michel, G.; Mine, N.; Gottesman, S.; Gerdes, K.; Van Melderen, L. Overproduction of the Lon protease triggers inhibition of translation in Escherichia coli: Involvement of the yefM-yoeB toxin–antitoxin system. Mol. Microbiol. 2004, 51, 1705–1717. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.; Datti, A.; Cossette, M.; Goodreid, J.; McCaw, S.E.; Mah, M.; Nakhamchik, A.; Ogata, K.; El Bakkouri, M.; Cheng, Y.Q.; et al. Activators of cylindrical proteases as antimicrobials: Identification and development of small molecule activators of ClpP protease. Chem. Biol. 2011, 18, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Sass, P.; Josten, M.; Famulla, K.; Schiffer, G.; Sahl, H.G.; Hamoen, L.; Brotz-Oesterhelt, H. Antibiotic acyldepsipeptides activate ClpP peptidase to degrade the cell division protein FtsZ. Proc. Natl. Acad. Sci. USA 2011, 108, 17474–17479. [Google Scholar] [CrossRef] [PubMed]
- Sayed, N.; Nonin-Lecomte, S.; Rety, S.; Felden, B. Functional and structural insights of a Staphylococcus aureus apoptotic-like membrane peptide from a toxin-antitoxin module. J. Biol. Chem. 2012, 287, 43454–43463. [Google Scholar] [CrossRef] [PubMed]
- Solecki, O.; Mosbah, A.; Baudy Floc’h, M.; Felden, B. Converting a Staphylococcus aureus toxin into effective cyclic pseudopeptide antibiotics. Chem. Biol. 2015, 22, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Huys, I.; Pirnay, J.P.; Lavigne, R.; Jennes, S.; De Vos, D.; Casteels, M.; Verbeken, G. Paving a regulatory pathway for phage therapy. Europe should muster the resources to financially, technically and legally support the introduction of phage therapy. EMBO Rep. 2013, 14, 951–954. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Mishra, N.K.; Bhatnagar, S.; Bhatnagar, R. PemK toxin of Bacillus anthracis is a ribonuclease: An insight into its active site, structure, and function. J. Biol. Chem. 2010, 285, 7254–7270. [Google Scholar] [CrossRef] [PubMed]
- Chopra, N.; Agarwal, S.; Verma, S.; Bhatnagar, S.; Bhatnagar, R. Modeling of the structure and interactions of the B. anthracis antitoxin, MoxX: Deletion mutant studies highlight its modular structure and repressor function. J. Comput. Aided Mol. Des. 2011, 25, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.A.; McClendon, C.L. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 2007, 450, 1001–1009. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | TA Module | Protein Toxin | Antitoxin (Molecular Species) | Toxin Activity | Inhibited Cellular Process | Ref. |
|---|---|---|---|---|---|---|
| I | istR-tisB | TisB | istR (RNA) | Depolarizes Cell Membrane | ATP Synthesis | [41,42] |
| I | sok-hok | Hok | sok (RNA) | Depolarizes Cell Membrane | ATP Synthesis | [43,44] |
| I | sibC-ibsC | IbsC | sibC (RNA) | Depolarizes Cell Membrane | ATP Synthesis | [45,46] |
| I | ratA-txpA | TxpA | ratA (RNA) | Lyses Cell Membrane | Not Applicable | [47,48] |
| I | fst–RNAI–RNAII | Fst | RNAII (RNA) | Damages Cell Membrane | Cell Division | [49,50] |
| I | symRE | SymE | symR (RNA) | mRNA Cleavage | Translation | [51] |
| II | mazEF | MazF | MazE (protein) | mRNA Cleavage | Translation | [52,53] |
| II | kis-kid | Kid | Kis (protein) | mRNA Cleavage | Translation | [54] |
| II | higBA | HigA | HigB (protein) | mRNA Cleavage | Translation | [55,56] |
| II | relBE | RelE | RelB (protein) | mRNA Cleavage | Translation | [57] |
| II | vapBC | VapC | VapB (protein) | RNA Cleavage | Translation | [58,59,60] |
| II | phd-doc | Doc | Phd (protein) | Phosphorylation of Translation Elongation Factor EF-Tu | Translation | [61,62] |
| II | hipBA | HipA | HipB (protein) | Phosphorylation of Glutamyl tRNA Synthetase | Translation | [63,64] |
| II | ccdAB | CcdB | CcdA (protein) | Poison of DNA gyrase | Transcription and Replication | [65,66] |
| II | parDE | ParE | ParD (protein) | Poison of DNA gyrase | Transcription and Replication | [65,66] |
| II | ω-ε-ζ | ζ | ε (protein) | Phosphorylation of UDP-N-acetylglucosamine | Peptidoglycan Synthesis | [67,68] |
| III | toxIN | ToxN | toxI (RNA) | RNA Cleavage | Translation | [69] |
| IV | yeeUV | YeeV | YeeU (protein) | Interacts with Cytoskeleton Proteins FtsZ and MreB | Cell Division | [70] |
| IV | cptBA | CptA | CptB (protein) | Interacts with Cytoskeleton Proteins FtsZ and MreB | Cell Division | [71] |
| V | ghoST | GhoT | GhoS (protein) | Damages Cell Membrane | Not Applicable | [72] |
| VI | socAB | SocB | SocA (protein) | Binds to the β sliding clamp DnaN | Replication | [73] |
| Pathogenic Bacteria | TA Protein | Oligomeric State (Stoichiometry) | Pfam Annotation/Accession ID | PDB ID/Deposit Date | Ref. |
|---|---|---|---|---|---|
| Streptococcus pyogenes | ε-ζ Complex | Hetero-tetramer (A2B2) | ε: Bacterial Epsilon Antitoxin Domain/PF08998 | 1GVN/2002-02-19 | [68] |
| ζ: Zeta Toxin Family/PF06414 | |||||
| Streptococcus pyogenes | ε-ζ Bound to the Substrate | Hetero-tetramer (A2B2) | ε: Bacterial Epsilon Antitoxin Domain/PF08998 | 3Q8X/2011-01-07 | [67] |
| ζ: Zeta Toxin Family/PF06414 | |||||
| Streptococcus pneumoniae | PezAT Complex | Hetero-tetramer (A2B2) | PezA: 3-layer (αβα) Sandwich Architecture and Rossmann Fold Topology* | 2P5T/2007-03-16 | [140] |
| PezT: Zeta Toxin Family/PF06414 | |||||
| Enterococcus faecalis | Fst Toxin | Monomer (A) | Toxin Fst Domain/PF13955 | 2KV5/2010-03-08 | [145] |
| Staphylococcus aureus | MazF Toxin | Homo-dimer (A2) | PemK-like Protein Family/PF02452 | 4MZM/2013-09-30 | [161] |
| Neisseria gonorrhoeae | FitB Toxin | Homo-dimer (A2) | FitB: PIN Domain/PF01850 | 2H1C/2006-05-16 | [162] |
| Neisseria gonorrhoeae | FitAB Bound to DNA | Hetero-octamer (A4B4) | FitB: PIN Domain/PF01850 | 2H1O/2006-05-16 | [162] |
| FitA: 3-layer (αβα) Sandwich Architecture and Rossmann Fold Topology * | |||||
| Rickettsia felis | VapBC2 Bound to DNA | Hetero-octamer (A4B4) | VapB2: Antitoxin MazE Domain/PF04014 | 3ZVK/2011-07-25 | [163] |
| VapC2: PIN Domain/PF01850 | |||||
| Shigella flexneri | YeeU Antitoxin | Monomer (A) | YagB/YeeU/YfjZ Family/PF06154 | 2INW/2006-10-09 | [125] |
| Shigella flexneri | VapBC Complex | Hetero-octamer (A4B4) | VapC: PIN Domain/PF01850 | 3TND/2011-09-01 | [164] |
| VapB: Antitoxin MazE Domain/PF04014 | |||||
| Escherichia coli O157 | PaaA2 Antitoxin | Monomer (A) | Not Applicable | 3ZBE/2012-11-08 | [165] |
| Escherichia coli O157 | PaaA2-ParE2 Complex | Hetero-heptadecamer (A8B8) | PaaA2: Not Applicable | 5CW7/2015-07-27 | [166] |
| ParE2: Plasmid Stabilization System Protein/PF05016 | |||||
| Escherichia coli K12 | MazEF Complex | Hetero-hexamer (A4B2) | MazE: Antidote-toxin Recognition MazE/PF04014 | 1UB4/2003-03-28 | [122] |
| MazF: PemK-like Protein/PF02452 | |||||
| Escherichia coli K12 | MazF-Substrate Complex | Homo-dimer (A2) | MazF: PemK-like Protein/PF02452 | 5CR2/2015-07-22 | [167] |
| Substrate Sequence: d(AUACAUA) | |||||
| Escherichia coli K12 | CcdB Toxin | Homo-dimer | CcdB Protein Domain/PF01845 | 1VUB/1998-04-17 | [168] |
| Escherichia coli K12 | CcdB Toxin Bound to Gyrase | Hetero-tetramer (A2B2) | CcdB Protein Domain/PF01845 | 1X75/2004-08-13 | [169] |
| Escherichia coli K12 | YefM-YoeB Complex | Hetero-trimer (A2B) | YefM: Antitoxin Phd_YefM Domain/PF02604 | 2A6Q/2005-07-04 | [124] |
| YoeB: Plasmid encoded Toxin Txe Family | |||||
| Vibrio fischeri | CcdB Toxin | Homo-dimer (A2) | CcdB Protein Domain/PF01845 | 3JSC/2009-09-10 | [170] |
| Vibrio fischeri | CcdB Toxin Bound to Gyrase | Hetero-tetramer (A2B2) | CcdB Protein Domain/PF01845 | 4ELZ/2012-04-11 | [171] |
| Brucella abortus | BrnT Toxin | Homo-tetramer (A4) | Protein of Unknown Function/PF04365 | 3U97/2011-10-18 | [172] |
| Salmonella typhimurium | TacT Toxin | Homo-dimer (A2) | Acetyltransferase (GNAT) Domain/PF13673 | 5FVJ/2016-02-08 | [159] |
| Mycobacterium tuberculosis | VapBC5 Complex | Hetero-dimer (AB) | VapB5: Antitoxin Phd_YefM Domain/PF02604 | 3DBO/2008-06-02 | [173] |
| VapC5: PIN Domain/PF01850 | |||||
| Mycobacterium tuberculosis | VapBC3 Complex | Hetero-octamer (A4B4) | VapB3: Ribbon-helix-helix Protein, copG Family/PF01402 | 3H87/2009-04-28 | [174] |
| VapC3: PIN Domain/PF01850 | |||||
| Mycobacterium tuberculosis | VapBC15 Complex | Hetero-tetramer (A2B2) | VapB15: Uncharacterized, Conserved Protein/PF09957 | 4CHG/2013-12-02 | [175] |
| VapC15: PIN Domain/PF01850 | |||||
| Mycobacterium tuberculosis | VapBC30 Complex | Hetero-tetramer (A2B2) | VapB30: Rv0623-like Transcription Factor/PF07704 | 4XGQ/2015-01-02 | [176] |
| VapC30: PIN Domain/PF01850 | |||||
| Mycobacterium tuberculosis | YefM Antitoxin | Homo-tetramer (A4) | Antitoxin Phd_YefM Domain/PF02604 | 3CTO/2008-04-14 | [177] |
| Bacillus subtilis | SpoIISA-SpoIISB Complex | Hetero-tetramer (A2B2) | SpoIISB: Antitoxin SpoIISB Family/PF14185 | 3O6Q/2010-07-29 | [178] |
| SpoIISA: Toxin SpoIISA Family/PF14171 | |||||
| Bacillus subtilis | MazEF Complex | Hetero-hexamer (A4B2) | MazE: Ribbon-helix-helix Protein, CopG family/PF01402 | 4ME7/2013-08-25 | [179] |
| MazF: PemK-like Protein/PF02452 | |||||
| Bacillus subtilis | MazF-Substrate Complex | Homo-dimer (A2) | MazF: PemK-like Protein/PF02452 | 4MDX/2013-08-23 | [179] |
| Substrate Sequence: UUdUACAUAA |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, K.-Y.; Lee, B.-J. Structure, Biology, and Therapeutic Application of Toxin–Antitoxin Systems in Pathogenic Bacteria. Toxins 2016, 8, 305. https://doi.org/10.3390/toxins8100305
Lee K-Y, Lee B-J. Structure, Biology, and Therapeutic Application of Toxin–Antitoxin Systems in Pathogenic Bacteria. Toxins. 2016; 8(10):305. https://doi.org/10.3390/toxins8100305
Chicago/Turabian StyleLee, Ki-Young, and Bong-Jin Lee. 2016. "Structure, Biology, and Therapeutic Application of Toxin–Antitoxin Systems in Pathogenic Bacteria" Toxins 8, no. 10: 305. https://doi.org/10.3390/toxins8100305
APA StyleLee, K.-Y., & Lee, B.-J. (2016). Structure, Biology, and Therapeutic Application of Toxin–Antitoxin Systems in Pathogenic Bacteria. Toxins, 8(10), 305. https://doi.org/10.3390/toxins8100305