
Increased Proinflammatory Cytokine Production and Decreased Cholesterol Efflux Due to Downregulation of ABCG1 in Macrophages Exposed to Indoxyl Sulfate

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
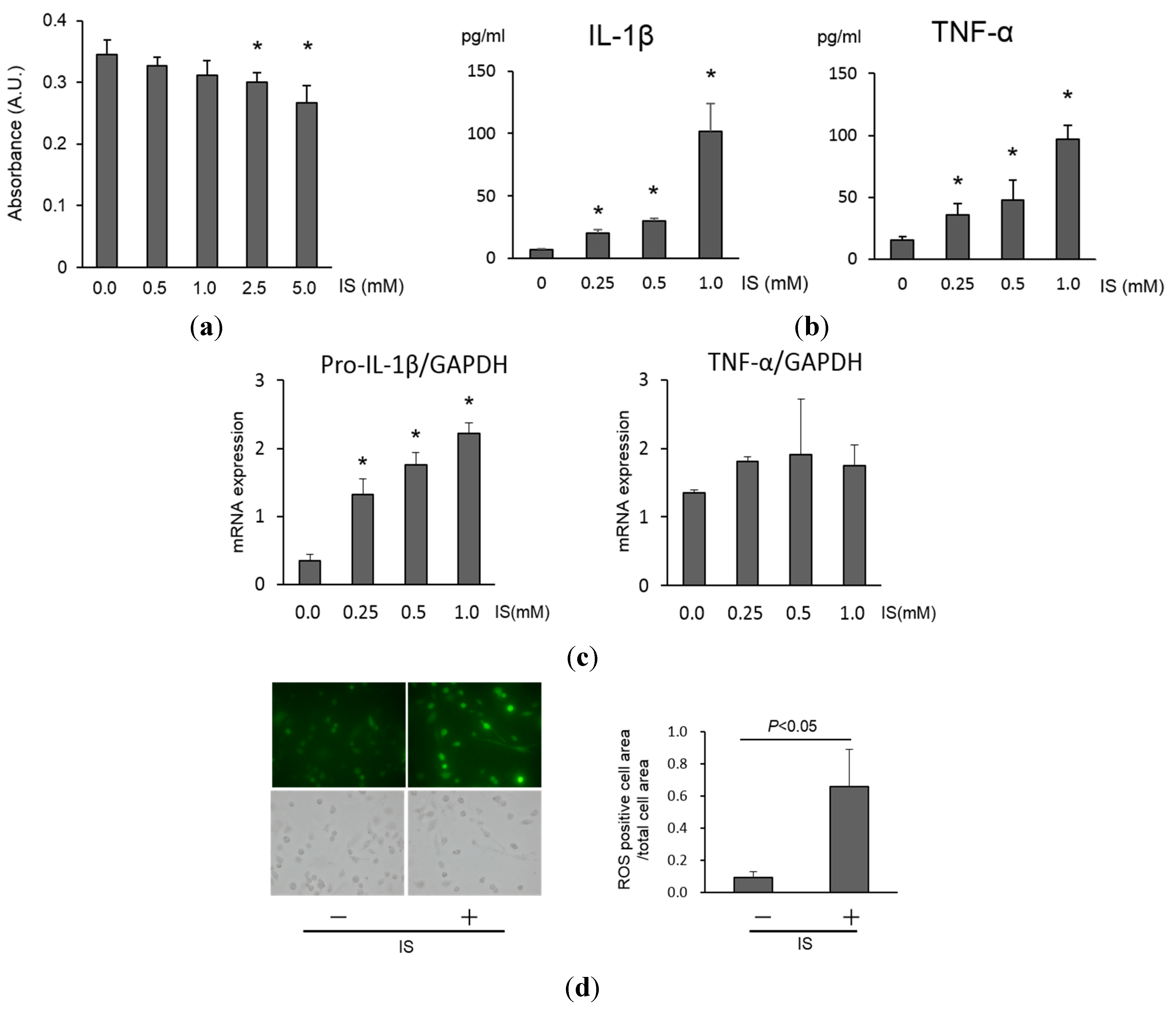
2.1. Cell Viability, Inflammation and Reactive Oxygen Species Production in Macrophages Exposed to IS

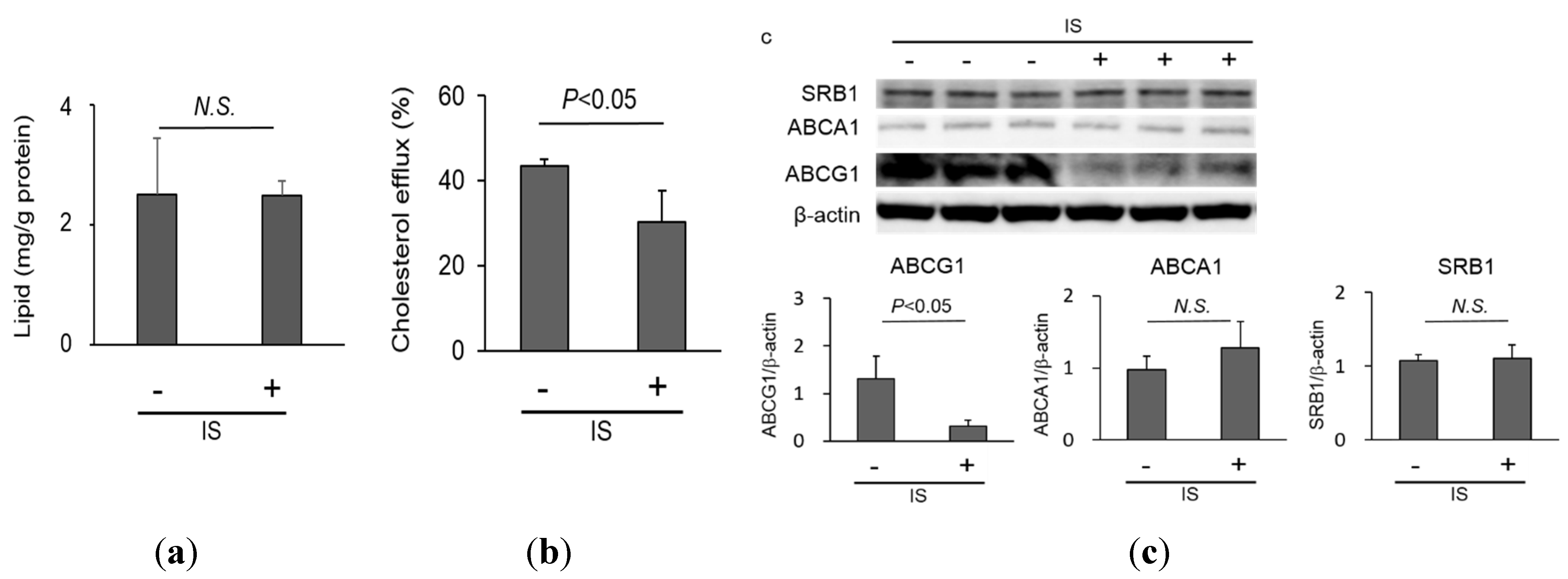
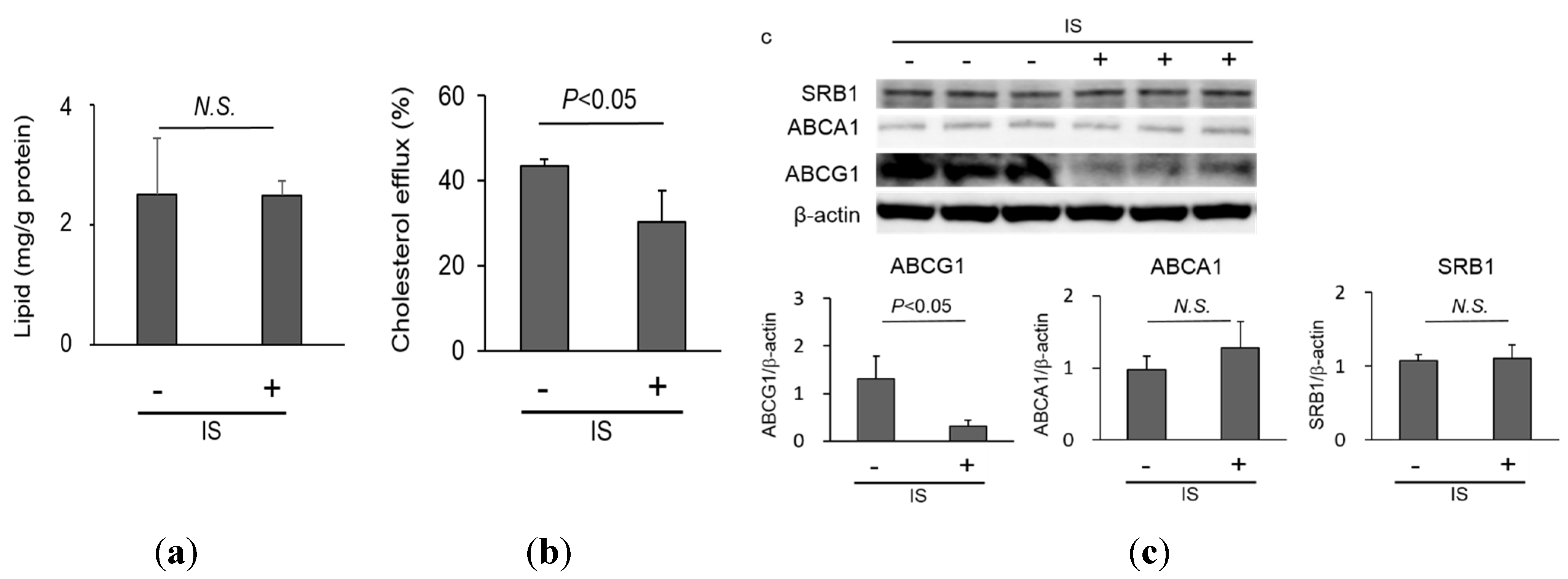
2.2. Lipid Homeostasis
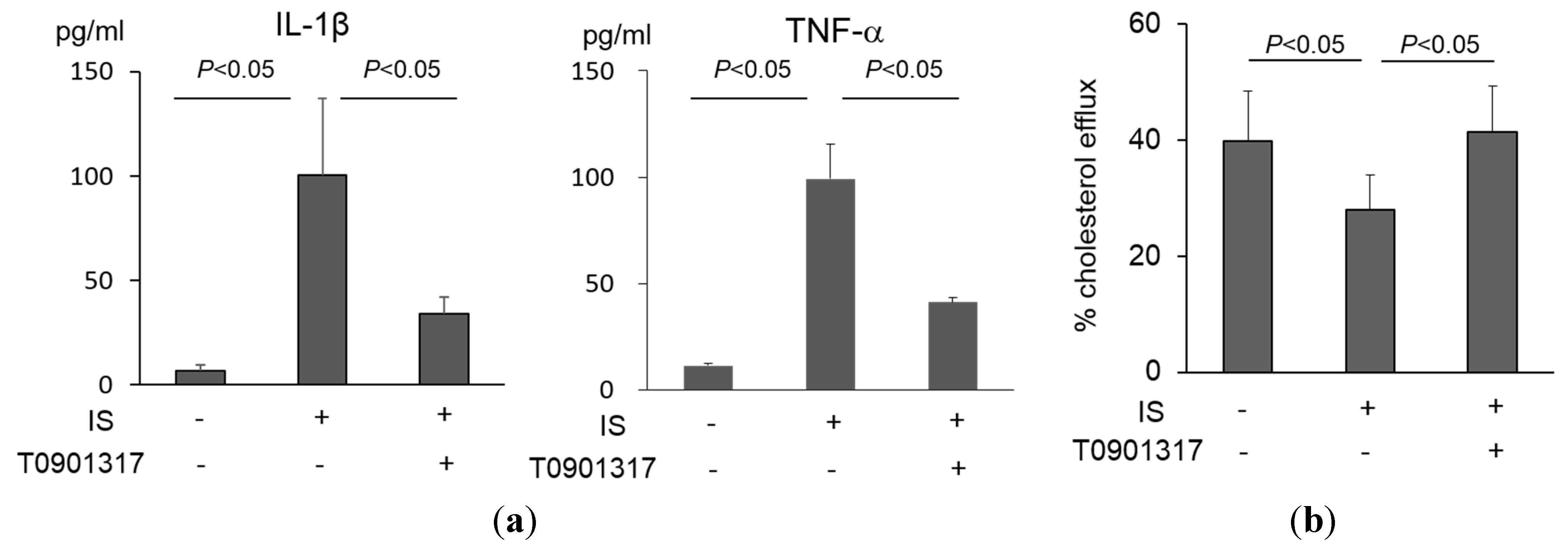
2.3. Intervention with a Liver X Receptor Agonist for IS-Induced Macrophage Dysfunction
3. Discussion
4. Experimental Section
4.1. Cell Culture
4.2. Cell Viability Assay
4.3. Measurement of Inflammatory Cytokines in Medium
4.4. Measurement of mRNA Expression
4.5. Measurement of Reactive Oxygen Species Production
4.6. Detection of Proteins in Cells
4.7. Lipid Accumulation and Cholesterol Efflux Study
4.8. Reaction with a Liver X Receptor Agonist
4.9. Statistics
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Muntner, P.; He, J.; Hamm, L.; Loria, C.; Whelton, P.K. Renal insufficiency and subsequent death resulting from cardiovascular disease in the united states. J. Am. Soc. Nephrol. 2002, 13, 745–753. [Google Scholar] [PubMed]
- Sarnak, M.J.; Levey, A.S.; Schoolwerth, A.C.; Coresh, J.; Culleton, B.; Hamm, L.L.; McCullough, P.A.; Kasiske, B.L.; Kelepouris, E.; Klag, M.J.; et al. Kidney disease as a risk factor for development of cardiovascular disease: A statement from the american heart association councils on kidney in cardiovascular disease, high blood pressure research, clinical cardiology, and epidemiology and prevention. Hypertension 2003, 42, 1050–1065. [Google Scholar] [CrossRef] [PubMed]
- Anavekar, N.S.; McMurray, J.J.; Velazquez, E.J.; Solomon, S.D.; Kober, L.; Rouleau, J.L.; White, H.D.; Nordlander, R.; Maggioni, A.; Dickstein, K.; et al. Relation between renal dysfunction and cardiovascular outcomes after myocardial infarction. N. Engl. J. Med. 2004, 351, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Colhoun, H.M.; Betteridge, D.J.; Durrington, P.N.; Hitman, G.A.; Neil, H.A.; Livingstone, S.J.; Thomason, M.J.; Mackness, M.I.; Charlton-Menys, V.; Fuller, J.H. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the collaborative atorvastatin diabetes study (cards): Multicentre randomised placebo-controlled trial. Lancet 2004, 364, 685–696. [Google Scholar] [CrossRef]
- Downs, J.R.; Clearfield, M.; Weis, S.; Whitney, E.; Shapiro, D.R.; Beere, P.A.; Langendorfer, A.; Stein, E.A.; Kruyer, W.; Gotto, A.M., Jr. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: Results of afcaps/texcaps. Air force/texas coronary atherosclerosis prevention study. J. Am. Med. Assoc. 1998, 279, 1615–1622. [Google Scholar] [CrossRef]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto, A.M., Jr.; Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to prevent vascular events in men and women with elevated c-reactive protein. N. Engl. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef] [PubMed]
- Horl, W.H.; Vanrenterghem, Y.; Canaud, B.; Mann, J.; Teatini, U.; Wanner, C.; Wikstrom, B. Optimal treatment of renal anaemia (opta): Improving the efficacy and efficiency of renal anaemia therapy in haemodialysis patients receiving intravenous epoetin. Nephrol. Dial. Transplant. 2005, 20 (Suppl. 3), 25–32. [Google Scholar] [CrossRef] [PubMed]
- Fellstrom, B.C.; Jardine, A.G.; Schmieder, R.E.; Holdaas, H.; Bannister, K.; Beutler, J.; Chae, D.W.; Chevaile, A.; Cobbe, S.M.; Gronhagen-Riska, C.; et al. Rosuvastatin and cardiovascular events in patients undergoing hemodialysis. N. Engl. J. Med. 2009, 360, 1395–1407. [Google Scholar] [CrossRef] [PubMed]
- Baigent, C.; Landray, M.J.; Reith, C.; Emberson, J.; Wheeler, D.C.; Tomson, C.; Wanner, C.; Krane, V.; Cass, A.; Craig, J.; et al. The effects of lowering ldl cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (study of heart and renal protection): A randomised placebo-controlled trial. Lancet 2011, 377, 2181–2192. [Google Scholar] [CrossRef] [PubMed]
- Kendrick, J.; Chonchol, M.B. Nontraditional risk factors for cardiovascular disease in patients with chronic kidney disease. Nat. Clin. Pract. Nephrol. 2008, 4, 672–681. [Google Scholar] [CrossRef] [PubMed]
- Lekawanvijit, S.; Kompa, A.R.; Wang, B.H.; Kelly, D.J.; Krum, H. Cardiorenal syndrome: The emerging role of protein-bound uremic toxins. Circ. Res. 2012, 111, 1470–1483. [Google Scholar] [CrossRef] [PubMed]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Zuo, Y.; Ma, J.; Yancey, P.G.; Hunley, T.E.; Motojima, M.; Fogo, A.B.; Linton, M.F.; Fazio, S.; Ichikawa, I.; et al. Oral activated charcoal adsorbent (ast-120) ameliorates extent and instability of atherosclerosis accelerated by kidney disease in apolipoprotein e-deficient mice. Nephrol. Dial. Transplant. 2011, 26, 2491–2497. [Google Scholar] [CrossRef] [PubMed]
- Im, S.S.; Osborne, T.F. Liver receptors in atherosclerosis and inflammation. Circ. Res. 2011, 108, 996–1001. [Google Scholar] [CrossRef] [PubMed]
- Ramezani, A.; Raj, D.S. The gut microbiome, kidney disease, and targeted interventions. J. Am. Soc. Nephrol. 2014, 25, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.J.; Andersen, K.; Stecher, B. The intestinal microbiota, a leaky gut, and abnormal immunity in kidney disease. Kidney Int. 2013, 83, 1010–1016. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Bertrand, E.; Cerini, C.; Faure, V.; Sampol, J.; Vanholder, R.; Berland, Y.; Brunet, P. The uremic solutes p-cresol and indoxyl sulfate inhibit endothelial proliferation and wound repair. Kidney Int. 2004, 65, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Yancey, P.G.; Zuo, Y.; Ma, L.J.; Kaseda, R.; Fogo, A.B.; Ichikawa, I.; Linton, M.F.; Fazio, S.; Kon, V. Macrophage polarization by angiotensin ii-type 1 receptor aggravates renal injury-acceleration of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2856–2864. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Ezawa, A.; Kikuchi, K.; Tsuruta, Y.; Niwa, T. Protein-bound uremic toxins in hemodialysis patients measured by liquid chromatography/tandem mass spectrometry and their effects on endothelial ros production. Anal. Bioanal. Chem. 2012, 403, 1841–1850. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Schepers, E.; Pletinck, A.; Nagler, E.V.; Glorieux, G. The uremic toxicity of indoxyl sulfate and p-cresyl sulfate: A systematic review. J. Am. Soc. Nephrol. 2014, 25, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Schepers, E.; Meert, N.; Glorieux, G.; Goeman, J.; van der Eycken, J.; Vanholder, R. P-cresylsulphate, the main in vivo metabolite of p-cresol, activates leucocyte free radical production. Nephrol. Dial. Transplant. 2007, 22, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.Y.; Chang, S.C.; Wu, M.S. Uremic toxins induce kidney fibrosis by activating intrarenal renin-angiotensin-aldosterone system associated epithelial-to-mesenchymal transition. PLoS ONE 2012, 7, e34026. [Google Scholar] [CrossRef] [PubMed]
- Trojanowicz, B.; Ulrich, C.; Seibert, E.; Fiedler, R.; Girndt, M. Uremic conditions drive human monocytes to pro-atherogenic differentiation via an angiotensin-dependent mechanism. PLoS One 2014, 9, e102137. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Saito, S.; Higashiyama, Y.; Nishijima, F.; Niwa, T. Creb, nf-kappab, and nadph oxidase coordinately upregulate indoxyl sulfate-induced angiotensinogen expression in proximal tubular cells. Am. J. Physiol. Cell Physiol. 2013, 304, C685–C692. [Google Scholar] [CrossRef] [PubMed]
- Muteliefu, G.; Shimizu, H.; Enomoto, A.; Nishijima, F.; Takahashi, M.; Niwa, T. Indoxyl sulfate promotes vascular smooth muscle cell senescence with upregulation of p53, p21, and prelamin a through oxidative stress. Am. J. Physiol. Cell Physiol. 2012, 303, C126–C134. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.Y.; Yisireyili, M.; Saito, S.; Lee, C.T.; Adelibieke, Y.; Nishijima, F.; Niwa, T. Indoxyl sulfate downregulates expression of mas receptor via oat3/ahr/stat3 pathway in proximal tubular cells. PLoS ONE 2014, 9, e91517. [Google Scholar] [CrossRef] [PubMed]
- Adesso, S.; Popolo, A.; Bianco, G.; Sorrentino, R.; Pinto, A.; Autore, G.; Marzocco, S. The uremic toxin indoxyl sulphate enhances macrophage response to lps. PLoS ONE 2013, 8, e76778. [Google Scholar] [CrossRef] [PubMed]
- Levin, N.; Bischoff, E.D.; Daige, C.L.; Thomas, D.; Vu, C.T.; Heyman, R.A.; Tangirala, R.K.; Schulman, I.G. Macrophage liver x receptor is required for antiatherogenic activity of LXR agonists. Arterioscler. Thrombosis Vasc. Biol. 2005, 25, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Tangirala, R.K.; Bischoff, E.D.; Joseph, S.B.; Wagner, B.L.; Walczak, R.; Laffitte, B.A.; Daige, C.L.; Thomas, D.; Heyman, R.A.; Mangelsdorf, D.J.; et al. Identification of macrophage liver x receptors as inhibitors of atherosclerosis. Proc. Natl. Acad. Sci. USA 2002, 99, 11896–11901. [Google Scholar] [CrossRef] [PubMed]
- Marathe, C.; Bradley, M.N.; Hong, C.; Lopez, F.; Ruiz de Galarreta, C.M.; Tontonoz, P.; Castrillo, A. The arginase ii gene is an anti-inflammatory target of liver x receptor in macrophages. J. Biol. Chem. 2006, 281, 32197–32206. [Google Scholar] [CrossRef] [PubMed]
- Holzer, M.; Birner-Gruenberger, R.; Stojakovic, T.; El-Gamal, D.; Binder, V.; Wadsack, C.; Heinemann, A.; Marsche, G. Uremia alters hdl composition and function. J. Am. Soc. Nephrol. 2011, 22, 1631–1641. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Yancey, P.G.; Ikizler, T.A.; Jerome, W.G.; Kaseda, R.; Cox, B.; Bian, A.; Shintani, A.; Fogo, A.B.; Linton, M.F.; et al. Dysfunctional high-density lipoprotein in patients on chronic hemodialysis. J. Am. Coll. Cardiol. 2012, 60, 2372–2379. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Yancey, P.; Castro, I.; Khan, W.N.; Motojima, M.; Ichikawa, I.; Fogo, A.B.; Linton, M.F.; Fazio, S.; Kon, V. Renal dysfunction potentiates foam cell formation by repressing abca1. Arterioscler. Thrombosis Vasc. Biol. 2009, 29, 1277–1282. [Google Scholar] [CrossRef] [PubMed]
- Ogura, M.; Ayaori, M.; Terao, Y.; Hisada, T.; Iizuka, M.; Takiguchi, S.; Uto-Kondo, H.; Yakushiji, E.; Nakaya, K.; Sasaki, M.; et al. Proteasomal inhibition promotes atp-binding cassette transporter a1 (abca1) and abcg1 expression and cholesterol efflux from macrophages in vitro and in vivo. Arterioscler. Thrombosis Vasc.Biol. 2011, 31, 1980–1987. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Wu, C.; Sun, L.; Zheng, J.; Guo, P. Sesamin enhances cholesterol efflux in raw 264.7 macrophages. Molecules 2014, 19, 7516–7527. [Google Scholar] [CrossRef] [PubMed]
- Hozoji, M.; Munehira, Y.; Ikeda, Y.; Makishima, M.; Matsuo, M.; Kioka, N.; Ueda, K. Direct interaction of nuclear liver x receptor-beta with abca1 modulates cholesterol efflux. J. Biol. Chem. 2008, 283, 30057–30063. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, R.L.; Medh, J.D. Down-regulation of lipoprotein lipase increases abca1-mediated cholesterol efflux in thp-1 macrophages. Biochem. Biophys. Res. Commun. 2014, 450, 1416–1421. [Google Scholar] [CrossRef] [PubMed]
- Kazawa, T.; Kawasaki, T.; Sakamoto, A.; Imamura, M.; Ohashi, R.; Jiang, S.; Tanaka, T.; Iwanari, H.; Hamakubo, T.; Sakai, J.; et al. Expression of liver x receptor alpha and lipid metabolism in granulocyte-macrophage colony-stimulating factor-induced human monocyte-derived macrophage. Pathol. Int. 2009, 59, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, T.; Inagi, R.; Wada, T.; Tanaka, T.; Fujita, T.; Nangaku, M. Indoxyl sulfate inhibits proliferation of human proximal tubular cells via endoplasmic reticulum stress. Am. J. Physiol. Ren. Physiol. 2010, 299, F568–F576. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsuo, K.; Yamamoto, S.; Wakamatsu, T.; Takahashi, Y.; Kawamura, K.; Kaneko, Y.; Goto, S.; Kazama, J.J.; Narita, I. Increased Proinflammatory Cytokine Production and Decreased Cholesterol Efflux Due to Downregulation of ABCG1 in Macrophages Exposed to Indoxyl Sulfate. Toxins 2015, 7, 3155-3166. https://doi.org/10.3390/toxins7083155
Matsuo K, Yamamoto S, Wakamatsu T, Takahashi Y, Kawamura K, Kaneko Y, Goto S, Kazama JJ, Narita I. Increased Proinflammatory Cytokine Production and Decreased Cholesterol Efflux Due to Downregulation of ABCG1 in Macrophages Exposed to Indoxyl Sulfate. Toxins. 2015; 7(8):3155-3166. https://doi.org/10.3390/toxins7083155
Chicago/Turabian StyleMatsuo, Koji, Suguru Yamamoto, Takuya Wakamatsu, Yoshimitsu Takahashi, Kazuko Kawamura, Yoshikatsu Kaneko, Shin Goto, Junichiro J. Kazama, and Ichiei Narita. 2015. "Increased Proinflammatory Cytokine Production and Decreased Cholesterol Efflux Due to Downregulation of ABCG1 in Macrophages Exposed to Indoxyl Sulfate" Toxins 7, no. 8: 3155-3166. https://doi.org/10.3390/toxins7083155
APA StyleMatsuo, K., Yamamoto, S., Wakamatsu, T., Takahashi, Y., Kawamura, K., Kaneko, Y., Goto, S., Kazama, J. J., & Narita, I. (2015). Increased Proinflammatory Cytokine Production and Decreased Cholesterol Efflux Due to Downregulation of ABCG1 in Macrophages Exposed to Indoxyl Sulfate. Toxins, 7(8), 3155-3166. https://doi.org/10.3390/toxins7083155