
Diverse Profiles of Ricin-Cell Interactions in the Lung Following Intranasal Exposure to Ricin

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
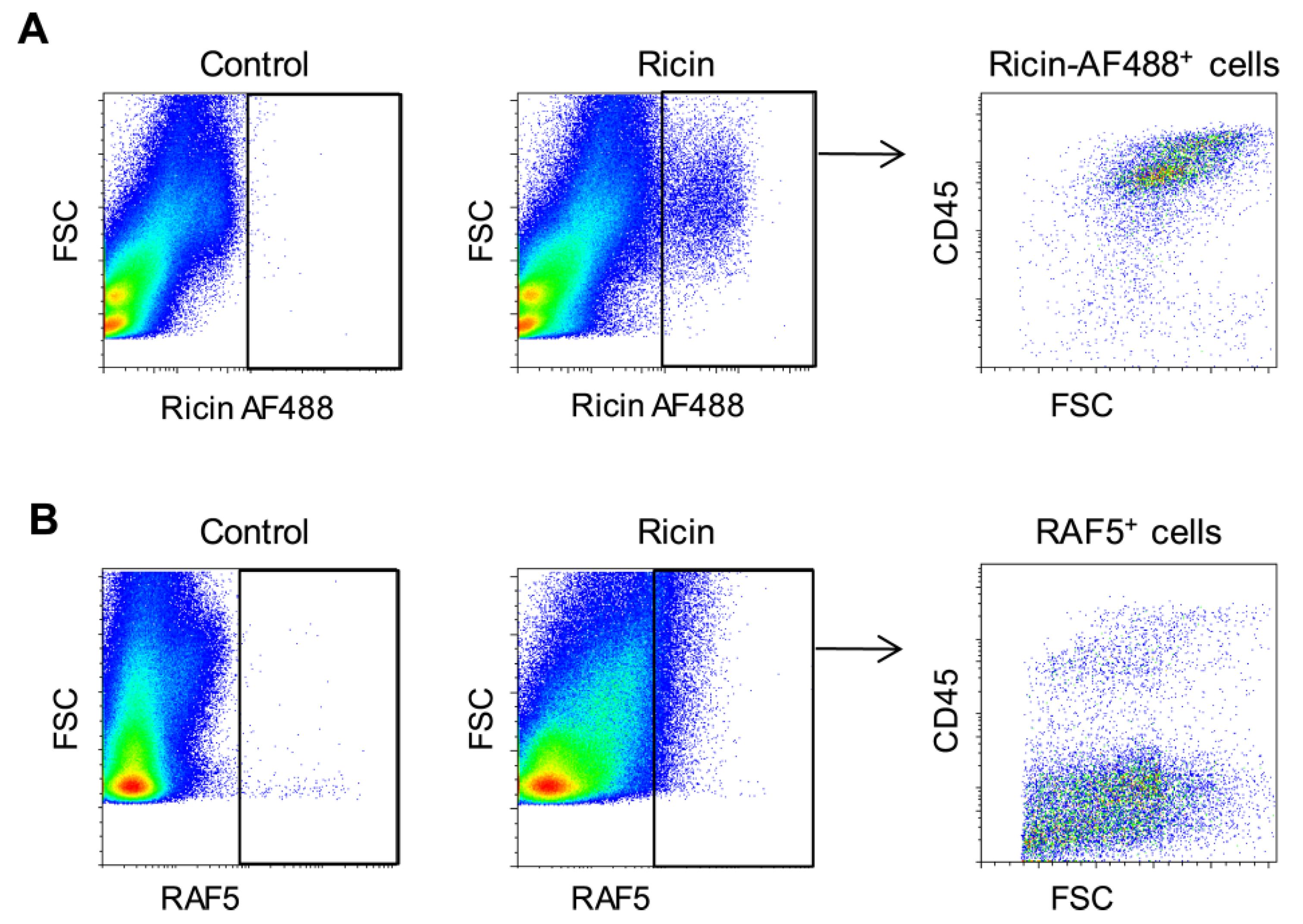
2.1. Differential Binding of Ricin to the Cells of the Mouse Lung
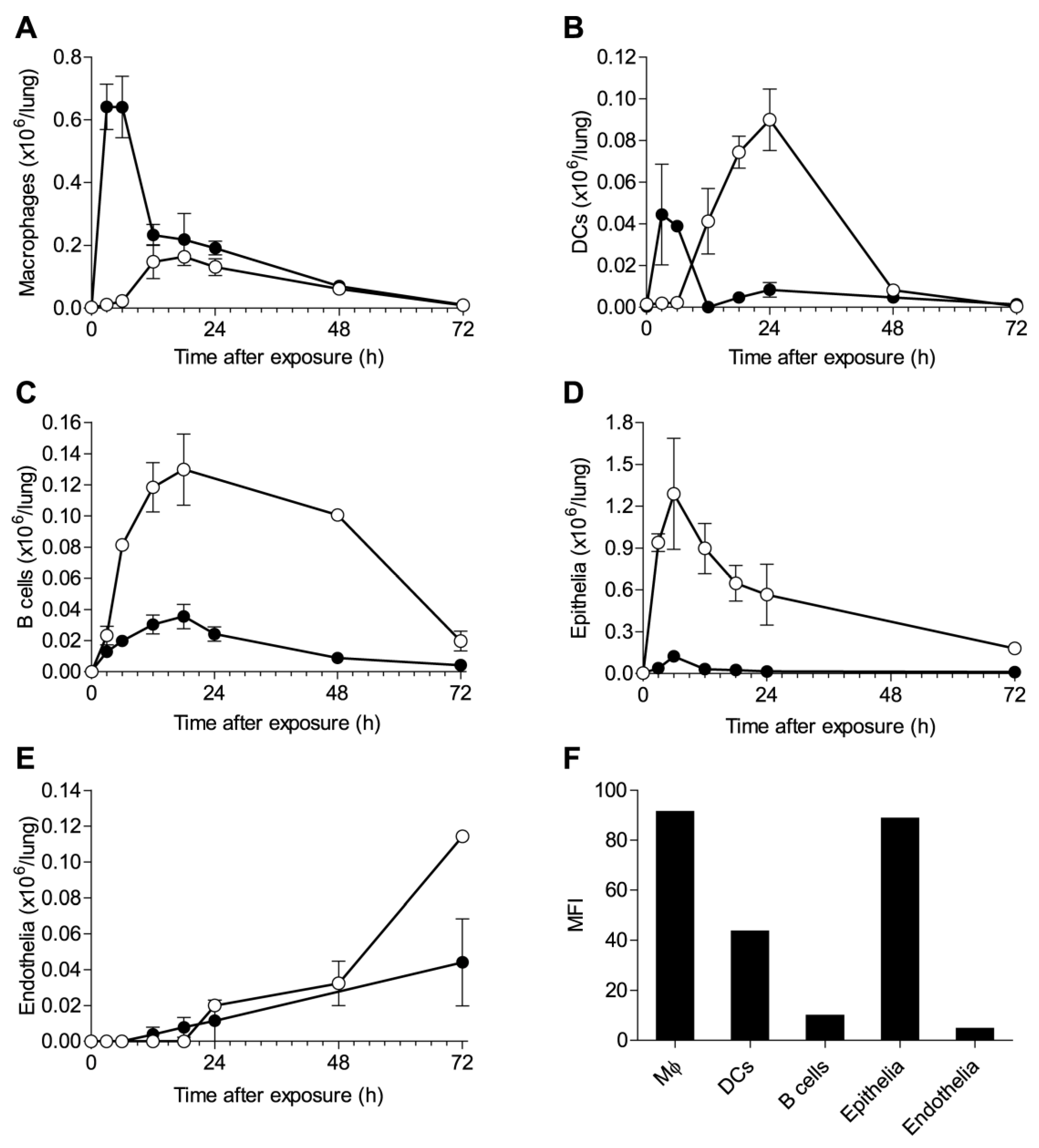
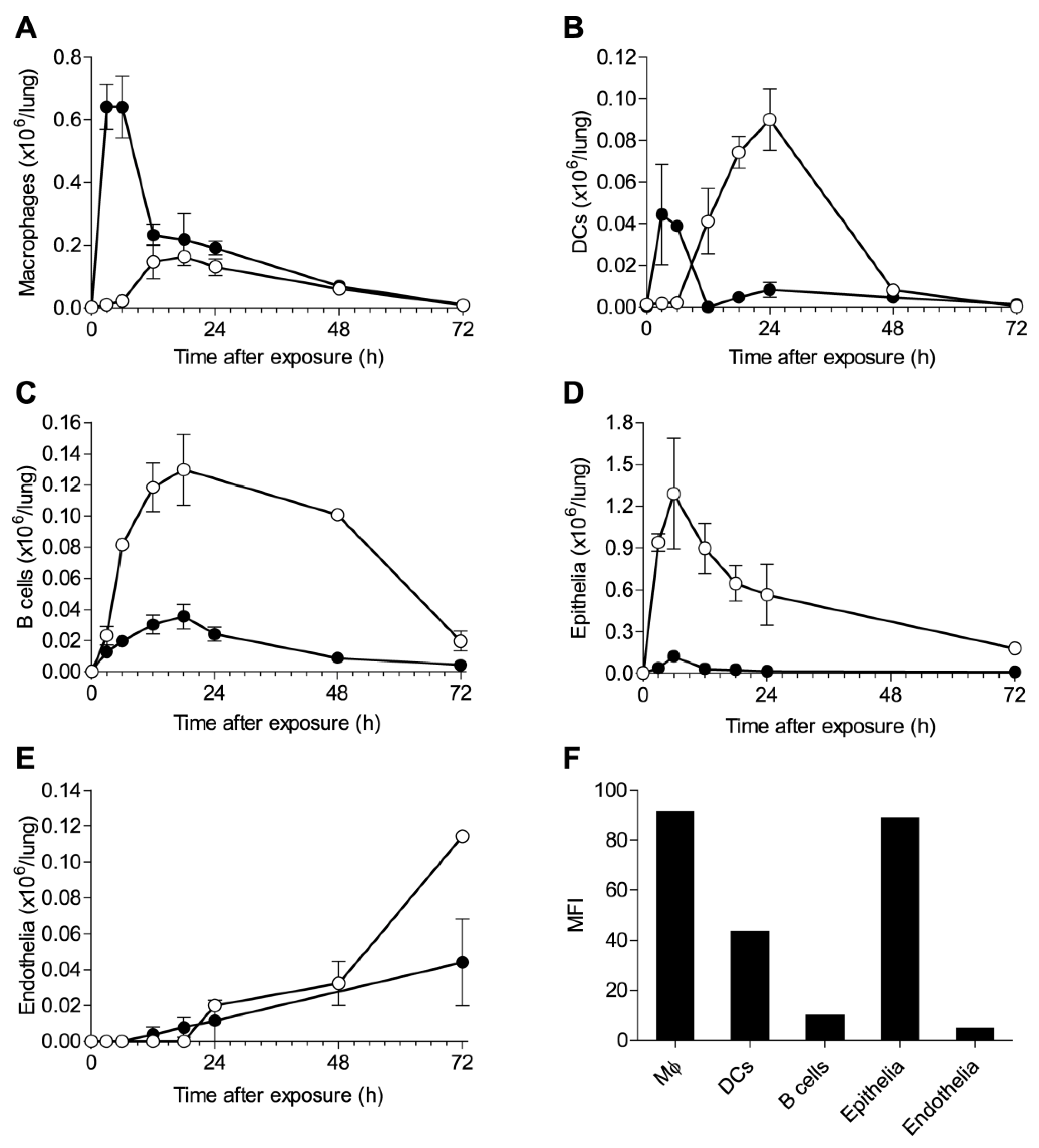
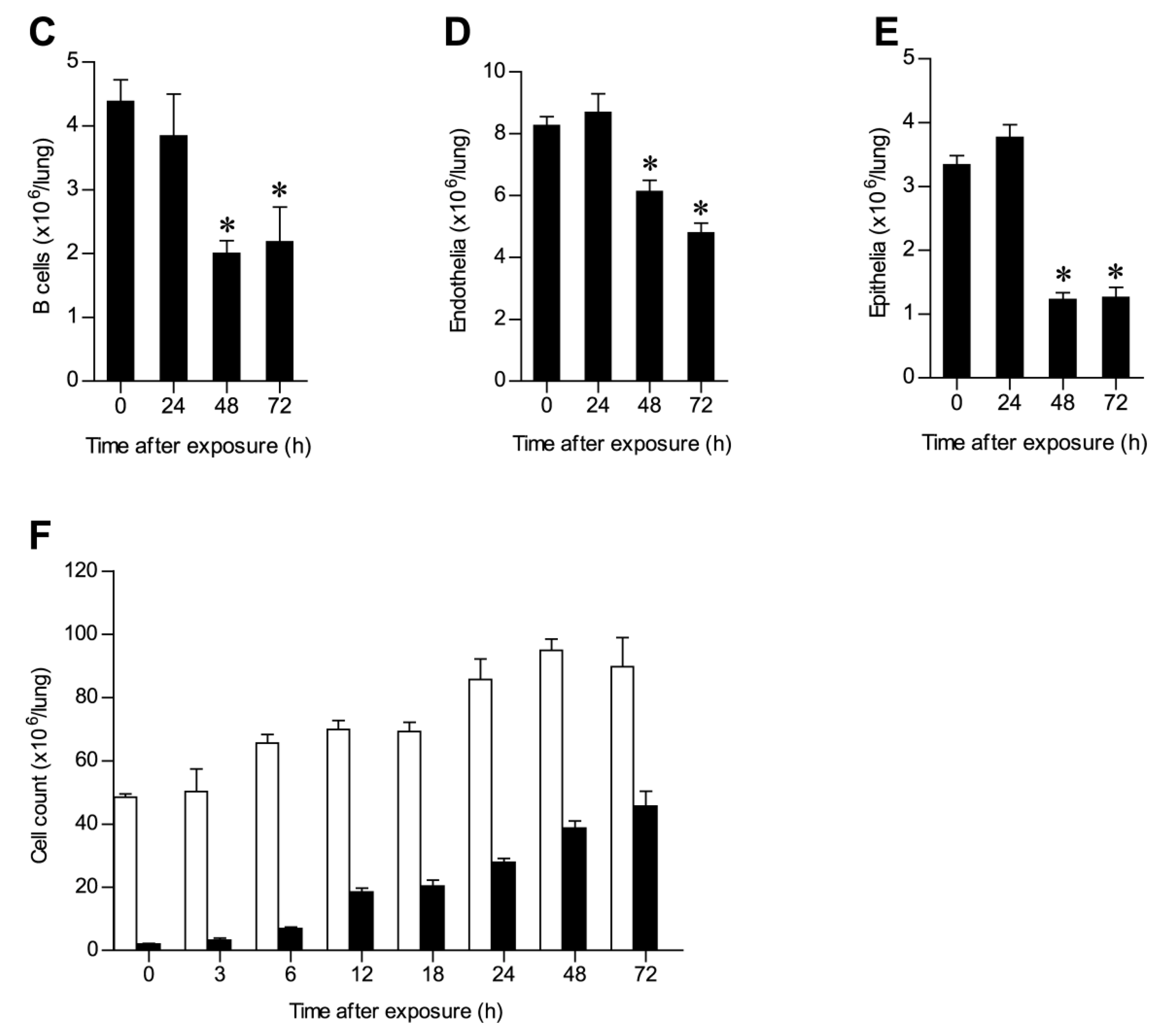
2.2. Alterations in Lung Cell Populations Following Ricin Intoxication
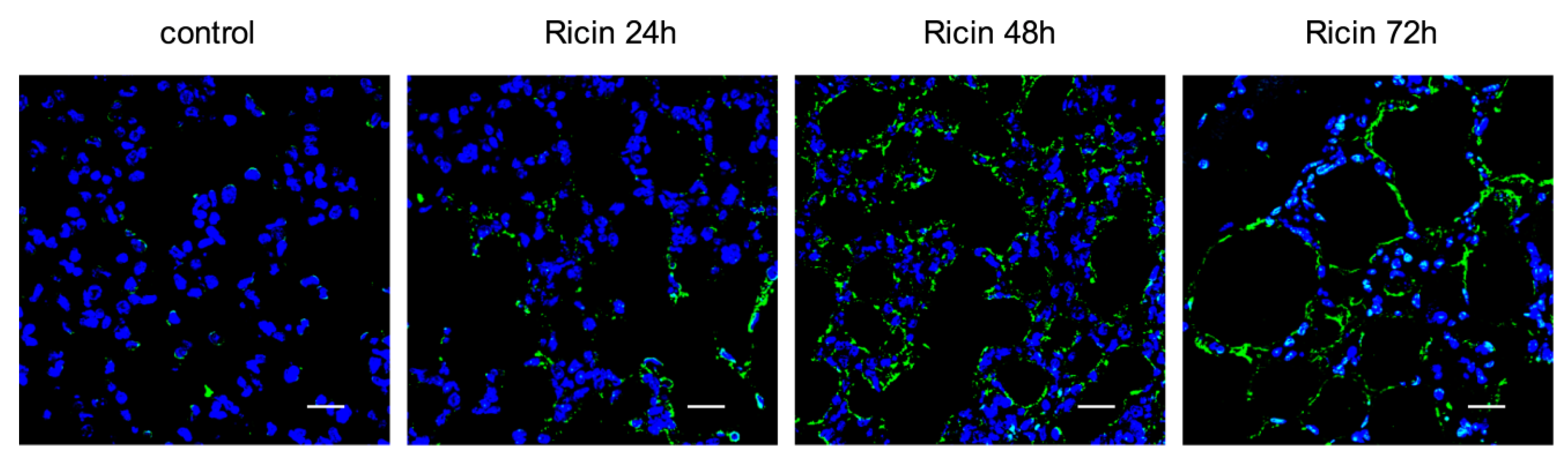
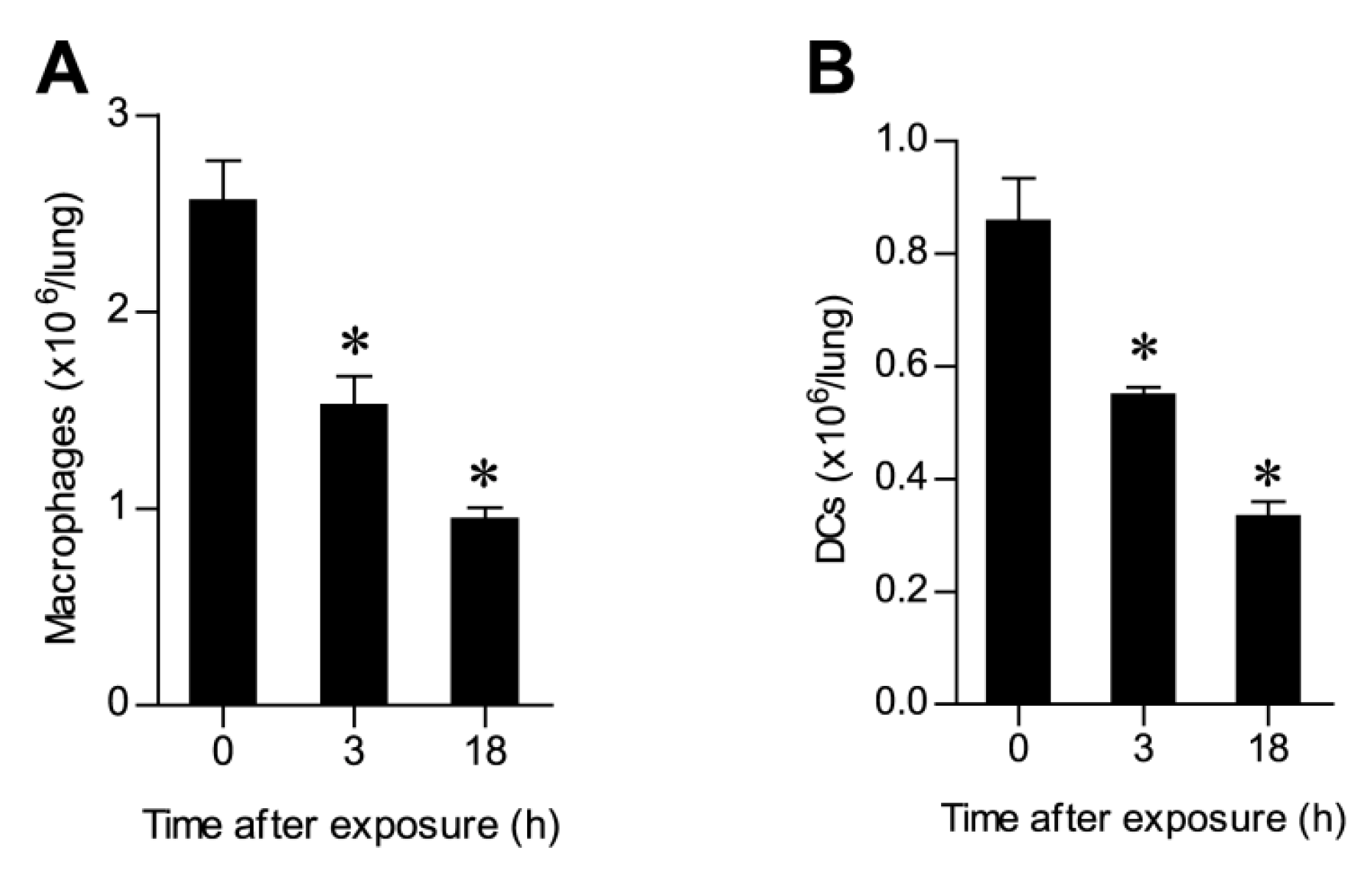
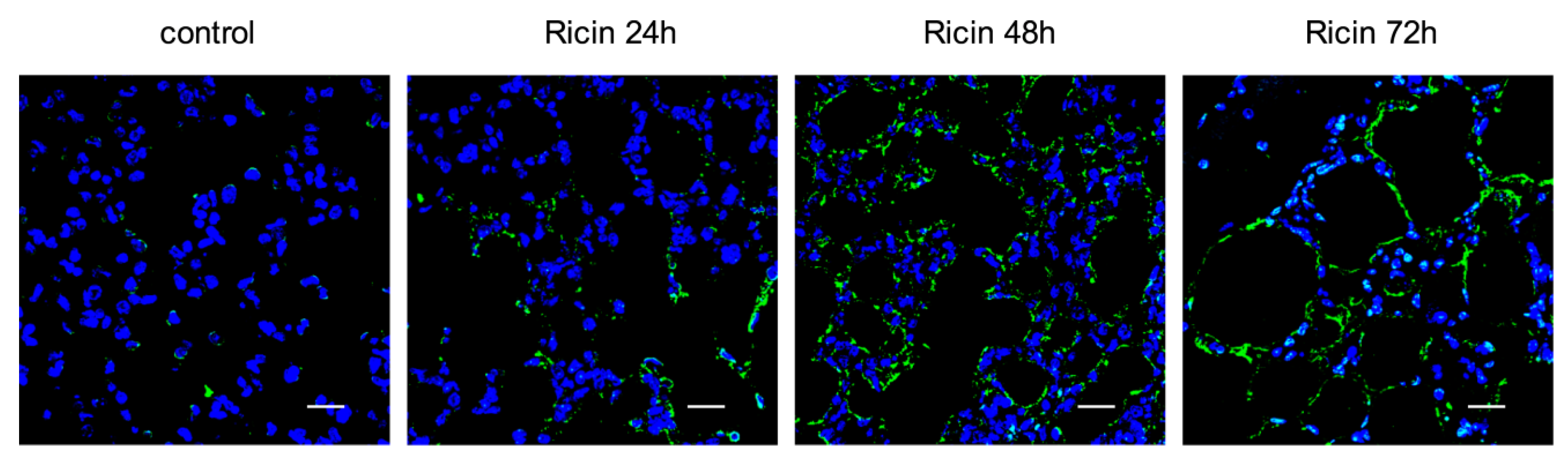
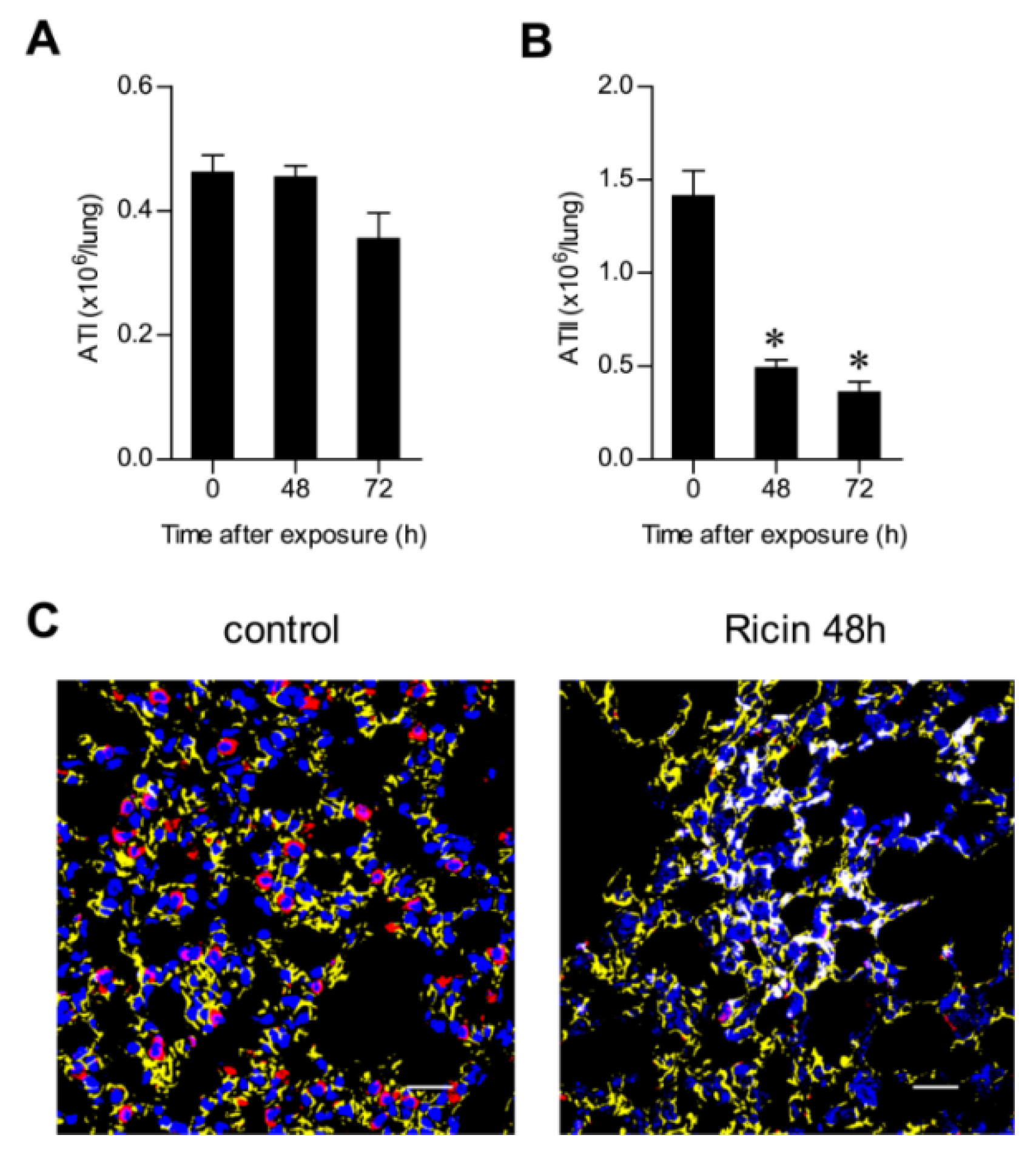
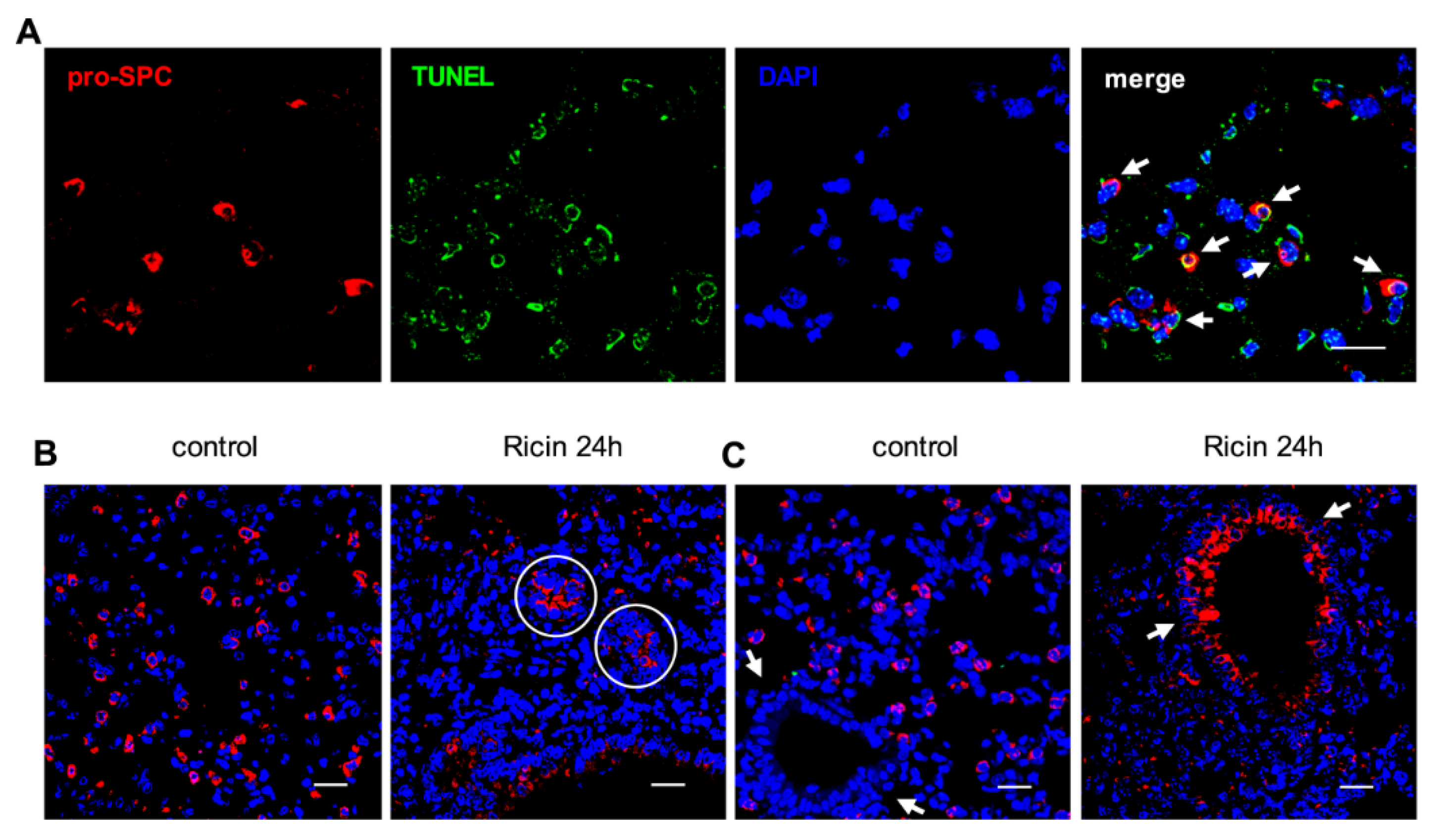
2.3. Ricin Intoxication Results in Specific Loss of Alveolar Type II Epithelia
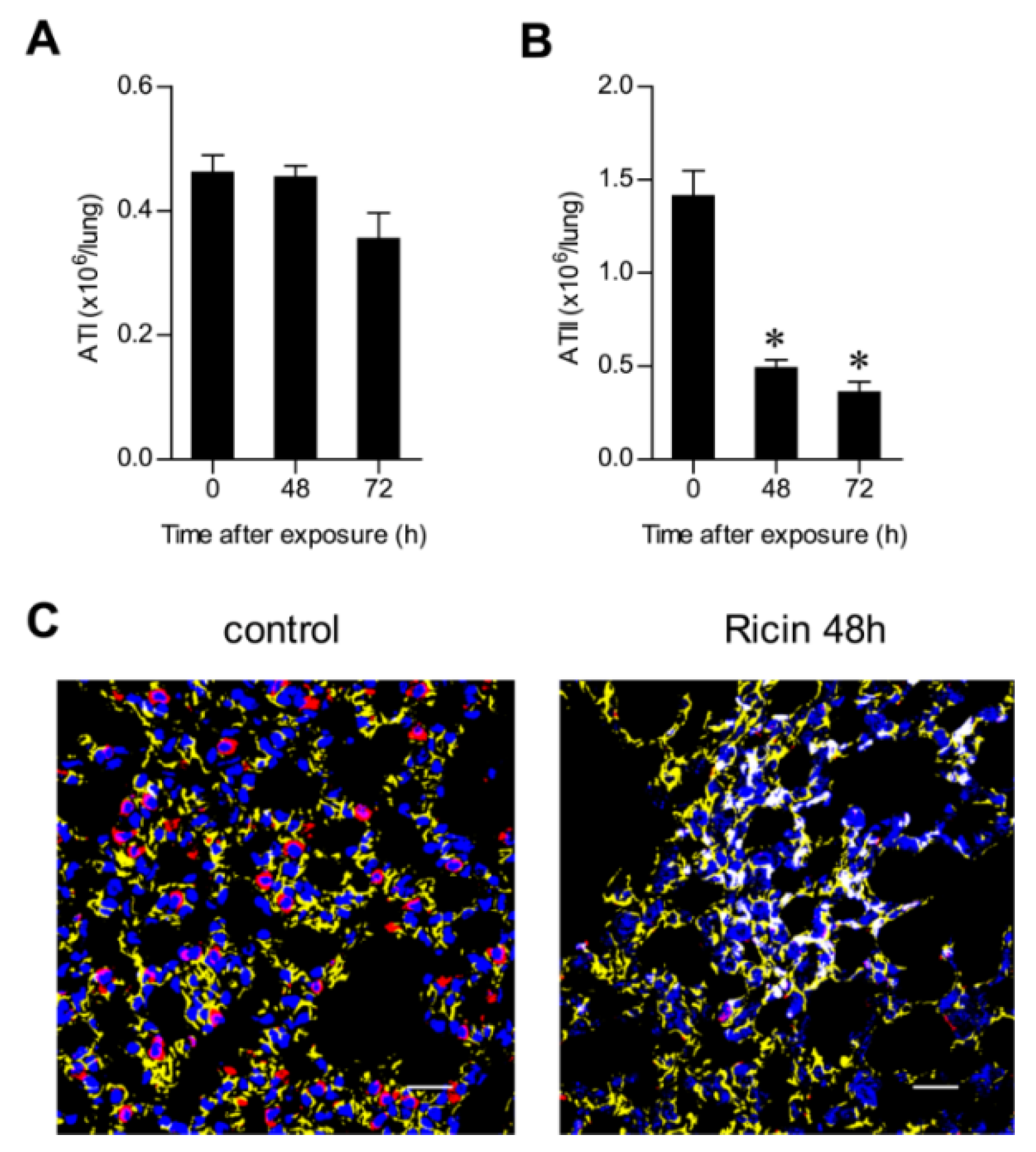
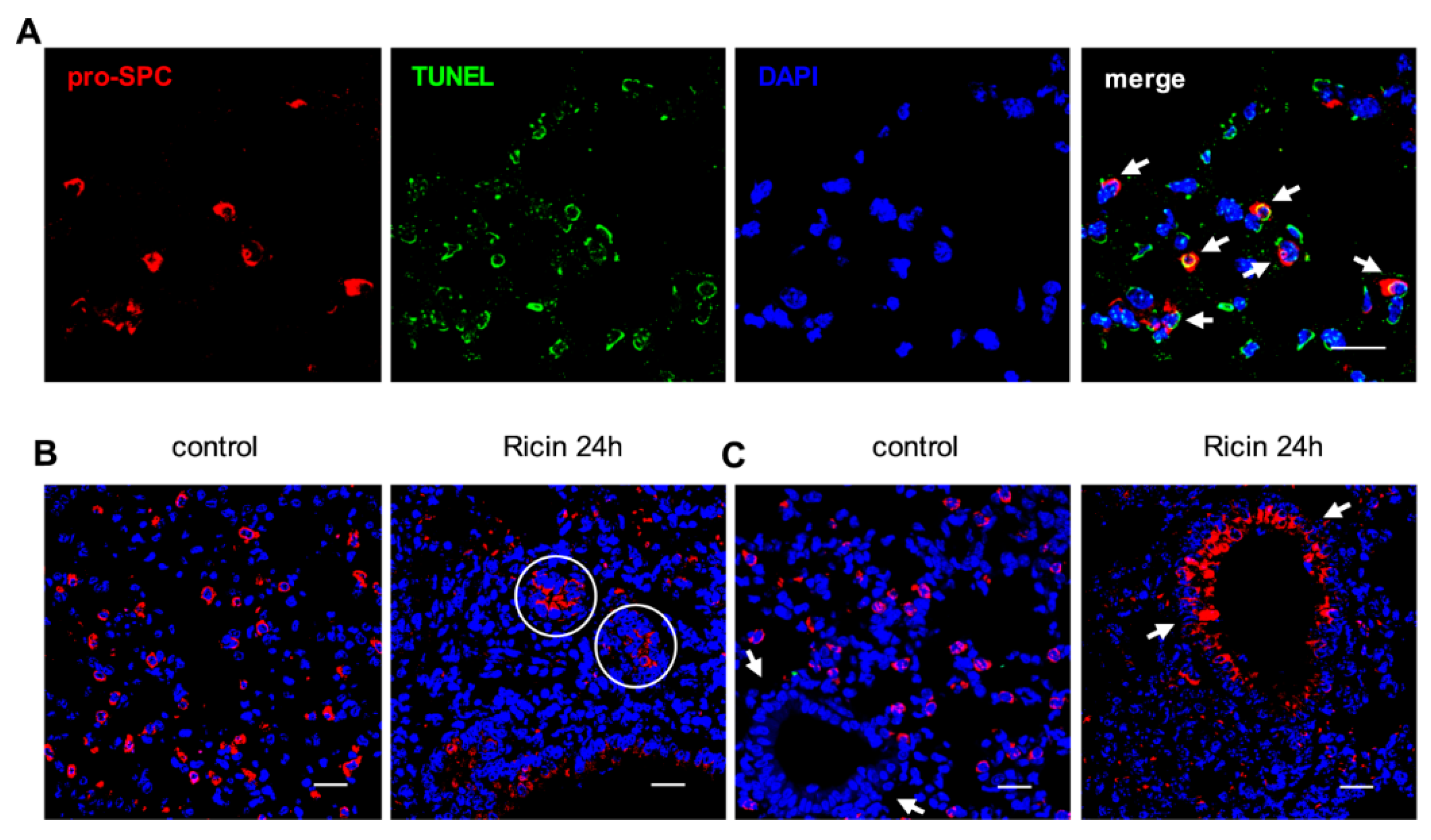
3. Discussion
4. Experimental Section
4.1. Animal Studies
4.2. Fluorescent Ricin Labeling and Intoxication
4.3. Purification of Anti-Ricin Specific Antibody RAF5
4.4. FACS Analysis
4.5. Immunohistochemistry
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Greenfield, R.A.; Brown, B.R.; Hutchins, J.B.; Iandolo, J.J.; Jackson, R.; Slater, L.N.; Bronze, M.S. Microbiological, biological and chemical weapons of warfare and terrorism. Am. J. Med. Sci. 2002, 323, 326–340. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Mitsui, K.; Motizuki, M.; Tsurugi, K. The mechanism of action of ricin and related toxic lectins on eukaryotic ribosomes. The site and the characteristics of the modification in 28S ribosomal RNA caused by the toxins. J. Biol. Chem. 1987, 262, 5908–5912. [Google Scholar] [PubMed]
- Endo, Y.; Tsurugi, K. RNA N-glycosidase activity of ricin A-chain. Mechanism of action of the toxic lectin ricin on eukaryotic ribosomes. J. Biol. Chem. 1987, 262, 8128–8130. [Google Scholar] [PubMed]
- Spooner, R.A.; Smith, D.C.; Easton, A.J.; Roberts, L.M.; Lord, J.M. Retrograde transport pathways utilized by viruses and protein toxins. Virol. J. 2006, 3, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; van Deurs, B. Endocytosis, intracellular transport, and cytotoxic action of Shiga toxin and ricin. Physiol. Rev. 1996, 76, 949–966. [Google Scholar] [PubMed]
- Sandvig, K.; van Deurs, B. Transport of protein toxins into cells: pathways used by ricin, cholera toxin and ricin. FEBS Lett. 2002, 529, 49–53. [Google Scholar] [CrossRef]
- Korcheva, V.; Wong, J.; Corless, C.; Iordanov, M.S.; Magun, B. Administration of ricin induces a severe inflammatory response via nonredundant stimulation of ERK, JNK and p38 MAPK and provides a mouse model of hemolytic uremic syndrome. Am. J. Pathol. 2005, 166, 323–339. [Google Scholar] [CrossRef]
- Lindauer, M.L.; Wong, J.; Magun, B. Ricin toxin activates the NALP3 Inflammasome. Toxins 2010, 2, 1500–1514. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, G.D. Understanding ricin from a defensive viewpoint. Toxins 2011, 3, 1373–1392. [Google Scholar] [CrossRef] [PubMed]
- Matute-Bello, G.; Downey, G.; Moore, B.B.; Groshong, S.D.; Matthay, M.A.; Slutsky, A.S.; Kuebler, W.M. An official American thoracic society workshop report: features and measurements of experimental acute lung injury in animals. Am. J. Respir. Cell Mol. Biol. 2011, 44, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Manicone, A.M. Role of the pulmonary epithelium and inflammatory signals in acute lung injury. Expert Rev. Clin. Immunol. 2009, 5, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Simmons, B.M.; Stahl, P.D.; Russel, J.H. Mannose receptor-mediated uptake of ricin toxin and ricin A chain by macrophages. Multiple intracellular pathways for A chain translocation. J. Biol. Chem. 1986, 261, 7912–7920. [Google Scholar] [PubMed]
- Barbieri, L.; Battelli, M.G.; Stirpe, F. Ribosome-inactivating proteins from plants. Biochim. Biophys. Acta 1993, 1154, 237–282. [Google Scholar] [CrossRef]
- Ware, L.B. Pathophysiology of acute lung injury and the acute respiratory distress syndrome. Semin. Respir. Crit. Care Med. 2006, 27, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Dobbs, L.G.; Williams, M.C.; Gonzalez, R. Monoclonal antibodies specific to apical surfaces of rat alveolar type I cells bind to surfaces of cultured, but not freshly isolated, type II cells. Biochim. Biophys. Acta 1988, 970, 146–156. [Google Scholar] [CrossRef]
- Glasser, S.W.; Korfhagen, T.R.; Bruno, M.D.; Dey, C.; Whitsett, J.A. Structure and expression of the pulmonary surfactant protein SP-C gene in the mouse. J. Biol. Chem. 1990, 265, 21986–21991. [Google Scholar] [PubMed]
- Arfilli, V.; Carnicelli, D.; Rocchi, L.; Ricci, F.; Pagliaro, P.; Tazzari, P.L.; Brigotti, M. Shiga toxin 1 and ricin A bind human polymorhonuclear leucocytes through a common receptor. Biochem. J. 2010, 432, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Mize, R.R.; Marrero, L.; Corti, M.; Kirk, J.M.; Pincus, S.H. Antibody to ricin A chain hinders intracellular routing of toxin and protects cells even after toxin has been internalized. PLoS ONE 2013, 8, e62417. [Google Scholar] [CrossRef] [PubMed]
- Lindauer, M.L.; Wong, J.; Iwakura, Y.; Magun, B. Pulmonary inflammation triggered by ricin toxin requires macrophages and IL-1 signalling. J. Immunol. 2009, 183, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Westphalen, K.; Gusarova, G.A.; Islam, M.N.; Subramanian, M.; Cohen, T.S.; Prince, A.S.; Bhattacharya, J. Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature 2014, 506, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Gage, E.; Hernandezm, M.O.; O’Hara, J.M.; McCarthy, E.A.; Mantis, N.J. Role of mannose receptor (CD206) in innate immunity to ricin toxin. Toxins 2011, 3, 1131–1145. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, S.; Faerevik, I.; Berg, T. Characterization of retroendocytosis in rat liver parenchymal cells and sinusoidal endothelial cells. Biochem. J. 1992, 287, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Battelli, M.G.; Musiani, S.; Buonamici, L.; Santi, S.; Riccio, M.; Maraldi, N.M.; Girbes, T.; Stirpe, F. Interactions of volkensin with HeLa cells: Binding, uptake, intracellular localization, degradation and exocytosis. Cell Mol. Life Sci. 2004, 61, 1975–1984. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Limmon, G.V.; Yin, L.; Leung, N.H.N.; Yu, H.; Chow, V.T.K.; Cheb, J. A cellular pathway involved in Clara cell to alveolar type II cell differentiation after severe lung injury. PLoS ONE 2013, 8, e71028. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Ware, L.B.; Zimmerman, G.A. The acute respiratory distress syndrome. J. Clin. Invest. 2012, 122, 2731–2740. [Google Scholar] [CrossRef] [PubMed]
- Groomes, J.; Soehnlein, O. Contribution of neutrophils to acute lung injury. Mol. Med. 2011, 17, 293–307. [Google Scholar] [PubMed]
- Abraham, E. Neutrophils and acute lung injury. Crit. Care Med. 2003, 31, S195–S199. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.; Carmody, A.; Shenkar, R.; Arcaroli, J. Neatrophils as early immunologic effectors in hemorrhage or endotoxemia-induced acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L1137–L1145. [Google Scholar] [PubMed]
- Gal, Y.; Mazor, O.; Alcalay, R.; Seliger, N.; Aftalion, M.; Sapoznikov, A.; Falach, R.; Kronman, C.; Sabo, T. Antibody/doxycycline combined therapy for pulmonary ricinosis: Attenuation of inflammation improves survival of ricin-intoxicated mice. Toxicol. Rep. 2014, 1, 496–504. [Google Scholar] [CrossRef]
- Sabo, T.; Gal, Y.; Elhanany, E.; Sapoznikov, A.; Falach, R.; Mazor, O.; Kronman, C. Antibody treatment against pulmonary exposure to abrin confers significantly higher levels of protection than treatment against ricin intoxication. Toxicol. Lett. 2015, 237, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Cohen, O.; Mechaly, A.; Sabo, T.; Alcalay, R.; Aloni-Grinstein, R.; Seliger, N.; Kronman, C.; Mazor, O. Characterization and epitope mapping of the polyclonal antibody repertoire elicited by ricin holotoxin-based vaccination. Clin. Vaccine Immunol. 2014, 21, 1534–1540. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Darzynkiewicz, Z. Detection of apoptosis and cell proliferation. Cell Prolif. 1995, 28, 572–579. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sapoznikov, A.; Falach, R.; Mazor, O.; Alcalay, R.; Gal, Y.; Seliger, N.; Sabo, T.; Kronman, C. Diverse Profiles of Ricin-Cell Interactions in the Lung Following Intranasal Exposure to Ricin. Toxins 2015, 7, 4817-4831. https://doi.org/10.3390/toxins7114817
Sapoznikov A, Falach R, Mazor O, Alcalay R, Gal Y, Seliger N, Sabo T, Kronman C. Diverse Profiles of Ricin-Cell Interactions in the Lung Following Intranasal Exposure to Ricin. Toxins. 2015; 7(11):4817-4831. https://doi.org/10.3390/toxins7114817
Chicago/Turabian StyleSapoznikov, Anita, Reut Falach, Ohad Mazor, Ron Alcalay, Yoav Gal, Nehama Seliger, Tamar Sabo, and Chanoch Kronman. 2015. "Diverse Profiles of Ricin-Cell Interactions in the Lung Following Intranasal Exposure to Ricin" Toxins 7, no. 11: 4817-4831. https://doi.org/10.3390/toxins7114817
APA StyleSapoznikov, A., Falach, R., Mazor, O., Alcalay, R., Gal, Y., Seliger, N., Sabo, T., & Kronman, C. (2015). Diverse Profiles of Ricin-Cell Interactions in the Lung Following Intranasal Exposure to Ricin. Toxins, 7(11), 4817-4831. https://doi.org/10.3390/toxins7114817