
Suppression of Aflatoxin Biosynthesis in Aspergillus flavus by 2-Phenylethanol Is Associated with Stimulated Growth and Decreased Degradation of Branched-Chain Amino Acids


Abstract
:1. Introduction
2. Results
2.1. Summary of RNA-Seq Datasets and Statistic Analysis
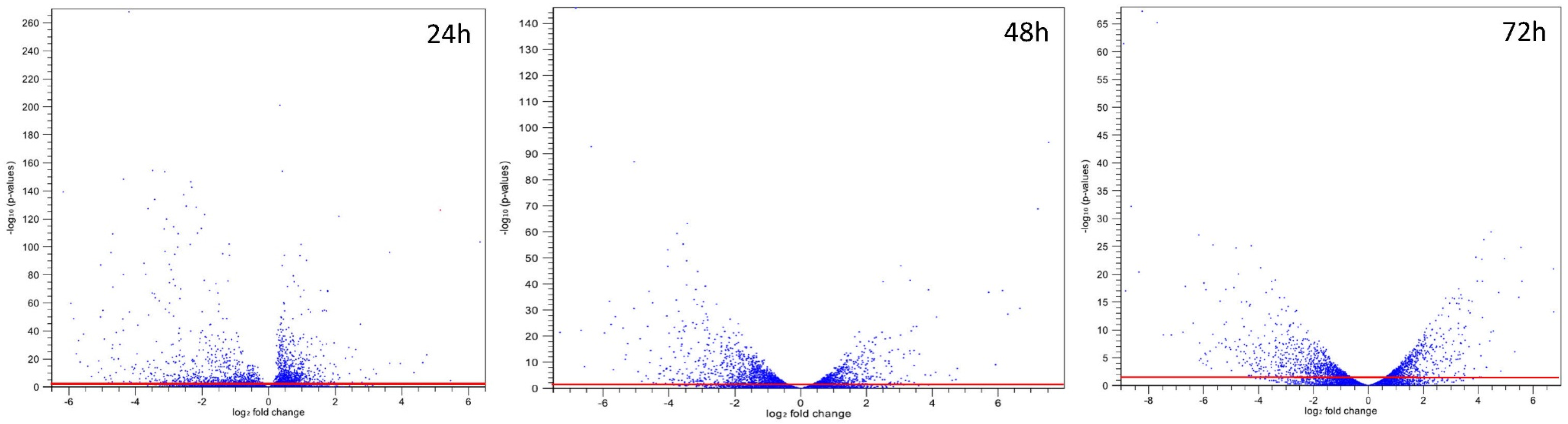

{kind=link}
{kind=link}
{kind=link}
| Expression a | Time | |||||
|---|---|---|---|---|---|---|
| 24 h | 48 h | 72 h | 24 & 48 h | 48 & 72 h | 24 & 48 & 72 h | |
| Decreased | 967 | 959 | 671 | 255 | 291 | 82 |
| Increased | 1200 | 593 | 527 | 163 | 197 | 49 |
2.2. Treatment by 2-PE Decreased Expression of All Genes in the Aflatoxin Gene Cluster of A. flavus
| Gene ID | Gene Name and Product | 24 h | FDR | 48 h | FDR | 72 h | FDR |
|---|---|---|---|---|---|---|---|
| AFLA_139140 | aflYa/nadA/NADH oxidase | −5.91 a | 1.00 | −1.89 | 0.00 | −2.67 a | 0.07 |
| AFLA_139150 | aflY/hypA/hypP/hypothetical protein | −10.32 | 0.00 | −2.39 | 0.00 | −2.17 a | 0.30 |
| AFLA_139160 | aflX/ordB/monooxygenase/oxidase | −7.33 | 0.00 | −1.56 | 0.00 | −2.07 a | 0.31 |
| AFLA_139170 | aflW/moxY/monooxygenase | −8.36 | 0.00 | −2.05 | 0.00 | −2.09 a | 0.35 |
| AFLA_139180 | aflV/cypX/P450 monooxygenase | −11.59 | 0.00 | −2.12 | 0.00 | −2.03 a | 0.40 |
| AFLA_139190 | aflK/vbs/VERB synthase | −5.67 | 0.00 | −2.98 | 0.00 | −2.50 a | 0.14 |
| AFLA_139200 | aflQ/ordA/P450 monooxygenase | −10.57 a | 0.07 | −1.87 | 0.00 | −2.11 a | 0.29 |
| AFLA_139210 | aflP/omtA/omt1/O-methyltransferase A | −14.69 | 0.00 | −2.27 | 0.00 | −2.03 a | 0.41 |
| AFLA_139220 | aflO/omtB/dmtA/O-methyltransferase B | −5.83 | 0.00 | −1.45 | 0.03 | −1.62 a | 0.74 |
| AFLA_139230 | aflI/avfA/P450 monooxygenase | −21.13 | 0.02 | −2.28 | 0.00 | −2.11 a | 0.21 |
| AFLA_139240 | aflLa/hypB/hypothetical protein | −13.51 | 0.00 | −2.46 | 0.00 | −2.37 a | 0.16 |
| AFLA_139250 | aflL/verB/desaturase/P450 monooxygenase | −8.64 | 0.00 | −2.44 | 0.00 | −2.36 a | 0.15 |
| AFLA_139260 | aflG/avnA/ord1/P450 monooxygenase | −5.09 a | 0.19 | −1.74 | 0.00 | −2.17 a | 0.20 |
| AFLA_139270 | aflNa/hypD/hypothetical protein | −3.32 | 0.00 | −1.09 a | 0.34 | −1.76 a | 0.46 |
| AFLA_139280 | aflN/verA/monooxygenase | −4.29 a | 0.11 | −1.58 | 0.00 | −2.27 a | 0.18 |
| AFLA_139290 | aflMa/hypE/hypothetical protein | −6.94 | 0.00 | −1.41 | 0.01 | −1.74 a | 0.56 |
| AFLA_139300 | aflM/ver1/dehydrogenase/ketoreductase | −18.07 | 0.00 | −1.79 | 0.00 | −1.83 a | 0.55 |
| AFLA_139310 | aflE/norA/aad/adh2/NOR reductase | −10.11 | 0.00 | −1.86 | 0.00 | −2.32 a | 0.19 |
| AFLA_139320 | aflJ/estA/esterase | −4.95 | 0.00 | −1.42 | 0.01 | −2.15 a | 0.29 |
| AFLA_139330 | aflH/adhA/short chain alcohol dehydrogenase | −4.75 | 0.00 | −1.78 | 0.00 | −2.22 a | 0.22 |
| AFLA_139340 | aflS/aflJ/pathway regulator | −2.67 | 0.00 | −1.12 a | 0.25 | −1.10 a | 1.00 |
| AFLA_139360 | aflR/apa2/afl2/C6 transcription factor | −2.00 | 0.02 | −1.10 a | 0.43 | −1.40 a | 0.72 |
| AFLA_139370 | aflB/fas1/fatty acid synthase beta subunit | −2.54 a | 0.24 | −1.79 | 0.00 | −1.58 a | 0.52 |
| AFLA_139380 | aflA/fas2/hexA/fatty acid synthase alpha subunit | −1.27 a | 1.00 | −2.13 | 0.00 | −2.29 a | 0.09 |
| AFLA_139390 | aflD/nor1/reductase | −3.46 | 0.00 | −2.33 | 0.00 | −2.29 a | 0.21 |
| AFLA_139400 | aflCa/hypC/hypothetical protein | −4.53 | 0.00 | −2.60 | 0.00 | −2.66 a | 0.08 |
| AFLA_139410 | aflC/pksA/pksL1/polyketide synthase | −2.01 a | 0.59 | −2.32 | 0.00 | −2.57 a | 0.12 |
| AFLA_139420 | aflT/transmembrane protein | −5.12 a | 0.62 | −1.53 | 0.00 | −1.79 a | 0.15 |
| AFLA_139430 | aflU/cypA/P450 monooxygenase | −4.92 a | 1.00 | −1.27 a | 0.59 | −1.29 a | 1.00 |
| AFLA_139440 | aflF/norB/dehydrogenase | −7.48 a | 1.00 | −1.18 a | 0.71 | −1.26 a | 1.00 |
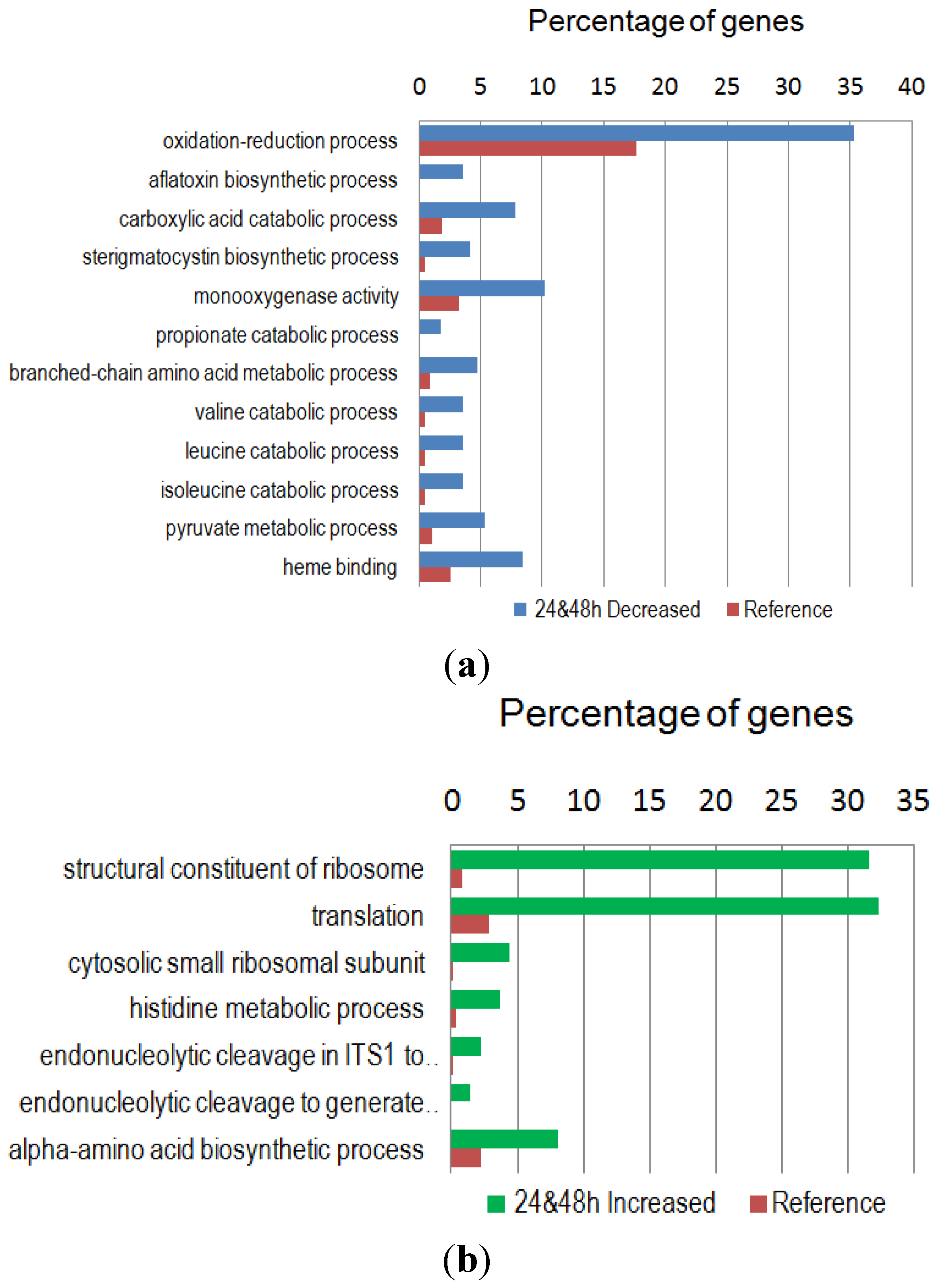
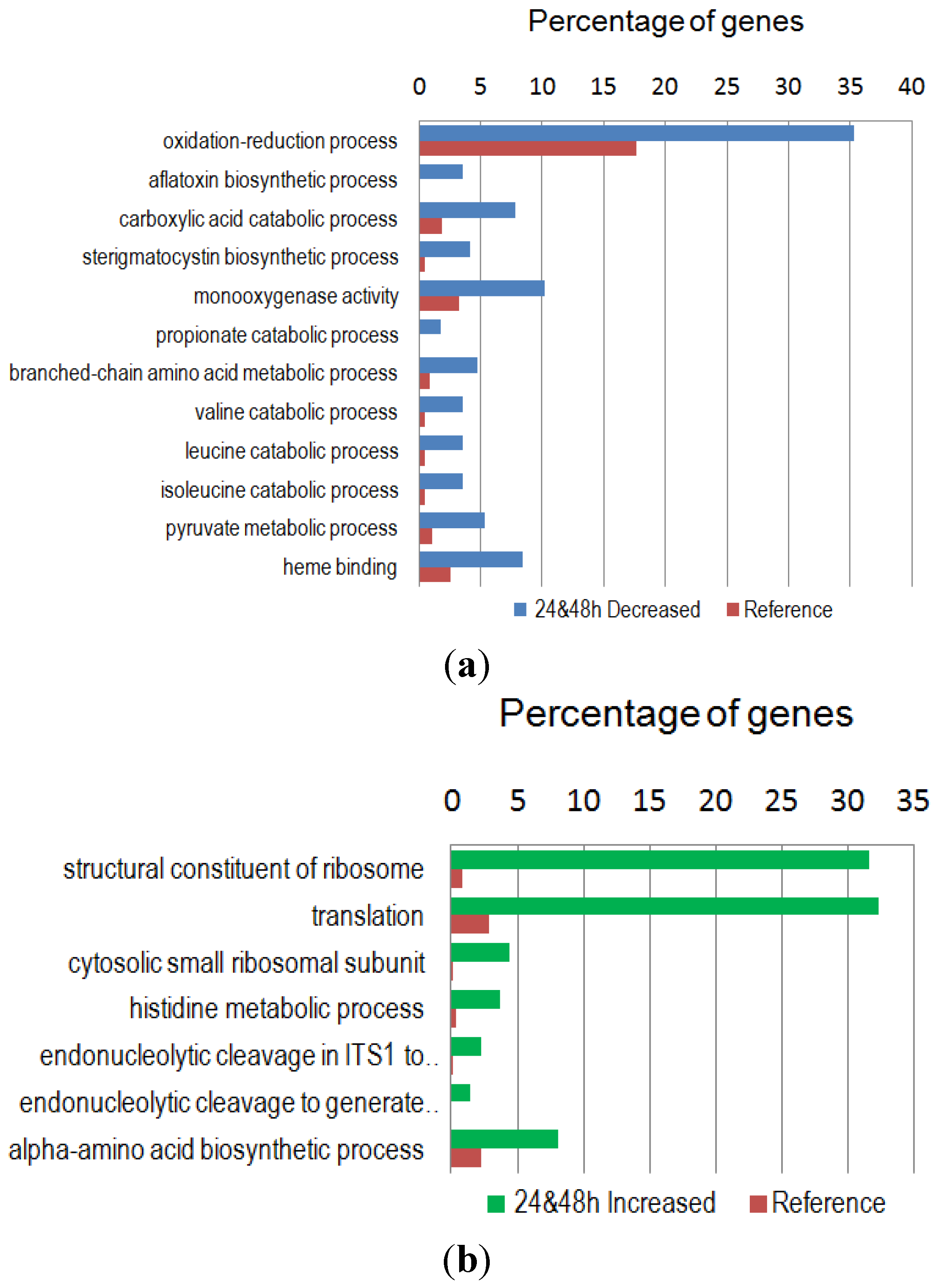
2.3. GO Functional Classification of Differentially Expressed Genes
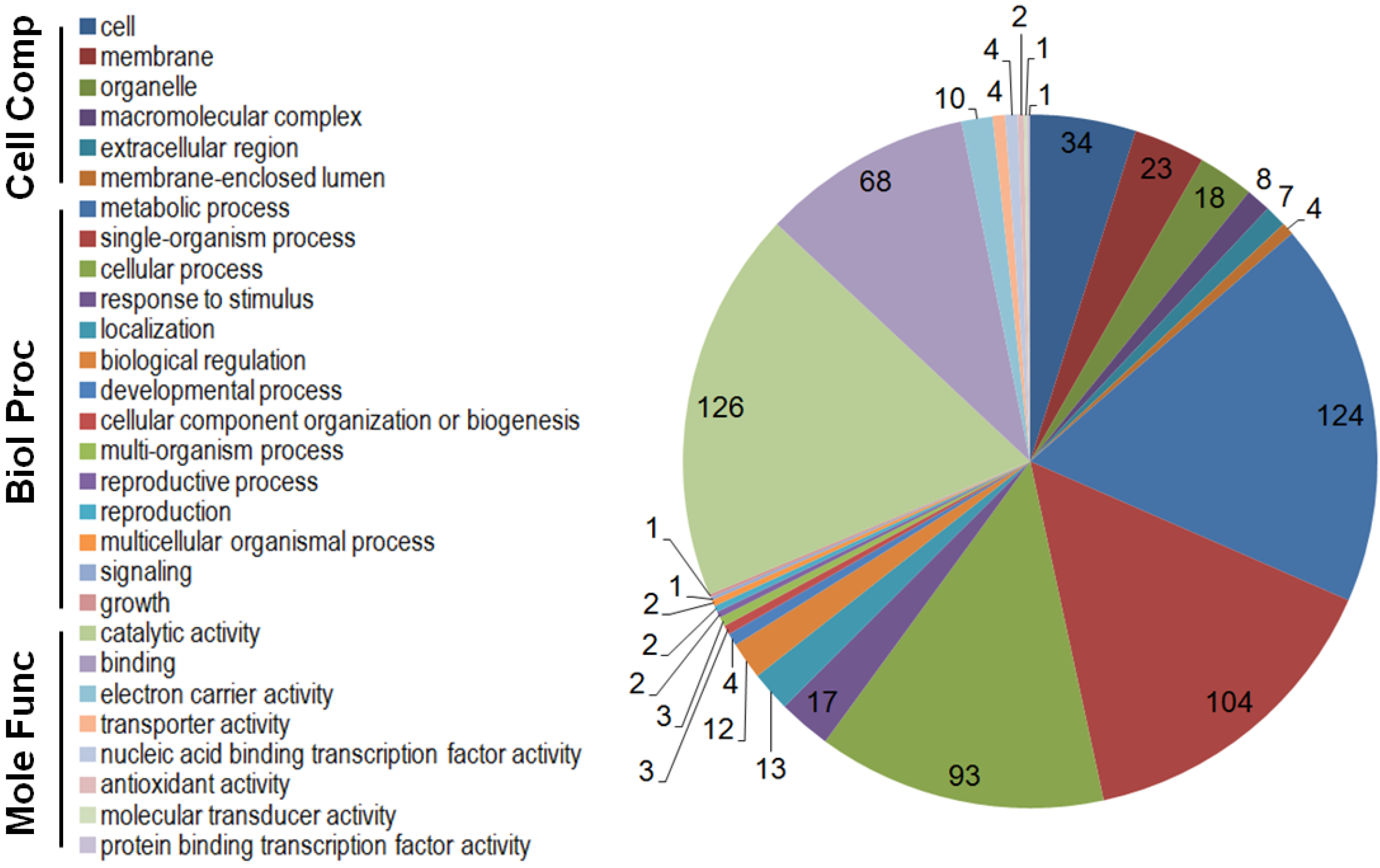
2.4. GO Enrichment and KEGG Metabolic Pathway Analysis of the Differentially Expressed Genes
| KEGG Metabolic Pathway a | #Seq/#Enz | Order of Abundance b |
|---|---|---|
| Down-Regulated Gene Group | ||
| Pyruvate metabolism | 7/8 | 18 |
| Valine, leucine and isoleucine degradation | 7/6 | 32 |
| Propanoate metabolism | 6/5 | 33 |
| Tryptophan metabolism | 5/4 | 7 |
| Glyoxylate and dicarboxylate metabolism | 5/4 | 28 |
| Phenylalanine metabolism | 4/3 | 8 |
| Fatty acid degradation | 4/4 | 10 |
| Glycine, serine and threonine metabolism | 4/3 | 3 |
| ß-Alanine metabolism | 4/4 | 48 |
| Up-Regulated Gene Group | ||
| Histidine metabolism | 4/6 | 53 |
| Arginine and proline metabolism | 3/5 | 5 |
| Pyruvate metabolism | 3/2 | 18 |
| Citrate cycle (TCA cycle) | 2/1 | 42 |
| Alanine, aspartate and glutamate metabolism | 2/2 | 21 |
| Phenylalanine, tyrosine and tryptophan biosynthesis | 2/5 | 44 |
3. Discussion
4. Experimental Section
4.1. Fungal Strain, Medium and Culture Growth
4.2. Preparation of Total RNA and Sequencing
4.3. Mapping Reads to A. flavus Reference Genome and Normalized Gene Expression Levels
4.4. Statistical Analysis of Digital Gene Expression
4.5. Gene Ontology, Functional Enrichment, and KEGG Metabolic Pathways
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Liu, Y.; Wu, F. Global burden of aflatoxin-induced hepatocellular carcinoma: A risk assessment. Environ. Health Perspect. 2010, 118, 818–824. [Google Scholar] [CrossRef] [PubMed]
- Cotty, P.J. Influence of field application of an atoxigenic strain of Aspergillus flavus on the populations of A. flavus infecting cotton bolls and on the aflatoxin content of cottonseed. Phytopathology 1994, 84, 1270–1277. [Google Scholar] [CrossRef]
- Abbas, H.K.; Zablotowicz, R.M.; Horn, B.W.; Phillips, N.A.; Johnson, B.J.; Jin, X.; Abel, C.A. Comparison of major biocontrol strains of non-aflatoxigenic Aspergillus flavus for the reduction of aflatoxins and cyclopiazonic acid in maize. Food Addit. Contam. 2011, 28, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Dorner, J.W. Development of biocontrol technology to manage aflatoxin contamination in peanuts. Peanut Sci. 2009, 36, 60–67. [Google Scholar] [CrossRef]
- Hua, S.S. Progress in prevention of aflatoxin contamination in food by preharvest application of Pichia anomala WRL-076. In Recent Advances in Multidisciplinary Applied Microbiology; Mendez-Vilas, A., Ed.; Wiley-VCH: Weinheim, Germany, 2006; pp. 322–326. [Google Scholar]
- Hua, S.S.; Baker, J.L.; Flores-Espiritu, M. Interactions of saprophytic yeasts with a nor mutant of Aspergillus flavus. Appl. Environ. Microbiol. 1999, 65, 2738–2740. [Google Scholar] [PubMed]
- Hua, S.S.; Brandl, M.T.; Hernlem, B.; Eng, J.G.; Sarreal, S.B. Fluorescent viability stains to probe the metabolic status of aflatoxigenic fungus in dual culture of Aspergillus flavus and Pichia anomala. Mycopathologia 2011, 171, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Hua, S.S.; Beck, J.J.; Sarreal, S.B.; Gee, W. The major volatile compound 2-phenylethanol from the biocontrol yeast, Pichia anomala, inhibits growth and expression of aflatoxin biosynthetic genes of Aspergillus flavus. Mycotoxin Res. 2015, 30, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Lingappa, B.T.; Prasad, M.; Lingappa, Y.; Hunt, D.F.; Biemann, K. Phenethyl alcohol and tryptophol: Autoantibiotics produced by the fungus Candida albicans. Science N.Y. 1969, 163, 192–194. [Google Scholar] [CrossRef]
- Gao, F.; Daugulis, A.J. Bioproduction of the aroma compound 2-phenylethanol in a solid-liquid two-phase partitioning bioreactor system by Kluyveromyces marxianus. Biotechnol. Bioeng. 2009, 104, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Dong, Q.; Meng, C.; Shi, X.A.; Guo, Y. A continuous and adsorptive bioprocess for efficient production of the natural aroma chemical 2-phenylethanol with yeast. Enzyme Microb. Technol. 2011, 48, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Cheng, Y.; Yang, M.; Liu, Y.; Chen, K.; Long, C.A.; Deng, X. Mechanisms of action for 2-phenylethanol isolated from Kloeckera apiculata in control of Penicillium molds of citrus fruits. BMC Microbiol. 2014, 14, 242. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, H.S.; Carr, H.S.; Rose, H.M. Phenethyl alcohol. I. Effect on macromolecular synthesis of Escherichia coli. J. Bacteriol. 1965, 89, 1354–1369. [Google Scholar] [PubMed]
- Chang, P.-K.; Scharfenstein, L.L. Aspergillus flavus Blast2GO gene ontology database: Elevated growth temperature alters amino acid metabolism. Genet. Genome Res. 2014, 1, 1. [Google Scholar]
- Berrah, G.; Konetzka, W.A. Selective and reversible inhibition of the synthesis of bacterial deoxyribonucleic acid by phenethyl alcohol. J. Bacteriol. 1962, 83, 738–744. [Google Scholar] [PubMed]
- Lester, G. Inhibition of growth, synthesis, and permeability in Neurospora crassa by phenethyl alcohol. J. Bacteriol. 1965, 90, 29–37. [Google Scholar] [PubMed]
- Fraud, S.; Rees, E.L.; Mahenthiralingam, E.; Russell, A.D.; Maillard, J.Y. Aromatic alcohols and their effect on Gram-negative bacteria, cocci and mycobacteria. J. Antimicrob. Chemother. 2003, 51, 1435–1436. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, H.S.; Carr, H.S.; Rose, H.M. Phenethyl alcohol. II. Effect on thymine-requiring Escherichia coli. J. Bacteriol. 1965, 89, 1370–1373. [Google Scholar] [PubMed]
- Chang, P.-K. The Aspergillus parasiticus protein AFLJ interacts with the aflatoxin pathway-specific regulator AFLR. Mol. Genet. Genom. 2003, 268, 711–719. [Google Scholar]
- Chang, P.-K.; Ehrlich, K.C.; Yu, J.; Bhatnagar, D.; Cleveland, T.E. Increased expression of Aspergillus parasiticus aflR, encoding a sequence-specific DNA-binding protein, relieves nitrate inhibition of aflatoxin biosynthesis. Appl. Environ. Microbiol. 1995, 61, 2372–2377. [Google Scholar] [PubMed]
- Calvo, A.M.; Bok, J.; Brooks, W.; Keller, N.P. veA is required for toxin and sclerotial production in Aspergillus parasiticus. Appl. Environ. Microbiol. 2004, 70, 4733–4739. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.-K.; Scharfenstein, L.L.; Ehrlich, K.C.; Wei, Q.; Bhatnagar, D.; Ingber, B.F. Effects of laeA deletion on Aspergillus flavus conidial development and hydrophobicity may contribute to loss of aflatoxin production. Fungal Biol. 2012, 116, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Fedorova, N.D.; Montalbano, B.G.; Bhatnagar, D.; Cleveland, T.E.; Bennett, J.W.; Nierman, W.C. Tight control of mycotoxin biosynthesis gene expression in Aspergillus flavus by temperature as revealed by RNA-Seq. FEMS Microbiol. Lett. 2011, 322, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Brock, M. Generation and phenotypic characterization of Aspergillus nidulans methylisocitrate lyase deletion mutants: Methylisocitrate inhibits growth and conidiation. Appl. Environ. Microbiol. 2005, 71, 5465–5475. [Google Scholar] [CrossRef] [PubMed]
- Upton, A.M.; McKinney, J.D. Role of the methylcitrate cycle in propionate metabolism and detoxification in Mycobacterium smegmatis. Microbiology Read. Engl. 2007, 153, 3973–3982. [Google Scholar] [CrossRef] [PubMed]
- Adye, J.; Mateles, R.I. Incorporation of labelled compounds into aflatoxins. Biochim. Biophys. Acta 1964, 86, 418–420. [Google Scholar] [CrossRef]
- Payne, G.A.; Hagler, W.M., Jr. Effect of specific amino acids on growth and aflatoxin production by Aspergillus parasiticus and Aspergillus flavus in defined media. Appl. Environ. Microbiol. 1983, 46, 805–812. [Google Scholar] [PubMed]
- Reddy, T.V.; Viswanathan, L.; Venkitasubramanian, T.A. Factors affecting aflatoxin production by Aspergillus parasiticus in a chemically defined medium. J. Gen. Microbiol. 1979, 114, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, J.R.; Yu, J.; Bland, J.M.; Nierman, W.C.; Bhatnagar, D.; Cleveland, T.E. Amino acid supplementation reveals differential regulation of aflatoxin biosynthesis in Aspergillus flavus NRRL 3357 and Aspergillus parasiticus SRRC 143. Appl. Microbiol. Biotechnol. 2007, 74, 1308–1319. [Google Scholar] [CrossRef] [PubMed]
- Treick, R.W.; Konetzka, W.A. Physiological state of Escherichia coli and the inhibition of deoxyribonucleic acid synthesis by phenethyl alcohol. J. Bacteriol. 1964, 88, 1580–1584. [Google Scholar] [PubMed]
- Anbazhagan, V.; Munz, C.; Tome, L.; Schneider, D. Fluidizing the membrane by a local anesthetic: Phenylethanol affects membrane protein oligomerization. J. Mol. Biol. 2010, 404, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Tai, P.C. Effects of antibiotics and other inhibitors on ATP-dependent protein translocation into membrane vesicles. J. Bacteriol. 1987, 169, 2373–2379. [Google Scholar] [PubMed]
- Greene-McDowelle, D.M.; Ingber, B.; Wright, M.S.; Zeringue, H.J., Jr.; Bhatnagar, D.; Cleveland, T.E. The effects of selected cotton-leaf volatiles on growth, development and aflatoxin production of Aspergillus parasiticus. Toxicon 1999, 37, 883–893. [Google Scholar] [CrossRef]
- Wright, M.S.; Greene-McDowelle, D.M.; Zeringue, H.J.; Bhatnagar, D.; Cleveland, T.E. Effects of volatile aldehydes from Aspergillus-resistant varieties of corn on Aspergillus parasiticus growth and aflatoxin biosynthesis. Toxicon 2000, 38, 1215–1223. [Google Scholar] [CrossRef]
- Cleveland, T.E.; Carter-Wientjes, C.H.; de Lucca, A.J.; Boue, S.M. Effect of soybean volatile compounds on Aspergillus flavus growth and aflatoxin production. J. Food. Sci. 2009, 74, H83–H87. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.-K.; Scharfenstein, L.L.; Mack, B.; Yu, J.; Ehrlich, K.C. Transcriptomic profiles of Aspergillus flavus CA42, a strain that produces small sclerotia, by decanal treatment and after recovery. Fungal Genet. Biol. 2014, 68, 39–47. [Google Scholar] [CrossRef] [PubMed]
- White, B.T.; Yanofsky, C. Structural characterization and expression analysis of the Neurospora conidiation gene con-6. Dev. Biol. 1993, 160, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Adams, T.H.; Hide, W.A.; Yager, L.N.; Lee, B.N. Isolation of a gene required for programmed initiation of development by Aspergillus nidulans. Mol. Cell Biol. 1992, 12, 3827–3833. [Google Scholar] [PubMed]
- Wieser, J.; Lee, B.N.; Fondon, J., 3rd; Adams, T.H. Genetic requirements for initiating asexual development in Aspergillus nidulans. Curr. Genet. 1994, 27, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.-K.; Scharfenstein, L.L.; Mack, B.; Ehrlich, K.C. Deletion of the Aspergillus flavus orthologue of A. nidulans fluG reduces conidiation and promotes production of sclerotia but does not abolish aflatoxin biosynthesis. Appl. Environ. Microbiol. 2012, 78, 7557–7563. [Google Scholar] [CrossRef] [PubMed]
- Keller, N.P.; Turner, G.; Bennett, J.W. Fungal secondary metabolism—From biochemistry to genomics. Nat. Rev. Microbiol. 2005, 3, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Li, R.W.; Rinaldi, M.; Capuco, A.V. Characterization of the abomasal transcriptome for mechanisms of resistance to gastrointestinal nematodes in cattle. Vet. Res. 2011, 42, 114. [Google Scholar] [CrossRef] [PubMed]
- CLC bio, a Qiagen Company. Available online: http://www.clcbio.com (accessed on 22 September 2015).
- Aspergillus flavus, Genome, NCBI. Available online: http://www.ncbi.nlm.nih.gov/genome/?term=aspergillus+flavus (accessed on 22 September 2015).
- Mortazav, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Word, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 7, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Smyth, G.K. Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics Oxford Engl. 2008, 9, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar]
- Conesa, A.; Götz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genom. 2008, 2008, 619832. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, P.-K.; Hua, S.S.T.; Sarreal, S.B.L.; Li, R.W. Suppression of Aflatoxin Biosynthesis in Aspergillus flavus by 2-Phenylethanol Is Associated with Stimulated Growth and Decreased Degradation of Branched-Chain Amino Acids. Toxins 2015, 7, 3887-3902. https://doi.org/10.3390/toxins7103887
Chang P-K, Hua SST, Sarreal SBL, Li RW. Suppression of Aflatoxin Biosynthesis in Aspergillus flavus by 2-Phenylethanol Is Associated with Stimulated Growth and Decreased Degradation of Branched-Chain Amino Acids. Toxins. 2015; 7(10):3887-3902. https://doi.org/10.3390/toxins7103887
Chicago/Turabian StyleChang, Perng-Kuang, Sui Sheng T. Hua, Siov Bouy L. Sarreal, and Robert W. Li. 2015. "Suppression of Aflatoxin Biosynthesis in Aspergillus flavus by 2-Phenylethanol Is Associated with Stimulated Growth and Decreased Degradation of Branched-Chain Amino Acids" Toxins 7, no. 10: 3887-3902. https://doi.org/10.3390/toxins7103887
APA StyleChang, P.-K., Hua, S. S. T., Sarreal, S. B. L., & Li, R. W. (2015). Suppression of Aflatoxin Biosynthesis in Aspergillus flavus by 2-Phenylethanol Is Associated with Stimulated Growth and Decreased Degradation of Branched-Chain Amino Acids. Toxins, 7(10), 3887-3902. https://doi.org/10.3390/toxins7103887