Abstract
Secretory phospholipasesA2 (sPLA2s) form a large family of structurally related enzymes widespread in nature. Herein, we studied the inhibitory effects of sPLA2s from Vipera lebetina (VLPLA2), Vipera berus berus (VBBPLA2), and Naja naja oxiana (NNOPLA2) venoms on (i) human platelets, (ii) four different bacterial strains (gram-negative Escherichia coli and Vibrio fischeri; gram-positive Staphylococcus aureus and Bacillus subtilis) and (iii) five types of cancer cells (PC-3, LNCaP, MCF-7, K-562 and B16-F10) in vitro. sPLA2s inhibited collagen-induced platelet aggregation: VBBPLA2 IC50 = 0.054, VLPLA2 IC50 = 0.072, NNOPLA2 IC50 = 0.814 μM. p-Bromophenacylbromide-inhibited sPLA2 had no inhibitory action on platelets. 36.17 μM VBBPLA2 completely inhibited the growth of gram-positive Bacillus subtilis whereas no growth inhibition was observed towards gram-negative Escherichia coli. The inhibitory action of sPLA2s (~0.7 μM and ~7 μM) towards cancer cells depended on both venom and cell type. VBBPLA2 (7.2 μM) inhibited significantly the viability of K-562 cells and the cell death appeared apoptotic. The sPLA2s exhibited no inhibitory effect towards LNCaP cells and some effect (8%–20%) towards other cells. Thus, already sub-μM concentrations of sPLA2s inhibited collagen-induced platelet aggregation and from the current suite of studied svPLA2s and test cells, VBBPLA2 was the most growth inhibitory towards Bacillus subtilis and K-562 cells.
Abbreviations
| svPLA2 | snake venom PLA2 |
| sPLA2 | secretory PLA2 |
| VLPLA2 | Vipera lebetina phospholipase A2 |
| VBBPLA2 | Vipera berus berus phospholipase A2 |
| NNOPLA2 | Naja naja oxiana phospholipase A2 |
| p-BPB | p-bromophenacylbromide |
| MALDI-TOF MS | matrix assisted laser desorption ionization time-of-flight mass spectrometry |
| lysoPC | lysophosphatidylcholine |
| PRP | platelet-rich plasma |
1. Introduction
Phospholipases A2 (E.C. 3.1.1.4) are enzymes that catalyze the hydrolysis of the sn-2 fatty acyl ester bond of sn-3 phosphoglycerides, liberating free fatty acids, and lysophospholipids. Phospholipases A2 (PLA2s) are a large family of proteins found in various mammalian tissues: arthropods, as well as in the venoms of snakes, scorpions and bees. Based on their source, catalytic activity, amino acid sequence, chain length and disulfide bond patterns, PLA2s are divided into 16 groups [1] including 10 groups of secretory PLA2s (sPLA2s) [2,3]. The variability of the structure of the conserved domains of sPLA2s from bacteria to mammals was recently investigated by Nevalainen et al. [4].
The sPLA2s are small-molecular-mass proteins (13–15 kDa) that require the presence of Ca2+ for their catalytic activity. In snake venoms, only two groups of sPLA2s (GI and GII) have been identified. Group I (GIA) includes the svPLA2s from Elapinae and Hydrophiinae venoms with 115–120 amino acid residues and these svPLA2s are homologous to mammalian pancreatic GIB sPLA2. Group II (GIIA and GIIB) comprises the svPLA2s from Crotalinae and Viperinae venoms with 120–125 amino acid residues and homologous to mammalian non-pancreatic Group II-A sPLA2 [3]. Group II PLA2s are in turn divided into different subgroups on the basis of amino acid residue in the 49th position: catalytically active D49 enzymes, catalytically inactive or with low activity K49, S49, N49 or R49 forms [5,6]. The above described subgroups exhibit a wide variety of physiological and pathological effects. In addition to their possible role in the digestion of prey, snake venom sPLA2s exhibit a wide spectrum of pharmacological effects such as neurotoxicity, cardiotoxicity, myotoxicity, anticoagulant, anticancer effects etc. [3,5,6,7,8,9,10,11,12].
Numerous snake venom sPLA2s that modulate platelet function have been characterized [13,14,15,16,17,18,19] and different mechanisms of action shown [6,15,20,21,22,23,24,25,26]. The sPLA2s effect on platelet aggregation can be independent or dependent on their catalytic activity. However, the mechanism of action of snake sPLA2s on platelet aggregation is not fully elucidated.
In addition, an increasing number of sPLA2s with antibacterial properties has been reported [27,28,29,30,31,32,33,34,35,36]. For example, sPLA2s have been shown to be inhibitory (bacteriostatic) or killing (bactericidal) to gram-positive bacteria Staphylococcus aureus [37]. In case of svPLA2 from Crotalus durissus collilineatus venom the bactericidal effect was entirely dependent on its enzymatic activity [38]. The effect of sPLA2s towards gram-positive and gram-negative bacteria and their role in the host defence against bacterial infections has been reviewed by Nevalainen et al. [39].
Different types of sPLA2s and synthetic peptides derived from sPLA2 homologues have been shown to possess antitumor and antiangiogenic activity against different cancer cells in vitro. The antitumor activities have been detected for the acidic BthA-I-PLA2 from Bothrops jararacussu venom [40], for RVV-7, a basic 7 kDa toxin from Russell’s viper venom [41], for two sPLA2s from Cerastes cerastes venom [42], for sPLA2 from Naja naja atra venom [43], for a Lys49 sPLA2 from Protobothrops flavoviridis venom [44], for a Drs-PLA2 from Daboia russelii siamensis venom [45]. Recent studies have shown that MVL-PLA2 from Macrovipera lebetina transmediterranea venom inhibited cell adhesion and migration of melanoma IGR39 cells and fibrosarcoma HT1080 cells in vitro [46,47]. Antitumor properties of different snake venom phospholipases A2 have been reviewed by Rodrigues et al. [12].
In the current study sPLAs from Vipera berus berus (common viper), Vipera lebetina (Levantine viper) and Naja naja oxiana (Middle-Asian cobra) venoms were studied for their biological effects using (i) human platelets, (ii) different gram-negative (Vibrio fischeri, Escherichia coli) and gram-positive (Bacillus subtilis, Staphylococcus aureus) bacterial strains and (iii) five different cancer cells lines (prostate cancer cell lines PC-3, LNCaP, breast cancer cell line MCF-7, chronic myeloid leukemic cell line K-562 and mouse melanoma cell line B16-F10).
2. Results
2.1. Purification and Characterization of sPLA2s
VLPLA2 (Vipera lebetina sPLA2) was purified as described by Vija et al. [18] and VBBPLA2 (Vipera berus berus sPLA2) according to Križaj et al. [48]. In the case of NNOPLA2 (Naja naja oxiana sPLA2), a new two-step purification scheme involving Sephadex G-50 sf and pentylagarose chromatography was used resulting in homogeneous sample.
The relative activity of studied svPLA2s was comparatively high: VLPLA2—882 μmol/min mg; VBBPLA2—1900 μmol/min mg and NNOPLA2—1200 μmol/min mg. The molecular masses of PLA2s after reduction with 2-mercaptoethanol detected by SDS-PAGE were about 14,000 Da. VLPLA2 had pI value in the acidic region (4.3), VBBPLA2 in the basic region (9.3) and NNOPLA2 in the neutral region (6.7). The activity of svPLA2 after isoelectric focusing in the gel was detected using egg-yolk overlay-technique (data not shown).
MALDI-TOF MS analysis confirmed the molecular masses estimates of native PLA2s revealing single peaks for enzymes with the actual molecular masses of 13,683 Da for VLPLA2, 13,824 Da for VBBPLA2 and 13,229 Da for NNOPLA2. To distinguish between the possible isoforms, PLA2s of different venoms were subjected to trypsinolysis and the masses of the resulting peptides were analysed by MALDI-TOF MS. The peptide mass fingerprinting results confirmed that VBBPLA2 was a close match with enzyme formerly sequenced by Križaj et al. [48], VLPLA2 matched with sequence (EU421953) [18] and NNOPLA2 with enzyme isoform 3 formerly sequenced by Ovchinnikov et al. [49] (Figure 1). MALDI-TOF analysis of tryptic peptides derived from NNOPLA2 is provided in Figure S1.

Figure 1.
Alignment of V. lebetina VLPLA2 (EU421953) [18], VBBPLA2 V. berus berus (P31854) [48] and NNOPLA2 isozyme E from N. naja oxiana (P25498) [49]. The alignment was performed using the program CLUSTAL W (1.83) multiple sequence alignment. “*” indicates positions which have a single, fully conserved residue; “:” indicates that one of the “strong” amino acid groups is fully conserved; “.” indicates that one of the “weaker” groups is fully conserved. Trypsin cleavage sites in NNOPLA2 are indicated as ↑. Cysteine residues are on red background, conserved catalytic network formed by four amino acid residues His48, Asp49, Tyr52 and Asp99 are on blue background.
2.2. Inhibition of Human Platelet Aggregation in Vitro
sPLA2s from all three venoms inhibited collagen-induced platelet aggregation in platelet-rich plasma in a concentration-dependent manner: the IC50 = 0.054 μM for VBBPLA2 (Figure 2A); IC50 = 0.072 μM for VLPLA2 [18] and IC50 = 0.814 μM for NNOPLA2 (Figure 2B).
In order to explore if the inhibitory effects of sPLA2s on platelet aggregation were related to their enzymatic activities, the native sPLA2s were treated by p-bromophenacylbromide (p-BPB) that modifies the histidine in the active center causing the inhibition of the catalytic activity. The p-BPB-treated enzymes were tested in the same conditions as the native vPLA2s. The treatment of all three svPLA2s by p-BPB resulted in complete loss of their catalytic activity that was accompanied by the loss of their inhibitory effect on collagen-induced platelet aggregation.
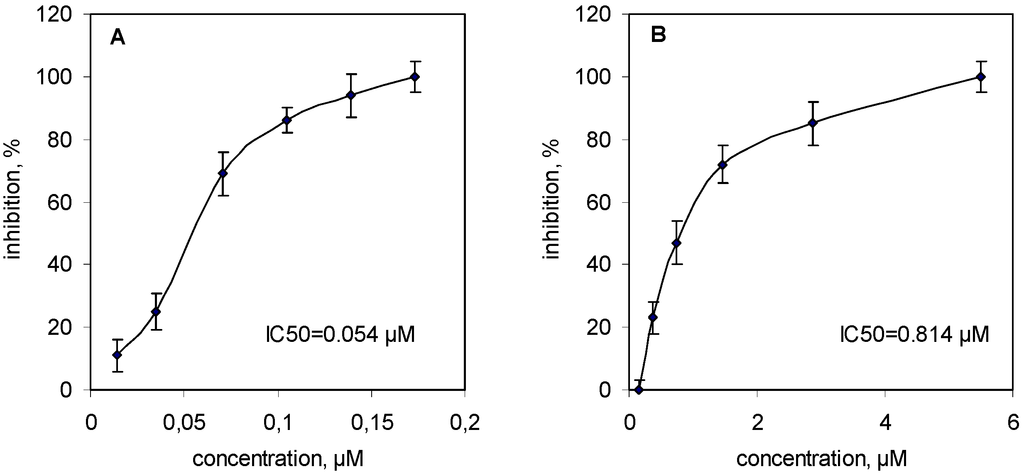
Figure 2.
Inhibitory effects of svPLA2s on collagen-induced human platelet aggregation. (A) human platelet rich plasma (PRP) samples were stirred for 2 min at 37 °C with VBBPLA2s (0.014–0.173 μM) and then 2 μg/mL of collagen (final concentration in the test) was added to induce platelet aggregation; (B) The PRP samples were preincubated with NNOPLA2s (0.148–5.510 μM) under the same conditions. Results are reported as means ± SD (n = 3).
2.3. Inhibitory Effect of Snake Venoms and Their sPLA2s on Bacteria
2.3.1. Acute Toxicity to Vibrio fischeri
For the evaluation of the acute toxicity of studied enzyme preparations, naturally luminescent gram-negative bacteria V. fischeri were used. In these bacteria, the exposure to toxicants causes rapid decrease of their bioluminescence whereas the effect is dose-dependent [50]. In the current study, in addition to svPLA2s also the effect of the whole venom was evaluated. As a toxicity endpoint, inhibition of bacterial bioluminescence after 15 min of exposure to the whole venom or sPLA2s was used. In general, the venoms and sPLA2s were not acutely toxic to V. fischeri. Also, the sPLA2s were not acutely toxic: only enzyme from V. lebetina inhibited the luminescence of bacteria at <100 μg/mL (<7.31 μM) level, the 15-min EC50 was 58 μg/mL, i.e., 4.24 μM; Table 1).

Table 1.
Acute toxicity (15-min EC50, μg/mL) of venoms and sPLA2s from different snakes to bacteria Vibrio fischeri. As a toxicity endpoint, inhibition of the bacterial bioluminescence was used.
| Tested item | Acute toxicity (15-min EC50, μg/mL) | |||
|---|---|---|---|---|
| 3,5-DCP * | V. b. berus | V. lebetina | N. n. oxiana | |
| Venom | 3–4 | 370 | 944 | >1315 |
| PLA2 | 3–4 | >909 (>65.76 μM) | 58 (4.24 μM) | >606 (>45.81 μM) |
* 3,5-dichlorophenol (a positive control).
2.3.2. Inhibitory Effect of the Snake Venom PLA2s on Bacterial Growth
The inhibitory effect of svPLA2s on bacterial growth (a chronic toxicity) was evaluated at 500 μg/mL (36.2 μM for VBBPLA2; 37.8 μM for NNOPLA2; 36.5 μM for VLPLA2) level of the enzymes. The effect of VBBPLA2 on the growth of gram-positive bacterial strains was studied in parallel for the native enzymes and p-bromophenacylbromide-inactivated VBBPLA2s. The results are shown in Table 2 and Figure 3. Although the tested concentration was relatively high, none of the svPLA2s inhibited the growth of gram-negative bacteria Escherichia coli but there were inhibitory effects in case of some enzyme preparations on gram-positive bacterial strains (Figure 3A–C). Specifically, the V. berus berus PLA2 was most potent and totally (100%) inhibited the growth of B. subtilis (Figure 3A). The total growth inhibition of B. subtilis was also observed in case of p-BPB-inactivated VBBPLA2 (Figure 3B) whereas the effect was dose-dependent (Figure 3C). PLA2 from V. lebetina showed also some inhibitory effect (13%) towards B. subtilis but this inhibitory effect was not observed in case of p-BPB-inactivated enzyme (Figure 3A). Intact VBBPLA2 preparations (Table 2) had no inhibitory effect on gram-positive bacteria S. aureus but there was some inhibitory effect in case of inactivated enzyme (Figure 3B; Table 2). The N. naja oxiana PLA2 was inhibitory (42%) towards S. aureus (Table 2).
Table 2.
Inhibition of the bacterial growth (incubation time 6 h) in LB medium at 30 °C supplemented by svPLA2s (500 μg/mL, i.e., 36.2 μM for VBBPLA2; 37.8 μM for NNOPLA2; 36.5 μM for VLPLA2) from three different snakes.
| Inhibition of the bacterial growth, % (t = 6 h) | ||||||
|---|---|---|---|---|---|---|
| Bacteria(Gram staining) | svPLA2 | |||||
| V. b. berus | V. b. berus * | V. lebetina | V. lebetina * | N. n. oxiana | ||
| Escherichia coli | Gram (−) | No effect | not tested | No effect | No effect | No effect |
| Bacillus subtilis | Gram (+) | 100% ** | 99% ** | 13% | No effect | Slight effect (6.5%) |
| Staphylococcus aureus | Gram (+) | No effect | 29% | No effect | No effect | 42% |
* histidine in PLA2 was modified by p-bromophenacylbromide, to inactivate its catalytic activity; ** growth was inhibited by 100% but the viability of bacteria remained unchanged (i.e., after the 6 h exposure to enzyme preparation, bacteria were able to grow on agarized LB-medium; data not shown).
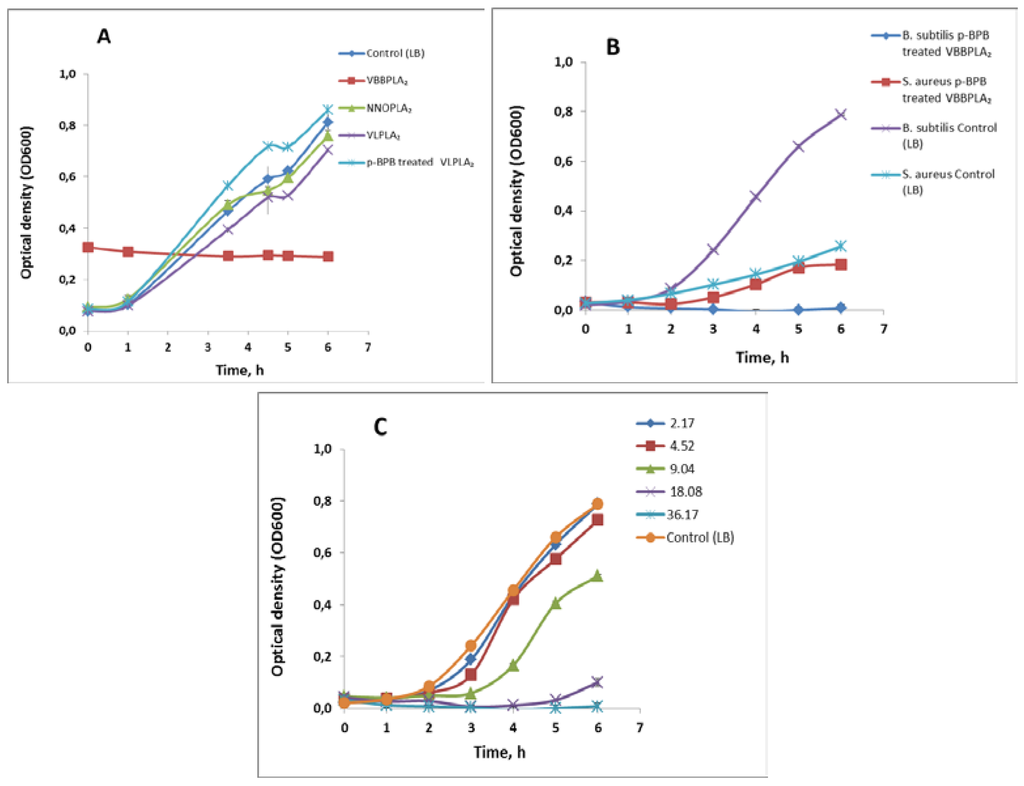
Figure 3.
The effect of different snake venom sPLA2s on the growth of bacteria in LB medium at 30 °C. (A) The effect of different snake venom sPLA2s (500 μg/mL, i.e., 36.2 μM for VBBPLA2; 37.8 μM for NNOPLA2; 36.5 μM for VLPLA2) on the growth of gram-positive bacteria Bacillus subtilis BR151. The different svPLA2s are indicated as data labels; (B) The effect of p-BPB-treated V. berus berus sPLA2 (500 μg/mL = 36.17 μM) on the growth of gram-positive bacteria Bacillus subtilis BR151 and Staphylococcus aureus. Growth of not treated bacteria is shown as data labels; (C) The effect of different concentrations of p-BPB-treated V. berus berus venom sPLA2 on the growth of Bacillus subtilis BR151; concentrations (μM) are shown as data labels. Results are reported as means ± SD (n = 3).
2.4. Effects of Snake Venom PLA2s on Cancer Cells Viability
Cancer cell lines (PC-3, LNCaP, MCF-7, B10-F16 and K-562) were exposed to PLA2s from V. lebetina, V. berus berus and N. naja oxiana at concentrations of 10 and 100 μg/mL (~0.7 and ~7 μM). The results are shown in Figure 4. There was no inhibitory effect of studied PLA2 preparations towards LNCaP cells in this concentration range (Figure 4A–C). The viability of PC-3 cells was not changed after treating with 7.31 μM of VLPLA2 (Figure 4B). NNOPLA2 had no cytotoxic effect on MCF-7 cells (Figure 4C), VBBPLA2 and VLPLA2 only slightly reduced the viability of MCF-7 cells (Figure 4A,B). VLPLA2 and NNOPLA2 decreased viability of B16-F10 cells about 17% (Figure 4B,C), VBBPLA2 had no effect (Figure 4A). All three enzymes inhibited the viability of K-562 cells (Figure 4A–C), although VLPLA2 had only slight effect (Figure 4B). The most potent inhibitory effect was observed in case of VBBPLA2. After 48 h treatment of K-562 cells with 7.23 μM of VBBPLA2, the cellular viability reduced to 20% (Figure 4D). p-BPB-treated VBBPLA2 inhibited the viability of K-562 cells by 27%. VBBPLA2 reduced the viability of K-562 cells in time- and dose-dependent manner.
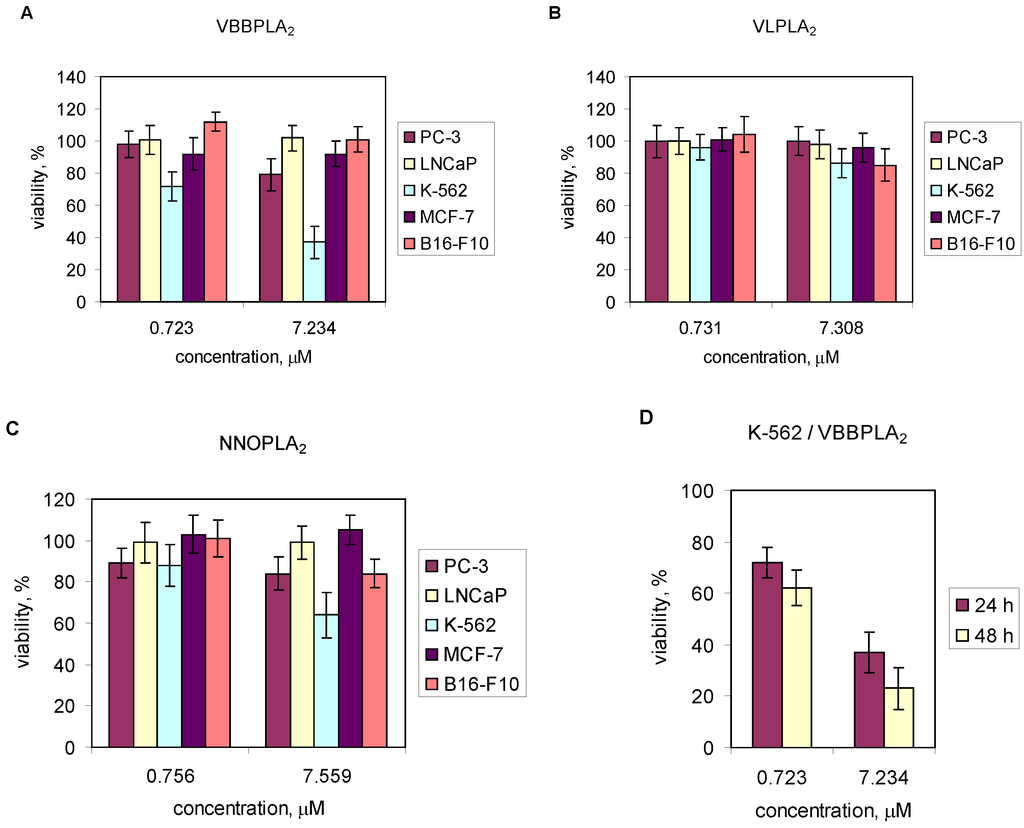
Figure 4.
Effect of svPLA2s on viability of PC-3, LNCaP, K-562, MCF-7 and B16-F10 cells in vitro. (A–C) Cells were seeded in 96-well plates at a density 105 cells/mL and incubated at 37 °C for at least 24 h. After treatment with snake venom PLA2s (~0.7 and ~7 μM) for 24 h, the viability of the cells was determined by MTT assay (PC-3) or by water-soluble tetrazolium salt WST-1 assay (LNCaP, K-562, MCF-7 and B16-F10); (D) K-562 cells were treated with VBBPLA2 (0.72 μM and 7.23μM) for 24 and 48 h. Data are means (±SD) from two independent experiments performed in triplicate.
To evaluate whether the cytotoxicity effect of VBBPLA2 on K-562 cells (Figure 4D) was necrotic or apoptotic, the treated cells were stained with Annexin-V-FITC and propidium iodide (PI) (Figure 5). One characteristic feature of apoptosis is the externalisation of the lipid phosphatidyl serine (PS) from the inner to the outer plasma membrane. Annexin-V is a calcium-dependent phospholipid-binding protein that specifically binds PS and hence stains apoptotic cells. When used in conjunction with a live/dead cell discriminator such as propidium iodide, which measures membrane integrity, the bright green early apoptotic cells (Annexin-V positive) can be distinguished from the red colored late apoptotic/necrotic cells (PI positive). PI stains the cells with ruptured plasma membrane as cells with intact membranes are not permeable to PI. Thus, PI stains both, the cells in the late stage of apoptosis and the cells in necrosis. The treatment of K-562 cells with 0.36 μM VBBPLA2 caused the loss of cell membrane’s asymmetry which is a sign of early apoptosis (Figure 5A).
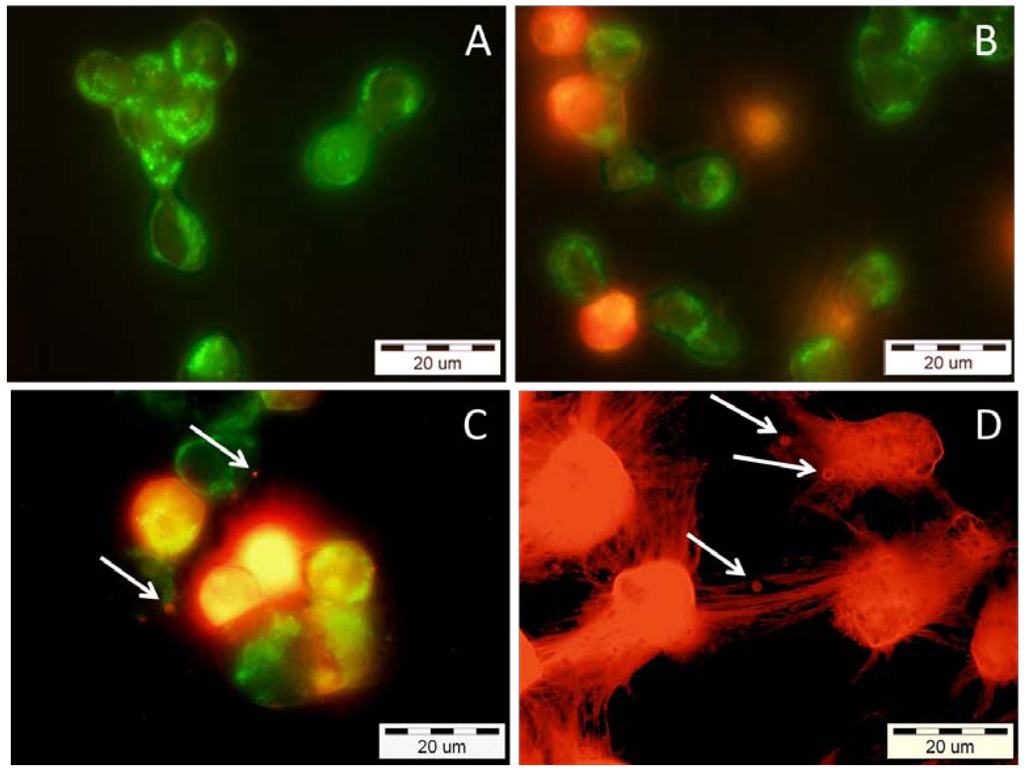
Figure 5.
Epifluorescence micrographs of human K-562 cells after incubation with different concentrations of VBBPLA2. After the exposure, the cells were stained with both Annexin V-FITC and propidium iodide, to visualize the early and late stage of apoptosis and/or necrosis of the cells, respectively. (A) 0.36 μM (expose 24 h)—early apoptotic cells with intact membranes—green; (B) 0.36 μM (expose 28 h)—mixture of early apoptotic cells (green) and cells which have already lost their membrane integrity (orange); (C) 0.72 μM (expose 24 h)—late apoptotic cells (orange to red) with blebbes (white arrows) and green membrane fragments; (D) 7.23 μM (expose 24 h)—totally destroyed necrotic (red) cells with membrane blebbes (white arrows).
The transition from apoptosis to necrosis is a loosely defined continuum that necessitates recognition of the various stages of the process. Therefore, we performed a time course experiment (the cells were photographed after 24 h and 28 h of incubation) to prove that the cells were traversing through early apoptosis before reaching the late apoptosis/necrosis (Figure 5A,B). In our study the bright green cells (Annexin-V positive early apoptotic cells) turned to orange (Annexin-V and PI positive late apoptotic cells) when VBBPLA2 concentration was increased from 0.36 μM (Figure 5A) to 0.72 μM (Figure 5C) but also in case of lower VBBPLA2 concentration (0.36 μM) if the incubation time was prolonged to 28 h (Figure 5B). The cells treated with 7.23 μM VBBPLA2 appeared totally destroyed, but it was still possible to detect the characteristic sign of apoptosis—membrane blebbing (Figure 5D, white arrows).
3. Discussion
Snake venom sPLA2s exhibit a large variety of pharmacological effects. In this work we compared the effects of sPLA2s originating from the venoms of three different snakes on human platelets, different bacteria and five types of cancer cells in vitro. Naja naja oxiana PLA2 belongs to PLA2 from old world snakes (group I) and has different disulfide bond pattern than PLA2s from new world’s snakes such as VBBPLA2 and VLPLA2 (group II).
Kini and Evans [15] divided snake venom PLA2s based on their effects on platelet function into three classes: class A involves PLA2s which initiate platelet aggregation, class B PLA2s cause only the inhibition of platelet aggregation induced by several physiological agonists such as collagen and class C involves PLA2s that have dual activity acting as inducer and inhibitor, depending of conditions. Classes B and C are both subdivided into two subgroups. Inhibitory activity of class B1 PLA2s (but not class B2) is dependent on their catalytic activity. Results of the current study show that VBBPLA2 and NNOPLA2 belong to class B1. In class B1 the inhibitory effects against platelets aggregation have been explained by hydrolysis of phospholipids from the plasma and/or from lipoproteins and the formation of lysophosphatidylcholine (lysoPC) [21,22,51]. The platelet aggregation inhibitory effects of PLA2s have shown to be dependent on plasma factor for several snake venom PLA2s, including VLPLA2 [18], the antiplatelet PLA2 purified from the venoms of Austrelaps superba [51], Lachesis muta [21,52], and Micropechis ikaheka [53]. Yuan et al. [51] showed that the formation of lysoPC after incubation with snake venom PLA2 correlated with the inhibition of platelet aggregation.
The isoelectric point values of snake venom PLA2s vary and therefore PLA2s are classified as acidic, neutral or basic. This property may affect the binding affinity and specificity of PLA2s to phospholipid membranes. However, pI values of PLA2s are not predictive for their effect on platelet aggregation: the acidic VLPLA2 [18], acidic PLA2s from the venoms of Trimeresurus gramineus [13] and Agkistrodon acutus [14] and basic PLA2s from V. berus berus venom (this work), from Acanthopis praelongus venom [16] and acanthins from Acanthopis antarcticus venom [22] are all potent platelet inhibitors. On the contrary, bothropstoxin-II (Bthtx-II), a basic Asp49 phospholipase A2 isolated from Bothrops jararacussu snake venom was able to induce platelet aggregation in a concentration-dependent manner [17]. NNOPLA2 with almost neutral pI (6.7) inhibited collagen induced platelet aggregation more slowly than VBBPLA2 and VLPLA2 (Figure 2).
Although only PLA2 from V. lebetina but not the PLA2s from V. berus berus and N. naja oxiana showed acute toxic effect on Vibrio fischeri at 4.24 μM level (Table 1), many snake venom phospholipases A2 have been shown antibacterial and antiparasitic properties. For example, the Lys49 protein from Bothrops asper venom showed bactericidal activity on both, gram-positive and gram-negative bacteria [27]. Contrarily, the Lys49 BmarPLA2 from Bothrops marajoensis showed no antibacterial and antiparasitic effects [36]. Two myotoxic Asp49 PLA2s from Bothrops neuwiedi pauloensis venom were bactericidal towards Escherichia coli and Staphylococcus aureus [31]. Myotoxin I Lys49 PLA2 from Bothrops atrox venom was weakly bactericidal against E. coli [30]. Myotoxin I Lys49 PLA2 and myotoxin II Asp49 PLA2from Bothrops jararacussu venom showed antibacterial effect against gram-negative bacteria Xanthomonas [54]. Myotoxic Asp49 PLA2 MTX-I and Lys49 PLA2 MTX-II isolated from Botrops brazili venom and cationic synthetic peptides derived from their 115–129 C-terminal region displayed toxic effects against E. coli, Candida albicans and Leishmania sp. and human T-cell leukemia (JURKAT) cell lines [55].
In the current study, the 36.17 μM VBBPLA2 totally inhibited the growth of gram-positive bacteria Bacillus subtilis (Table 2, Figure 3A) but did not inhibit the growth of other bacterial strains analyzed (Table 2). VBBPLA2 has highly cationic nature as it contains numerous positively charged Arg and Lys residues that may promote its binding to negatively-charged outer surface of bacteria. The majority of antimicrobial peptides are positively charged at physiological pH, and prevailing view is that their selectivity stems from electrostatic attraction of the cationic peptide to the anionic bacterial membranes [56]. However, to another gram-positive bacterium, Staphylococcus aureus, native VBBPLA2 had no inhibitory effect (Table 2).
The activity and expression of several PLA2 isoforms are increased in several human cancers, including breast, pancreatic and prostate cancers, suggesting that these enzymes may have a central role in both tumor development and progression and thus can be targets for anticancer drugs [12,57]. On the other hand, some snake venom PLA2s may have antitumoral activity [12]. Crotoxin, a noncovalent complex (formed by two nonidentical subunits: a basic PLA2 crotoxinB and a nonenzymatic acidic crotoxinA) isolated from the venom of Crotalus durissus terrificus, exhibits a preferential cytotoxic activity against various types of tumor cells including K-562 cells [58], MCF-7 cells [59] and lung adenocarcinoma A549 cells. Treatment of A549 cells with crotoxin significantly inhibited the cell growth in a dose-dependent manner and displayed anti-angiogenic effects in vitro [60]. Crotoxin has been used in the treatment of different advanced carcinomas [61]. It has been shown that blD- PLA2 from Bothrops leucurus snake venom reduced K-562 cellular viability in a dose-dependent manner causing disruption of cellular membrane integrity [62]. Several secreted PLA2s were found to play role in apoptosis [63]. PLA2 from Naja naja atra venom induced apoptotic cell death of K-562 cells [43]. A Lys49 phospholipase A2 from Protobothrops flavoviridis venom induced caspase-independent apoptotic cell death accompanied by rapid plasma-membrane rupture in human leukaemia cells. However, Asp49 PLA2 from the same venom failed to induce death of JURKAT cells [1].
In this study, different cancer cell lines (PC-3, LNCaP, K-562, MCF-7, B10-F16) were exposed to different PLA2s from V. lebetina, V. berus berus and N. naja oxiana. At the highest concentration tested (~7 μM), there was no inhibitory effect of studied PLA2 preparations towards LNCaP cells (Figure 4A–C). This is coherent with the data of Sved et al. [64] on the consistent and dose-dependent stimulatory effect of human recombinant sPLA2-IIA on LNCaP cell growth. In the current study, the most potent inhibitory effect of studied svPLA2s was observed for VBBPLA2 towards human chronic myeloid leukemic cell line K-562 (Figure 4D). In addition, p-BPB-treated inactive VBBPLA2 yielded 27% loss of viability in K-562 cells. Thus, VBBPLA2-induced cell death is dependent not only of enzymatic activity.
4. Materials and Methods
4.1. Materials
The venoms of V. lebetina and N. n. oxiana were commercial preparations from Tashkent Integrated Zoo Plant (Uzbekistan), V. b. berus venom was obtained from Khimki Serpentarium (Moscow, Russia). Sephadex G-100 (superfine) was product of Pharmacia (Uppsala, Sweden). 2,5-dihydroxybenzoic acid (DHB), 3,5-dichlorophenol, bovine serum albumin (BSA), ovalbumin, carboanhydrase, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), soybean trypsin inhibitor, Substance P, Cytochrome C, insulin B chain, p-bromophenacylbromide (p-BPB) and camptothecin were from Sigma (St. Louis, MO, USA), trypsin (Promega, Madison, WI, USA). WST-1 was from Roche Diagnostics, collagen from Chronolog. Annexin V/Dead Cell Apoptosis Kit with FITC annexin V and propidium iodide (PI) were from Invitrogen, Eugene, OR, USA. All other reagents used were of analytical grade.
4.2. Purification of Enzymes
Vipera lebetina PLA2 was purified according to Vija et al. [18], Vipera berus berus PLA2 (VBBPLA2) was separated from the venom as described by Križaj et al [48]. Naja naja oxiana venom PLA2 (NNOPLA2) was purified by gel filtration on Sephadex G-50 sf. and hydrophobic chromatography on pentylagarose. Purity and molecular masses of enzymes were detected by SDS-PAGE and MALDI-TOF MS (see 4.6.).
4.3. PLA2 Assay
Phospholipase A2 activity was assayed by titrimetric method using egg yolk phosphatidylcholine as a substrate [65]. Briefly, one egg yolk was added to 100 mL of bidistilled water and aqueous emulsion was prepared by homogenisation. Per assay, 1.5 mL of the egg yolk emulsion was diluted with 3 mL of Triton X-100 and CaCl2 being 0.75% and 0.15 mM, respectively. The pH was set at 8.0; 10 μL (0.1 mg/mL) of PLA2 sample was added and the fatty acids released were titrated with 10 mM KOH using a pH-stat (TTT80/pHM84/ABU80, Radiometer, Copenhagen, Denmark) at 25 °C.
4.4. PLA2 Activity Inhibition with p-bromophenacylbromide (p-BPB)
PLA2s (0.4 mg) were dissolved in 0.4 mL of 0.1 M ammonium acetate (pH 7.4) containing 0.4 mM of p-BPB and incubated for 24 h at room temperature. Excess of the reagents was removed by ultrafiltration through the microspin filter (cut-off 5000 MW, Cole-Parmer, Vernon Hills, IL, USA), the protein fraction was washed with 0.1 M ammonium acetate (pH 7.4) and lyophilized.
4.5. Protein Quantification
Protein concentrations were determined using the Pierce micro BCA kit. Bovine serum albumin was used as a standard. During the process of column chromatography, the elution profile of proteins was followed by the absorbance at 280 nm.
4.6. Molecular Mass Detection and Isoelectric Focusing of Proteins
The molecular masses of the purified proteins were determined by SDS-PAGE on 12.5% polyacrylamide gels using the method of Laemmli [66]. Molecular mass standards for SDS-PAGE were albumin—66 kDa, ovalbumin—45 kDa, carboanhydrase—29 kDa, soybean trypsin inhibitor—20 kDa, cytochrome C—12.3 kDa.
The molecular masses of the fractions were also determined using a home-built matrix-assisted laser desorption/ionization-time of flight mass spectrometer (MALDI-TOF MS) (National Institute of Chemical Physics and Biophysics, Tallinn, Estonia). Before the analysis the freeze-dried samples of protein fractions were dissolved in 5 μL of 50% acetonitrile containing 0.1% trifluoroacetic acid. Aliquots of 0.5 μL were applied onto the target, allowed to air dry and 0.5 μL of the matrix solution (2,5-dihydroxybenzoic acid) was applied to the target and allowed to dry in air. The mass calibration standards were cytochrome C, insulin B chain. A nitrogen 337 nm laser (4 ns pulse) was used and at least 30–40 shots were summarized.
Analytical isoelectric focusing was performed on 5% polyacrylamide gel plates according to the method of Vesterberg [67] in Multiphor 2117 (LKB, Bromma, Sweden) apparatus in the pH range of 3.6–9.3. Isoelectric focusing markers were amyloglucosidase (pI 3.60), soybean trypsin inhibitor (pI 4.55), β-lactoglobulin A (pI 5.20), bovine carbonic anhydrase B (pI 5.85), human carbonic anhydrase B (pI 6.55), horse myoglobin-acidic band (pI 6.85), horse myoglobin-basic band (pI 7.35) lentil lectin-acidic band (pI 8.15), lentil lectin-middle band (pI 8.45), lentil lectin-basic band (pI 8.65) and trypsinogen (pI 9.30). The gels were stained for proteins with Coomassie Brilliant Blue R250.
4.7. In-Gel Tryptic Digestion and Mass Fingerprinting of Proteins
After visualization with Coomassie Blue the gel-electrophoresis bands of protein in interest (native or reduced) were excised from SDS-PAGE gels, each gel slice cut into small pieces (1 mm2), placed into eppendorf tubes and treated as described earlier [68]. Equal volumes (0.5 μL) of the peptide mixture and the matrix (2,5-dihydroxybenzoic acid, or α-cyano-4-hydroxycinnamic acid) were mixed on the MALDI-TOF plate. The mass calibration standards were substance P and angiotensin II.
4.8. Preparation of Human Platelet Suspension and Collagen-Induced Platelet Aggregation Assay
Collagen-induced platelet aggregation assays were performed in human platelet-rich plasma (PRP). Blood was collected from healthy adult volunteers who had not taken any medication for at least two weeks prior to sampling. The blood was collected according to the permissions LO2354 (14.12.2010) and LO2513 (21.07.2011).
In order to obtain PRP the blood was dispensed into polystyrene tubes containing 0.129 M sodium citrate (9:1 v/v) as anticoagulant and after centrifugation at 180 × g at room temperature for 10 min platelet suspensions were prepared according to the previously described protocol [69]. Platelet aggregation was measured photometrically in a Whole-Blood aggregometer (Chronolog Corporation, Havertown, PA, USA) under continuous stirring at 900 rpm at 37 °C. Control experiments were done using collagen (platelet agonist) alone.
4.9. Antibacterial Activity
4.9.1. Bacterial Strains
Altogether, four different bacterial strains were used. Naturally luminescent Vibrio fischeri NRRL-B-11177 was purchased from Aboatox (Turku, Finland). Constitutively luminescent Escherichia coli MC1061(pSLlux) and Staphylococcus aureus RN4220(p602/22lux) were constructed earlier by Ivask et al. [70]. Bacillus subtilis BR151 was obtained from Turku University (Finland). Two former strains are gram-negative and two latter ones gram-positive bacteria.
4.9.2. Analysis of Antibacterial Activity of PLA2s
Antibacterial activity of sPLA2s was analyzed using two different methods: (i) inhibition of the luminescence of naturally luminescent gram-negative bacterium Vibrio fischeri after 15 minutes of exposure and (ii) inhibition of the growth of gram-negative bacteria Escherichia coli and Staphylococcus aureus and gram-positive bacteria Bacillus subtilis upon 6 hour exposure to PLA2s of various snakes.
4.9.2.1. Bioluminescence Inhibition Assay Using Vibrio fischeri
The Vibrio fischeri test bacteria were prepared as described in Kurvet et al. [71]. Briefly, V. fischeri bacterial suspension was obtained by rehydration of freeze-dried V. fischeri Reagent (Aboatox, Turku, Finland) using 2% NaCl, stabilized for 40 min at 4 °C and then at 20 °C for 40 min and then used for testing. 2% NaCl served as a test diluent and as a negative control. 3,5-dichlorophenol was used as a positive control. The assay was performed at 20 °C instead of 15 °C recommended by standard operational procedure of Microtox™ (AZUR Environmental, Carlsbad, CA, USA) as most luminometers do not allow the temperature adjustment to 15 °C.
Testing was performed essentially as described in Kahru [50] using 1253 Luminometer and respective software for the data reduction (both BioOrbit, Turku, Finland). Toxicity (15-min EC50), i.e., the concentration of svPLA2 causing a 50% reduction in light output of bacteria after 15-min contact time, was determined from respective concentration-effect curves.
4.9.2.2. Bacterial Growth Inhibition Assays
E. coli, S. aureus and B. subtilis were maintained in LB agar plates (LabM, Lancashire, UK) supplemented with respective antibiotics (see below) at +4 °C. For the toxicity tests, bacteria were cultivated (on a shaker at 200 rpm, 30 °C) overnight in 3 mL of LB medium. As a test medium for the growth inhibition assays and as a diluent for svPLA2s LB medium without NaCl was used. Ampicillin (100 μg/mL) in case of E. coli and kanamycin (50 μg/mL) in case of S. aureus were added to LB medium. No antibiotics were added to B. subtilis culture medium. For the assay, overnight bacterial culture was diluted 1:25 in LB medium containing respective antibiotics (see above). Then, 100 μL of test bacteria was added to 100 μL of the svPLA2 dilution. Each svPLA2 was tested in following concentrations: 500, 250, 125, 62.5 and 31.25 μg/mL. Each svPLA2 concentration was tested in three and the controls in ten replicates. 96-well polystyrene microplates with transparent bottoms and not-transparent sides of the wells (Greiner Bio-One, Frickenhausen, Germany) were used. Optical density of the bacterial suspensions at 600 nm (OD600) was measured using Multiscan Spectrum spectrophotometer (Thermo Scientific, Vantaa, Finland). The measurements were performed in 1 h intervals till 6 h and then also 24 h data were registered. Between the measurements till 6 h the plates were incubated at 30 °C on a shaker (Heidolph Titramax 1000, Schwabach, Germany) at 750 rpm and then statically overnight in the incubator at 30 °C. The inhibition of the growth of bacteria was calculated as percentage of the non-exposed control.
To evaluate the ability of svPLA2-exposed bacteria (after 6 h and 24 h incubation) to grow on solid media, 1 μL of bacterial suspension was streaked onto Petri dishes with LB agar containing no antibiotics. The growth of bacteria was visually checked after incubation of Petri plates at 30 °C for 48 h.
4.10. Human Cell Lines and Toxicity Testing of sPLA2s
The human prostate cancer cell lines PC-3, LNCaP, human chronic myeloid leukemic cell line K-562, breast cancer cell line MCF-7 and mouse melanoma cell line B16-F10 were purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA). PC-3 cells were maintained in DMEM/F-12 medium (Gibco, Grand Island, NY, USA), LNCaP, K-562, MCF-7 and B16-F10 cells in RPMI 1640 medium (Gibco, Grand Island, NY, USA), supplemented with 10% fetal bovine serum (Gibco) and antibiotics (100 units/mL penicillin and 100 μg/mL streptomycin) at 37 °C and 5% CO2 in a fully humidified atmosphere.
4.10.1. Analysis of the Viability of the Cells
The viability was determined by the MTT assay (PC-3 cells) and WST-1 assay (LNCaP, K-562, MCF-7 and B16-F10) based on the reduction of MTT or WST-1 by viable cells, respectively.
4.10.1.1. MTT Assay
Human prostate cancer PC-3 cells were seeded in 96-well plates (Sarstedt, Germany) at a density of 1–2 × 105 cells/ml. After 24 h of incubation 37 °C the cells were incubated with svPLA2s diluted with medium and added to the wells at final concentrations of 10 and 100 μg/mL. The cells not treated with sPLA2 served as a control. After certain time intervals, MTT solution was added to each well at a final concentration of 0.5 mg/mL and the plates were incubated at 37 °C for 4 h. The MTT formazan product was dissolved by addition of 110 μL acidified 2-propanol (in 0.04 N HCl) to each well. The absorbance was detected in micro-plate reader (Multiskan Spectrum, Thermo, Vantaa, Finland) at 540 nm. Cell survival rate was calculated as (absorbance of the treated wells)/(absorbance of the control wells) × 100%.
4.10.1.2. WST-1 Assay
Human LNCaP, K-562, MCF-7 and B16-F10 cells were seeded in 96-well plates at a density of 1–2 × 105 cells/ml. After 24 h of growth cells were incubated with svPLA2s diluted with medium and added to the wells at the desired final concentrations (10 and 100 μg/mL). The cells that were not treated with protein served as control cells. After various time intervals 10 μL/well WST-1 solution was added to each well and the plates were incubated for 1–2 h at 37 °C and 5% CO2. The absorbance of the WST-1 formazan salt was detected in micro-plate reader at 450 nm. Cell survival rate was calculated as (absorbance of the treated wells)/(absorbance of the control wells) × 100%.
4.10.2. Apoptosis Detection Using Annexin V-FITC and Propidium Iodide (PI)
The detection of K-562 cells apoptosis was performed according to the instructions of FITC Annexin-V/Dead Cell Apoptosis Kit with FITC Annexin-V and PI (Invitrogen, Eugene, OR, USA). The suspension of K-562 cells was seeded into 24-well plates (2 × 105 cells/well) on round cover slips and incubated at 37 °C with 5% CO2 for 24 h. After this period, the cells were treated with VBBPLA2 (0.36, 0.72 and 7.23 μM) for 24 h. In case of 0.36 μM the treatment was prolonged to up to 28 h. 4 μM camptothecin-treated cells (4 h) were used as a positive control for apoptosis. The cells were washed twice with cold phosphate-buffered saline (PBS) and 200 μL of Annexin-V binding buffer, 10 μL of Annexin-V-FITC and 10 μL of PI working solution were added. After incubation in the dark for 15 min at room temperature the reaction mixture was removed and the cells were washed with Annexin-V binding buffer. Then, the cover slips with cells were taken out from the wells and the mounted preparations were made. The viability of the treated and non-treated (control) K-562 cells was observed under an epifluorescence microscope Olympus CX41 with a 100× oil immersion objective lens and fluorescence optics (excitation at 488 nm, >515 nm emission for Annexin V-FITC (green) and for propidium iodide (red)). The pictures were taken using an Olympus U-CMAD3 real time colour digital DP71 camera (Tokyo, Japan) using the CellB Software (Olympus Soft Imaging Solutions GmbH, Münster, Germany).
5. Conclusions
The adverse effects of PLA2s from Vipera lebetina, Vipera berus berus and Naja naja oxiana venom depended on venom (snake) as well as on target cells (platelets, different cancer cell types and bacteria). As a rule, the observed biological effects on platelets were observed already at 1 μg/mL level (<0.1 μM) and all three PLA2s were dose-dependently inhibiting the collagen-induced platelet aggregation. The chemical modification of histidine in studied PLA2s by p-bromophenacylbromide resulted in complete loss of their catalytic activity and inhibitory action on collagen-induced platelet aggregation. VBBPLA2 (but not the PLA2s from V. lebetina and N. naja oxiana) was totally inhibiting the growth of gram-positive Bacillus subtilis at 500 μg/mL (36.2 μM) whereas the inhibitory effect was not due to its catalytic activity but to other properties of the protein. To another gram-positive bacterium, S. aureus, native sPLA2 from N. naja oxiana inhibited the growth of bacteria by 42% but caused only slight inhibition of growth of B. subtilis. None of the studied svPLA2s was inhibitory to the growth of gram-negative bacteria E. coli even at 500 μg/mL (~37 μM) level.
The viability of the most sensitive cancer cell type (K-562) was reduced upon exposure of the cells to 7.2 μM VBBPLA2 and to some extent also by PLA2s from V. lebetina and N. naja oxiana. There was no inhibitory effect of all studied svPLA2 preparations towards LNCaP cells and low inhibitory effect (8%–20%) towards the PC-3, MCF-7 and B10-F16 cells. Thus, from the current suite of studied svPLA2s and test cells, VBBPLA2 was most growth inhibitory towards gram positive bacteria B. subtilis and K-562 cells in vitro.
Acknowledgements
The work is financially supported by Estonian Science Foundation Grant No. 8899 and by the Estonian Ministry of Education and Research Target Financing Grant No.SF0690063s08.
Conflict of interest
The authors have no conflict of interest.
References
- Murakami, M.; Taketomi, Y.; Miki, Y.; Sato, H.; Hirabayashi, T.; Yamamoto, K. Recent progress in phospholipase A2 research: From cells to animals to humans. Progr. Lipid Res. 2011, 50, 152–192. [Google Scholar] [CrossRef]
- Schaloske, R.H.; Dennis, E.A. The phospholipase A2 superfamily and its group numbering system. Biochim. Biophys. Acta 2006, 1761, 1246–1259. [Google Scholar]
- Burke, J.E.; Dennis, E.A. Phospholipase A2 structure/function, mechanism, and signaling. J. Lipid Res. 2009, 59, S237–S242. [Google Scholar]
- Nevalainen, T.J.; Cardoso, J.C.; Riikonen, P.T. Conserved domains and evolution of secreted phospholipases A(2). FEBS J. 2012, 279, 636–649. [Google Scholar] [CrossRef]
- Lomonte, B.; Angulo, Y.; Sasa, M.; Gutiérrez, J.M. The phospholipase A2 homologues of snake venoms: Biological activities and their possible adaptive roles. Protein Pept. Lett. 2009, 16, 860–876. [Google Scholar] [CrossRef]
- Doley, R.; Zhou, X.; Kini, R.M. Snake Venom Phospholipase A2 Enzymes. In Handbook of Venoms and Toxins of Reptiles; Mackessy, S.P., Ed.; CRC Press: Boca Raton, FL, USA, 2010; pp. 173–205. [Google Scholar]
- Kini, R.M. Excitement ahead: Structure, function and mechanism of snake venom phospholipase A2 enzymes. Toxicon 2003, 42, 827–840. [Google Scholar] [CrossRef]
- Kini, R.M. Structure-function relationships and mechanism of anticoagulant phospholipase A2 enzymes from snake venoms. Toxicon 2005, 45, 1147–1161. [Google Scholar] [CrossRef]
- Lambeau, G.; Lazdunski, M. Receptors for a growing family of secreted phospholipases A2. Trends Pharmacol. Sci. 1999, 20, 162–170. [Google Scholar] [CrossRef]
- Soares, A.M.; Giglio, J.R. Chemical modifications of phospholipases A2 from snake venoms: Effects on catalytic and pharmacological properties. Toxicon 2003, 42, 855–868. [Google Scholar] [CrossRef]
- Montecucco, C.; Gutiérrez, J.M.; Lomonte, B. Cellular pathology induced by snake venom phospholipase A2 myotoxins and neurotoxins: Common aspects of their mechanism of action. Cell. Mol. Life Sci. 2008, 65, 2897–2912. [Google Scholar] [CrossRef]
- Rodrigues, R.S.; Izidoro, L.F.; de Oliveira, R.J., Jr.; Sampaio, S.V.; Soares, A.M.; Rodrigues, V.M. Snake venom phospholipases A2: A new class of antitumor agents. Protein Pept. Lett. 2009, 16, 894–898. [Google Scholar] [CrossRef]
- Ouyang, C.; Huang, T.F. Potent platelet aggregation inhibitor from Trimeresurus gramineus snake venom. Biochim. Biophys. Acta 1983, 757, 332–341. [Google Scholar] [CrossRef]
- Chen, R.H.; Chen, Y.C. Isolation of an acidic phospholipase A2 from the venom of Agkistrodon acutus (five pace snake) and its effect on platelet aggregation. Toxicon 1989, 27, 675–682. [Google Scholar] [CrossRef]
- Kini, R.M.; Evans, H.J. Effects of Phospholipase A2 Rnzymes on Platelet Sggregation. In Venom phospholipase A2 Rnzymes: Dtructure, Gunction and Mechanism; Kini, R.M., Ed.; John Wiley & Sons: Chichester, UK, 1997; pp. 369–387. [Google Scholar]
- Sim, K.L. Purification and preliminary characterisation of praelongin phospholipases, antiplatelet agents from the snake venom of Acanthophis praelongus. Biochim. Biophys. Acta 1998, 1379, 198–206. [Google Scholar]
- Fuly, A.L.; Soares, A.M.; Marcussi, S.; Giglio, J.R.; Guimarães, J.A. Signal transduction pathways involved in the platelet aggregation induced by a D-49 phospholipase A2 isolated from Bothrops jararacussu snake venom. Biochimie 2004, 86, 731–739. [Google Scholar] [CrossRef]
- Vija, H.; Samel, M.; Siigur, E.; Aaspõllu, A.; Trummal, K.; Tõnismägi, K.; Subbi, J.; Siigur, J. Purification, characterization, and cDNA cloning of acidic platelet aggregation inhibiting phospholipases A2 from the snake venom of Vipera lebetina (Levantine viper). Toxicon 2009, 54, 429–439. [Google Scholar] [CrossRef]
- Tsai, I.H.; Wang, Y.M.; Cheng, A.C.; Starkov, V.; Osipov, A.; Nikitin, I.; Makarova, Y.; Ziganshin, R.; Utkin, Y. cDNA cloning, structural, and functional analyses of venom phospholipases A2 and a Kunitz-type protease inhibitor from steppe viper Vipera ursinii renardi. Toxicon 2011, 57, 332–341. [Google Scholar] [CrossRef]
- Prasad, B.N.; Kemparaju, K.; Bhatt, K.G.; Gowda, T.V. A platelet aggregation inhibitor phospholipase A2 from Russell’s viper (Vipera russelli) venom: Isolation and characterization. Toxicon 1996, 34, 1173–1185. [Google Scholar] [CrossRef]
- Fuly, A.L.; Machado, O.L.T.; Alves, E.W.; Carlini, C.R. Mechanism of inhibitory action on platelet activation of a phospholipase A2 isolated from Lachesis muta (Bushmaster) snake venom. Thromb. Haemost. 1997, 78, 1372–1380. [Google Scholar]
- Chow, G.; Subburaju, S.; Kini, R.M. Purification, characterization, and amino acid sequence determination of acanthins, potent inhibitors of platelet aggregation from Acanthophis antarcticus (common death adder) venom. Arch. Biochem. Biophys. 1998, 354, 232–238. [Google Scholar] [CrossRef]
- Kemparaiu, K.; Krishnakanth, T.P.; Veerabasappa Gowda, T. Purification and characterization of a platelet aggregation inhibitor acidic phospholipase A2 from Indian saw-scaled viper (Echis carinatus) venom. Toxicon 1999, 37, 1659–1671. [Google Scholar] [CrossRef]
- Serrano, S.M.T.; Reichl, A.P.; Mentele, R.; Auerswald, E.A.; Santoro, M.L.; Sampaio, C.A.M.; Camargo, A.C.M.; Assakura, M.T. A novel phospholipase A2, BJ-PLA2, from the venom of the snake Bothrops jararaca: Purification, primary structure analysis, and its characterization as a platelet-aggregation-inhibiting factor. Arch. Biochem. Biophys. 1999, 367, 26–32. [Google Scholar] [CrossRef]
- Roberto, P.G.; Kashima, S.; Marcussi, S.; Pereira, J.O.; Astolfi-Filho, S.; Nomizo, A.; Giglio, J.R.; Fontes, M.R.; Soares, A.M.; Franca, S.C. Cloning and identification of a complete cDNA coding for a bactericidal and antitumoral acidic phospholipase A2 from Bothrops jararacussu venom. Protein J. 2004, 23, 273–285. [Google Scholar] [CrossRef]
- Satish, S.; Tejaswini, J.; Krishnakantha, T.P.; Gowda, T.V. Purification of a Class B1 platelet aggregation inhibitor phospholipase A2 from Indian cobra (Naja Naja) venom. Biochimie 2004, 86, 203–210. [Google Scholar] [CrossRef]
- Páramo, L.; Lomonte, B.; Pizarro-Cerdá, J.; Bengoechea, J.A.; Gorvel, J.P.; Moreno, E. Bactericidal activity of Lys49 and Asp49 myotoxic phospholipases A2 from Bothrops asper snake venom—Synthetic Lys49 myotoxin II-(115–129)-peptide identifies its bactericidal region. Eur. J. Biochem. 1998, 253, 452–461. [Google Scholar]
- Koduri, R.S.; Gronroos, J.O.; Laine, V.J.; le Calvez, C.; Lambeau, G.; Nevalainen, T.J.; Gelb, M.H. Bactericidal properties of human and murine groups I, II, V, X, and XII secreted phospholipases A2. J. Biol. Chem. 2002, 277, 5849–5857. [Google Scholar]
- Oliveira, D.G.; Toyama, M.H.; Novello, J.C; Beriam, L.O.; Marangoni, S. Structural and functional characterization of basic PLA2 isolated from Crotalus durissus terrificus venom. J. Protein Chem. 2002, 21, 161–168. [Google Scholar] [CrossRef]
- Núñez, V.; Arce, V.; Gutiérrez, J.M.; Lomonte, B. Structural and functional characterization of myotoxin I, a Lys49 phospholipase A2 homologue from the venom of the snake Bothrops atrox. Toxicon 2004, 44, 91–101. [Google Scholar] [CrossRef]
- Rodrigues, V.M.; Marcussi, S.; Cambraia, R.S.; de Araújo, A.L.; Malta-Neto, N.R.; Hamaguchi, A.; Ferro, E.A.; Homsi-Brandeburgo, M.I.; Giglio, J.R.; Soares, A.M. Bactericidal and neurotoxic activities of two myotoxic phospholipases A2 from Bothrops neuwiedi pauloensis snake venom. Toxicon 2004, 44, 305–314. [Google Scholar] [CrossRef]
- Santamaría, C.; Larios, S.; Angulo, Y.; Pizarro-Cerda, J.; Gorvel, J.P.; Moreno, E.; Lomonte, B. Antimicrobial activity of myotoxic phospholipases A2 from crotalid snake venoms and synthetic peptide variants derived from their C-terminal region. Toxicon 2005, 45, 807–815. [Google Scholar] [CrossRef]
- Xu, C.; Ma, D.; Yu, H.; Li, Z.; Liang, J.; Lin, G.; Zhang, Y.; Lai, R. A bactericidal homodimeric phospholipases A2 from Bungarus fasciatus venom. Peptides 2007, 28, 969–973. [Google Scholar] [CrossRef]
- Perumal Samy, R.; Gopalakrishnakone, P.; Ho, B.; Chow, V.T. Purification, characterization and bactericidal activities of basic phospholipase A2 from the venom of Agkistrodon halys (Chinese pallas). Biochimie 2008, 90, 1372–1388. [Google Scholar] [CrossRef]
- Perumal Samy, R.; Gopalakrishnakone, P.; Bow, H.; Puspharaj, P.N.; Chow, V.T.K. Identification and characterization of a phospholipase A2 from the venom of the Saw-scaled viper: Novel bactericidal and membrane damaging activities. Biochimie 2010, 92, 1854–1866. [Google Scholar] [CrossRef]
- Costa Torres, A.F.; Dantas, R.T.; Toyama, M.H.; Diz Filho, E.; Zara, F.J.; Rodrigues de Queiroz, M.G.; Pinto Nogueira, N.A.; Rosa de Oliveira, M.; de Oliveira Toyama, D.; Monteiro, H.S.; Martins, A.M. Antibacterial and antiparasitic effects of Bothrops marajoensis venom and its fractions: Phospholipase A2 and L-amino acid oxidase. Toxicon 2010, 55, 795–804. [Google Scholar] [CrossRef]
- Samy, R.P.; Stiles, B.G.; Gopalakrishnakone, P.; Chow, V.T. Antimicrobial proteins from snake venoms: Direct bacterial damage and activation of innate immunity against Staphylococcus aureus skin infection. Curr. Med. Chem. 2011, 18, 5104–5113. [Google Scholar] [CrossRef]
- Toyama, M.H.; Toyama, D.O.; Joazeiro, P.P.; Carneiro, E.M.; Beriam, L.O.; Marangoni, L.S.; Boschero, A.C. Biological and structural characterization of a new PLA2 from the Crotalus durissus collilineatus venom. Protein J. 2005, 24, 103–112. [Google Scholar] [CrossRef]
- Nevalainen, T.J.; Graham, G.G.; Scott, K.F. Antibacterial actions of secreted phospholipases A2. Biochim. Biophys. Acta 2008, 1781, 1–9. [Google Scholar]
- Roberto, P.G.; Kashima, S.; Soares, A.M.; Chioato, L.; Faca, V.M.; Fuly, A.L.; Astolfi-Filho, S.; Pereira, J.O.; Franca, S.C. Cloning and expression of an acidic platelet aggregation inhibitor phospholipase A2 cDNA from Bothrops jararacussu venom gland. Protein Expr. Purif. 2004, 37, 102–108. [Google Scholar] [CrossRef]
- Maity, G.; Mandal, S.; Chatterjee, A.; Bhattacharyya, D. Purification and characterization of a low molecular weight multifunctional cytotoxic phospholipase A2 from Russell's viper venom. J. Chromatogr. B 2007, 845, 232–243. [Google Scholar] [CrossRef]
- Zouari-Kessentini, R.; Luis, J.; Karray, A.; Kallech-Ziri, O.; Srairi-Abid, N.; Bazaa, A.; Loret, E.; Bezzine, S.; el Ayeb, M.; Marrakchi, N. Two purified and characterized phospholipases A2 from Cerastes cerastes venom, that inhibit cancerous cell adhesion and migration. Toxicon 2009, 53, 444–453. [Google Scholar] [CrossRef]
- Chen, K.C.; Liu, W.H.; Chang, L.S. Taiwan cobra phospholipase A2-elicited JNK activation is responsible for autocrine fas-mediated cell death and modulating Bcl-2 and Bax protein expression in human leukemia K562 cells. J. Cell Biochem. 2010, 109, 245–254. [Google Scholar]
- Murakami, T.; Kamikado, N.; Fujimoto, R.; Hamaguchi, K.; Nakamura, H.; Chijiwa, T.; Ohno, M.; Oda-Ueda, N. A [Lys49] phospholipase A2 from Protobothrops flavoviridis venom induces caspase-independent apoptotic cell death accompanied by rapid plasma-membrane rupture in human leukemia cells. Biosci. Biotechnol. Biochem. 2011, 75, 864–870. [Google Scholar] [CrossRef]
- Khunsap, S.; Pakmanee, N.; Khow, O.; Chanhome, L.; Sitprija, V.; Suntravat, M.; Lucena, S.E.; Perez, J.C.; Sánchez, E.E.J. Purification of a phospholipase A(2) from Daboia russelii siamensis venom with anticancer effects. Venom Res. 2011, 2, 42–51. [Google Scholar]
- Bazaa, A.; Luis, J.; Srairi-Abid, N.; Kallech-Ziri, O.; Kessentini-Zouari, R.; Defilles, C.; Lissitzky, J.C.; el Ayeb, M.; Marrakchi, N. MVL-PLA2, a phospholipase A2 from Macrovipera lebetina transmediterranea venom, inhibits tumor cells adhesion and migratio. Matrix Biol. 2009, 28, 188–193. [Google Scholar] [CrossRef]
- Bazaa, A.; Pasquier, E.; Defilles, C.; Limam, I.; Kessentini-Zouari, R.; Kallech-Ziri, O.; el Battari, A.; Braguer, D.; el Ayeb, M.; Marrakchi, N.; Luis, J. MVL-PLA2, a snake venom phospholipase A2, inhibits angiogenesis through an increase in microtubule dynamics and disorganization of focal adhesions. PLoS One 2010, 5, e10124. [Google Scholar]
- Križaj, I.; Siigur, J.; Samel, M.; Cotiĉ, V.; Gubenšek, F. Isolation, partial characterization and complete amino acid sequence of the toxic phospholipase A2 from the venom of the common viper, Vipera berus berus. Biochim. Biophys. Acta 1993, 1157, 81–85. [Google Scholar]
- Ovchinnikov, Y.A.; Miroshnikov, A.I.; Nazimov, I.V.; Apsalaon, U.R.; Soldatova, L.N. Complete amino acid sequence of phospholipase A2 (isozyme E3) from the venom of middle Asian cobra Naja naja oxiana. J. Bioorg. Khim. 1979, 5, 805–813. [Google Scholar]
- Kahru, A. In vitro toxicity testing using marine luminescent bacteria Photobacterium phosphoreum: The BiotoxTM test. Atla-Altern. Lab. Anim. 1993, 21, 210–215. [Google Scholar]
- Yuan, Y.; Jackson, S.P.; Newnham, H.H.; Mitchell, C.A.; Salem, H.H. An essential role for lysophosphatidylcholine in the inhibition of platelet aggregation by secretory phospholipase A2. Blood 1995, 86, 4166–4174. [Google Scholar]
- Fuly, A.L.; de Miranda, A.L.P.; Zingali, R.B.; Guimarães, J.A. Purification and characterization of a phospholipase A2 isoenzyme isolated from Lachesis muta snake venom. Biochem. Pharmacol. 2002, 63, 1589–1597. [Google Scholar] [CrossRef]
- Gao, R.; Kini, R.M.; Gopalakhrishnakone, P. Purification, properties, and amino acid sequence of a hemoglobinuria-inducing phospholipase A2 MiPLA-1, from micropechis ikeheka venom. Arch. Biochem. Biophys. 1999, 369, 181–192. [Google Scholar] [CrossRef]
- Barbosa, P.S.; Martins, A.M.; Havt, A.; Toyama, D.O.; Evangelista, J.S.; Ferreira, D.P.; Joazeiro, P.P.; Beriam, L.O.; Toyama, M.H.; Fonteles, M.C.; Monteiro, H.S. Renal and antibacterial effects induced by myotoxin I and II isolated from Bothrops jararacussu venom. Toxicon 2005, 46, 376–386. [Google Scholar] [CrossRef]
- Costa, T.R.; Menaldo, D.L.; Oliveira, C.Z.; Santos-Filho, N.A.; Teixeira, S.S.; Nomizo, A.; Fuly, A.L.; Monteiro, M.C.; de Souza, B.M.; Palma, M.S.; et al. Myotoxic phospholipases A(2) isolated from Bothrops brazili snake venom and synthetic peptides derived from their C-terminal region: Cytotoxic effect on microorganism and tumor cells. Peptides 2008, 29, 1645–1656. [Google Scholar] [CrossRef]
- Glukhov, E.; Stark, M.; Burrows, L.L.; Deber, C.M. Basis for selectivity of cationic antimicrobial peptides for bacterial versus mammalian membranes. J. Biol. Chem. 2005, 280, 33960–33967. [Google Scholar]
- Cummings, B.S. Phospholipase A2 as targets for anti-cancer drugs. Biochem. Pharmacol. 2007, 74, 949–959. [Google Scholar] [CrossRef]
- Yan, C.H.; Liang, Z.Q.; Gu, Z.L.; Yang, Y.P.; Reid, P.; Qin, Z.H. Contributions of autophagic and apoptotic mechanisms to CrTX-induced death of K562 cells. Toxicon 2006, 47, 521–530. [Google Scholar] [CrossRef]
- Yan, C.H.; Yang, Y.P.; Qin, Z.H.; Gu, Z.L.; Reid, P.; Liang, Z.Q. Autophagy is involved in cytotoxic effects of crotoxin in human breast cancer cell line MCF-7 cells. Acta Pharmacol. Sin. 2007, 28, 540–548. [Google Scholar] [CrossRef]
- Ye, B.; Xie, Y.; Qin, Z.H.; Wu, J.C.; Han, R.; He, J.K. Anti-tumor activity of CrTX in human lung adenocarcinoma cell line A549. Acta Pharmacol. Sin. 2011, 32, 1397–1401. [Google Scholar] [CrossRef]
- Cura, J.E.; Blanzaco, D.P.; Brisson, C.; Cura, M.A.; Carbol, R.; Larrateguy, L.; Mendez, C.; Sechi, J.C.; Silveira, J.S.; Theiller, E.; de Roodt, A.R.; Vidal, J.C. Phase I and pharmacokinetics study crotoxin (cytotoxic PLA2 NSC-624 244) in patients with advanced cancer. Clin. Cancer Res. 2002, 8, 1033–1041. [Google Scholar]
- Higuchi, D.A.; Barbosa, C.M.V.; Bincoletto, C.; Chagas, J.R.; Magalhaes, A.; Richardson, M.; Sanches, E.F.; Pesquero, J.B.; Araujo, R.C.; Pesquero, J.L. Purification and partial characterization of two phospholipases A2 from Bothrops leucurus venom (white-tailed-jararaca) snake venom. Biochimie 2007, 89, 319–328. [Google Scholar] [CrossRef]
- Taketo, M.M.; Sanoshita, M. Phospholipase A2 and apoptosis. Biochim. Biophys. Acta 2002, 1585, 72–76. [Google Scholar]
- Sved, P.; Scott, K.F.; McLeod, D.; King, N.J.; Singh, J.; Tsatralis, T.; Nikolov, B.; Boulas, J.; Nallan, L.; Gelb, M.H.; et al. Oncogenic action of secreted phospholipase A2 in prostate cancer. Cancer Res. 2004, 64, 6934–6940. [Google Scholar] [CrossRef]
- De Haas, G.H.; Postema, N.M.; Nieuwenhuizen, W.; van Deenen, L.L.M. Purification and properties of phospholipase A from porcine pancreas. Biochim. Biophys. Acta 1968, 159, 103–117. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Vesterberg, O. Isoelectric focusing of proteins in polyacrylamide gels. Biochim. Biophys. Acta 1972, 257, 11–19. [Google Scholar] [CrossRef]
- Siigur, E.; Aaspõllu, A.; Trummal, K.; Tõnismägi, K.; Tammiste, I.; Kalkkinen, N.; Siigur, J. Factor X activator from Vipera lebetina venom is synthesized from different genes. Biochim. Biophys. Acta 2004, 1702, 41–51. [Google Scholar]
- Siigur, J.; Samel, M.; Tõnismägi, K.; Subbi, J.; Siigur, E.; Tu, A.T. Biochemical characterization of lebetase, a direct-acting fibrinolytic enzyme from Vipera lebetina snake venom. Thromb. Res. 1998, 90, 39–49. [Google Scholar] [CrossRef]
- Ivask, A.; Rõlova, T.; Kahru, A. A suite of recombinant luminescent bacterial strains for the quantification of bioavailable heavy metals and toxicity testing. BMC Biotechnol. 2009, 9, 41. [Google Scholar]
- Kurvet, I.; Ivask, A.; Bondarenko, O.; Sihtmäe, M.; Kahru, A. LuxCDABE—Transformed Constitutively Bioluminescent Escherichia coli for toxicity Screening: Comparison with naturally luminous Vibrio fischeri. Sensors 2011, 11, 7865–7878. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).