Animal Toxins Can Alter the Function of Nav1.8 and Nav1.9

{kind=link}
Abstract
:1. Introduction
3. Current Approaches for Studying Nav1.8 and Nav1.9 Function
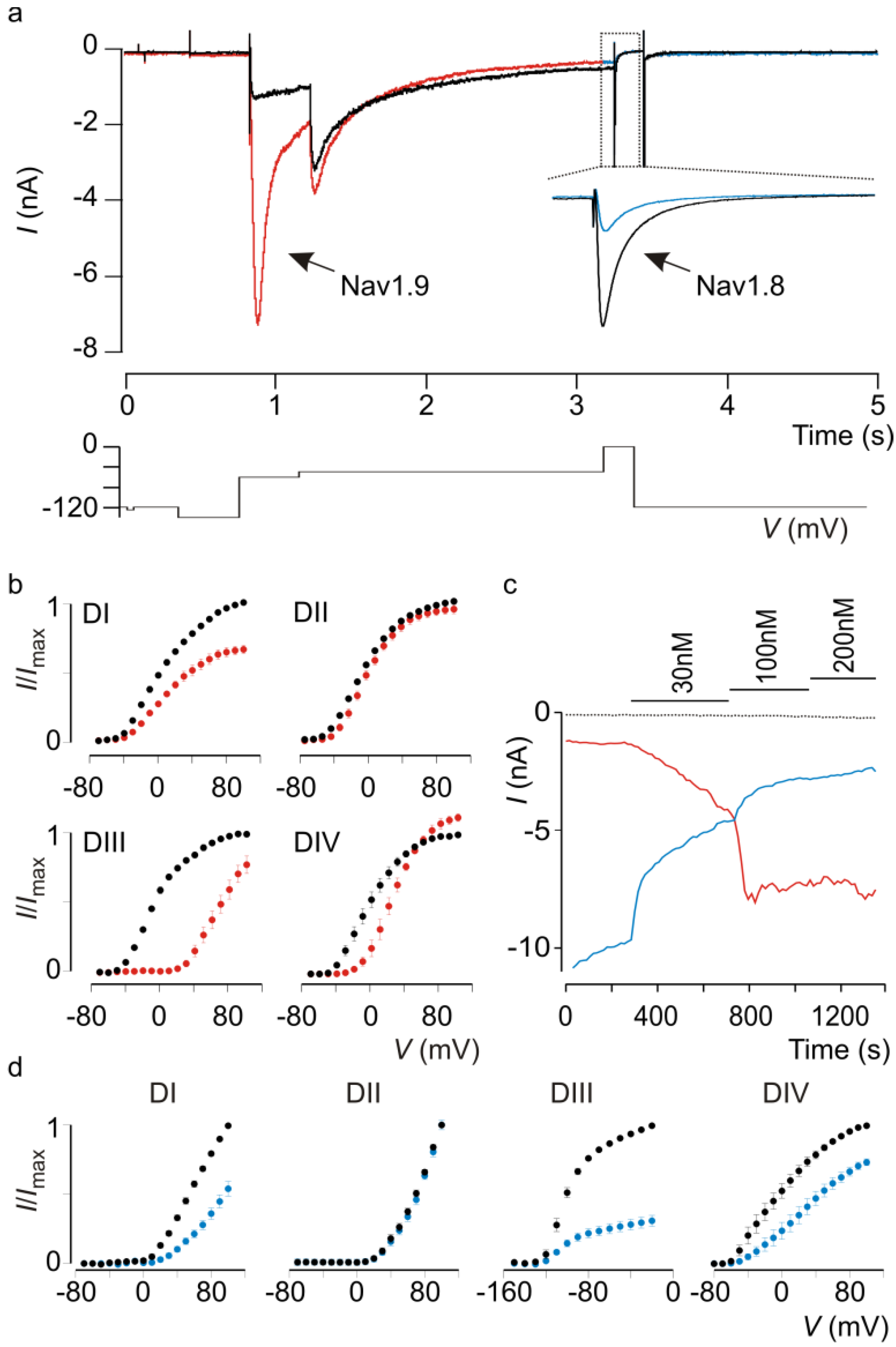
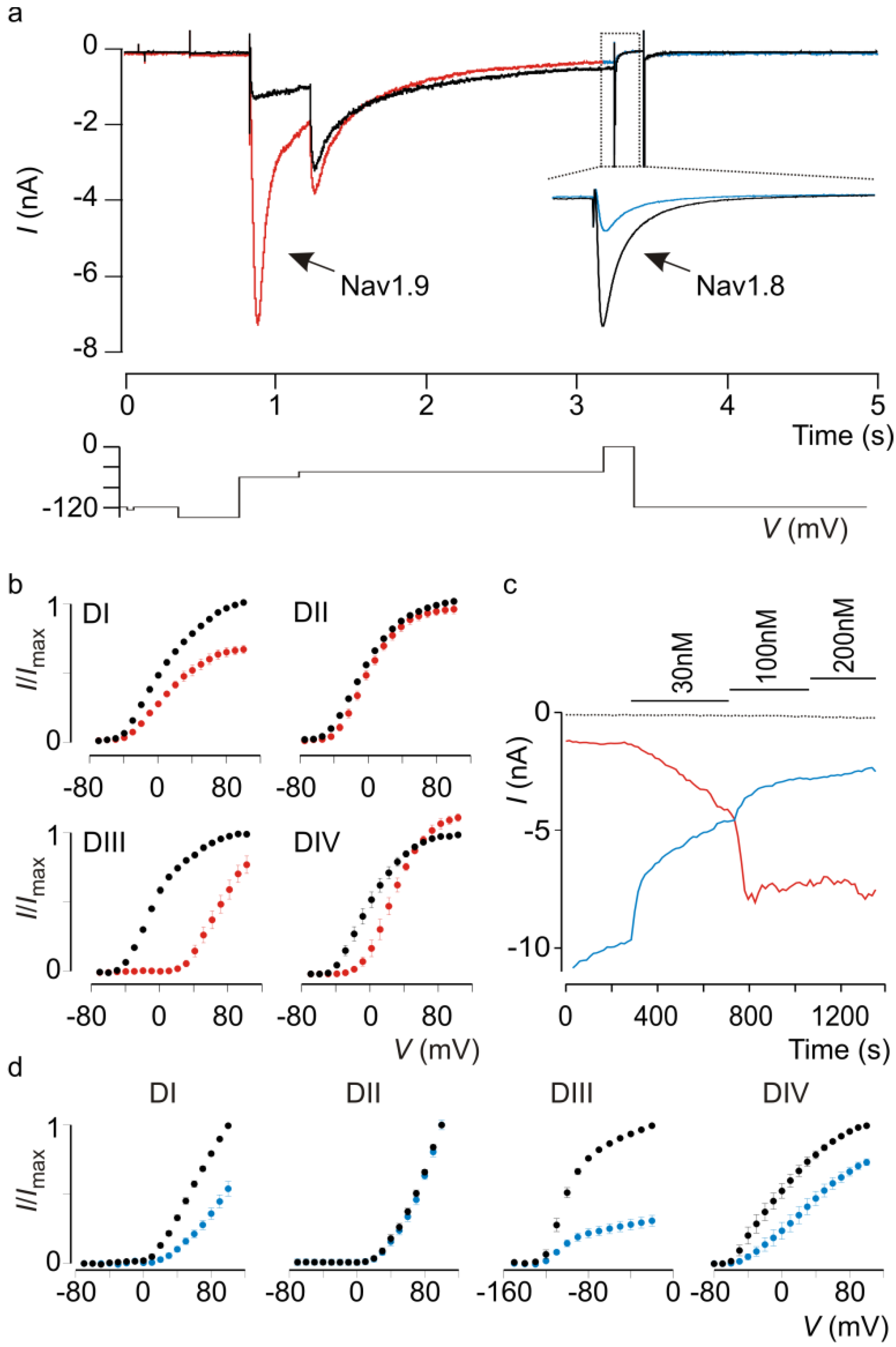
5. Future Challenges
Acknowledgments
Conflict of Interest
References
- Catterall, W.A.; Goldin, A.L.; Waxman, S.G. International union of pharmacology. XlVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 2005, 57, 397–409. [Google Scholar] [CrossRef]
- Cummins, T.R.; Dib-Hajj, S.D.; Black, J.A.; Akopian, A.N.; Wood, J.N.; Waxman, S.G. A novel persistent tetrodotoxin-resistant sodium current in sns-null and wild-type small primary sensory neurons. J. Neurosci. 1999, 19, RC43. [Google Scholar]
- Maingret, F.; Coste, B.; Padilla, F.; Clerc, N.; Crest, M.; Korogod, S.M.; Delmas, P. Inflammatory mediators increase Nav1.9 current and excitability in nociceptors through a coincident detection mechanism. J. Gen. Physiol. 2008, 131, 211–225. [Google Scholar] [CrossRef]
- Maruyama, H.; Yamamoto, M.; Matsutomi, T.; Zheng, T.; Nakata, Y.; Wood, J.N.; Ogata, N. Electrophysiological characterization of the tetrodotoxin-resistant Na+ channel, Na(v)1.9, in mouse dorsal root ganglion neurons. Pflugers Arch. Eur. J. Physiol. 2004, 449, 76–87. [Google Scholar] [CrossRef]
- Ostman, J.A.; Nassar, M.A.; Wood, J.N.; Baker, M.D. Gtp up-regulated persistent Na+ current and enhanced nociceptor excitability require Nav1.9. J. Physiol. 2008, 586, 1077–1087. [Google Scholar]
- Priest, B.T.; Murphy, B.A.; Lindia, J.A.; Diaz, C.; Abbadie, C.; Ritter, A.M.; Liberator, P.; Iyer, L.M.; Kash, S.F.; Kohler, M.G.; et al. Contribution of the tetrodotoxin-resistant voltage-gated sodium channel Nav1.9 to sensory transmission and nociceptive behavior. Proc. Natl. Acad. Sci. USA 2005, 102, 9382–9387. [Google Scholar]
- Rugiero, F.; Mistry, M.; Sage, D.; Black, J.A.; Waxman, S.G.; Crest, M.; Clerc, N.; Delmas, P.; Gola, M. Selective expression of a persistent tetrodotoxin-resistant Na+ current and Nav1.9 subunit in myenteric sensory neurons. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 2715–2725. [Google Scholar]
- Hillsley, K.; Lin, J.H.; Stanisz, A.; Grundy, D.; Aerssens, J.; Peeters, P.J.; Moechars, D.; Coulie, B.; Stead, R.H. Dissecting the role of sodium currents in visceral sensory neurons in a model of chronic hyperexcitability using Nav1.8 and Nav1.9 null mice. J. Physiol. 2006, 576, 257–267. [Google Scholar] [CrossRef]
- Verkerk, A.O.; Remme, C.A.; Schumacher, C.A.; Scicluna, B.P.; Wolswinkel, R.; de Jonge, B.; Bezzina, C.R.; Veldkamp, M.W. Functional Nav1.8 channels in intracardiac neurons: The link between scn10a and cardiac electrophysiology. Circ. Res. 2012, 111, 333–343. [Google Scholar] [CrossRef]
- O’Brien, B.J.; Caldwell, J.H.; Ehring, G.R.; Bumsted O’Brien, K.M.; Luo, S.; Levinson, S.R. Tetrodotoxin-resistant voltage-gated sodium channels Na(v)1.8 and Na(v)1.9 are expressed in the retina. J. Comp. Neurol. 2008, 508, 940–951. [Google Scholar] [CrossRef]
- Wells, J.E.; Bingham, V.; Rowland, K.C.; Hatton, J. Expression of Nav1.9 channels in human dental pulp and trigeminal ganglion. J. Endod. 2007, 33, 1172–1176. [Google Scholar] [CrossRef]
- Padilla, F.; Couble, M.L.; Coste, B.; Maingret, F.; Clerc, N.; Crest, M.; Ritter, A.M.; Magloire, H.; Delmas, P. Expression and localization of the Nav1.9 sodium channel in enteric neurons and in trigeminal sensory endings: Implication for intestinal reflex function and orofacial pain. Mol. Cell. Neurosci. 2007, 35, 138–152. [Google Scholar] [CrossRef]
- Akopian, A.N.; Souslova, V.; England, S.; Okuse, K.; Ogata, N.; Ure, J.; Smith, A.; Kerr, B.J.; McMahon, S.B.; Boyce, S.; et al. The tetrodotoxin-resistant sodium channel sns has a specialized function in pain pathways. Nat. Neurosci. 1999, 2, 541–548. [Google Scholar] [CrossRef]
- Amaya, F.; Wang, H.; Costigan, M.; Allchorne, A.J.; Hatcher, J.P.; Egerton, J.; Stean, T.; Morisset, V.; Grose, D.; Gunthorpe, M.J.; et al. The voltage-gated sodium channel Na(v)1.9 is an effector of peripheral inflammatory pain hypersensitivity. J. Neurosci. 2006, 26, 12852–12860. [Google Scholar] [CrossRef]
- Dib-Hajj, S.; Black, J.A.; Cummins, T.R.; Waxman, S.G. NaN/Nav1.9: A sodium channel with unique properties. Trends Neurosci. 2002, 25, 253–259. [Google Scholar]
- Dib-Hajj, S.D.; Cummins, T.R.; Black, J.A.; Waxman, S.G. Sodium channels in normal and pathological pain. Annu. Rev. Neurosci. 2010, 33, 325–347. [Google Scholar] [CrossRef]
- Leo, S.; D’Hooge, R.; Meert, T. Exploring the role of nociceptor-specific sodium channels in pain transmission using Nav1.8 and Nav1.9 knockout mice. Behav. Brain Res. 2010, 208, 149–157. [Google Scholar] [CrossRef]
- Zimmermann, K.; Leffler, A.; Babes, A.; Cendan, C.M.; Carr, R.W.; Kobayashi, J.; Nau, C.; Wood, J.N.; Reeh, P.W. Sensory neuron sodium channel Nav1.8 is essential for pain at low temperatures. Nature 2007, 447, 855–858. [Google Scholar]
- Baker, M.D.; Wood, J.N. Involvement of Na+ channels in pain pathways. Trends Pharmacol. Sci. 2001, 22, 27–31. [Google Scholar] [CrossRef]
- Gurdon, J.B.; Lane, C.D.; Woodland, H.R.; Marbaix, G. Use of frog eggs and oocytes for the study of messenger RNA and its translation in living cells. Nature 1971, 233, 177–182. [Google Scholar] [CrossRef]
- Neher, E.; Sakmann, B.; Steinbach, J.H. The extracellular patch clamp: A method for resolving currents through individual open channels in biological membranes. Pflugers Arch. Eur. J. Physiol. 1978, 375, 219–228. [Google Scholar] [CrossRef]
- Hamill, O.P.; Marty, A.; Neher, E.; Sakmann, B.; Sigworth, F.J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. Eur. J. Physiol. 1981, 391, 85–100. [Google Scholar] [CrossRef]
- Barnard, E.A.; Miledi, R.; Sumikawa, K. Translation of exogenous messenger rna coding for nicotinic acetylcholine receptors produces functional receptors in xenopus oocytes. Proc. R. Soc. Lond. B Biol. Sci. 1982, 215, 241–246. [Google Scholar] [CrossRef]
- Akopian, A.N.; Sivilotti, L.; Wood, J.N. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature 1996, 379, 257–262. [Google Scholar]
- Bosmans, F.; Puopolo, M.; Martin-Eauclaire, M.F.; Bean, B.P.; Swartz, K.J. Functional properties and toxin pharmacology of a dorsal root ganglion sodium channel viewed through its voltage sensors. J. Gen. Physiol. 2011, 138, 59–72. [Google Scholar] [CrossRef]
- Blair, N.T.; Bean, B.P. Roles of tetrodotoxin (TTX)-sensitive Na+ current, TTX-resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. J. Neurosci. 2002, 22, 10277–10290. [Google Scholar]
- Coste, B.; Crest, M.; Delmas, P. Pharmacological dissection and distribution of NaN/Nav1.9, T-type Ca2+ currents, and mechanically activated cation currents in different populations of DRG neurons. J. Gen. Physiol. 2007, 129, 57–77. [Google Scholar]
- Kerr, B.J.; Souslova, V.; McMahon, S.B.; Wood, J.N. A role for the TTX-resistant sodium channel Nav 1.8 in NGF-induced hyperalgesia, but not neuropathic pain. Neuroreport 2001, 12, 3077–3080. [Google Scholar] [CrossRef]
- Dong, X.W.; Goregoaker, S.; Engler, H.; Zhou, X.; Mark, L.; Crona, J.; Terry, R.; Hunter, J.; Priestley, T. Small interfering RNA-mediated selective knockdown of Na(v)1.8 tetrodotoxin-resistant sodium channel reverses mechanical allodynia in neuropathic rats. Neuroscience 2007, 146, 812–821. [Google Scholar] [CrossRef]
- Joshi, S.K.; Mikusa, J.P.; Hernandez, G.; Baker, S.; Shieh, C.C.; Neelands, T.; Zhang, X.F.; Niforatos, W.; Kage, K.; Han, P.; et al. Involvement of the TTX-resistant sodium channel Nav 1.8 in inflammatory and neuropathic, but not post-operative, pain states. Pain 2006, 123, 75–82. [Google Scholar] [CrossRef]
- Porreca, F.; Lai, J.; Bian, D.; Wegert, S.; Ossipov, M.H.; Eglen, R.M.; Kassotakis, L.; Novakovic, S.; Rabert, D.K.; Sangameswaran, L.; et al. A ccomparison of the potential role of the tetrodotoxin-insensitive sodium channels, PN3/SNS and NaN/SNS2, in rat models of chronic pain. Proc. Natl. Acad. Sci. USA 1999, 96, 7640–7644. [Google Scholar]
- Ritter, A.M.; Martin, W.J.; Thorneloe, K.S. The voltage-gated sodium channel Nav1.9 is required for inflammation-based urinary bladder dysfunction. Neurosci. Lett. 2009, 452, 28–32. [Google Scholar] [CrossRef]
- Martinez, V.; Melgar, S. Lack of colonic-inflammation-induced acute visceral hypersensitivity to colorectal distension in Na(v)1.9 knockout mice. Eur. J. Pain 2008, 12, 934–944. [Google Scholar] [CrossRef]
- Lolignier, S.; Amsalem, M.; Maingret, F.; Padilla, F.; Gabriac, M.; Chapuy, E.; Eschalier, A.; Delmas, P.; Busserolles, J. Nav1.9 channel contributes to mechanical and heat pain hypersensitivity induced by subacute and chronic inflammation. PLoS One 2011, 6, e23083. [Google Scholar]
- Blum, R.; Kafitz, K.W.; Konnerth, A. Neurotrophin-evoked depolarization requires the sodium channel Na(v)1.9. Nature 2002, 419, 687–693. [Google Scholar]
- Tate, S.; Benn, S.; Hick, C.; Trezise, D.; John, V.; Mannion, R.J.; Costigan, M.; Plumpton, C.; Grose, D.; Gladwell, Z.; et al. Two sodium channels contribute to the TTX-R sodium current in primary sensory neurons. Nat. Neurosci. 1998, 1, 653–655. [Google Scholar] [CrossRef]
- Vanoye, C.G.; Ehring, G.R.; George, A.L. Functional analysis of stably expressed human Nav1.9. Biophys. J. 2012, 102, 527. [Google Scholar]
- Fang, X.; Djouhri, L.; Black, J.A.; Dib-Hajj, S.D.; Waxman, S.G.; Lawson, S.N. The presence and role of the tetrodotoxin-resistant sodium channel Na(v)1.9 (NaN) in nociceptive primary afferent neurons. J. Neurosci. 2002, 22, 7425–7433. [Google Scholar]
- Binshtok, A.M.; Bean, B.P.; Woolf, C.J. Inhibition of nociceptors by TRPV1-mediated entry of impermeant sodium channel blockers. Nature 2007, 449, 607–610. [Google Scholar] [CrossRef]
- Coste, B.; Osorio, N.; Padilla, F.; Crest, M.; Delmas, P. Gating and modulation of presumptive Nav1.9 channels in enteric and spinal sensory neurons. Mol. Cell. Neurosci. 2004, 26, 123–134. [Google Scholar] [CrossRef]
- Herzog, R.I.; Cummins, T.R.; Waxman, S.G. Persistent TTX-resistant Na+ current affects resting potential and response to depolarization in simulated spinal sensory neurons. J. Neurophysiol. 2001, 86, 1351–1364. [Google Scholar]
- Gaudioso, C.; Hao, J.; Martin-Eauclaire, M.F.; Gabriac, M.; Delmas, P. Menthol pain relief through cumulative inactivation of voltage-gated sodium channels. Pain 2012, 153, 473–484. [Google Scholar] [CrossRef]
- Leffler, A.; Reiprich, A.; Mohapatra, D.P.; Nau, C. Use-dependent block by lidocaine but not amitriptyline is more pronounced in tetrodotoxin (TTX)-resistant Nav1.8 than in TTX-sensitive Na+ channels. J. Pharmacol. Exp. Ther. 2007, 320, 354–364. [Google Scholar]
- John, V.H.; Main, M.J.; Powell, A.J.; Gladwell, Z.M.; Hick, C.; Sidhu, H.S.; Clare, J.J.; Tate, S.; Trezise, D.J. Heterologous expression and functional analysis of rat Nav1.8 (SNS) voltage-gated sodium channels in the dorsal root ganglion neuroblastoma cell line ND7-23. Neuropharmacology 2004, 46, 425–438. [Google Scholar] [CrossRef]
- Zhou, X.; Dong, X.W.; Crona, J.; Maguire, M.; Priestley, T. Vinpocetine is a potent blocker of rat Nav1.8 tetrodotoxin-resistant sodium channels. J. Pharmacol. Exp. Ther. 2003, 306, 498–504. [Google Scholar] [CrossRef]
- Gaida, W.; Klinder, K.; Arndt, K.; Weiser, T. Ambroxol, a Nav1.8-preferring Na(+) channel blocker, effectively suppresses pain symptoms in animal models of chronic, neuropathic and inflammatory pain. Neuropharmacology 2005, 49, 1220–1227. [Google Scholar] [CrossRef]
- Dekker, L.V.; Daniels, Z.; Hick, C.; Elsegood, K.; Bowden, S.; Szestak, T.; Burley, J.R.; Southan, A.; Cronk, D.; James, I.F. Analysis of human Nav1.8 expressed in SH-SY5Y neuroblastoma cells. Eur. J. Pharmacol. 2005, 528, 52–58. [Google Scholar] [CrossRef]
- Farrag, K.J.; Bhattacharjee, A.; Docherty, R.J. A comparison of the effects of veratridine on tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels in isolated rat dorsal root ganglion neurons. Pflugers Arch. Eur. J. Physiol. 2008, 455, 929–938. [Google Scholar] [CrossRef]
- Jarvis, M.F.; Honore, P.; Shieh, C.C.; Chapman, M.; Joshi, S.; Zhang, X.F.; Kort, M.; Carroll, W.; Marron, B.; Atkinson, R.; et al. A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc. Natl. Acad. Sci. USA 2007, 104, 8520–8525. [Google Scholar]
- Middleton, R.E.; Warren, V.A.; Kraus, R.L.; Hwang, J.C.; Liu, C.J.; Dai, G.; Brochu, R.M.; Kohler, M.G.; Gao, Y.D.; Garsky, V.M.; et al. Two tarantula peptides inhibit activation of multiple sodium channels. Biochemistry 2002, 41, 14734–14747. [Google Scholar]
- Bosmans, F.; Martin-Eauclaire, M.F.; Swartz, K.J. Deconstructing voltage sensor function and pharmacology in sodium channels. Nature 2008, 456, 202–208. [Google Scholar] [CrossRef]
- Bosmans, F.; Swartz, K.J. Targeting voltage sensors in sodium channels with spider toxins. Trends Pharmacol. Sci. 2010, 31, 175–182. [Google Scholar] [CrossRef]
- Bosmans, F.; Rash, L.; Zhu, S.; Diochot, S.; Lazdunski, M.; Escoubas, P.; Tytgat, J. Four novel tarantula toxins as selective modulators of voltage-gated sodium channel subtypes. Mol. Pharmacol. 2006, 69, 419–429. [Google Scholar]
- Zeng, X.; Deng, M.; Lin, Y.; Yuan, C.; Pi, J.; Liang, S. Isolation and characterization of Jingzhaotoxin-V, a novel neurotoxin from the venom of the spider Chilobrachys jingzhao. Toxicon 2007, 49, 388–399. [Google Scholar] [CrossRef]
- Deng, M.; Kuang, F.; Sun, Z.; Tao, H.; Cai, T.; Zhong, L.; Chen, Z.; Xiao, Y.; Liang, S. Jingzhaotoxin-IX, a novel gating modifier of both sodium and potassium channels from Chinese tarantula Chilobrachys jingzhao. Neuropharmacology 2009, 57, 77–87. [Google Scholar] [CrossRef]
- Bulaj, G.; Zhang, M.M.; Green, B.R.; Fiedler, B.; Layer, R.T.; Wei, S.; Nielsen, J.S.; Low, S.J.; Klein, B.D.; Wagstaff, J.D.; et al. Synthetic muO-conotoxin MrVIB blocks TTX-resistant sodium channel Nav1.8 and has a long-lasting analgesic activity. Biochemistry 2006, 45, 7404–7414. [Google Scholar]
- Ekberg, J.; Jayamanne, A.; Vaughan, C.W.; Aslan, S.; Thomas, L.; Mould, J.; Drinkwater, R.; Baker, M.D.; Abrahamsen, B.; Wood, J.N.; et al. MuO-conotoxin MrVIB selectively blocks Nav1.8 sensory neuron specific sodium channels and chronic pain behavior without motor deficits. Proc. Natl. Acad. Sci. USA 2006, 103, 17030–17035. [Google Scholar]
- Knapp, O.; Nevin, S.T.; Yasuda, T.; Lawrence, N.; Lewis, R.J.; Adams, D.J. Biophysical properties of Na(v) 1.8/Na(v) 1.2 chimeras and inhibition by microO-conotoxin MrVIB. Br. J. Pharmacol. 2012, 166, 2148–2160. [Google Scholar] [CrossRef]
- Leipold, E.; DeBie, H.; Zorn, S.; Borges, A.; Olivera, B.M.; Terlau, H.; Heinemann, S.H. MuO conotoxins inhibit NaV channels by interfering with their voltage sensors in domain-2. Channels (Austin) 2007, 1, 253–262. [Google Scholar]
- Bosmans, F.; Maertens, C.; Verdonck, F.; Tytgat, J. The poison dart frog’s batrachotoxin modulates Nav1.8. FEBS Lett. 2004, 577, 245–248. [Google Scholar] [CrossRef]
- Du, Y.; Garden, D.P.; Wang, L.; Zhorov, B.S.; Dong, K. Identification of new batrachotoxin-sensing residues in segment iiis6 of the sodium channel. J. Biol. Chem. 2011, 286, 13151–13160. [Google Scholar]
- Wang, S.Y.; Wang, G.K. Batrachotoxin-resistant Na+ channels derived from point mutations in transmembrane segment d4-s6. Biophys. J. 1999, 76, 3141–3149. [Google Scholar] [CrossRef]
- Rush, A.M.; Waxman, S.G. PGE2 increases the tetrodotoxin-resistant Nav1.9 sodium current in mouse DRG neurons via G-proteins. Brain Res. 2004, 1023, 264–271. [Google Scholar] [CrossRef]
- Cummins, T.R.; Black, J.A.; Dib-Hajj, S.D.; Waxman, S.G. Glial-derived neurotrophic factor upregulates expression of functional SNS and NaN sodium channels and their currents in axotomized dorsal root ganglion neurons. J. Neurosci. 2000, 20, 8754–8761. [Google Scholar]
- Fjell, J.; Cummins, T.R.; Dib-Hajj, S.D.; Fried, K.; Black, J.A.; Waxman, S.G. Differential role of GDNF and NGF in the maintenance of two TTX-resistant sodium channels in adult DRG neurons. Brain Res. Mol. Brain Res. 1999, 67, 267–282. [Google Scholar]
- Copel, C.; Osorio, N.; Crest, M.; Gola, M.; Delmas, P.; Clerc, N. Activation of neurokinin 3 receptor increases Na(v)1.9 current in enteric neurons. J. Physiol. 2009, 587, 1461–1479. [Google Scholar] [CrossRef]
- Bosmans, F.; Escoubas, P.; Nicholson, G.M. Animal Toxins: State of the Art – Perspectives in Health and Biotechnology; Editora UFMG: Belo Horizonte, Brazil, 2009. [Google Scholar]
- Fozzard, H.A.; Lipkind, G.M. The tetrodotoxin binding site is within the outer vestibule of the sodium channel. Mar. Drugs 2010, 8, 219–234. [Google Scholar] [CrossRef]
- Zhang, M.M.; Gruszczynski, P.; Walewska, A.; Bulaj, G.; Olivera, B.M.; Yoshikami, D. Cooccupancy of the outer vestibule of voltage-gated sodium channels by micro-conotoxin KIIIA and saxitoxin or tetrodotoxin. J. Neurophysiol. 2010, 104, 88–97. [Google Scholar] [CrossRef]
- Bulaj, G.; West, P.J.; Garrett, J.E.; Watkins, M.; Zhang, M.M.; Norton, R.S.; Smith, B.J.; Yoshikami, D.; Olivera, B.M. Novel conotoxins from Conus striatus and Conus kinoshitai selectively block TTX-resistant sodium channels. Biochemistry 2005, 44, 7259–7265. [Google Scholar] [CrossRef]
- Wang, C.Z.; Zhang, H.; Jiang, H.; Lu, W.; Zhao, Z.Q.; Chi, C.W. A novel conotoxin from Conus striatus, mu-SIIIA, selectively blocking rat tetrodotoxin-resistant sodium channels. Toxicon 2006, 47, 122–132. [Google Scholar]
- West, P.J.; Bulaj, G.; Garrett, J.E.; Olivera, B.M.; Yoshikami, D. Mu-conotoxin SMIIIA, a potent inhibitor of tetrodotoxin-resistant sodium channels in amphibian sympathetic and sensory neurons. Biochemistry 2002, 41, 15388–15393. [Google Scholar]
- Swartz, K.J. Tarantula toxins interacting with voltage sensors in potassium channels. Toxicon 2007, 49, 213–230. [Google Scholar] [CrossRef]
- Alabi, A.A.; Bahamonde, M.I.; Jung, H.J.; Kim, J.I.; Swartz, K.J. Portability of paddle motif function and pharmacology in voltage sensors. Nature 2007, 450, 370–375. [Google Scholar] [CrossRef]
- Chakrapani, S.; Cuello, L.G.; Cortes, D.M.; Perozo, E. Structural dynamics of an isolated voltage-sensor domain in a lipid bilayer. Structure 2008, 16, 398–409. [Google Scholar] [CrossRef]
- Jiang, Y.; Lee, A.; Chen, J.; Ruta, V.; Cadene, M.; Chait, B.T.; MacKinnon, R. X-ray structure of a voltage-dependent K+ channel. Nature 2003, 423, 33–41. [Google Scholar] [CrossRef]
- Jiang, Y.; Ruta, V.; Chen, J.; Lee, A.; MacKinnon, R. The principle of gating charge movement in a voltage-dependent K+ channel. Nature 2003, 423, 42–48. [Google Scholar] [CrossRef]
- Long, S.B.; Tao, X.; Campbell, E.B.; MacKinnon, R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 2007, 450, 376–382. [Google Scholar] [CrossRef]
- Phillips, L.R.; Milescu, M.; Li-Smerin, Y.; Mindell, J.A.; Kim, J.I.; Swartz, K.J. Voltage-sensor activation with a tarantula toxin as cargo. Nature 2005, 436, 857–860. [Google Scholar]
- Milescu, M.; Bosmans, F.; Lee, S.; Alabi, A.A.; Kim, J.I.; Swartz, K.J. Interactions between lipids and voltage sensor paddles detected with tarantula toxins. Nat. Struct. Mol. Biol. 2009, 16, 1080–1085. [Google Scholar] [CrossRef]
- Milescu, M.; Vobecky, J.; Roh, S.H.; Kim, S.H.; Jung, H.J.; Kim, J.I.; Swartz, K.J. Tarantula toxins interact with voltage sensors within lipid membranes. J. Gen. Physiol. 2007, 130, 497–511. [Google Scholar] [CrossRef]
- Lee, S.Y.; MacKinnon, R. A membrane-access mechanism of ion channel inhibition by voltage sensor toxins from spider venom. Nature 2004, 430, 232–235. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gilchrist, J.; Bosmans, F. Animal Toxins Can Alter the Function of Nav1.8 and Nav1.9. Toxins 2012, 4, 620-632. https://doi.org/10.3390/toxins4080620
Gilchrist J, Bosmans F. Animal Toxins Can Alter the Function of Nav1.8 and Nav1.9. Toxins. 2012; 4(8):620-632. https://doi.org/10.3390/toxins4080620
Chicago/Turabian StyleGilchrist, John, and Frank Bosmans. 2012. "Animal Toxins Can Alter the Function of Nav1.8 and Nav1.9" Toxins 4, no. 8: 620-632. https://doi.org/10.3390/toxins4080620
APA StyleGilchrist, J., & Bosmans, F. (2012). Animal Toxins Can Alter the Function of Nav1.8 and Nav1.9. Toxins, 4(8), 620-632. https://doi.org/10.3390/toxins4080620