Immunochemical Methods for Ochratoxin A Detection: A Review

Abstract
:1. Introduction
2. Antibodies
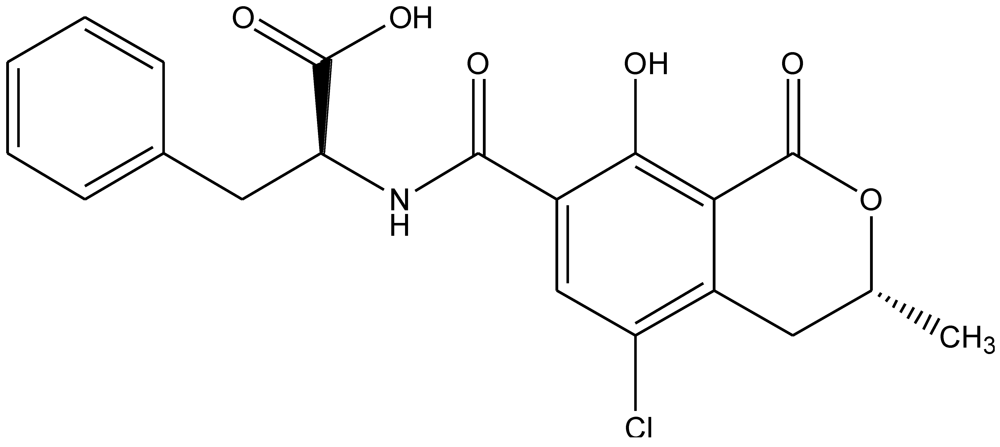
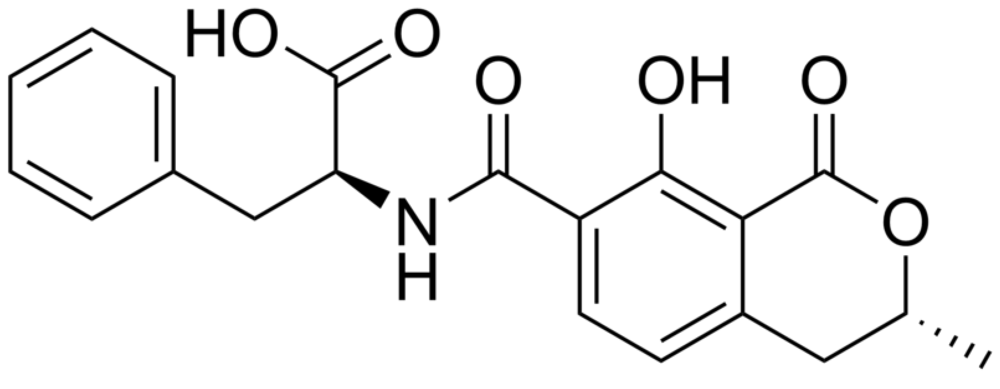
3. Immunoassay Formats
3.1. RIA (Radioimmunoassay)
3.2. EIA (Enzyme Immunoassay)/ELISA (Enzyme-Linked Immuno Sorbent Assay)

{kind=link}
{kind=link}
| Year | Antibody | Format | Matrix | D.L. | Validation | Reference |
|---|---|---|---|---|---|---|
| 1981 | PAb 1 | dc | buffer | 25 pg | cr | [69] |
| 1988 | MAb 1 | ic | barley | 5 µg/kg | − | [70] |
| 1989 | MAb 1 | ic | various | 50 pg/mL | + | [71] |
| 1990 | MAb 1 | dc | barley | 1 ng/mL | + | [72] |
| 1991 | MAb 1 | dc | barley | 1 ng/mL | + | [73] |
| 1996 | MAb 1 | dc | cereals | 0.5 ng/g | + | [74] |
| 1996 | MAb 1 | dc | buffer | 45 pg/mL | cr | [75] |
| 1997 | MAb 1 | dc | serum | 0.2 ng/mL | + | [34] |
| 1998 | MAb 1 | dc | plasma | 4–20 pg/mL | − | [38] |
| 1999 | MAb 2 | m | wheat | 0.4 ng/mL | +/− | [76] |
| 2000 | MAb 1 | ic | chilies | 5 ng/mLa | + | [77] |
| 2002 | MAb 1 | dc | plasma/tissue | 0.5 ng/g | + | [43] |
| 2002 | MAb 2 | m | coffee | 4 µg/kg | cr | [78] |
| 2006 | MAb 1 | ic | coffee | 50 pg/mL | + | [28] |
| 2007 | MAb 1 | ic | coffee | 3.73 ng/g | + | [79] |
| 2007 | PAb 1 | m/dc | chilies | 10 µg/kg | + | [80] |
| 2007 | PAb 1 | dc | various | 1 ng/mL | +/− | [81] |
| 2009 | MAb | str | corn | 2.5 ng/mL | − | [82] |
| 2011 | PAb 1 | str | grains | 1.5 µg/kg | + | [83] |
| 2011 | MAb 1 | ic | cereals | 1.70 ng/mL a | − | [84] |
3.3. Sandwich ELISA
3.4. Chemiluminenscent Immunoassay (CL-IA)
3.5. Fluorescent Immunoassay (FIA)
3.5.1. Time-Resolved Fluorescent Immunoassay (TR-FIA)
3.5.2. Fluorescence-Polarization Immunoassay (FP-IA)
3.5.3. FRET (Fluorescence Resonance Energy Transfer) Immunoassay
3.5.4. Fluorescent Micro-Array Immunoassay
3.6. Lateral Flow Immunoassay (LF-IA)
3.7. Flow-Through Immunoassay
4. Immunosensors
5. Synthetic/Chemical Antibodies
6. Immunoaffinity Chromatography
7. Summary
Conflict of Interest
References
- van Egmond, H.P. Methods for determing ochratoxin A and other nephrotoxic mycotoxins. IARC Sci. Publ. 1991, 115, 57–70. [Google Scholar]
- van Egmond, H.P. Analytical methodology and regulations for ochratoxin A. Food Addit. Contam. 1996, 13 (Suppl.), 11–13. [Google Scholar]
- van Egmond, H.P.; Schothorst, R.C.; Jonker, M.A. Regulations relating to mycotoxins in food. perspectives in a global and European context. Anal. Bioanal. Chem. 2007, 389, 147–157. [Google Scholar] [CrossRef]
- Bazin, I.; Nabais, E.; Lopez-Ferber, M. Rapid visual tests: Fast and reliable detection of ochratoxin A. Toxins 2010, 2, 2230–2241. [Google Scholar] [CrossRef]
- Crosby, N.T. Determination of Veterinary Residues in Food; Woodhead Publishing Series in Food Science: Cambridge, UK, 1984; p. 177. [Google Scholar]
- Gilbert, J.; Anklam, E. Validation of analytical methods for determining mycotoxins in foodstuffs. Trends Anal. Chem. 2002, 21, 468–486. [Google Scholar] [CrossRef]
- Shephard, G.S.; Berthiller, F.; Burdaspal, P.; Crews, C.; Jonker, M.A.; Krska, R.; MacDonald, S.; Malone, B.; Maragos, C.; Sabino, M.; et al. Developments in mycotoxins analysis: An update for 2009–2010. World Mycotoxin J. 2011, 4, 3–28. [Google Scholar] [CrossRef]
- Krska, R.; Schubert-Ullrich, P.; Molinelli, A.; Sulyok, M.; Macdonald, S.; Crews, C. Mycotoxin analysis: An update. Food Addit. Contamin. 2008, 25, 152–163. [Google Scholar] [CrossRef]
- Turner, N.W.; Subrahmanyam, S.; Pieletsky, S.A. Analytical methods for determination of mycotoxins: A review. Anal. Chim. Acta 2009, 632, 168–180. [Google Scholar] [CrossRef]
- Fukal, L.; Reisnerova, H. Monitoring of aflatoxins and ochratoxin A in Czechoslovac human sera by immunoassay. Bull. Environ. Contam. Toxicol. 1990, 44, 345–349. [Google Scholar]
- Jorgensen, K.; Rasmussen, G.; Thorup, I. Ochratoxin A in Danish cereals 1986–1992 and daily intake by the Danish population. Food Addit. Contam. 1996, 13, 95–104. [Google Scholar] [CrossRef]
- Ominski, K.H.; Frohlich, A.A.; Marquardt, R.R.; Crow, G.H.; Abrahmson, D. The incidence and distribution of ochratoxin A in western Canadian swine. Food Addit. Contam. 1996, 13, 185–198. [Google Scholar] [CrossRef]
- Valenta, H.; Goll, M. Determination of ochratoxin A in regional samples of cows milk from Germany. Food Addit. Contam. 1996, 13, 669–676. [Google Scholar] [CrossRef]
- Zimmerli, B.; Dick, R. Ochratoxin A in table wine and grapejuice: Occurrence and risk assessment. Food Addit. Contam. 1996, 13, 655–668. [Google Scholar] [CrossRef]
- Scudamore, K.A.; Hetmanski, M.T.; Nawaz, S.; Naylor, J.; Rainbird, S. Determination of mycotoxins in pet foods sold for domestic pets and wild birds using linked-column immunoassay clean-up and HPLC. Food Addit. Contam. 1997, 14, 175–186. [Google Scholar] [CrossRef]
- Thellmann, A.; Weber, W. Bestimmung von Ochratoxin A in Getreide, Malz und Bier nach Anreicherung und Separation an Immunaffinitätssäulen und anschließender Hochleistungflüssigkeitschromatographie mit Fluoreszenzdetektion. Deutsche Lebensmittel-Rundschau 1997, 93, 1–3. [Google Scholar]
- Skaug, M.A. Analysis of Norwegian milk and infant formulas for ochratoxin A. Food Addit. Contam. 1999, 16, 75–78. [Google Scholar] [CrossRef] [Green Version]
- Pietri, A.; Bertuzzi, T.; Pallaroni, L.; Piva, G. Occurrence of ochratoxin A in Italian wines. Food Addit. Contam. 2001, 18, 647–654. [Google Scholar]
- Thuvander, A.; Paulsen, J.E.; Axberg, K.; Johansson, N.; Vidnes, A.; Enghardt-Barbien, H.; Trygg, K.; Lund-Larsen, K.; Jahn, S.; Widenfalk, A.; et al. Levels of ochratoxin A in blood from Norwegian and Swedish blood donors and their possible correlation with food consumption. Food Chem. Toxicol. 2001, 39, 1145–1151. [Google Scholar] [CrossRef]
- Belli, N.; Marin, S.; Sanchis, V.; Ramos, A.J. Review: Ochratoxin A (OTA) in wines, musts and grapejuices: Occurrence, regulations and methods of analysis. Food Sci. Technol. Int. 2002, 8, 325–335. [Google Scholar]
- Fazekas, B.; Tar, A.K.; Zomborszki-Covács, M. Ochratoxin A contamination of cereal grains and coffee in Hungary in the year 2001. Acta Vet. Hungarica 2002, 50, 177–188. [Google Scholar] [CrossRef]
- Rizzo, A.; Eskola, M.; Atroshi, F. Ochratoxin A in cereals, foodstuffs and human plasma. Eur. J. Plant Pathol. 2002, 108, 631–637. [Google Scholar] [CrossRef]
- Lombaert, G.A.; Pellaers, P.; Roscoe, V.; Mankotia, M.; Neil, R.; Scott, P.M. Mycotoxins in infant cereal foods from the Canadian retail market. Food Addit. Contan. 2003, 20, 494–504. [Google Scholar] [CrossRef]
- Battilani, P.; Magan, N.; Logrico, A. European research on ochratoxin A in grapes and wine. Int. J. Food Microbiol. 2006, 111, S2–S4. [Google Scholar] [CrossRef]
- Aydin, A.; Gunsen, U.; Demirel, S. Total aflatoxin, aflatoxin B1 and ochratoxin A levels in Turkish wheat flour. J. Food Drug Anal. 2008, 16, 48–53. [Google Scholar]
- Martins, H.M.; Marques, M.; Almeida, I.; Guerra, M.M.; Bernardo, F. Mycotoxins in feedstuffs in Portugal: An overview. Mycotoxin Res. 2008, 24, 19–23. [Google Scholar] [CrossRef]
- Flajs, D.; Domijan, A.M.; Ivic, D.; Svjetkovic, B.; Peraica, M. ELISA and HPLC analysis of ochratoxin A in red wines of Croatia. Food Control 2009, 20, 590–592. [Google Scholar] [CrossRef]
- Fujii, S.; Ono, E.Y.S.; Fungaro, M.H.P.; Itano, E.N.; Oliviera, T.C.R.N.; Prete, C.E.C.; Taniwaki, M.H.; Kawamura, O.; Ueno, Y.; Hirooka, E.Y. Indirect competitive ELISA for ochratoxin A detection in coffee and molecular identification of ochratoxin A producing Aspergillus strains. In Mycotoxins and Phycotoxins: Advances in Determination, Toxicology and Exposure Management; Njapau, H., Trujilo, S., van Egmond, H., Park, D., Eds.; Wageningen Academic Press: New York, NY, USA, 2009; pp. 61–71. [Google Scholar]
- Prieto-Simon, B.; Campas, M. Immunochemical tools for mycotoxins detection in food. Monatsh. Chem. 2009, 140, 915–920. [Google Scholar] [CrossRef]
- Santos, L.; Marin, S.; Sanchis, V.; Ramos, A.J. Screening of mycotoxins multicontamination in medicinal and aromatic herbs sampled in Spain. J. Sci. Food Agric. 2009, 89, 1802–1807. [Google Scholar] [CrossRef]
- Breitholz, A.; Olsen, M.; Dahlbäck, A.; Hult, K. Plasma ochratoxin A levels in three Swedish populations surveyed using an ion-pair HPLC technique. Food Addit. Contam. 1991, 8, 183–192. [Google Scholar] [CrossRef]
- Turconi, G.; Guarcello, M.; Livieri, C.; Comizzoli, S.; Maccarini, L.; Castelazzi, A.M.; Pietri, A.; Piva, G.; Roggi, C. Evaluation of xenobiotics in human milk and ingestion by the newborn. An epidemiological survey in Lombardy (Northern Italy). Eur. J. Nutr. 1994, 43, 191–197. [Google Scholar]
- Micco, C.; Miraglia, M.; Brera, C.; Corneli, S.; Ambruzzi, A. Evaluation of ochratoxin A level in human milk in Italy. Food Addit. Contam. 1995, 12, 351–354. [Google Scholar] [CrossRef]
- Miraglia, M.; de Dominicis, A.; Brera, C.; Corneli, S.; Cava, E.; Menghetti, E.; Miraslia, E. Ochratoxin A levels in human milk and related food samples: An exposure assessment. Nat. Toxins 1995, 3, 436–444. [Google Scholar] [CrossRef]
- Solti, L.; Salamon, F.; Barna-Vetro, I.; Gyöngyösi, A.; Szabo, E.; Wölfling, A. Ochratoxin A content of human sera determined by a sensitive ELISA. J. Anal. Toxicol. 1997, 21, 44–48. [Google Scholar]
- Gareis, M.; Märtlbauer, E.; Bauer, J.; Gedek, B. Bestimmung von Ochratoxin A in Muttermilch. Zeitschr. Lebensm. Forsch. 1988, 186, 114–117. [Google Scholar]
- Jimenez, A.M.; Lopez de Cerain, A.; Gonzalez-Penas, E.; Bello, J.; Betbeder, A.N.; Creppy, E.E. Exposure to ochratoxin A in Europe: Comparison with the region of northern Spain. Toxin Rev. 1998, 17, 479–491. [Google Scholar] [CrossRef]
- Scott, P.M.; Kanhere, S.R.; Lau, B.P.-Y.; Levvis, D.A.; Hayward, S.; Ryan, J.J.; Kuiper-Goodman, T. Survey of Canadian human blood plasma for ochratoxin A. Food Addit. Contam. 1998, 15, 555–562. [Google Scholar] [CrossRef]
- Ueno, Y.; Maki, S.; Lin, J.; Furuya, M.; Sugiura, Y.; Kawamura, O. A four-year study of plasma ochratoxin A in a selected population in Tokyo by immunoassay and immunoaffinity column-linked HPLC. Food Chem. Toxicol. 1998, 36, 445–449. [Google Scholar] [CrossRef]
- Gilbert, J.; Brereton, P.; MacDonald, S. Assessment of dietary exposure to ochratoxin A in the UK using a duplicate diet approach and analysis of urine and plasma samples. Food Addit. Contam. 2001, 18, 1088–1093. [Google Scholar] [CrossRef]
- Peraica, M.; Dornijan, A.M.; Matasin, M.; Lucic, A.; Radic, B.; Delas, F.; Horvat, M.; Bosanac, I.; Balija, M.; Grgicevic, D. Variations of ochratoxin A concentration in the blood of healthy populations in some Croatian cities. Arch. Toxicol. 2001, 75, 410–414. [Google Scholar] [CrossRef]
- Skaug, M.A.; Helland, I.; Solvoll, K.; Saugstad, O.D. Presence of ochratoxin A in human milk in relation to dietary intake. Food Addit. Contam. 2001, 18, 321–327. [Google Scholar] [Green Version]
- Chiavaro, E.; Lepiani, A.; Colla, F.; Bettoni, P.; Pari, E.; Spotti, E. Ochratoxin A determination in ham by immunoaffinity clean-up and a quick fluorometric method. Food Addit. Contam. 2002, 19, 575–581. [Google Scholar] [CrossRef]
- Biro, K.; Solti, L.; Barna-Vetro, I; Bago, G.; Glavits, R.; Szabo, E.; Fink-Gremmels, J. Tissue distribution of ochratoxin A as determend by HPLC and ELISA and histopathological effects in chickens. Avian Pathol. 2002, 31, 141–148. [Google Scholar] [CrossRef]
- Filali, A.; Betbeder, A.M.; Baudrimot, I.; Benayada, A.; Soulaymani, R.; Creppy, E.E. Ochratoxin A in human plasma in Morocco: A preliminary survey. Human Exp. Toxicol. 2002, 21, 241–245. [Google Scholar] [CrossRef]
- Stander, M.A.; Steyn, P.S. Survey of ochratoxin A in South African wines. S. Afr. J. Enol. Vitic. 2002, 23, 9–13. [Google Scholar]
- Pacin, A.M.; Ciancio Bovier, E.V.; Motta, E.; Resnik, S.L.; Villa, D.; Olsen, M. Survey of Argentinian human plasma for ochratoxin A. Food Addit. Contam. 2008, 25, 635–641. [Google Scholar] [CrossRef]
- Coronel, M.B.; Marin, S.; Tarragó, M.; Cano-Sancho, G.; Ramos, A.J.; Sanchis, V. Ochratoxin A and its metabolite ochratoxin alpha in urine and assessment of the exposure of inhabitants of Lleida, Spain. Food Chem. Toxicol. 49, 1436–1442.
- Coronel, M.B.; Sanchis, V.; Ramos, A.J.; Marin, S. Ochratoxin A in adult population of Lleida, Spain: Presence in blood plasma and consumption in different regions and seasons. Food Chem. Toxicol. 49, 2697–2705.
- Hooper, D.G.; Bolton, V.E.; Guilford, F.T.; Straus, D.C. Mycotoxin detection in human samples from patients exposed to environmental molds. Int. J. Mol. Sci. 2009, 10, 1465–1475. [Google Scholar] [CrossRef]
- Märtlbauer, E.; Usleber, E.; Dietrich, R.; Schneider, E. Ochratoxin A in human blood serum—Retrospective long-term data. Mycotoxin Res. 2009, 25, 175–186. [Google Scholar] [CrossRef]
- Zheng, M.Z.; Richard, J.L.; Binder, J. A review of rapid methods for the analysis of mycotoxins. Mycopathologia 2006, 161, 261–273. [Google Scholar] [CrossRef]
- Goryacheva, I.Y.; Rusanova, T.Y.; Burmistrova, N.A.; De Saeger, S. Immunochemical methods for the determination of mycotoxins. J. Anal. Chem. 2009, 64, 768–785. [Google Scholar] [CrossRef]
- Huybrechts, B.; Tangni, E. Evaluation of Immunoassay Kits for Ochratoxin A Determination in Cereals; CODA-CERVA Veterinary and Agrochemical Research Center: Tervuren, Belgium, 2010. [Google Scholar]
- Heussner, A.H.; Moeller, I.; Day, B.W.; Dietrich, D.R.; O’Brian, E. Production and characterization of monoclonal antibodies against ochratoxin B. Food Chem. Toxicol. 2007, 45, 827–833. [Google Scholar] [CrossRef]
- Eyer, L.; Franek, M. Production of antibodies for immunoanalytical methods. In Antibodies: Applications and New Developments; Meulenberg, E.P., Ed.; Bentham eBooks: Bussum, The Netherlands, 2012; pp. 29–47. [Google Scholar]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells producing antibody of predefined specificity. Nature London 1975, 256, 495. [Google Scholar]
- Benjouad, A. Antibody biotechnology. Afr. J. Biotechnol. 2009, 8, 2911–2915. [Google Scholar]
- Altshuler, E.P.; Serebryanaya, D.V.; Katrukha, A.G. Generation of recombinant antibodies and means for increasing their affinity. Biochemistry (Moscow) 2010, 75, 1584–1605. [Google Scholar] [CrossRef]
- Huang, L.; Muyldersmans, S.; Saerens, D. Nanobodies: Proficient tools in diagnostics. Exp. Rev. Mol. Diag. 2010, 10, 777–785. [Google Scholar]
- de Muynck, B.; Navarre, C.; Boutry, M. Production of antibodies in plants: Status after 20 years. Plant Biotechnol. J. 2010, 8, 529–563. [Google Scholar] [CrossRef]
- Xie, X.; Richard, G.; Hall, J.C. Antibody fragment engineering and applications in diagnostics and therapies. In Antibodies: Applications and New Developments; Meulenberg, E.P., Ed.; Bentham eBooks: Bussum, The Netherlands, 2012; pp. 225–279. [Google Scholar]
- Aalund, O.; Branfeldt, K.; Hald, B.; Krogh, P.; Poulsen, K. A radioimmunoassay for ochratoxin A: A preliminary investigation. Acta Pathol. Microbiol. Scand. Section C Immunol. 1975, 83C, 390–392. [Google Scholar]
- Chu, F.S.; Chang, F.C.C.; Hinsdill, R.D. Production of antibody against ochratoxin A. Appl. Environ. Microbiol. 1976, 31, 831–835. [Google Scholar]
- Rousseau, D.M.; Slegers, G.A.; van Peteghem, C.H. Radioimmunoassay of ochratoxin A in barley. Appl. Environ. Microbiol. 1985, 50, 529–531. [Google Scholar]
- Rousseau, D.M.; Slegers, G.A.; van Peteghem, C.H. Solid phase radioimmunoassay of ochratoxin A in serum. J. Agric. Food Chem. 1986, 34, 862–865. [Google Scholar] [CrossRef]
- Fukal, L. A survey of cereals, cereal products, feedstuff and porcine kidney for ochratoxin A by radioimmunoassa. Food Addit. Contam. 1990, 7, 253–258. [Google Scholar] [CrossRef]
- Morris, H.A. Standardization of immunoassays. In Antibodies: Applications and New Developments; Meulenberg, E.P., Ed.; Bentham eBooks: Bussum, The Netherlands, 2012; pp. 48–57. [Google Scholar]
- Pestka, J.J.; Steinert, B.W.; Shu, F.S. Enzyme-linked immunosorbent assay for detection of ochratoxin A. Appl. Environ. Microbiol. 1981, 41, 1472–1474. [Google Scholar]
- Candlish, A.A.; Stimson, W.H.; Smith, J.E. Determination of ochratoxin A by monoclonal antibody-based enzyme immunoassay. J. Assoc. Anal. Chem. 1988, 71, 961–964. [Google Scholar]
- Kawamura, O.; Sato, S.; Kajii, H.; Nagayama, S.; Ohtanie, K.; Shiba, J.; Ueno, Y. A sensitive enzyme-linked immunosorbent assay of ochratoxin A based on monoclonal antibodies. Toxicon 1989, 27, 887–897. [Google Scholar] [CrossRef]
- Ramakrishna, N.; Lacey, J.; Candlish, A.A.; Smith, J.E.; Goodbrand, I.A. Monoclonal antibody-based enzyme-linked immunosorbent assay of aflatoxin B1, T-2 toxin and ochratoxin A in barley. J. Assoc. Anal. Chem. 1990, 73, 1–6. [Google Scholar]
- Lacey, J.; Ramakrishna, N.; Candlish, A.A.; Smith, J.E. Immunoassay of ochratoxin and other mycotoxins from a single extract of cereal grains utilizing monoclonal antibodies. IARC Sci. Publ. 1991, 115, 97–103. [Google Scholar]
- Barna-Vetro, I.; Solti, L.; Téren, J.; Gyöngyösi, Á.; Szabó, E.; Wölfling, A. Sensitive ELISA test for determination of ochratoxin A. J. Agric. Food Chem. 1996, 44, 4071–4074. [Google Scholar]
- Barna-Vetro, I.; Solti, L. A new monoclonal antibody detecting ochratoxin A at the pictogram level. Lett. Appl. Microbiol. 1996, 22, 103–105. [Google Scholar] [CrossRef]
- de Saeger, S.; van Peteghem, C. Flow-through membrane-based enzyme immunoassay for rapid detection of ochratoxin A in wheat. J. Food Prot. 1999, 62, 65–69. [Google Scholar]
- Thirumala-Devi, K.; Mayo, M.A.; Reddy, G.; Reddy, S.V.; Delfosse, P.; Reddy, D.V.R. Production of polyclonal antibodies against ochratoxin A and its detection in chilies by ELISA. J. Agric. Food Chem. 2000, 48, 5079–5082. [Google Scholar]
- Sibanda, L.; de Saeger, S.; Barna-Vetro, I.; van Peteghem, C. Development of a solid-phase cleanup and portable rapid flow-through enzyme immunoassay for the detection of ochratoxin A in roased coffee. J. Agric. Food Chem. 2002, 50, 6964–6967. [Google Scholar]
- Fujii, S.; Ono, E.Y.S.; Ribeiro, R.M.R.; Asuncao, F.G.A.; Takabayashi, C.R.; de Oliviera, T.C.R.M.; Itano, E.N.; Ueno, Y.; Kawamura, O.; Hirooka, E.Y. A comparison between enzyme immunoassay and HPLC for ochratoxin A detection in green, roasted and instant coffee. Braz. Ach. Biol. Technol. 2007, 50, 349–359. [Google Scholar]
- Saha, D.; Acharya, D.; Roy, D.; Shrestha, D.; Dhar, T.K. Simultaneous enzyme immunoassay for the screening of aflatoxin B1 and ochratoxin A in chili samples. Anal. Chim. Acta 2007, 584, 343–349. [Google Scholar] [CrossRef]
- Wang, X.H.; Liu, T.; Xu, N.; Zhang, Y.; Wang, S. Enzyme-linked immunosorbent assay and colloidal gold immunoassay for ochratoxin A: Investigation of analytical conditions and sample matrix on assay performance. Anal. Bioanal. Chem. 2007, 389, 903–911. [Google Scholar] [CrossRef]
- Shim, W.B.; Dzantiev, B.B.; Eremin, S.A.; Chung, D.H. One-step simultaneous immunochromatographic strip test for multianalysis of ochratoxin A and zearalenone. J. Microbiol. Biotechnol. 2009, 19, 83–92. [Google Scholar]
- Anfossi, L.; D’Arco, G.; Baggiano, C.; Giovannoli, C.; Gianfranco, G. A lateral flow immunoassay for measuring ochratoxin A: Development of a single system for maize, wheat and durum wheat. Food Control 2011, 22, 1965–1970. [Google Scholar] [CrossRef]
- Zhang, A.; Ma, Y.; Feng, L.; Wang, Y.; He, C.; Wang, X.; Zhang, H. Development of a sensitive competitive indirect ELISA method for determination of ochratoxin A levels in cereals originating from Nanjing, China. Food Control 2011, 22, 1723–1728. [Google Scholar] [CrossRef]
- Kwak, B.Y.; Kwon, B.J.; Kweon, C.H.; Shon, D.H. Detection of Aspergillus, Penicillium, and Fusarium species by sandwich enzyme-linked immunosorbent assay using mixed monoclonal antibodies. J. Microbiol. Biotechnol. 2004, 14, 385–389. [Google Scholar]
- Punyatong, M.; Pongpiachan, P.; Pongpiachan, P. Monoclonal antibodies production and quantification of ochratoxin A in feedstuffs by enzyme-linked immunosorbent assay. Kasetsart J. (Nat. Sci.) 2003, 37, 137–144. [Google Scholar]
- Yu, F.Y.; Vdovenko, M.M.; Wang, J.J.; Sakharov, I.Y. Comparison of enzyme-linked immunosorbent assays with chemiluminescent and colorimetric detection for the determination of ochratoxin A in food. J. Agric. Food Chem. 2011, 59, 809–813. [Google Scholar]
- Qui, Y.Q.; Wang, W.; Li, F.Q. Detection of ochratoxin A in food by chemiluminescent immunoassay. Food Sci. 2010, 31, 432–435. [Google Scholar]
- Hemmilá, I.; Mukkala, V.M. Time-resolution in fluorometry: Technologies, labels, and applications in bioanalytical assay. Crit. Rev. Clin. Lab. Sci. 2001, 38, 441–519. [Google Scholar] [CrossRef]
- PerkinElmer, Delphia® TRF Assays. Available online: www.Perkinelmer.com accessed on 15 February 2012.
- Hagan, A.K.; Zuchner, T. Lanthanide-based time-resolved luminescence immunoassays. Anal. Bioanal. Chem. 2011, 400, 2847–2864. [Google Scholar] [CrossRef]
- Huang, B.; Xiao, H.; Zhang, J.; Zhang, L.; Yang, H.; Zhang, Y.; Jin, J. Dual-label time-resolved fluoroimmunoassay for simultaneous detection of aflatoxin A and ochratoxin A. Arch. Toxicol. 2009, 83, 619–624. [Google Scholar] [CrossRef]
- Maragos, C. Fluorescence polarization immunoassay of mycotoxins: A review. Toxins 2009, 1, 196–207. [Google Scholar] [CrossRef]
- Zezza, F.; Longobardi, F.; Pascale, M.; Eremin, S.A.; Visconti, A. Fluorescence polarization immunoassay for rapid screening of ochratoxin A in red wine. Anal. Bioanal. Chem. 2009, 395, 1317–1323. [Google Scholar] [CrossRef]
- Kreissig, T.; Hoffmann, R.; Zuchner, T. Homogeneous fluorescence-based immunoassay detects antigens within 90 seconds. Anal. Chem. 2011, 83, 4281–4287. [Google Scholar]
- Li, T.; Jeon, K.S. A label-free, direct and noncompetitive FRET immunoassay for ochratoxin A based on intrinsic fluorescence of an antigen and antibody complex. Chem. Comm. 2011, 47, 9098–9100. [Google Scholar]
- Ngundi, M.M.; Shriver-Lake, L.C.; Moore, M.H.; Lassman, M.E.; Ligler, F.S.; Taitt, C.R. Array biosensor for detection of ochratoxin A in cereals in beverages. Anal. Chem. 2005, 77, 148–154. [Google Scholar]
- Lamberti, H.; Tanzarella, C.; Solinas, I.; Padula, C.; Mosiello, L. An antibody-based microarray assay for the simultaneous detection of aflatoxin B1 and fumonisin B1. Mycotox. Res. 2009, 25, 193–200. [Google Scholar] [CrossRef]
- Posthuma-Trumpie, G.A.; van Amerongen, A. Lateral flow assays. In Antibodies: Applications and New Developments; Meulenberg, E.P., Ed.; Bentham eBooks: Bussum, The Netherlands, 2012; pp. 175–183. [Google Scholar]
- Peters, J.; Bienemann-Ploum, M.; de Rijk, T.; Haasnoot, W. Development of a multiplex flow cytometric microsphere immunoassay for mycotoxins and evaluation of its application in feed. Mycotox. Res. 2011, 27, 63–72. [Google Scholar] [CrossRef]
- Anderson, G.P.; Kowtha, V.A.; Taitt, C.R. Detection of fumonisin B1 and ochratoxin A in grain products using microsphere-based fluid array immunoassays. Toxins 2010, 2, 297–309. [Google Scholar] [CrossRef]
- Aqai, P.; Peters, J.; Gerssen, A.; Haasnoot, W.; Nielen, M.W.F. Immunomagnetic microbeads for screening with flow cytocytometry and identification with nano-liquid chromatography mass spectrometry of ochratoxins in wheat and cereal. Anal. Bioanal. Chem. 2011, 400, 3085–3096. [Google Scholar] [CrossRef]
- Sheikh, S.; Blaszykowski, C.; Thompson, M. Acoustic wave-based detection in bioanalytical chemistry: Competition for surface plasmon resonance? Anal. Lett. 2008, 41, 2525–2538. [Google Scholar] [CrossRef]
- Wolfbeis, O.S. Fiber-optic chemical sensors and biosensors. Anal. Chem. 2008, 80, 4269–4283. [Google Scholar] [CrossRef]
- Karyankin, A.A. Biosensors. In Sensors for Environment, Health and Security; Baraton, M.I., Ed.; Springer Science + Business Medi: New York, NY, USA, 2009; pp. 255–265. [Google Scholar]
- Ligler, F.S. Fluorescence-based optical biosensors. In Biophotonics, Biological and Medical Physics, Biomedical Engineering; Pavesi, L., Fauchet, P.M., Eds.; Springer: New York, NY, USA, 2008; pp. 199–215. [Google Scholar]
- Manco, G.; Nucci, R.; Febbraio, F. Use of esterase activities for the detection of chemical neurotoxic agents. Prot. Pept. Lett. 2009, 16, 1225–1234. [Google Scholar] [CrossRef]
- Viswanathan, S.; Radecka, H.; Radecki, J. Electrochemical biosensors for food analysis. Monatsh. Chem. 2009, 140, 891–899. [Google Scholar] [CrossRef]
- van der Gaag, B.; Spath, S.; Dietrich, H.; Stigter, E.; Boonzaayer, G.; van Osenbruggen, T.; Koopal, K. Biosensors and multiple mycotoxin analysis. Food Control 2003, 14, 251–254. [Google Scholar] [CrossRef]
- Hodnik, V.; Anderluh, G. Toxin detection by surface plasmon resonance. Sensors 2009, 9, 1339–1354. [Google Scholar] [CrossRef]
- Adányi, N.; Levkovets, I.A.; Rodriguez-Gil, S.; Ronald, A.; Váradi, M.; Szendró, I. Development of immunosensor based on OWLS technique for determining aflatoxin B1 and ochratoxin A. Biosensors Bioelectr. 2007, 22, 797–802. [Google Scholar] [CrossRef]
- Yuan, J.; Deng, D.; Lauren, D.R.; Aguilar, M.I.; Wu, Y. Surface plasmon resonance biosensor for the detection of ochratoxin A in cereals and beverages. Anal. Chim. Acta 2009, 656, 63–71. [Google Scholar] [CrossRef]
- Prietó-Simon, B.; Campas, M.; Marty, J.L.; Noguer, T. Novel high-performing immunosensors-based strategy for ochratoxin A detection in wine samples. Biosensors Bioelectr. 2008, 23, 995–1002. [Google Scholar] [CrossRef]
- Liu, X.P.; Deng, Y.J.; Jin, X.Y.; Chen, L.G.; Jiang, J.H.; Shen, G.L.; Yu, R.Q. Ultrasensitive electrochemical immunosensors for ochratoxin A using gold colloid-mediated hapten immobilization. Anal. Biochem. 2009, 389, 63–68. [Google Scholar]
- Radi, A.E.; Munoz-Berbel, X.; Cortina-Puig, M.; Marty, J.L. An electrochemical immunosensors for ochratoxin A based on immobilization of antibodies on diazonium-functionalized gold electrode. Electrochim. Acta 2009, 54, 2180–2184. [Google Scholar] [CrossRef]
- Vidal, J.C.; Bonel, L.; Duato, P.; Castillo, J.R. Improved electrochemical competitive immunosensors for ochratoxin A with a biotinylated monoclonal antibody capture probe and colloidal gold nanostructuring. Anal. Meth. 2011, 3, 977–984. [Google Scholar]
- Urusov, A.E.; Kostenko, S.N.; Sveshnikov, P.G.; Sherdev, A.V.; Dzantiev, B.B. Ochratoxin A immunoassay with surface plasmon resonance registration: Lowering limit of detection by the use of colloidal gold immunoconjugates. Sensors Actuat. B. 2011, 156, 343–349. [Google Scholar] [CrossRef]
- Zamfir, L.G.; Geana, I.; Bourigua, S.; Rotariu, L.; Bala, C.; Errachid, A.; Jaffrezic-Renault, N. Highly sensitive label-free immunosensors for ochratoxin A based on functionalized magnetic nanoparticles and EIS/SPR detection. Sensors Actuat. 2011, 159, 178–184. [Google Scholar] [CrossRef]
- Whitcombe, M.J.; Vulfson, E.N. Imprinted polymers. Adv. Mater. 2001, 13, 467–478. [Google Scholar] [CrossRef]
- Lavingnac, N.; Allender, C.J.; Brain, K.R. Current status of molecularly imprinted polymers as alternatives to antibodies in sorbent assays. Anal. Chim. Acta 2004, 510, 139–145. [Google Scholar] [CrossRef] [Green Version]
- Pichon, V.; Chappuis-Hugon, F. Role of molecularly imprinted polymers for selective determination of environmental pollutants—A review. Anal. Chim. Acta 2008, 622, 48–61. [Google Scholar] [CrossRef]
- Mairal, T.; Özalp, V.C.; Sánchez, P.L.; Mir, M.; Katakis, I.; O’Sullivan, C.K. Aptamers: Molecular tools for analytical applications. Anal. Bioanal. Chem. 2008, 390, 989–1007. [Google Scholar] [CrossRef]
- Zhou, J.; Battig, M.R.; Wang, Y. Aptamer-based molecular recognition for biosensor development. Anal. Bioanal. Chem. 2010, 398, 2471–2480. [Google Scholar] [CrossRef]
- Velasco-Garcia, M.N.; Missailidis, S. New trends in aptamer-based electrochemical biosensors. Gene Ther. Mol. Biol. 2009, 13, 1–10. [Google Scholar]
- Tombelli, S.; Mascini, M. Aptamers as molecular tools for bioanalytical methods. Curr. Opin. Mol. Ther. 2009, 11, 179–188. [Google Scholar]
- Tombelli, S.; Mascini, M. Aptamers biosensors for pharmaceutical compounds. Combinat. Chem. High Throughput Screen. 2010, 13, 641–649. [Google Scholar]
- Houwen, F.P.; Kage, A. Aptamers: The chemical antbodies. In Antibodies: Applications and New Developments; Meulenberg, E.P., Ed.; Bentham eBooks: Bussum, The Netherlands, 2012; pp. 300–314. [Google Scholar]
- Sassolas, A.; Blum, L.J.; Leca-Bouvier, B.D. Homogeneous assays using aptamers. Analyst 2011, 136, 257–274. [Google Scholar] [CrossRef]
- Wang, R.E.; Zhang, Y.; Cai, J.; Cai, W.; Gao, T. Aptamer-based fluorescent biosensors. Curr. Med. Chem. 2011, 18, 1–10. [Google Scholar] [CrossRef]
- Wang, L.; Chen, W.; Ma, W.; Liu, L.; Ma, W.; Zhao, Y.; Zhu, Y.; Xu, L.; Kuang, H.; Xu, C. Fluorescent strip sensor for rapid determination of toxins. Chem. Commun. 2011, 47, 1574–1576. [Google Scholar]
- Cruz-Aguado, J.A.; Penner, G. Determination of ochratoxin A with a DNA aptamer. J. Agric. Food Chem. 2008, 56, 10456–10461. [Google Scholar] [CrossRef]
- Barthelmebs, L.; Jonca, J.; Hayat, A.; Prieto-Simon, B.; Marty, J.L. Enzyme-linked aptamer assays (ELAAs), based on a competition format for a rapid and sensitive detection of ochratoxin A in wine. Food Control 2011, 22, 737–743. [Google Scholar] [CrossRef]
- Kuang, H.; Chen, W.; Xu, D.; Xu, L.; Zhu, Y.; Liu, L.; Chu, H.; Peng, C.; Xu, C.; Chu, S. Fabricated aptamer-based electrochemical “signal-off” sensor of ochratoxin A. Biosensors Bioelectr. 2010, 26, 710–716. [Google Scholar] [CrossRef]
- Bonel, L.; Vidal, J.C.; Duato, P.; Castillo, J.R. An electrochemical competitive biosensor for ochratoxin A based on a DNA biotinylated aptamer. Biosensors Bioelectr. 2011, 26, 3254–3259. [Google Scholar] [CrossRef]
- Tong, P.; Zhang, L.; Xu, J.J.; Chen, H.Y. Simply amplified electrochemical aptasensor of ochratoxin A based on exonuclease-catalyzed target recycling. Biosensors Bioelectr. 2011, 29, 97–101. [Google Scholar] [CrossRef]
- Yang, C.; Wang, Y.; Marty, J.L.; Yang, X. Aptamer-based colorimetric biosensing of ochratoxin A using unmodified gold nanoparticles indicator. Biosensors Bioelectr. 2011, 26, 2724–2727. [Google Scholar] [CrossRef]
- Yang, C.; Lates, V.; Prieto-Simón, B.; Marty, J.L.; Yang, X. Aptamer-DNAzyme hairpins for biosensing of ochratoxin A. Biosensors Bioelectr. 2012, 32, 208–212. [Google Scholar] [CrossRef]
- Lobeau, M.; de Saeger, S.; Sibanda, L.; Barna-Vetró, I.; van Peteghem, C. Development of a new clean-up tandem assay column for the detection of ochratoxin A in roasted coffee. Anal. Chim. Acta 2005, 538, 57–61. [Google Scholar] [CrossRef]
- Goryacheva, I.Y.; de Saeger, S.; Lobeau, M.; Eremin, S.A.; Barna-Vetró, I.; van Peteghem, C. Approach for ochratoxin A fast screening in spices using clean-up tandem immunoassay columns with confirmation by high performance liquid chromatography—tandem mass spectrometry (HPLC-MS/MS). Anal. Chim. Acta 2006, 577, 38–45. [Google Scholar] [CrossRef]
- Goryacheva, I.Y.; de Saeger, S.; Nesterenko, I.S.; Eremin, S.A.; van Peteghem, C. Rapid all-in-one three-step immunoassay for non-instrumental detection of ochratoxin A in high-coloured herbs and spices. Talanta 2007, 72, 1230–1234. [Google Scholar] [CrossRef]
- Rusanova, T.Y.; Beloglazova, N.V.; Goryacheva, I.Y.; Lobeau, M.; van Peteghem, C.; de Saeger, S. Non-instrumental immunochemical tests for rapid ochratoxin A detection in red wine. Anal. Chim. Acta 2009, 653, 97–102. [Google Scholar] [CrossRef]
- Beloglazova, N.V.; Goryacheva, I.Y.; Rusanova, T.Y.; Yurasov, N.A.; Galve, R.; Marco, M.P.; de Saeger, S. Gel-based immunotest for simultaneous detection of 2,4,6-trichlorophenol and ochratoxin A in red wine. Anal. Chim. Acta 2010, 672, 3–8. [Google Scholar] [CrossRef]
- Goryacheva, I.Y.; Rusanova, T.Y.; Beloglazova, N.V.; Voronov, I.I.; de Saeger, S. Determination of ochratoxin A in colored food products: Sample preparation and an immunoassay test method. J. Anal. Chem. 2010, 65, 760–766. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Meulenberg, E.P. Immunochemical Methods for Ochratoxin A Detection: A Review. Toxins 2012, 4, 244-266. https://doi.org/10.3390/toxins4040244
Meulenberg EP. Immunochemical Methods for Ochratoxin A Detection: A Review. Toxins. 2012; 4(4):244-266. https://doi.org/10.3390/toxins4040244
Chicago/Turabian StyleMeulenberg, Eline P. 2012. "Immunochemical Methods for Ochratoxin A Detection: A Review" Toxins 4, no. 4: 244-266. https://doi.org/10.3390/toxins4040244
APA StyleMeulenberg, E. P. (2012). Immunochemical Methods for Ochratoxin A Detection: A Review. Toxins, 4(4), 244-266. https://doi.org/10.3390/toxins4040244