The Use of Convection-Enhanced Delivery with Liposomal Toxins in Neurooncology

Abstract
:1. Introduction
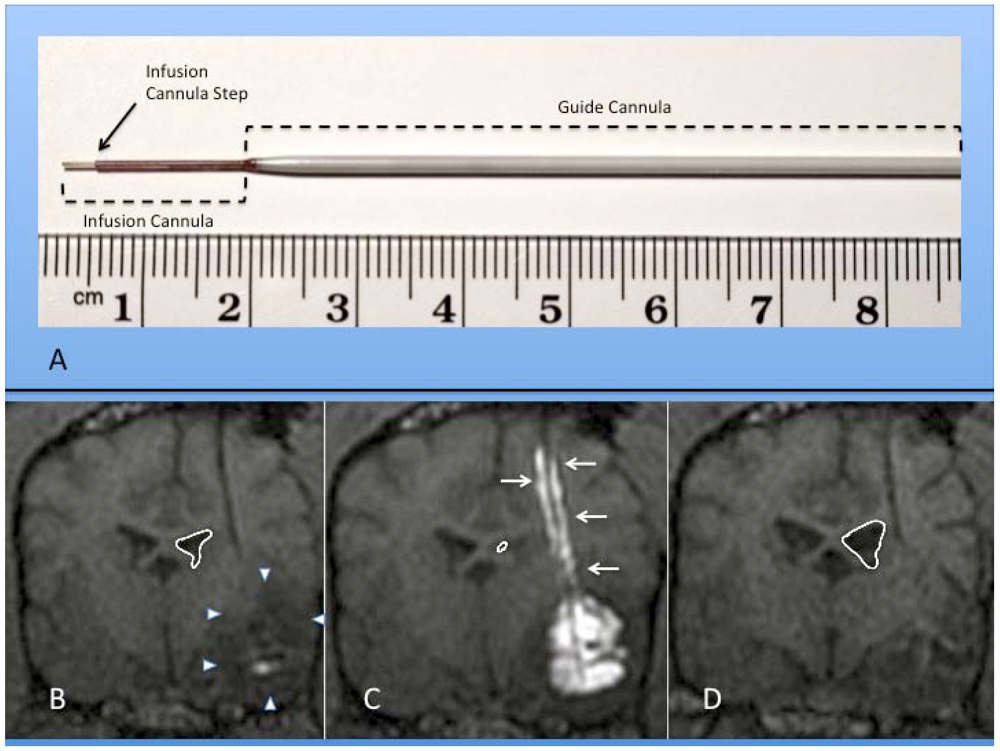
2. Diffusion versus Convection-Enhanced Delivery (CED)

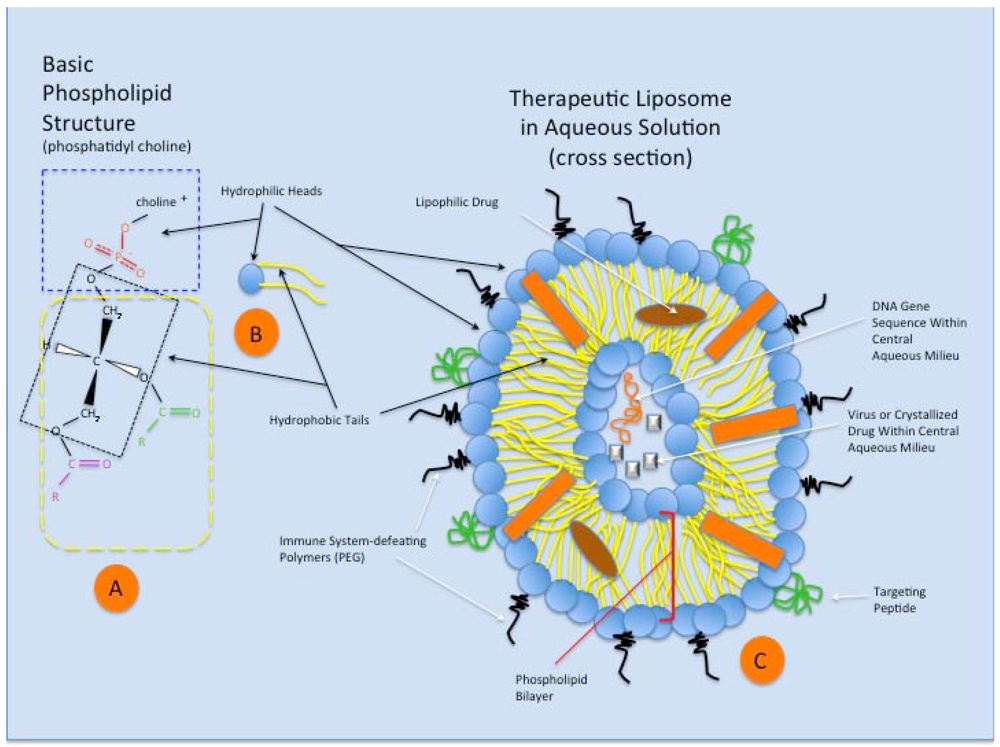
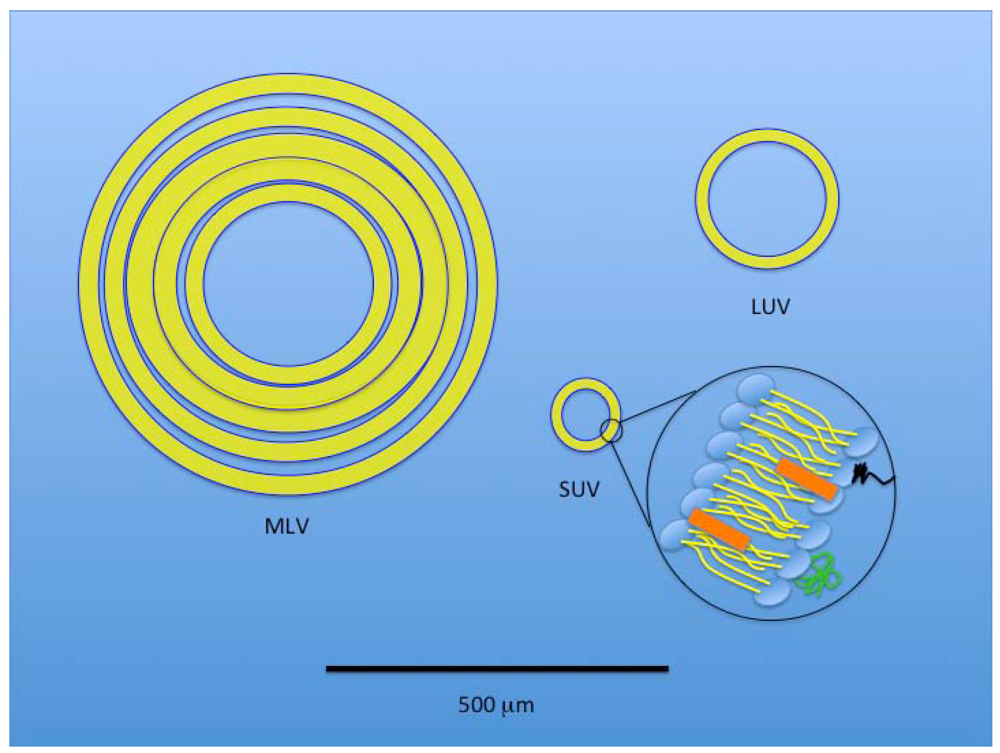
3. Liposomes
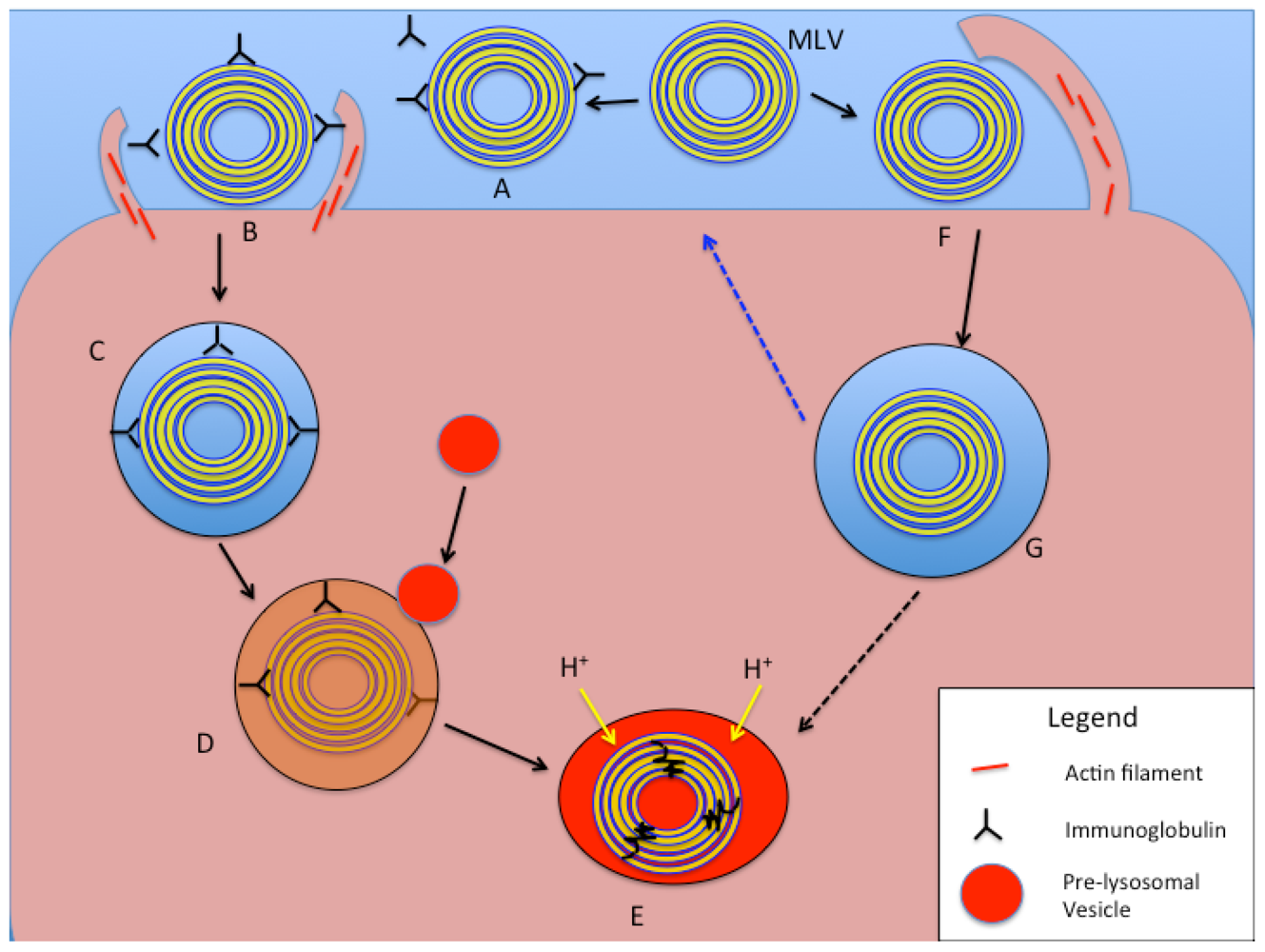
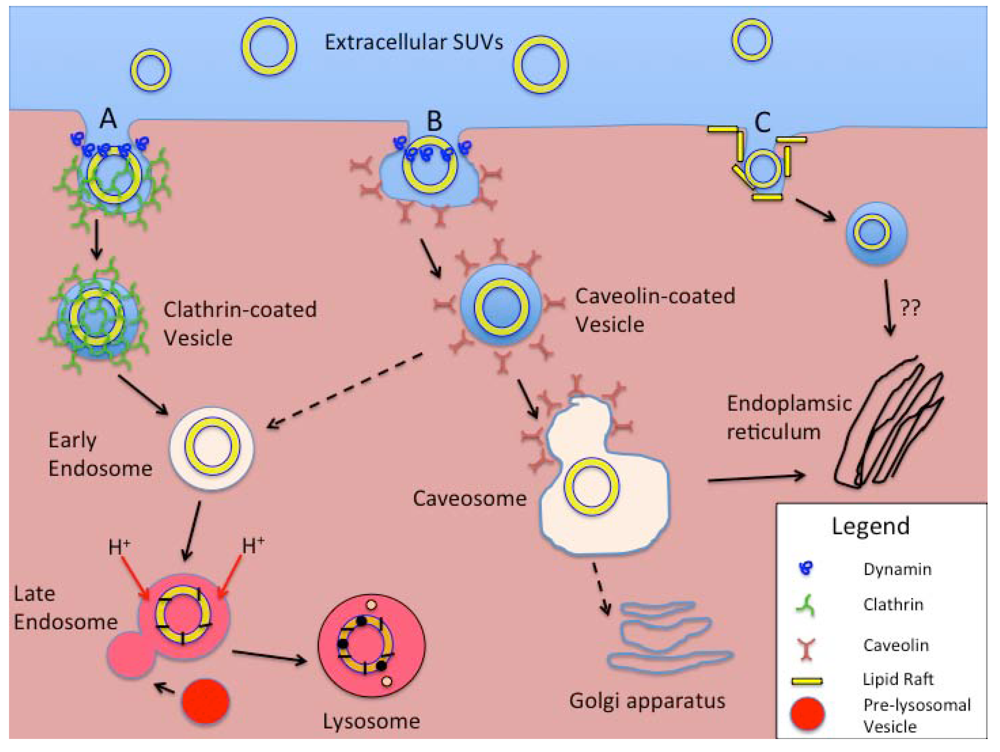
4. Cellular Uptake of Liposomes
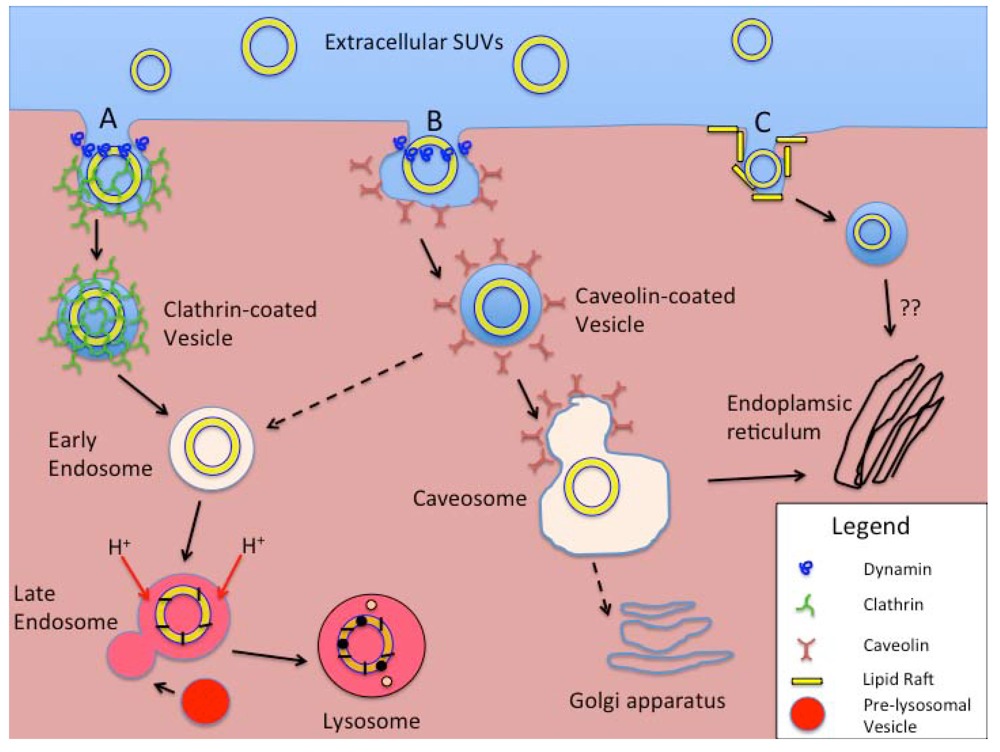

{kind=link}
{kind=link}
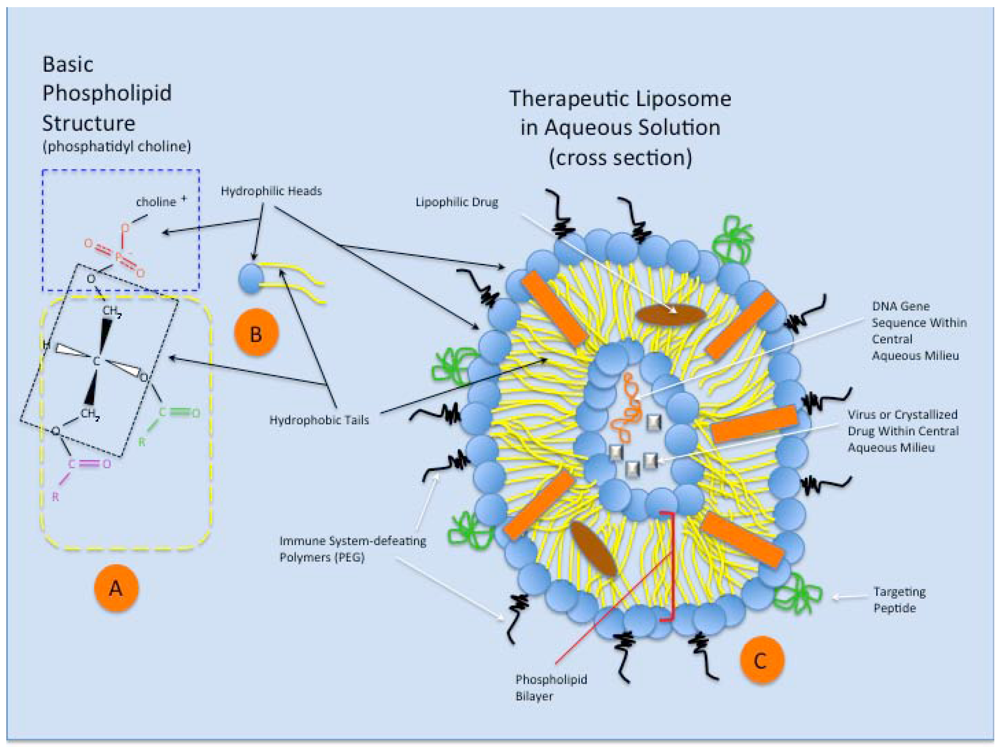{kind=link}
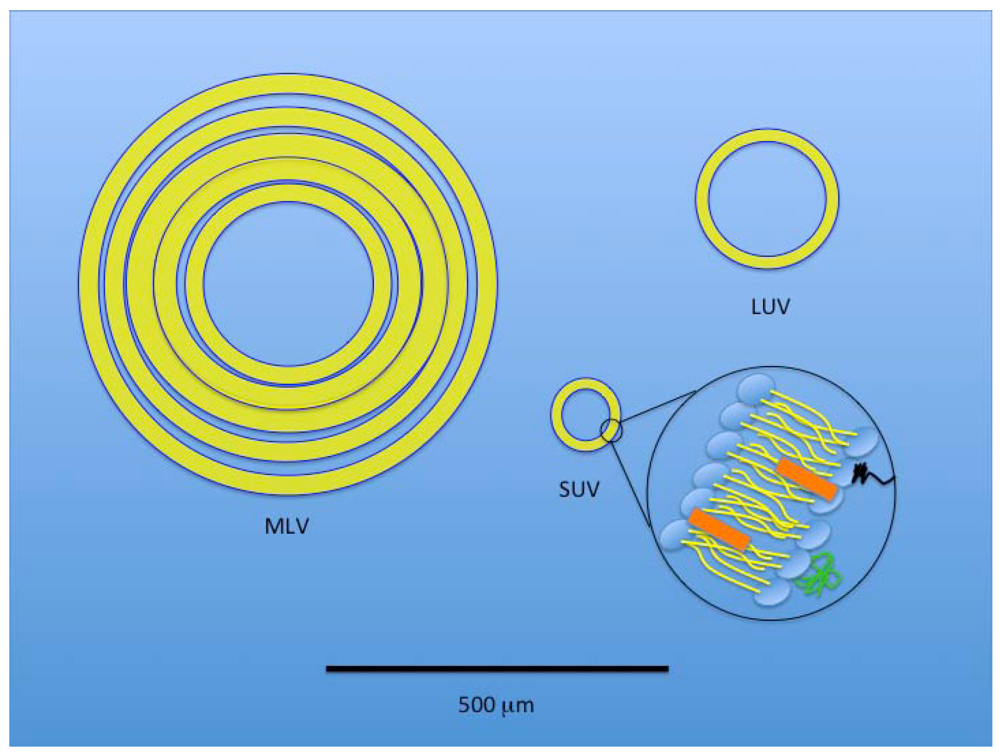{kind=link}
{kind=link}
{kind=link}
| Mechanisms of Endocytosis | Primary Cell Types Involved | Opsonization Dependent | Surface Feature-Dependent | Length of Process to Processing | Vesicle Size | Typical Cellular Processing | Other Factors |
|---|---|---|---|---|---|---|---|
| Phagocytosis | Macrophages Monocytes Neutrophils Dendritic cells | Yes, usually | Yes, increased for both cationic and anionic particles | 30 min to 2 h | >250 nm | Acidified, enzyme-rich phagolyso-some. | Increased with hydrophobic, rigid, non-spherical particles. Actin-dependent. |
| Macropinocytosis | All cells | No | No | - | 1–5 μm | Degradative lysosome | Actin-dependent |
| CME and fluid-phase endocytosis | All cells | No | Enhanced by specific ligands | 5–10 min for receptor-mediated. 45–90 min for fluid phase. | <150 nm | Early and late endosomes and eventually degradative lysosomes | Receptor-mediated and non-specific uptake exists. Clathrin- and dynamin-dependent. |
| CvME | All cells, but especially endothelial cells. | No | Receptor-ligand trafficking on cell surface | 20–40 min | <80 nm | Caveosome, avoiding acidic- and enzyme-rich processing. | Caveolin- and dynamin- dependent |
| CME- and CvME- independent endocytosis | All cells | No | Selected by using targeting ligands specific for “rafts”. | - | <50 nm | Non-lysosomal pathways | Still being investigated. |
5. Liposomal Toxins
6. CED of Liposomal Toxins: Pre-clinical Neurooncologic Studies
7. CED of Liposomal Toxins: Clinical Neurooncologic Studies
8. Future Directions
9. Summary and Conclusions
| Treatment Modality | Essential Components |
|---|---|
| CED | Thorough understanding and implementation of parameters to optimize convection. |
| Cannula size and shape | |
| Infusion flow rates | |
| Specific infusion volumes | |
| Safe use of contrast agents (free vs. liposomal) to visualize the CED process (e.g., RCD). | |
| Avoid reflux or leakage | |
| Document Vd, and specific coverage of tumor | |
| Liposomes | Effective use of liposomal technology for improved formulation of LTs. |
| Improved understanding of the cellular processing of LTs based on particle size. | |
| Effective use of lysosomal or non-lysosomal pathways based on delivered therapeutic agent. | |
| Better appreciation of ultrasound-induced release dynamics and efficacy. | |
| Effective tumor-specific targeting based on surface ligands. |
Acknowledgements
References
- Scherrmann, J.M. Drug delivery to brain via the blood-brain barrier. Vasc. Pharmacol. 2002, 38, 349–354. [Google Scholar] [CrossRef]
- Ballabh, P.; Braun, A.; Nedergaard, M. The blood–brain barrier: An overview. Structure, regulation, and clinical implications. Neurobiol. Dis. 2004, 16, 1–13. [Google Scholar] [CrossRef]
- Persidsky, Y.; Ramirez, S.H.; Haorah, J.; Kanmongne, G.D. Blood-brain barrier: Structural components and function under physiologic and pathologic conditions. J. Neuroimmune Pharmacol. 2006, 1, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Greib, P.; Forster, R.E.; Strome, D.; Goodwin, C.W.; Pape, P.C. O2 exchange between blood and brain tissues studied with 18O2 indicator-dilution technique. J. Appl. Physiol. 1985, 58, 1929–1941. [Google Scholar] [PubMed]
- Pardridge, W.M.; Eisenberg, J.; Yang, J. Human blood-brain barrier insulin receptor. J. Neurochem. 1985, 44, 1771–1778. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Pardridge, W.M. Rapid transferrin efflux from brain to blood across the blood-brain barrier. J. Neurochem. 2001, 76, 1597–1600. [Google Scholar] [CrossRef] [PubMed]
- Rejman, J.; Oberle, V.; Zuhorn, I.S.; Hoekstra, D. Size-dependent internalization of particles via the pathways of clathrinand caveolae-mediated endocytosis. Biochem. J. 2004, 377, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Brightman, M.W.; Broadwell, R.D. The morphological approach to the study of normal and abnormal brain permeability. Adv. Exp. Med. Biol. 1976, 69, 41–54. [Google Scholar] [PubMed]
- Shivers, R.R.; Edmonds, C.L.; Del Maestro, R.F. Microvascular permeability in induced astrocytomas and peritumor neuropil of rat brain. A high-voltage electron microscope-protein tracer study. Acta Neuropathol. 1984, 64, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Vick, N.A.; Khandekar, J.D.; Bigner, D.D. Chemotherapy of brain tumors: The “blood-brain barrier” is not a factor. Arch. Neurol. 1977, 34, 523–526. [Google Scholar] [PubMed]
- Donelli, M.G.; Zucchetti, M.; D’Incalci, M. Do anticancer agents reach the tumor target in the human brain? Cancer Chemother. Pharmacol. 1992, 30, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.J. A critique of the role of the blood-brain barrier in the chemotherapy of human brain tumors. J. Neuro-Oncol. 1994, 20, 121–139. [Google Scholar] [CrossRef]
- Groothuis, D.R. The blood-brain and blood-tumor barriers: A review of strategies for increasing drug delivery. Neuro-Oncology 2000, 2, 45–59. [Google Scholar] [PubMed]
- Groothuis, D.R.; Lippitz, B.E.; Fekete, I.; Schlageter, K.E.; Molnar, P.; Colvin, O.M.; Roe, C.R.; Bigner, D.D.; Friedman, H.S. The effect of an amino acid-lowering diet on the rate of melphalan entry into brain and xenotransplanted glioma. Cancer Res. 1992, 52, 5590–5596. [Google Scholar] [PubMed]
- Pardridge, W.M.; Boado, R.J.; Kang, Y.S. Vector-mediated delivery of a polyamide (“peptide”) nucleic acid analogue through the blood-brain barrier in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 5592–5596. [Google Scholar] [CrossRef]
- van de Waterbeemd, H.; Camenish, G.; Folkers, G.; Chretien, J.R.; Raevsky, O.A. Estimation of blood-brain barrier crossing of drugs using molecular size and shape, and H-bonding descriptors. J. Drug Target. 1998, 6, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, S.J.; Thompson, H.K. Osmotic opening of the blood-brain barrier in the monkey without associated neurological de cits. Science 1973, 180, 971. [Google Scholar] [PubMed]
- Neuwelt, E.A.; Dahlborg, S.A. Blood-brain barrier disruption in the treatment of brain tumors: Clinical implications. In Implications of the Blood-Brain Barrier and Its Manipulation; Neuwelt, E.A., Ed.; Plenum Medical Book Company: New York, NY, USA, 1989; Volume 2, pp. 195–262. [Google Scholar]
- Black, K.L.; Cloughesy, T.; Huang, S.C.; Gobin, Y.P.; Zhou, Y.; Grous, J.; Nelson, G.; Farahani, K.; Hoh, C.K.; Phelps, M. Intracarotid infusion of RMP-7, a bradykinin analog, and transport of gallium-68 ethylenediamine tetraacetic acid into human gliomas. J. Neurosurg. 1997, 86, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.T.; Coleman, R.E.; Friedman, A.H.; Friedman, H.S.; McLendon, R.E.; Reiman, R.; Felsberg, G.J.; Tien, R.D.; Bigner, S.H.; Zalutsky, M.R.; et al. Intrathecal 131I-labeled antitenascin monoclonal antibody 81C6 treatment of patients with leptomeningeal neoplasms or primary brain tumor resection cavities with subarachnoid communication: Phase I trial results. Clin. Cancer Res. 1996, 2, 963–972. [Google Scholar] [PubMed]
- Blasberg, R.G. Methotrexate, cytosine arabinoside, and BCNU concentration in brain after ventriculocisternal perfusion. Cancer Treat. Rep. 1977, 61, 625–631. [Google Scholar] [PubMed]
- Groothuis, D.R.; Levy, R.M. The entry of antiviral and antiretroviral drugs into the central nervous system. J. Neurovirol. 1997, 3, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Patlak, C.S.; Fenstermacher, J.D. Measurements of dog bloodbrain transfer constants by ventriculocisternal perfusion. Amer. J. Physiol. 1975, 229, 877–884. [Google Scholar]
- Tomita, T. Interstitial chemotherapy for brain tumors: Review. J. Neuro-Oncol. 1991, 10, 57–74. [Google Scholar] [CrossRef]
- de Lange, E.C.M.; Danhof, M.; de Boer, A.G.; Breimer, D.D. Methodological considerations of intracerebral microdialysis in pharmacokinetic studies on drug transport across the blood-brain barrier. Brain Res. Rev. 1997, 25, 27–49. [Google Scholar] [CrossRef]
- Parsons, L.H.; Justice, J.B., Jr. Quantitative approaches to in vivo brain microdialysis. Crit. Rev. Neurobiol. 1994, 8, 189–220. [Google Scholar] [PubMed]
- de Lange, E.C.M.; de Boer, A.G.; Breimer, D.D. Microdialysis for pharmacokinetic analysis of drug transport to the brain. Adv. Drug. Deliv. Rev. 1999, 36, 211–227. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Shoichet, M.S. Delivering neuroactive molecules from biodegradable microspheres for application in central nervous system disorders. Biomaterials 1999, 20, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Olivi, A.; Brem, H. Interstitial chemotherapy with sustained release polymer systems for the treatment of malignant gliomas. Recent Results Canc. Res. 1994, 135, 149–154. [Google Scholar]
- Groothuis, D.R.; Benalcazar, H.; Allen, C.V.; Wise, R.M.; Dills, C.; Dobrescu, C.; Rothholtz, V.; Levy, R.M. Comparison of cytosine arabinoside delivery to rat brain by intravenous, intrathecal, intraventricular and intraparenchymal routes of administration. Brain Res. 2000, 856, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.M.; Goyal, B.R.; Bhadada, S.V.; Bhatt, J.D.; Amin, A.F. Getting into the brain: Approaches to enhance brain drug delivery. CNS Drugs 2009, 23, 35–58. [Google Scholar]
- Schantz, E.J.; Lauffer, M.A. Diffusion measurements in agar gel. Biochemistry 1962, 1, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Transport of molecules in the tumor interstitium: A review. Cancer Res. 1987, 47, 3039–3051. [Google Scholar] [PubMed]
- Jain, R.K. Delivery of novel therapeutic agents in tumors: Physiological barriers and strategies. J. Natl. Cancer Inst. 1989, 81, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Fung, L.K.; Ewend, M.G.; Sills, A.; Sipos, E.P.; Thompson, R.; Watts, M.; Colvin, O.M.; Brem, H.; Saltzman, W.M. Pharmacokinetics of interstitial delivery of carmustine, 4-hydroperoxycyclophosphamide, and paclitaxel from a biodegradable polymer implant in the monkey brain. Cancer Res. 1998, 58, 672–684. [Google Scholar] [PubMed]
- Kroin, J.S.; Penn, R.D. Intracerebral chemotherapy: Chronic microinfusion of cisplatin. Neurosurgery 1982, 10, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Sendelbeck, S.L.; Urquhart, J. Spatial distribution of dopamine, methotrexate and antipyrine during continuous intracerebral microperfusion. Brain Res. 1985, 328, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Morrison, P.F.; Dedrick, R.L. Transport of cisplatin in rat brain following microinfusion: An analysis. J. Pharm. Sci. 1986, 75, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Fiandaca, M.S.; Forsayeth, J.R.; Dickinson, P.J.; Bankiewicz, K.S. Image-guided convection-enhanced delivery platform in the treatment of neurological diseases. Neurotherapeutics 2008, 5, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Sykova, E.; Nicholson, C. Diffusion in brain extracellular space. Physiol. Rev. 2008, 88, 1277–1340. [Google Scholar] [CrossRef] [PubMed]
- Vargova, L.; Homola, A.; Zamecnik, J.; Tichy, M.; Benes, V.; Sykova, E. Diffusion parameters of the extracellular space in human gliomas. Glia 2003, 42, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Zamecnik, J.; Vargova, L.; Homola, A.; Kodet, R.; Sykova, E. Extracellular matrix glycoproteins and diffusion barriers in human astrocytic tumours. Neuropathol. Appl. Neurobiol. 2004, 30, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Sykova, E. Diffusion properties of the brain in health and disease. Neurochem. Int. 2004, 45, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Bobo, R.H.; Laske, D.W.; Akbasak, A.; Morrison, P.F.; Dedrick, R.L.; Oldfield, E.H. Convection-enhanced delivery of macromolecules in the brain. Proc. Natl. Acad. Sci. USA 1994, 91, 2076–2080. [Google Scholar]
- Morrison, P.F.; Laske, D.W.; Bobo, H.; Oldfield, E.H.; Dedrick, R.L. High-flow microinfusion: Tissue penetration and pharmacodynamics. Amer. J. Physiol. 1994, 266, 292–305. [Google Scholar]
- Szerlip, N.J.; Walbridge, S.; Yang, L.; Morrison, P.F.; Degen, J.W.; Jarrell, S.T.; Kouri, J.; Kerr, P.B.; Kotin, R.; Oldfield, E.H.; et al. Real-time imaging of convection-enhanced delivery of viruses and virus-sized particles. J. Neurosurg. 2007, 107, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Krauze, M.T.; Forsayeth, J.; Park, J.W.; Bankiewicz, K.S. Real-time imaging and quantification of brain delivery of liposomes. Pharm. Res. 2006, 23, 2493–2504. [Google Scholar] [CrossRef] [PubMed]
- Hadaczek, P.; Yamashita, Y.; Mirek, H.; Tamas, L.; Bohn, M.C.; Noble, C.; Park, J.W.; Bankiewicz, K. The “perivascular pump” driven by arterial pulsation is a powerful mechanism for the distribution of therapeutic molecules within the brain. Mol. Ther. 2006, 14, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.F.; Morrison, P.F.; Chen, M.Y.; Harvey-White, J.; Pernaute, R.S.; Phillips, H.; Oldfield, E.; Bankiewicz, K.S. Heparin coinfusion during convection-enhanced delivery (CED) increases the distribution of the glial-derived neurotrophic factor (GDNF) ligand family in rat striatum and enhances the pharmacological activity of neurturin. Exp. Neurol. 2001, 168, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Neeves, K.B.; Sawyer, A.J.; Foley, C.P.; Saltzman, W.M.; Olbricht, W.L. Dilation and degradation of the brain extracellular matrix enhances penetration of infused polymer nanoparticles. Brain Res. 2007, 1180, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.B.; Sanchez-Pernaute, R.; Cunningham, J.; Bankiewicz, K.S. Convection-enhanced delivery of AAV-2 combined with heparin increases TK gene transfer in the rat brain. Neuroreport 2001, 12, 1961–1964. [Google Scholar] [CrossRef] [PubMed]
- Morrison, P.F.; Chen, M.Y.; Chadwick, R.S.; Lonser, R.R.; Oldfield, E.H. Focal delivery during direct infusion to brain: role of flow rate, catheter diameter, and tissue mechanics. Amer. J. Physiol. 1999, 277, 1218–1229. [Google Scholar]
- Chen, M.Y.; Lonser, R.R.; Morrison, P.F.; Governale, L.S.; Oldfield, E.H. Variables affecting convection-enhanced delivery to the striatum: a systematic examination of rate of infusion, cannula size, infusate concentration, and tissue-cannula sealing time. J. Neurosurg. 1999, 90, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Krauze, M.T.; Saito, R.; Noble, C.; Tamas, M.; Bringas, J.; Park, J.W.; Berger, M.S.; Bankiewicz, K.S. Reflux-free cannula for convection-enhanced high-speed delivery of therapeutic agents. J. Neurosurg. 2005, 103, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Fiandaca, M.S.; Varenika, V.; Eberling, J.; McKnight, T.R.; Bringas, J.; Pivirotto, P.; Beyer, J.; Hadaczek, P.; Forsayeth, J.; Bowers, W.J.; et al. Real-time MR imaging of adeno-associated viral vector delivery to the primate brain. Neuroimage 2009, 47, 27–35. [Google Scholar]
- Nguyen, T.T.; Pannu, Y.S.; Sung, C.; Dedrick, R.L.; Walbridge, S.; Brechbiel, M.W.; Garmestani, K.; Beitzel, M.; Yordanov, A.T.; Oldfield, E.H. Convective distribution of macromolecules in the primate brain demonstrated using computerized tomography and magnetic resonance imaging. J. Neurosurg. 2003, 98, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Krauze, M.T.; Bringas, J.R.; Noble, C.; McKnight, T.R.; Jackson, P.; Wendland, M.F.; Mamot, C.; Drummond, D.C.; Kirpotin, D.B.; et al. Gadolinium-loaded liposomes allow for real-time magnetic resonance imaging of convection-enhanced delivery in the primate brain. Exp. Neurol. 2005, 196, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Krauze, M.T.; McKnight, T.R.; Yamashita, Y.; Bringas, J.; Noble, C.O.; Saito, R.; Geletneky, K.; Forsayeth, J.; Berger, M.S.; Jackson, P.; et al. Real-time visualization and characterization of liposomal delivery into the monkey brain by magnetic resonance imaging. Brain Res. Protoc. 2005, 16, 20–26. [Google Scholar] [CrossRef]
- Murad, G.J.; Walbridge, S.; Morrison, P.F.; Garmestani, K.; Degen, J.W.; Brechbiel, M.W.; Oldfield, E.H.; Lonser, R.R. Real-time, image-guided, convection-enhanced delivery of interleukin 13 bound to pseudomonas exotoxin. Clin. Cancer Res. 2006, 12, 145–151. [Google Scholar]
- Lonser, R.R.; Schiffman, R.; Robison, R.A.; Butman, J.A.; Quezado, Z.; Walker, M.L.; Morrison, P.F.; Walbridge, S.; Murray, G.J.; Park, D.M.; et al. Image-guided, direct convective delivery of glucocerebrosidase for neuronopathic Gaucher disease. Neurology 2007, 68, 254–261. [Google Scholar] [PubMed]
- Lonser, R.R.; Warren, K.E.; Butman, J.A.; Quezado, Z.; Robison, R.A.; Walbridge, S.; Schiffman, R.; Merrill, M.; Walker, M.L.; Park, D.M.; et al. Real-time image-guided direct convective perfusion of intrinsic brainstem lesions. Technical note. J. Neurosurg. 2007, 107, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Varenika, V.; Dickenson, P.; Bringas, J.; LeCouteur, R.; Higgins, R.; Park, J.; Fiandaca, M.; Berger, M.; Sampson, J.; Bankiewicz, K.S. Detection of infusate leakage in the brain using real-time imaging of convection-enhanced delivery. J. Neurosurg. 2008, 109, 874–880. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.S.; Patel, N.K.; Hotton, G.R.; O’Sullivan, K.; McCarter, R.; Bunnage, M.; Brooks, D.J.; Svendsen, C.N.; Heywood, P. Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat. Med. 2003, 9, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.K.; Bunnage, M.; Plaha, P.; Svendsen, C.N.; Heywood, P.; Gill, S.S. Intraputamenal infusion of glial cell line-derived neurotrophic factor in PD: a two-year outcome study. Ann. Neurol. 2005, 57, 298–302. [Google Scholar]
- Love, S.; Plaha, P.; Patel, N.K.; Hotton, G.R.; Brooks, D.J.; Gill, S.S. Glial cell line-derived neurotrophic factor induces neuronal sprouting in human brain. Nat. Med. 2005, 11, 703–705. [Google Scholar] [PubMed]
- Marks, W.J.; Bartus, R.T.; Siffert, J.; Davis, C.S.; Lozano, A.; Boulis, N.; Vitek, J.; Stacy, M.; Turner, D.; Verhagen, L.; et al. Gene delivery of AAV2-neurturin for Parkinson’s disease: A double-blind, randomised, controlled trial. Lancet Neurol. 2010, 9, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Kunwar, S.; Pai, L.H.; Pastan, I. Cytotoxicity and antitumor effects of growth factor-toxin fusion proteins on human glioblastoma multiforme cells. J. Neurosurg. 1993, 79, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Laske, D.W.; Youle, R.J.; Oldfield, E.H. Tumor regression with regional distribution of the targeted toxin TF-CRM107 in patients with malignant brain tumors. Nat. Med. 1997, 3, 1362–1368. [Google Scholar] [CrossRef] [PubMed]
- Rand, R.W.; Kreitman, R.J.; Patronas, N.; Varricchio, F.; Pastan, I.; Puri, R.K. Intratumoral administration of recombinant circularly permuted interleukin-4-Pseudomonas exotoxin in patients with high-grade glioma. Exp. Neurol. 2000, 6, 2157–2165. [Google Scholar]
- Sampson, J.H.; Akabani, G.; Archer, G.E.; Bigner, D.D.; Berger, M.S.; Friedman, A.H.; Friedman, H.S.; Herndon, J.E., II; Kunwar, S.; Marcus, S.; McLendon, R.E.; Paolino, A.; Penne, K.; Provenzale, J.; Quinn, J.; Reardon, D.A.; Rich, J.; Stenzel, T.; Tourt-Uhlig, S.; Wikstrand, C.; Wong, T.; Williams, R.; Yuan, F.; Zalutsky, M.R.; Pastan, I. Progress report of a Phase I study of the intracerebral microinfusion of a recombinant chimeric protein composed of transforming growth factor (TGF)-alpha and a mutated form of the Pseudomonas exotoxin termed PE-38 (TP-38) for the treatment of malignant brain tumors. J. Neuro-Oncol. 2003, 65, 27–35. [Google Scholar] [CrossRef]
- Weber, F.; Asher, A.; Bucholz, R.; Berger, M.; Prados, M.; Chang, S.; Bruce, J.; Hall, W.; Rainov, N.G.; Westphal, M.; et al. Safety, tolerability, and tumor response of IL4-Pseudomonas exotoxin (NBI-3001) in patients with recurrent malignant glioma. J. Neuro-Oncol. 2003, 64, 125–137. [Google Scholar]
- Weber, F.W.; Floeth, F.; Asher, A.; Bucholz, R.; Berger, M.; Prados, M.; Chang, S.; Bruce, J.; Hall, W.; Rainov, N.G.; et al. Local convection enhanced delivery of IL4-Pseudomonas exotoxin (NBI-3001) for treatment of patients with recurrent malignant glioma. Acta Neurochir. 2003, 88, 93–103. [Google Scholar]
- Patel, S.J.; Shapiro, W.R.; Laske, D.W.; Jensen, R.L.; Asher, A.L.; Wessels, B.W.; Carpenter, S.P.; Shan, J.S. Safety and feasibility of convection-enhanced delivery of Cotara for the treatment of malignant glioma: initial experience in 51 patients. Neurosurgery 2005, 56, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Fiandaca, M.S.; Bankiewicz, K.S. Gene therapy for Parkinson’s disease: From nonhuman primates to humans. Curr. Opin. Mol. Ther. 2010, 12, 519–529. [Google Scholar] [PubMed]
- Sampson, J.H.; Archer, G.; Pedain, C.; Wembacher-Schröder, E.; Westphal, M.; Kunwar, S.; Vogelbaum, M.A.; Coan, A.; Herndon, J.E.; Raghavan, R.; et al. Poor drug distribution as a possible explanation for the results of the PRECISE trial. J. Neurosurg. 2010, 113, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Valles, F.; Fiandaca, M.S.; Bringas, J.; Dickinson, P.J.; LeCouteur, R.A.; Higgins, R.J.; Berger, M.; Forsayeth, J.; Bankiewicz, K.S. Anatomical compression caused by high-volume convection-enhanced delivery to the brain. Neurosurgery 2009, 65, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Behin, A.; Hoang-Xuan, K.; Carpentier, A.F.; Delattre, J.Y. Primary brain tumours in adults. Lancet 2003, 361, 323–331. [Google Scholar] [PubMed]
- Zamecnik, J. The extracellular space and matrix of gliomas. Acta Neuropathol. 2005, 110, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Baxter, L.T.; Jain, R.K. Transport of fluid and macromolecules in tumors. I. Role of interstitial pressure and convection. Microvasc. Res. 1989, 37, 77–104. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.H.; Humphrey, J.A. Interstitial transport and transvascular fluid exchange during infusion into brain and tumor tissue. Microvasc. Res. 2007, 73, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.J.; Navlitloha, Y.; Vavra, M.W.; Kang, E.W.Y.; Itskovitch, A.C.; Molnar, P.; Levy, R.M.; Groothuis, D.R. Isolation of drug delivery from drug effect: Problems of optimizing drug delivery parameters. Neuro Oncol. 2006, 8, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Gregoriadis, G. Engineering liposomes for drug delivery: Progress and problems. TrendsBiotechnol. 1995, 13, 527–537. [Google Scholar]
- Torchilin, V.P. Structure and design of polymeric surfactant-based drug delivery systems. J. Control. Release 2001, 73, 137–172. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.Y.; Wang, Y.; Powell, R.; Chan, P. Polymeric core-shell nanoparticles for therapeutics. Clin. Exp. Pharmacol. Physiol. 2006, 33, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Bangham, A.D.; Standish, M.M.; Miller, N. Cation permeability of phospholipid model membranes: Effect of narcotics. Nature 1965, 208, 1295–1297. [Google Scholar] [PubMed]
- Bangham, A.D.; Standish, M.M.; Watkins, J.C. Diffusion of univalent ions across the lamellae of swollen phospholipids. J. Mol. Biol. 1965, 13, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Bangham, A.D. Lipid bilayers and biomembranes. Annu. Rev. Biochem. 1972, 41, 753–776. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V.P. Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Discov. 2005, 4, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Chapman, D. Lipid dynamics in cell membranes. In Cell Membranes: Biochemistry, Cell Biology and Pathology; Weissmann, G., Claiborne, R., Eds.; Hospital Practice Publishing Company: New York, NY, USA, 1975; pp. 13–22. [Google Scholar]
- Chapman, D.; Benga, G. Biomembrane fluidity-studies of model and natural membranes. In Biological Membranes; Chapman, D., Ed.; Academic Press: London, UK, 1984; Volume 5, pp. 1–56. [Google Scholar]
- Kates, M.; Manson, L.A. Membrane Fluidity. In Biomembranes; Plenum Press: New York, NY, USA, 1984; Volume 12. [Google Scholar]
- Haines, T.H. Do sterols reduce proton and sodium leaks through lipid bilayers? Prog. Lipid Res. 2001, 40, 299–324. [Google Scholar] [CrossRef] [PubMed]
- Panwar, P.; Pandey, B.; Lakhera, P.C.; Singh, K.P. Preparation, characterization, and in vitro release study of albendazole-encapsulated nanosize liposomes. Int. J. Nanomed. 2010, 5, 101–108. [Google Scholar]
- Szoka, F.J.; Papahadjopoulos, D. Comparative properties and methods of preparation of lipid vesicles (liposomes). Annu. Rev. Biophys. Bioeng. 1980, 9, 467–508. [Google Scholar] [CrossRef] [PubMed]
- Penwar, P.; Pandey, B.; Lakhera, P.C.; Singh, K.P. Preparation, characterization, and in vitro release study of albendazole-encapsulated nanosize liposomes. Int. J. Nanomed. 2010, 9, 101–108. [Google Scholar]
- Wang, H.; Zhao, P.; Liang, X.; Gong, X.; Song, T.; Niu, R.; Chang, J. Folate-PEG coated cationic modified chitosan-Cholesterol liposomes for tumor-targeted drug delivery. Biomaterials 2010, 31, 4129–4138. [Google Scholar] [CrossRef] [PubMed]
- Pagano, R.E.; Weinstein, J.N. Interaction of liposomes with mammalian cells. Annu. Rev. Biophys. Bioeng. 1978, 7, 435–468. [Google Scholar] [CrossRef] [PubMed]
- Samad, A.; Sultana, Y.; Aqil, M. Liposomal drug delivery systems: An update review. Curr. Drug Deliv. 2007, 4, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Deamer, D.; Bangham, A.D. Large volume liposomes by an ether vaporization method. Biochim. Biophys. Acta 1976, 443, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Schieren, H.; Rudolph, S.; Finkelstein, M.; Coleman, P.; Weissmann, G. Comparison of large unilamellar vesicles prepared by a petroleum ether vaporization method with multilamellar vesicles: ESR, diffusion and entrapment analyses. Biochim. Biophys. Acta 1978, 542, 137–153. [Google Scholar] [PubMed]
- Szoka, F.J.; Papahadjopoulos, D. Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaporation. Proc. Natl. Acad. Sci.USA 1978, 75, 4194–4198. [Google Scholar]
- Pick, U. Liposomes with a large trapping capacity prepared by freezing and thawing of sonicated phospholipid mixtures. Arch. Biochem. Biophys. 1981, 212, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Lasic, D.D. The spontaneous formation of unilamellar vesicles. J. Colloid Interf. Sci. 1988, 124, 428–435. [Google Scholar] [CrossRef]
- Gould-Fogerite, S.; Mannino, R.J. Preparation of large unilamellar liposomes with high entrapment yield by rotary dialysis or agarose plug diffusion. In Liposome Technology. Liposome Preparation and Related Techniques; Gregoriadis, G., Ed.; CRC Press: Boca Raton, FL, USA, 1993; Volume 1, pp. 67–79. [Google Scholar]
- Kikuchi, H.; Yamauchi, H.; Hirota, S. A polyol dilution method for mass production of liposomes. J. Liposome Res. 1994, 4, 71–91. [Google Scholar] [CrossRef]
- Talsma, H.; van Steenbergen, M.J.; Borchert, J.C.H.; Crommelin, D.J.A. A novel technique for the one-step preparation of liposomes and nonionic surfactant vesicles without the use of organic solvents. Liposome formation in a continuous gas stream: The bubble method. J. Pharm. Sci. 1994, 83, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Vemuri, S.; Rhodes, C.T. Preparation and characterization of liposomes as therapeutic delivery systems: A review. Pharm. Acta Helv. 1995, 70, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Lasic, D.D.; Joannic, R.; Keller, B.C.; Frederik, P.M.; Auvray, L. Spontaneous vesiculation. Adv. Colloid Interface Sci. 2001, 89-90, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Mozafari, M.R.; Reed, C.J.; Rostron, C. Development of non-toxic liposomal formulations for gene and drug delivery to the lung. Technol. Health Care 2002, 10, 342–344. [Google Scholar]
- Mozafari, M.R.; Reed, C.J.; Rostron, C.; Kocum, C.; Piskin, E. Construction of stable anionic liposome-plasmid particles using the heating method: A preliminary investigation. Cell. Mol. Biol. Lett. 2002, 7, 923–927. [Google Scholar] [PubMed]
- Mozafari, M.R.; Reed, C.J.; Rostron, C.; Martin, D.S. Transfection of human airway epithelial cells using a lipid-based vector prepared by the heating method. J. Aerosol Med. 2004, 17, 100. [Google Scholar]
- Mozafari, M.R. Liposomes: An overview of manufacturing techniques. Cell. Mol. Biol. Lett. 2005, 10, 711–719. [Google Scholar] [PubMed]
- Barenholz, Y.; Gibbes, D.; Litman, B.J.; Goll, J.; Thompson, T.E.; Carlson, R.D. A simple method for the preparation of homogeneous phospholipid vesicles. Biochemistry 1977, 16, 2806–2810. [Google Scholar] [PubMed]
- Hope, M.J.; Bally, M.B.; Webb, G.; Cullis, P.R. Production of large unilamellar vesicles by a rapid extrusion procedure. Characterization of size distribution, trapped volume and ability to maintain a membrane potential. Biochim. Biophys. Acta 1985, 812, 55–65. [Google Scholar] [CrossRef]
- MacDonald, R.C.; MacDonald, R.I.; Menco, B.P.M.; Takeshita, K.; Subbarao, N.K.; Hu, L. Small volume extrusion apparatus for preparation of large unilamellar vesicles. Biochim. Biophys. Acta 1991, 1061, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Klibanov, A.L.; Murayama, K.; Torchilin, V.P.; Huang, L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett. 1990, 268, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Murayama, K.; Kennel, S.J.; Huang, L. Lipid composition is important for highly efficient target binding and retention of immunoliposomes. Proc. Natl. Acad. Sci. USA 1990, 87, 5744–5748. [Google Scholar] [CrossRef]
- Murayama, K. PEG-Immunoliposoome. Biosci. Rep. 2002, 22, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V.P.; Omelyanenko, V.G.; Papisov, M.I.; Bogdanov, A.A.J.; Trubetskoy, V.S.; Herron, J.N.; Gentry, C.A. Poly(ethylene glycol) on the liposome surface: on the mechanism of polymer-coated liposome longevity. Biochim. Biophys. Acta 1994, 1195, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Pedroso de Lima, M.C.; Girao da Cruz, M.T.; Cardoso, A.L.C.; Simoes, S.; Pereira de Almeida, L. Liposomal and viral vectors for gene therapy of the central nervous system. Curr. Drug Targets CNS Neurol. Disord. 2005, 4, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Black, C.; Gregoriadis, G. Intracellular fate and effect of liposome-entrapped actinomycin-d injected into rats. Biochem. Soc. Trans. 1974, 2, 869–871. [Google Scholar]
- Hillaireau, H.; Couvreur, P. Nanocarriers’ entry into the cell: relevance to drug delivery. Cell. Mol. Life Sci. 2009, 66, 2873–2896. [Google Scholar] [CrossRef] [PubMed]
- Aderem, A.; Underhill, D. Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 1999, 17, 593–623. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitch, M. Professional and nonprofessional phagocytes-an introduction. Trends Cell Biol. 1995, 5, 85–87. [Google Scholar] [CrossRef] [PubMed]
- Owens, D.; Peppas, N. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm. 2006, 307, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Vonarbourg, A.; Passirani, C.; Saulnier, P.; Benoit, J. Parameters influencing the stealthiness of colloidal drug delivery systems. Biomaterials 2006, 27, 4356–4373. [Google Scholar] [PubMed]
- Korn, E.D.; Weisman, R.A. Phagocytosis of latex beads by Acanthamoeba. II. Electron microscopic study of the initial events. J. Cell Biol. 1967, 34, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, H.; Nagasaki, Y.; Kataoka, K. PEGylated nanoparticles for biological and pharmaceutical applications. Adv. Drug Deliv. Rev. 2003, 55, 403–419. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Ghosh, R.N.; Maxfield, F.R. Endocytosis. Physiol. Rev. 1997, 77, 759–803. [Google Scholar] [PubMed]
- Conner, S.D.; Schmid, S.L. Regulated portals of entry into the cell. Nature 2003, 422, 37–44. [Google Scholar] [PubMed]
- Swanson, J.A.; Watts, C. Macropinocytosis. Trends Cell Biol. 1995, 5, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Kanaseki, T.; Kadota, K. The “vesicle in a basket”. A morphological study of the coated vesicle isolated from the nerve endings of the guinea pig brain, with special reference to the mechanism of membrane movements. J. Cell Biol. 1969, 42, 202–220. [Google Scholar] [CrossRef] [PubMed]
- Woodward, M.P.; Roth, T.F. Coated vesicles: Characterization, selective dissociation, and reassembly. Proc. Natl. Acad. Sci. USA 1978, 75, 4394–4398. [Google Scholar] [CrossRef]
- Bareford, L.M.; Swaan, P.W. Endocytic mechanisms for targeted drug delivery. Adv. Drug Deliv. Rev. 2007, 59, 748–758. [Google Scholar] [CrossRef] [PubMed]
- Marsh, M.; Helenius, A. Virus entry: Open sesame. Cell 2006, 124, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Mayor, S.; Pagano, R.E. Pathways of clathrin-independent endocytosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 602–612. [Google Scholar]
- Drummond, D.C.; Meyer, O.; Hong, K.; Kirpotin, D.B.; Papahadjopoulos, D. Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharmacol. Rev. 1999, 51, 691–743. [Google Scholar] [PubMed]
- Chonn, A.; Cullis, P.R. Recent advances in liposomal drug-delivery systems. Curr. Opin. Biotechnol. 1995, 6, 698–708. [Google Scholar] [CrossRef] [PubMed]
- Mayer, L.D.; Madden, T.M.; Bally, M.B.; Cullis, P.R. pH gradient-mediated drug entrapment in Uposomes. In Liposome Technology, 2nd; Gregoriadis, G., Ed.; CRC Press: Boca Raton, FL, USA, 1993; Volume 2, pp. 27–44. [Google Scholar]
- Abdiche, Y.N.; Myszka, D.G. Probing the mechanism of drug/lipid membrane interactions using Biacore. Anal. Biochem. 2004, 328, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Krauze, M.T.; Noble, C.O.; Drummond, D.C.; Kirpotin, D.B.; Berger, M.S.; Park, J.W.; Bankiewicz, K.S. Convection-enhanced delivery of Ls-TPT enables an effective, continuous, low-dose chemotherapy against malignant glioma xenograft model. Neuro Oncol. 2006, 8, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Ostro, M.J. Liposome from biophysics to therapeutics; Marcel Dekker: New York, NY, USA, 1987. [Google Scholar]
- Allen, T.M. Liposomal drug formulations. Drugs 1998, 56, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Iinuma, H.; Murayama, K.; Okinaga, K.; Sasaki, K.; Sekine, T.; Ishida, O.; Ogiwara, N.; Johkura, K.; Yonemura, Y. Intracellular targeting therapy of cisplatin-encapsulated transferring-polyethyleneglycol liposome on peritoneal dissemination of gastric cancer. Int. J. Cancer 2002, 99, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Bringas, J.R.; McKnight, T.R.; Wendland, M.F.; Mamot, C.; Drummond, D.C.; Kirpotin, D.B.; Park, J.W.; Berger, M.S.; Bankiewicz, K.S. Distribution of liposomes into brain and rat brain tumor models by convection-enhanced delivery monitored with magnetic resonance imaging. Cancer Res. 2004, 64, 2572–2579. [Google Scholar] [PubMed]
- Noble, C.O.; Krauze, M.T.; Drummond, D.C.; Yamashita, Y.; Saito, R.; Berger, M.S.; Kirpotin, D.B.; Bankiewicz, K.S.; Park, J.W. Novel nanoliposomal CPT-11 infused by convection-enhanced delivery in intracranial tumors: pharmacology and efficacy. Cancer Res. 2006, 66, 2801–2806. [Google Scholar] [PubMed]
- Saito, R.; Krauze, M.T.; Noble, C.O.; Tamas, M.; Drummond, D.C.; Kirpotin, D.B.; Berger, M.S.; Park, J.W.; Bankiewicz, K.S. Tissue affinity of the infusate affects the distribution volume during convection-enhanced delivery into rodent brains: implications for local drug delivery. J. Neurosci. Meth. 2006, 154, 225–232. [Google Scholar] [CrossRef]
- Krauze, M.T.; Noble, C.O.; Kawaguchi, T.; Drummond, D.; Kirpotin, D.B.; Yamashita, Y.; Kullberg, E.; Forsayeth, J.; Park, J.W.; Bankiewicz, K.S. Convection-enhanced delivery of nanoliposomal CPT-11 (irinotecan) and PEGylated liposomal doxorubicin (Doxil) in rodent intracranial brain tumor xenografts. Neuro Oncol. 2007, 9, 393–403. [Google Scholar] [PubMed]
- Yamashita, Y.; Krauze, M.T.; Kawaguchi, T.; Noble, C.O.; Drummond, D.C.; Park, J.W.; Bankiewicz, K.S. Convection-enhanced delivery of a topoisomerase I inhibitor (nanoliposomal topotecan) and a topoisomerase II inhibitor (pegylated liposomal doxorubicin) in intracranial brain tumor xenografts. Neuro Oncol. 2007, 9, 20–28. [Google Scholar] [PubMed]
- Dickinson, P.J.; LeCouteur, R.A.; Higgins, R.J.; Bringas, J.R.; Roberts, B.; Larson, R.F.; Yamashita, Y.; Krauze, M.; Noble, C.O.; Drummond, D.; et al. Canine model of convection-enhanced delivery of liposomes containing CPT-11 monitored with real-time magnetic resonance imaging: laboratory investigation. J. Neurosurg. 2008, 108, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, P.J.; LeCouteur, R.A.; Higgins, R.J.; Bringas, J.R.; Larson, R.F.; Yamashita, Y.; Krauze, M.T.; Forsayeth, J.; Noble, C.O.; Drummond, D.C.; et al. Canine spontaneous glioma: A translational model system for convection-enhanced delivery. Neuro Oncol. 2010, 12, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Krauze, M.T.; Vandenberg, S.R.; Yamashita, Y.; Saito, R.; Forsayeth, J.; Noble, C.; Park, J.; Bankiewicz, K.S. Safety of real-time convection-enhanced delivery of liposomes to primate brain: A long-term retrospective. Exp. Neurol. 2008, 210, 638–644. [Google Scholar] [CrossRef]
- Krauze, M.; Forsayeth, J.; Yin, D.; Bankiewicz, K.S. Convection-enhanced delivery of liposomes to primate brain. Method Enzymol. 2009, 465, 349–362. [Google Scholar]
- Krauze, M.T.; Saito, R.; Noble, C.; Bringas, J.; Forsayeth, J.; McKnight, T.R.; Park, J.; Bankiewicz, K.S. Effects of the perivascular space on convection-enhanced delivery of liposomes in primate putamen. Exp. Neurol. 2005, 196, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Grahn, A.Y.; Bankiewicz, K.S.; Dugich-Djordjevic, M.; Bringas, J.R.; Hadaczek, P.; Johnson, G.A.; Eastman, S.; Luz, M. Non-PEGylated liposomes for convection-enhanced delivery of topotecan and gadodiamide in malignant glioma: initial experience. J. Neuro-Oncol. 2009, 95, 185–197. [Google Scholar] [CrossRef]
- Hadjipanayis, C.G.; Machaidze, R.; Kaluzova, M.; Wang, L.; Schuette, A.J.; Chen, H.; Wu, X.; Mao, H. EGFRvIII antibody-conjugated iron oxide nanoparticles for magnetic resonance imaging-guided convection-enhanced delivery and targeted therapy of glioblastoma. Cancer Res. 2010, 70, 6303–6312. [Google Scholar] [CrossRef] [PubMed]
- MacKay, J.A.; Deen, D.F.; Szoka, F.C., Jr. Distribution in brain of liposomes after convection enhanced delivery; modulation by particle charge, particle diameter, and presence of steric coating. Brain Res. 2005, 1035, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Gabizon, A.; Peretz, T.; Sulkes, A.; Amselem, S.; Ben-Yosef, R.; Ben-Baruch, N.; Catane, R.; Biran, S.; Barenholz, Y. Systemic administration of doxorubicin-containing liposomes in cancer patients: a phase I study. Eur. J. Clin. Oncol. 1989, 25, 1795–1803. [Google Scholar] [CrossRef]
- Noble, C.O.; Kirpotin, D.B.; Hayes, M.E.; Mamot, C.; Hong, K.; Park, J.W.; Benz, C.C.; Marks, J.D.; Drummond, D.C. Development of ligand-targeted liposomes for cancer therapy. Expert Opin. Ther. Targets 2004, 8, 335–353. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Hansen, C.B.; Stuart, D.D. Targeted sterically stabilized liposomal drug delivery. In Medical Applications of Liposomes; Lasic, D., Papahadjopoulos, D., Eds.; Elsevier Science B.V.: New York, NY, USA, 1998; pp. 297–323. [Google Scholar]
- Barenholz, Y. Liposome application: Problems and prospects. Curr. Opin. Colloid Interface Sci. 2001, 6, 66–77. [Google Scholar] [CrossRef]
- Drummond, D.C.; Noble, C.O.; Guo, Z.; Hong, K.; Park, J.W.; Kirpotin, D.B. Development of a highly active nanoliposomal irinotecan using a novel intraliposomal stabilization strategy. Cancer Res. 2006, 66, 3271–3277. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Kiwada, H. Accelerated blood clearance (ABC) phenomenon upon repeated injection of PEGylated liposomes. Int. J. Pharm. 2008, 354, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J.; Baranyi, L.; Savay, S.; Milosevits, J.; Bunger, R.; Laverman, P.; Metselaar, J.M.; Storm, G.; Chanan-Khan, A.; Liebes, L.; et al. Role of complement activation in hypersensitivity reactions to doxil and hynic PEG liposomes: Experimental and clinical studies. J. Liposome Res. 2002, 12, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J. Complement activation-related pseudoallergy: A new class of drug-induced acute immune toxicity. Toxicology 2005, 216, 106–121. [Google Scholar]
- Harrington, K.J.; Lewanski, C.R.; Stewart, J.S. Liposomes as vehicles for targeted therapy of cancer. Part 1: Preclinical development. Clin. Oncol. 2000, 12, 2–15. [Google Scholar]
- Jacobs, A.; Voges, J.; Reszka, R.; Lercher, M.; Gossmann, A.; Kracht, L.; Kaestle, C.; Wagner, R.; Wienhard, K.; Heiss, W.D. Positron-emission tomography of vector-mediated gene expression in gene therapy for gliomas. Lancet 2001, 358, 727–729. [Google Scholar] [PubMed]
- Voges, J.; Reszka, R.; Gossmann, A.; Dittmar, C.; Richter, R.; Garlip, G.; Kracht, L.; Coenen, H.H.; Sturm, V.; Wienhard, K.; et al. Image-guided convection-enhanced delivery and gene therapy of glioblastoma. Ann. Neurol. 2003, 54, 479–487. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, B.W. Suicide gene therapy. In Gene Therapy for Cancer: Therapeutic Mechanisms and Strategies; Templeton, N., Ed.; Marcel Dekker: New York, NY, USA, 2000; pp. 353–370. [Google Scholar]
- Fillat, C.; Carrio, M.; Cascante, A.; Sangro, B. Suicide gene therapy mediated by the Herpes Simplex virus thymidine kinase gene/Ganciclovir system: fifteen years of application. Curr. Gene Ther. 2003, 3, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Boulikas, T.; Soling, A.; Warnke, P.C.; Rainov, N.G. Immunogene therapy of recurrent glioblastoma multiforme with a liposomally encapsulated replication-incompetent Semliki forest virus vector carrying the human interleukin-12 gene—a phase I/II clinical protocol. J. Neuro-Oncol. 2003, 64, 147–154. [Google Scholar]
- Chiu, T.L.; Lin, S.Z.; Hsieh, W.H.; Peng, C.W. AAV2-mediated interleukin-12 in the treatment of malignant brain tumors through activation of NK cells. Int. J. Oncol. 2009, 35, 1361–1367. [Google Scholar] [PubMed]
- Roche, F.P.; Sheahan, B.J.; O'Mara, S.M.; Atkins, G.J. Semliki Forest virus-mediated gene therapy of the RG2 rat glioma. Neuropathol. Appl. Neurobiol. 2010, 36, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Huynh, G.H.; Ozawa, T.; Deen, D.F.; Tihan, T.; Szoka, F.C., Jr. Retro-convection enhanced delivery to increase blood to brain transfer of macromolecules. Brain Res. 2007, 1128, 181–190. [Google Scholar] [PubMed]
- Myers, R.D.; Rezvani, A.H.; Gurley-Orkin, L.A. New double-lumen polyethylene cannula for push-pull perfusion of brain tissue in vivo. J. Neurosci. Meth. 1985, 12, 205–218. [Google Scholar] [CrossRef]
- Miyata, S.; Kawabata, S.; Hiramatsu, R.; Doi, A.; Ikeda, N.; Yamashita, T.; Kuroiwa, T.; Kasaoka, S.; Maruyama, K.; Miyatake, S.I. CT imaging of transferrin targeting liposomes encapsulating both boron and iodine contrast agent by CED to F98 rat glioma for boron neutron capture therapy. Neurosurgery 2011. [Google Scholar]
- Doi, A.; Kawabata, S.; Iida, K.; Yokoyama, K.; Kajimoto, Y.; Kuroiwa, T.; Shirakawa, T.; Kirihata, M.; Kasaoka, S.; Maruyama, K.; et al. Tumor-specific targeting of sodium borocaptate (BSH) to malignant glioma by transferrin-PEG liposomes: A modality for boron neutron capture therapy. J. Neuro-Oncol. 2008, 87, 287–294. [Google Scholar] [CrossRef]
- Wu, G.; Barth, R.F.; Yang, W.; Lee, R.J.; Tjarks, W.; Backer, M.V.; Backer, J.M. Boron containing macromolecules and nanovehicles as delivery agents for neutron capture therapy. Anti-Cancer Agents Med. Chem. 2006, 6, 167–184. [Google Scholar] [CrossRef]
- Schroeder, A.; Kost, J.; Barenholz, Y. Ultrasound, liposomes, and drug delivery: Principles for using ultrasound to control the release of drugs from liposomes. Chem. Phys. Lipids 2009, 162, 1–16. [Google Scholar] [CrossRef] [PubMed]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fiandaca, M.S.; Berger, M.S.; Bankiewicz, K.S. The Use of Convection-Enhanced Delivery with Liposomal Toxins in Neurooncology. Toxins 2011, 3, 369-397. https://doi.org/10.3390/toxins3040369
Fiandaca MS, Berger MS, Bankiewicz KS. The Use of Convection-Enhanced Delivery with Liposomal Toxins in Neurooncology. Toxins. 2011; 3(4):369-397. https://doi.org/10.3390/toxins3040369
Chicago/Turabian StyleFiandaca, Massimo S., Mitchel S. Berger, and Krystof S. Bankiewicz. 2011. "The Use of Convection-Enhanced Delivery with Liposomal Toxins in Neurooncology" Toxins 3, no. 4: 369-397. https://doi.org/10.3390/toxins3040369
APA StyleFiandaca, M. S., Berger, M. S., & Bankiewicz, K. S. (2011). The Use of Convection-Enhanced Delivery with Liposomal Toxins in Neurooncology. Toxins, 3(4), 369-397. https://doi.org/10.3390/toxins3040369