Current and Future Experimental Strategies for Structural Analysis of Trichothecene Mycotoxins—A Prospectus

Abstract
:1. Introduction
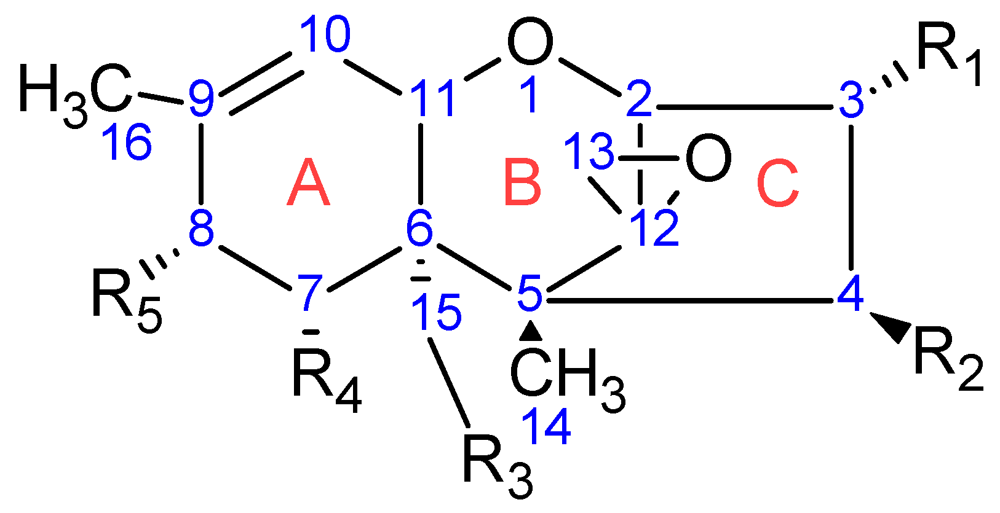
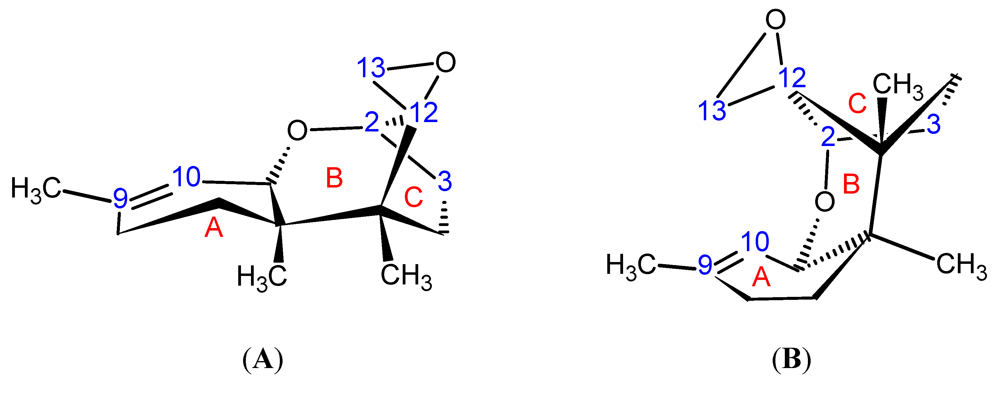
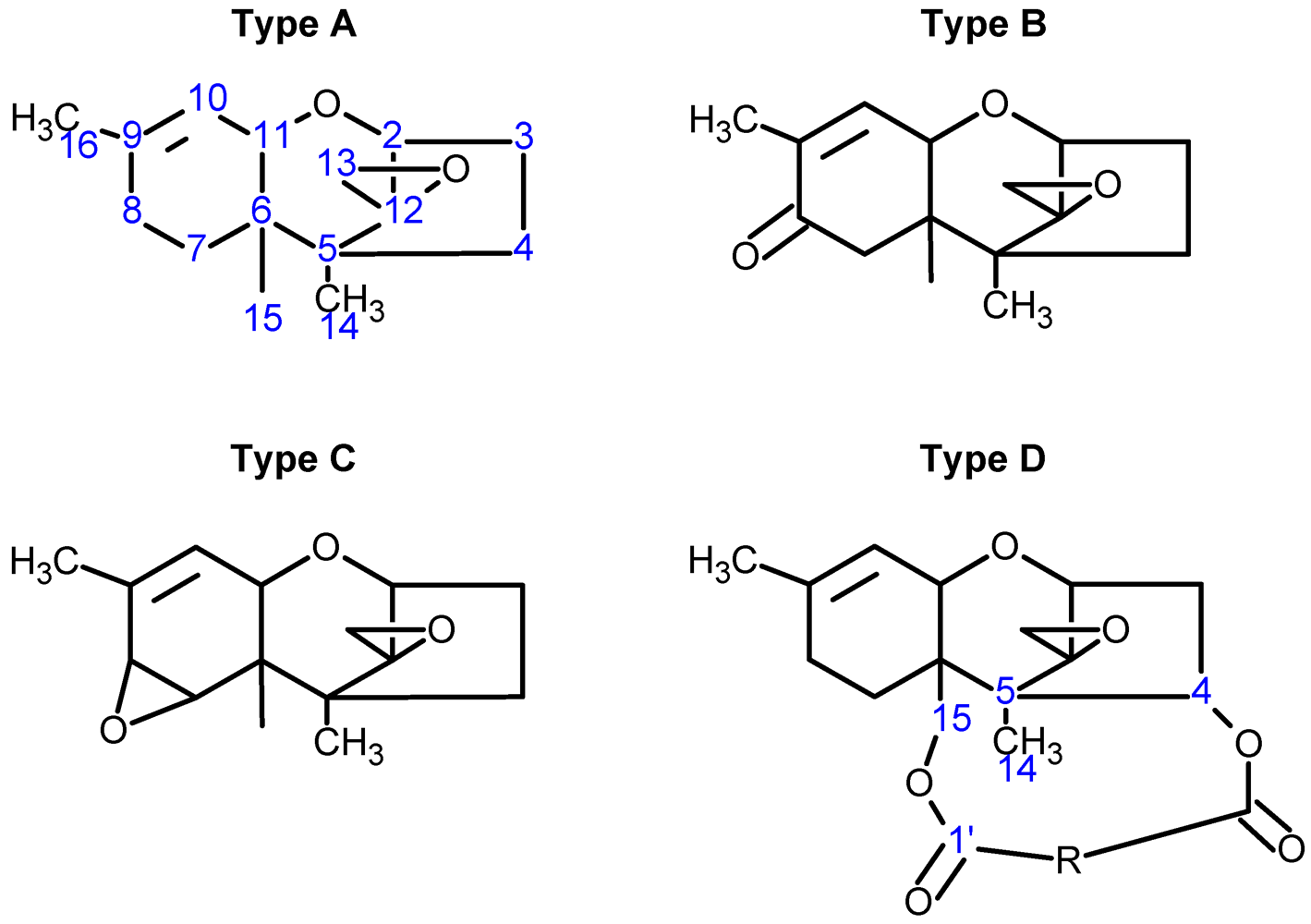

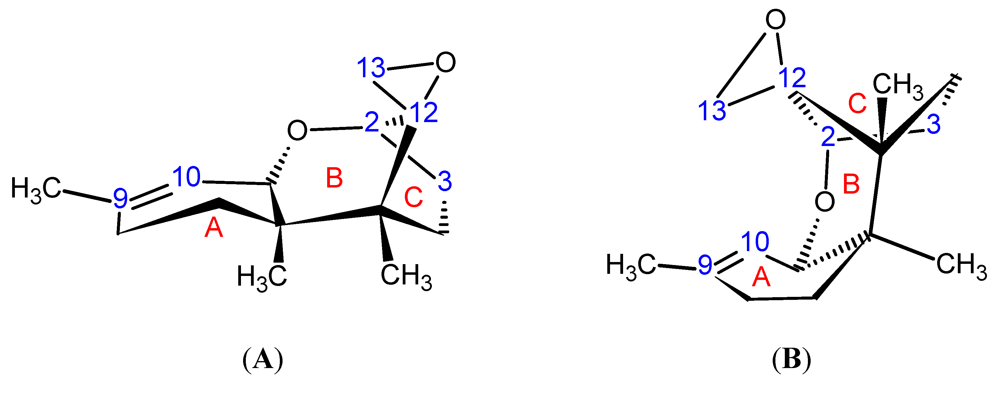
{kind=link}
{kind=link}
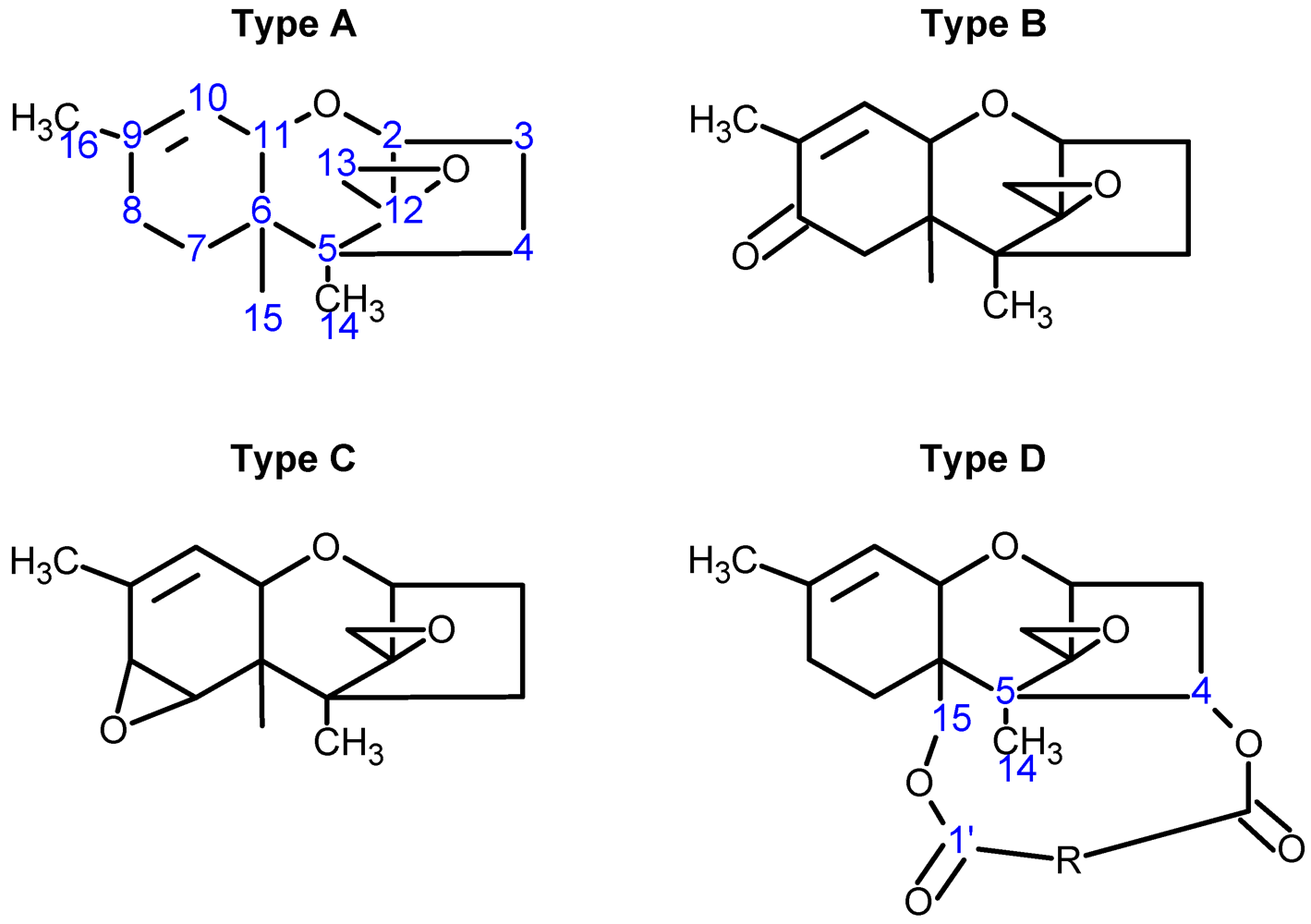{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Toxin Name | Type | R1 | R2 | R3 | R4 | R5 |
|---|---|---|---|---|---|---|
| T-2 toxin | A | OH | 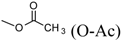 (O-Ac) (O-Ac) | O-Ac | H | 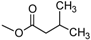 |
| Diacetoxyscirpenol (DAS) | A | OH | O-Ac | O-Ac | H | H |
| Trichodermin | A | H | O-Ac | H | H | H |
| Decalonectrin | A | O-Ac | H | OH | H | H |
| Deoxynivalenol (DON) | B | OH | H | OH | OH | =O |
| 3-acetyldeoxynivalenol (3-ADON) | B | O-Ac | H | OH | OH | =O |
| Nivalenol (NIV) | B | OH | OH | OH | OH | =O |
| Trichothecin | B | H |  | H | H | =O |
| Crotocin | C | H | 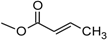 | H | Epoxide | |
| Isororidin A | D | H | 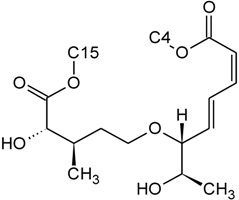 | H | H | |
| Roridin E | D | H | 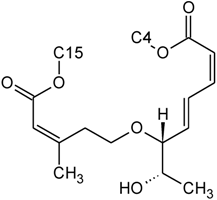 | H | H | |
| Roridin H (Verrucarin H) | D | H |  | H | H | |
| Baccharinoid B1 | D | H | 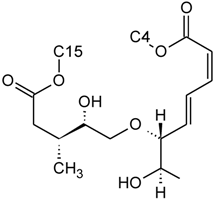 | H | H | |
| Baccharinoid B2 | D | H | 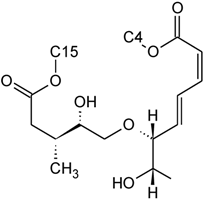 | H | H | |
| Baccharinoid B3 | D | H | 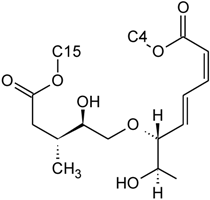 | H | H | |
| Baccharinoid B7 | D | H |  | H | H | |
| Verrucarin A | D | H |  | H | H | |
| Verrucarin B | D | H | 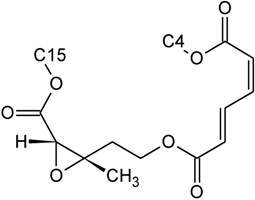 | H | H | |
| Myrotoxin A | D | H | 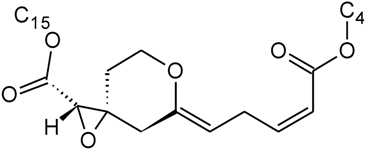 | H | H | |
| Myrotoxin B | D | H | 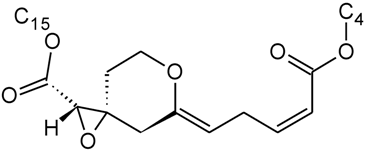 | H | OH | |
2. Structure-Function Relationships
Trichothecene Toxicity
3. Methods to Study Structure
3.1. Crystallography
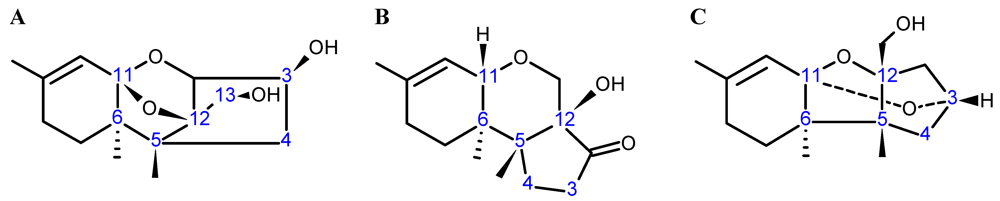
3.2. Nuclear Magnetic Resonance (NMR)
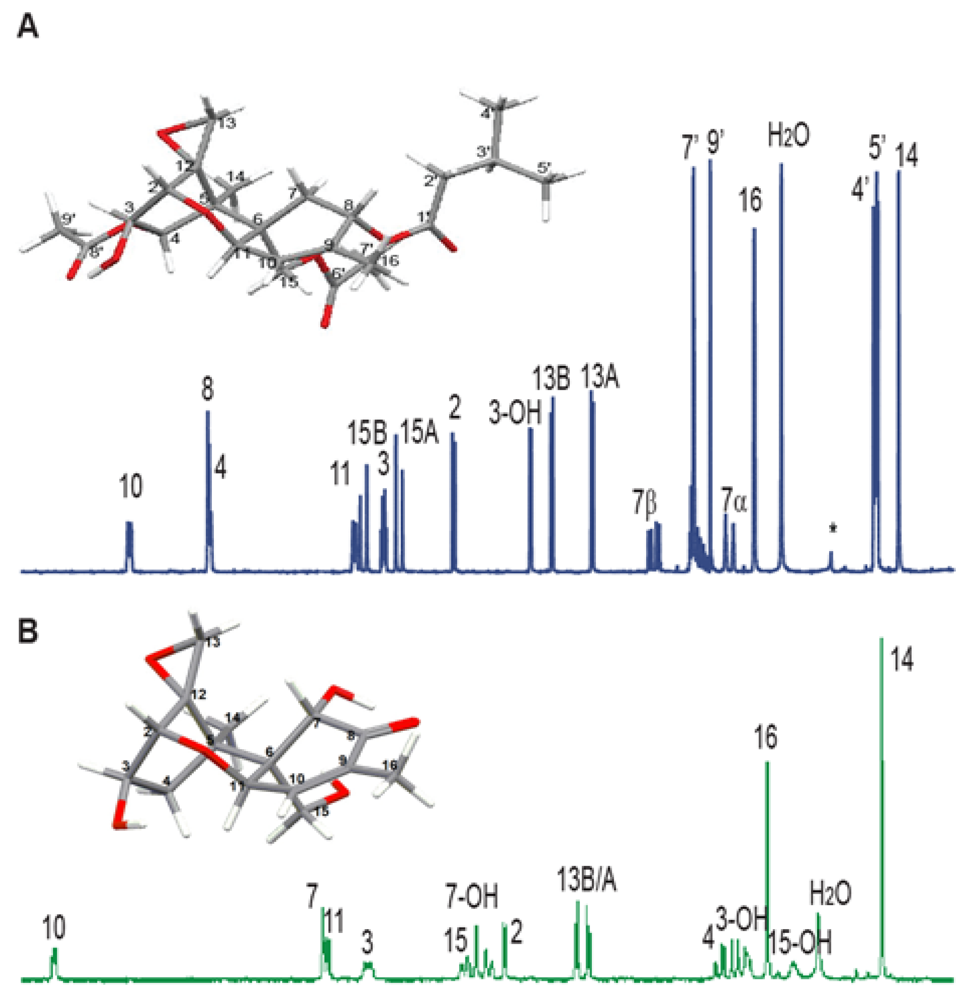
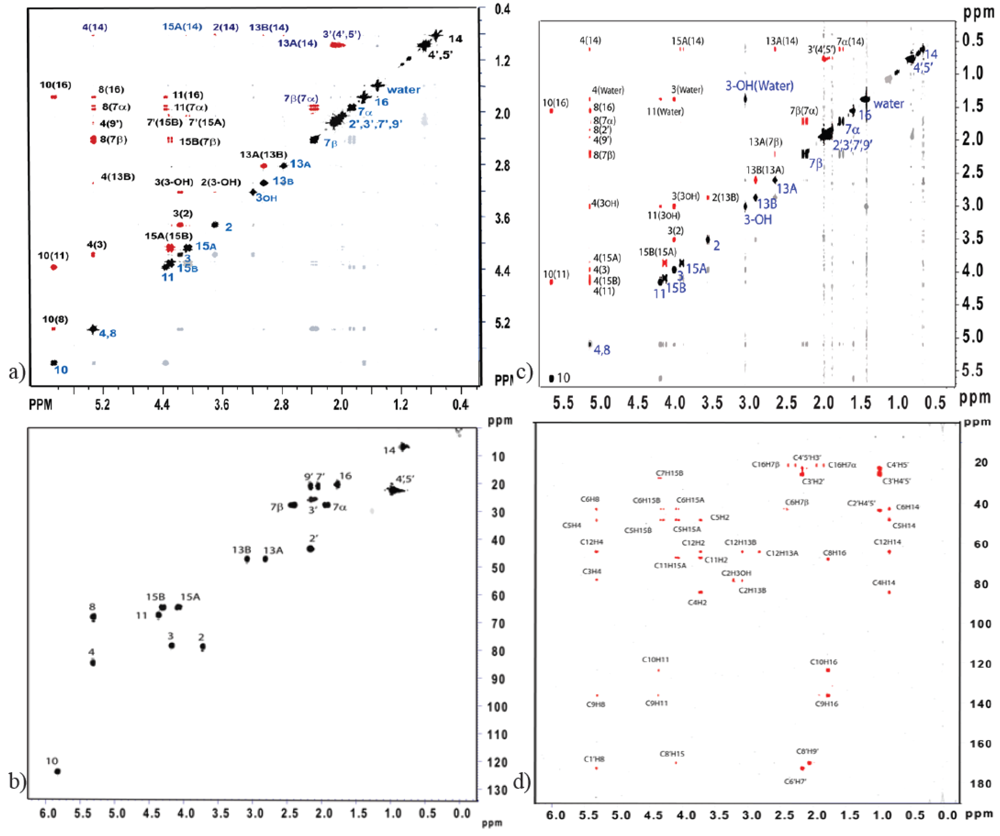
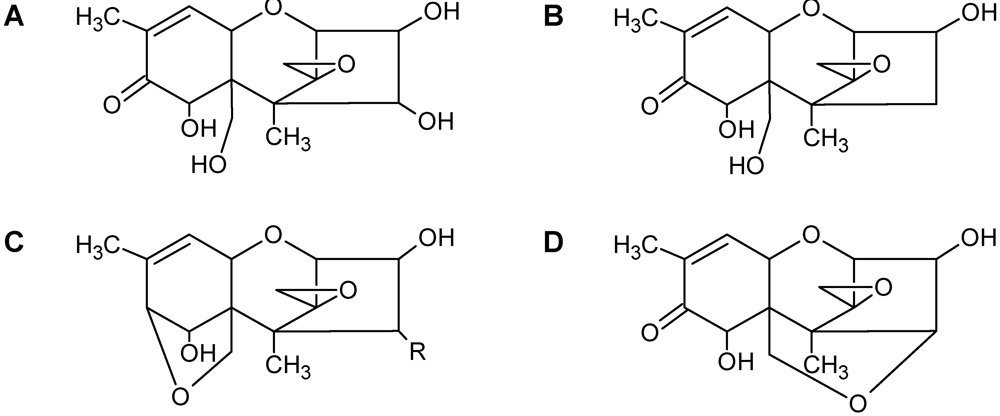
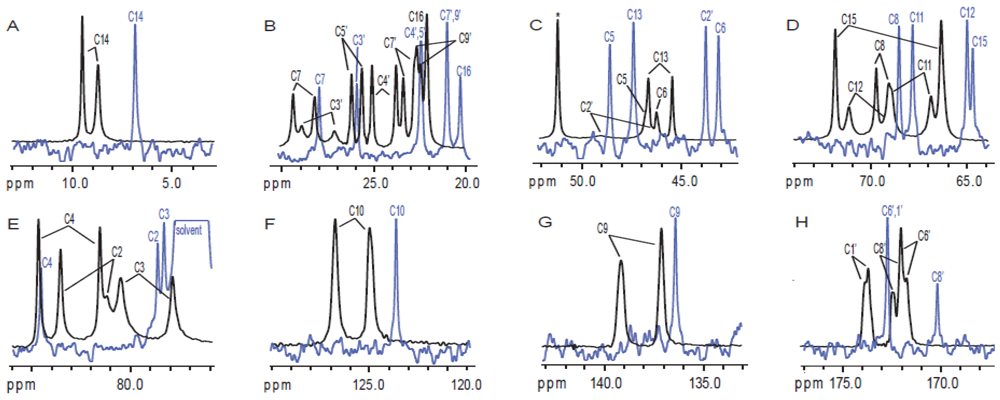
3.3. Solution NMR and the Structure Determination of Trichothecenes


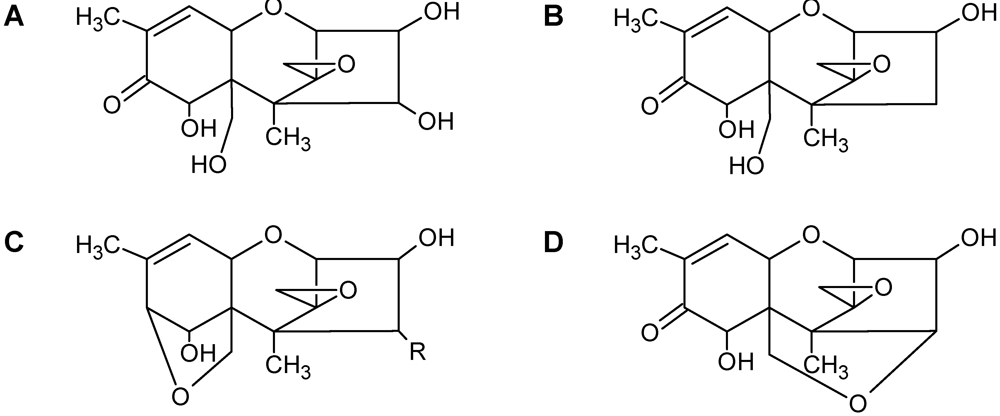
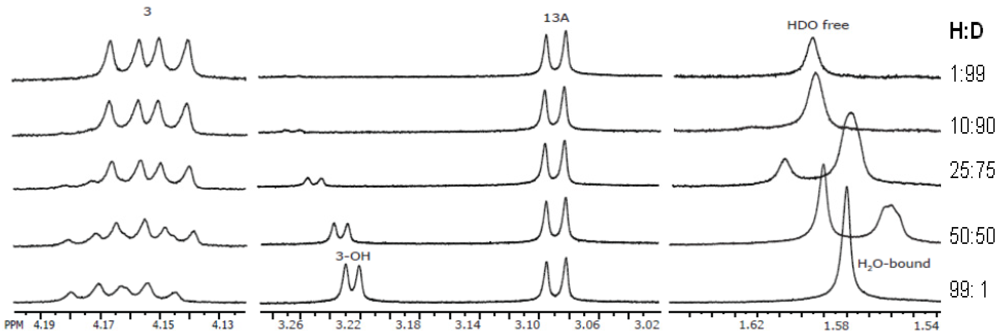
3.4. Spectroscopic Determination of Trichothecenes Through Solid-State NMR
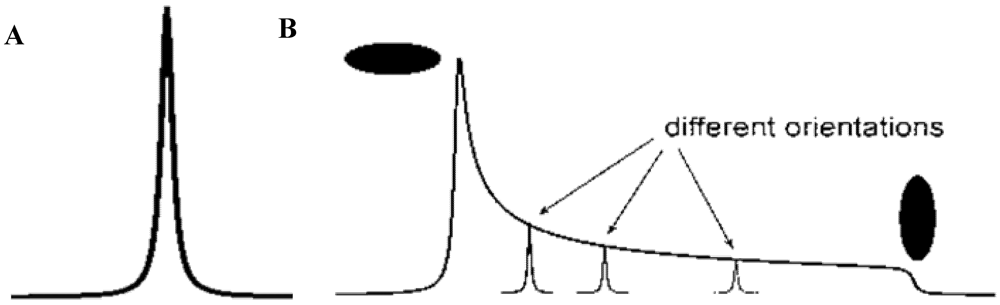

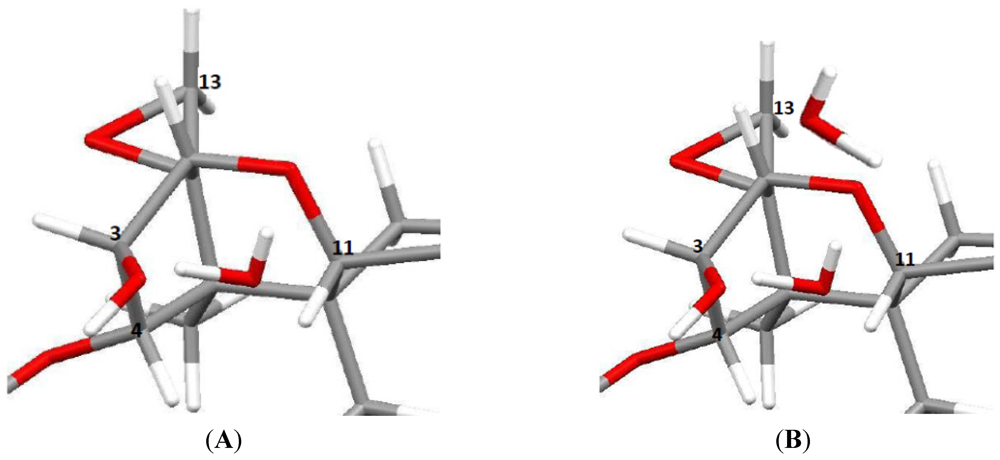
3.5. NMR Crystallography
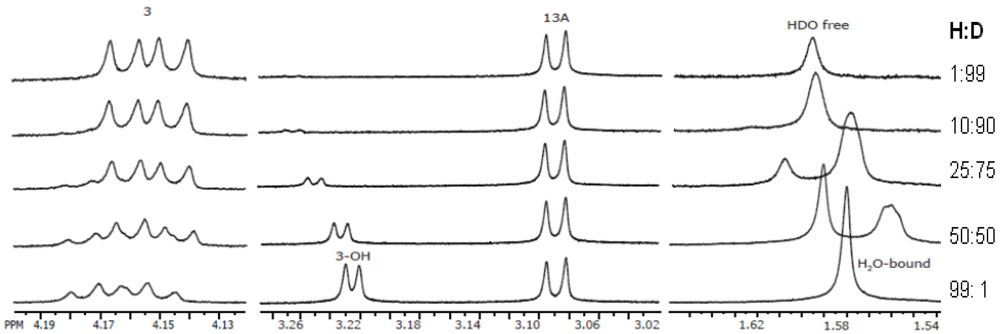
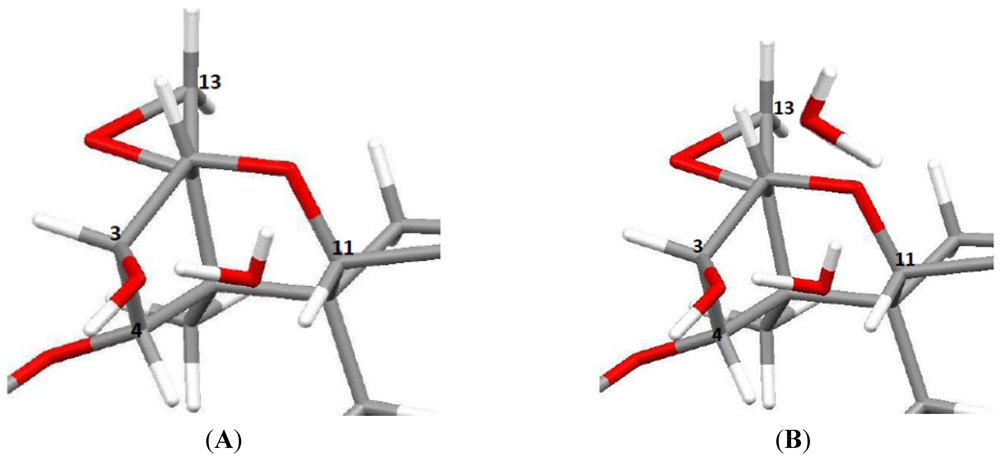
3.5.1. Hydrogen Positions
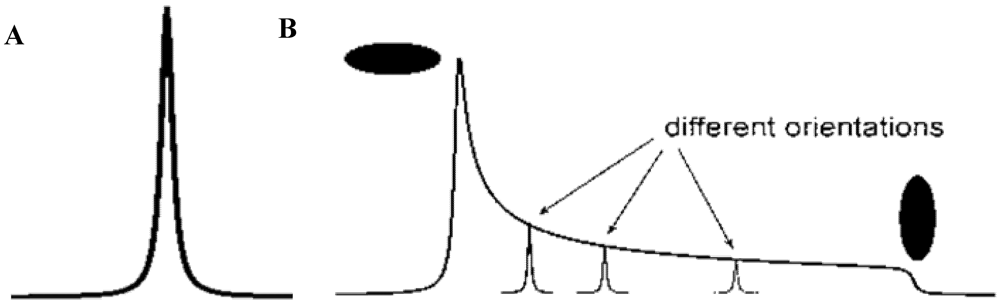
3.5.2. Powder Samples and Polycrystallinity

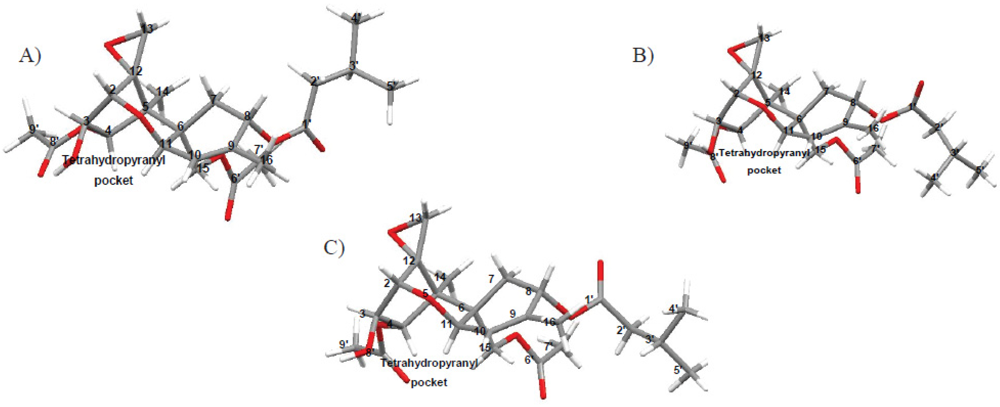
3.5.3. Inclusion and Dynamic Disorder
3.5.4. Absolute Configurations
4. Conclusions
References
- Cole, R.J.; Cox, R.H. The Trichothecenes. In Handbook of Toxic Fungal Metabolites; Academic Press: Toronto, ON, Canada, 1981; pp. 152–263. [Google Scholar]
- Cole, R.J.; Jarvis, B.B.; Schweikert, M.A. Handbook of Secondary Fungal Metabolites; Academic Press: San Diego, CA, USA, 2003. [Google Scholar]
- Wilkins, K.; Nielsen, K.; Din, S. Patterns of volatile metabolites and nonvolatile trichothecenes produced by isolates of Stachybotrys, Fusarium, Trichoderma, Trichothecium and Memnoniel. Environ. Sci. Pollut. Res. 2003, 10, 162–166. [Google Scholar] [CrossRef]
- Trapp, S.C.; Hohn, T.M.; McCormick, S.; Jarvis, B.B. Characterization of the gene cluster for biosynthesis of macrocyclic trichothecenes in Myrothecium roridum. Mol. Gen. Genet. 1998, 257, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, B.B.; Mazzola, E.P. Macrocyclic and other novel trichothecenes: Their structure, synthesis, and biological significance. Acc. Chem. Res. 1982, 15, 388–395. [Google Scholar] [CrossRef]
- Gilly, M.; Benson, N.R.; Pellegrini, M. Affinity labeling the ribosome with eukaryotic-specific antibiotics: (bromoacetyl)trichodermin. Biochemistry 1985, 24, 5787–5792. [Google Scholar]
- Jarvis, B.B.; Stahly, G.P.; Pavanasasivam, G.; Mazzola, E.P. Antileukemic compounds derived from the chemical modification of macrocyclic trichothecenes. 1. Derivatives of verrucarin A. J. Med. Chem. 1980, 23, 1054–1058. [Google Scholar] [PubMed]
- Jarvis, B.B.; Midiwo, J.O.; Mazzola, E.P. Antileukemic compounds derived by chemical modification of macrocyclic trichothecenes. 2. Derivatives of roridins A and H and verrucarins A and J. J. Med. Chem. 1984, 27, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.D.; ApSimon, J.W.; Blackwell, B.A.; Greenhalgh, R.; Taylor, A. Deoxynivalenol: A 25 Year Perspective on a Trichothecene of Agricultural Importance. In Fusarium, Paul E. Nelson Memorial Symposium; Summerell, B.E., Leslie, J.F., Backhouse, D., Bryden, W.L., Eds.; APS Press: St. Paul, MI, USA, 2001; pp. 310–320. [Google Scholar]
- Jarvis, B.B.; Lee, Y.W.; Çömezoğlu, S.N.; Yatawara, C.S. Trichothecenes produced by Stachybotrys atra from Eastern Europe. Appl. Environ. Microbiol. 1986, 51, 915–918. [Google Scholar] [PubMed]
- Bennett, J.W.; Klich, M. Mycotoxins. Clin. Microbiol. Rev. 2003, 16, 497–516. [Google Scholar]
- Foroud, N.A.; Eudes, F. Trichothecenes in cereal grains. Int. J. Mol. Sci. 2009, 10, 147–173. [Google Scholar]
- Backhouse, D.; Abubakar, A.; Burgess, L.; Dennisc, J.; Hollaway, G.; Wildermuth, G.; Wallwork, H.; Henry, F. Survey of Fusarium species associated with crown rot of wheat and barley in eastern Australia. Australas. Plant Pathol. 2004, 33, 255–261. [Google Scholar] [CrossRef]
- Thompson, A.H.; Narayanin, C.D.; Smith, M.F.; Slabbert, M.M. A disease survey of Fusarium wilt and Alternaria blight on sweet potato in South Africa. Crop Prot. 2011, 30, 1409–1413. [Google Scholar]
- Sánchez-Peña, P.; Cauich-Pech, S.O.; Núñez-Farfán, J.; Núñez-Cebreros, R.D.; Hernández-Verdugo, S.; Parra-Terraza, S.; Villarreal-Romero, M. Incidence of Fusarium oxysporum f. sp. lycopersici Races in Tomato in Sinaloa, Mexico. Plant Dis. 2010, 94, 1376–1376. [Google Scholar]
- Pestka, J. Toxicological mechanisms and potential health effects of deoxynivalenol and nivalenol. World Mycotoxin J. 2010, 3, 323–347. [Google Scholar]
- Desjardins, A.E. From yellow rain to green wheat: 25 years of trichothecene biosynthesis research. J. Agric. Food Chem. 2009, 57, 4478–4484. [Google Scholar]
- Freeman, G.G.; Morrison, R.I. Trichothecin: An antifungal metabolic product of Trichothecium roseum Link. Nature 1948, 162. [Google Scholar] [CrossRef]
- Brian, P.W.; Dawkins, A.W.; Grove, J.F.; Hemming, H.G.; Lowe, D.; Norris, G.L.F. Phytotoxic compounds produced by Fusarium equiseti. J. Exp. Bot. 1961, 12, 1–12. [Google Scholar] [CrossRef]
- Tatsuno, T.M.; Saito, M.; Enomoto, M.; Tsunoda, H. Nivalenol, a toxic principle of Fusarium nivale. Chem. Pharm. Bull. 1968, 16, 2519–2520. [Google Scholar] [CrossRef] [PubMed]
- Bamburg, J.R.; Riggs, N.V.; Strong, F.M. The structures of toxins from two strains of Fusarium Tricinctum. Tetrahedron 1968, 24, 3329. [Google Scholar]
- Marasus, W.F.O.; Nelson, P.E.; Toussoun, T.A. Toxigenic Fusarium Species; Pennsylvania State University Press: University Park, PA, USA, 1984. [Google Scholar]
- Vesonder, R.F.; Hesseltine, C.W. Vomitoxin: Natural occurrence on cereal grains and significance as a refusal and emetic factor to swine. Process Biochem. 1980, 44, 12–14. [Google Scholar]
- Savard, M.E.; Blackwell, B.A.; Greenhalgh, R. An 1H nuclear magnetic resonance study of derivatives of 3-hydroxy-12,13-epoxytrichothec-9-enes. Can. J. Chem. 1987, 65, 2254–2262. [Google Scholar]
- Savard, M.E.; Blackwell, B.A. Spectral Characteristics of Secondary Metabolites from Fusarium Fungi. In Mycotoxins in Grain; Miller, J.D., Trenholm, H.L., Eds.; Eagen Press: St. Paul, MI, USA, 1994; pp. 59–228. [Google Scholar]
- Jarvis, B.B.; Mazzocchi, D.B.; Ammon, H.L.; Mazzola, E.P.; Flippen-Anderson, J.L.; Gilardi, R.D.; George, C.F. Conformational effects in trichothecenes: Structures of 15-hydroxy C4 and C8 ketones. J. Org. Chem. 1990, 55, 3660–3662. [Google Scholar]
- Greenhalgh, R.; Blackwell, B.A.; Savard, M.E. The NMR spectra of trichothecenes and related fungal metabolites. Tetrahedron 1989, 45, 2373–2383. [Google Scholar]
- Schollenberger, M.; Drochner, W.; Müller, H.-M. Fusarium toxins of the scirpentriol subgroup: A review. Mycopathologia 2007, 164, 101–118. [Google Scholar] [CrossRef] [PubMed]
- Ueno, Y. Toxicological features of T-2 toxin and related trichothecenes. Toxicol. Sci. 1984, 4, S124–S132. [Google Scholar]
- Jarvis, B.B.; Çömezoğlu, S.N.; Rao, M.M.; Pena, N.B.; Boettner, F.E.; Williams, T.M.; Forsyth, G.; Epling, B. Isolation of macrocyclic trichothecenes from a large-scale extract of Baccharis megapotamica. J. Org. Chem. 1987, 52, 45–56. [Google Scholar]
- Jarvis, B.B.; Midiwo, J.O.; Bean, G.A.; Aboul-Nasr, M.B.; Barnos, C.S. The mystery of trichothecene antibiotics in Baccharis species. J. Nat. Prod. 1988, 51, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Bertoni, M.D.; Romero, N.; Reddy, P.V.; White, J.F., Jr. A hypocrealean epibiont on meristems of Baccharis coridifolia. Mycologia 1997, 89, 375–382. [Google Scholar] [CrossRef]
- Zamir, L.O.; Nikolakakis, A.; Sauriol, F.; Mamer, O. Biosynthesis of trichothecenes and apotrichothecenes. J. Agric. Food Chem. 1999, 47, 1823–1835. [Google Scholar]
- Greenhalgh, R.; Fielder, D.A.; Blackwell, B.A.; Miller, J.D.; Charland, J.P.; ApSimon, J.W. Some minor secondary metabolites of Fusarium sporotrichioides DAOM 165006. J. Agric. Food Chem. 1990, 38, 1978–1984. [Google Scholar]
- ApSimon, J.W.; Blackwell, B.; Blais, L.; Fielder, D.A.; Greenhalgh, R.; Kasitu, G.; Kendall, J.E.; Miller, J.D.; Savard, M. The Fusarium Genus: A Versatile Biosynthetic Engine. In Studies in Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 1991. [Google Scholar]
- Rocha, O.; Ansari, K.; Doohan, F.M. Effects of trichothecene mycotoxins on eukaryotic cells: A review. Food. Addit. Contam. 2005, 22, 369–378. [Google Scholar]
- Schindler, D.; Grant, P.; Davies, J. Trichodermin resistance-Mutation affecting eukaryotic ribosomes. Nature 1974, 248, 535–536. [Google Scholar]
- Carter, C.J.; Cannon, M. Structural requirements for the inhibitory action of 12,13-epoxytrichothecenes on protein synthesis in eukaryotes. Biochem. J. 1977, 166, 399–409. [Google Scholar]
- McLaughlin, C.S.; Vaughn, M.H.; Campbell, J.M.; Wei, C.M.; Stafford, M.E. hibition of Protein Synthesis by Trichothecenes. In Mycotoxins in Human and Health; Rodricks, J.V., Hesseltine, C.W., Mehlman, M.A., Eds.; Pathotox Publishers: Park Forest South, IL, USA, 1977; pp. 263–275. [Google Scholar]
- Cundliffe, E.; Davies, J.E. Inhibition of initiation, elongation, and termination of eukaryotic protein synthesis by trichothecene fungal toxin. Antimicrob. Agents Chemother. 1977, 11, 491–499. [Google Scholar]
- Matsumoto, M.; Minato, H.; Uotani, N.; Matsumoto, K.; Kondo, E. New antibiotics from Cylindrocarpon sp. J. Antibiot. 1977, 30, 681–682. [Google Scholar] [PubMed]
- Ehrlich, K.C.; Daigle, K.W. Protein synthesis inhibition by 8 oxo-12,13-epoxytrichothecenes. Biochim. Biophys. Acta 1987, 923, 206–213. [Google Scholar]
- McLean, M. The phytotoxicity of Fusarium metabolites: An update since 1989. Mycopathologia 1996, 133, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, T.; Sakamoto, T.; Okamoto, K. In vitro formation of 3'-hydroxy T-2 and 3'-hydroxy HT-2 toxins from T-2 toxin by liver homogenates from mice and monkeys. Appl. Environ. Microbiol. 1984, 47, 130–134. [Google Scholar] [PubMed]
- Völkl, A.; Vogler, B.; Schollenberger, M.; Karlovsky, P. Microbial detoxification of mycotoxin deoxynivalenol. J. Basic Microbiol. 2004, 44, 147–156. [Google Scholar]
- Madhyastha, M.S.; Marquardt, R.R.; Masi, A.; Borsa, J.; Frohllch, A.A. Comparison of toxicity of different mycotoxins to several species of bacteria and yeasts: Use of Bacillus brevis in a disc diffusion assay. J. Food Prot. 1994, 57, 48–53. [Google Scholar]
- Cheng, B.; Wan, C.; Yang, S.; Xu, H.; Wei, H.U.A.; Liu, J.; Tian, W.; Zeng, M. Detoxification of deoxynivalenol by Bacillus strains. J. Food Saf. 2010, 30, 599–614. [Google Scholar]
- Kupchan, S.M.; Streelman, D.R.; Jarvis, B.B.; Dailey, R.G.; Sneden, A.T. Isolation of potent new antileukemic trichothecenes from Baccharis megapotamica. J. Org. Chem. 1977, 42, 4221–4225. [Google Scholar]
- Zamir, L.O.; Gauthier, M.J.; Devor, K.A.; Nadeau, Y.; Sauriol, F. Trichodiene is a precursor to trichothecenes. J. Chem. Soc. Chem. Commun. 1989, 598–600. [Google Scholar]
- Dohnal, V.; Jezkova, A.; Jun, D.; Kuca, K. Metabolic pathways of T-2 toxin. Curr. Drug Metab. 2008, 9, 77–82. [Google Scholar]
- Yagen, B.; Jarvis, B.B. Synthesis of tritium labelled verrucarol and verrucarin A. J. Label. Comp. Radiopharm. 1989, 27, 675–681. [Google Scholar]
- Jarvis, B.B.; Yatawara, C.S.; Greene, S.L.; Vrudhula, V.M. Production of verrucarol. Appl. Environ. Microbiol. 1984, 48, 673–674. [Google Scholar]
- Esmond, R.; Fraser-Reid, B.; Jarvis, B.B. Synthesis of trichoverrin B and its conversion to verrucarin J. J. Org. Chem. 1982, 47, 3358–3360. [Google Scholar]
- Wei, R.D.; Chu, F.S. Modification of in vitro metabolism of T-2 toxin by esterase inhibitors. Appl. Environ. Microbiol. 1985, 50, 115–119. [Google Scholar] [PubMed]
- Garvey, G.S.; McCormick, S.P.; Rayment, I. Structural and functional characterization of the TRI101 trichothecene 3-O-acetyltransferase from Fusarium sporotrichioides and Fusarium graminearum. J. Biol. Chem. 2008, 283, 1660–1669. [Google Scholar] [PubMed]
- Garvey, G.S.; McCormick, S.P.; Alexander, N.J.; Rayment, I. Structural and functional characterization of TRI3 trichothecene 15-O-acetyltransferase from Fusarium sporotrichioides. Protein Sci. 2009, 18, 747–761. [Google Scholar]
- Chaudhary, P.; Shank, R.A.; Montina, T.; Goettel, J.T.; Foroud, N.A.; Hazendonk, P.; Eudes, F. Hydrogen-bonding interactions in T-2 toxin studied using solution and solid-state NMR. Toxins 2011, 3, 1310–1331. [Google Scholar]
- Fried, H.M.; Warner, J.R. Cloning of yeast gene for trichodermin resistance and ribosomal protein L3. Proc. Natl.Acad. Sci. USA 1981, 78, 238–242. [Google Scholar]
- Grant, P.G.; Schindler, D.; Davies, J.E. Mapping of trichodermin resistance in Saccharomyces cerevisiae: A genetic locus for a component of the 60S ribsomal subunit. Genetics 1976, 83, 667–673. [Google Scholar] [PubMed]
- Harris, L.J.; Gleddie, S.C. A modified rpl3 gene from rice confers tolerance of the Fusarium graminearum mycotoxin deoxynivalenol to transgenic tobacco. Physiol. Mol. Plant Pathol. 2001, 58, 173–181. [Google Scholar] [CrossRef]
- Mitterbauer, R.; Poppenberger, B.; Raditschnig, A.; Lucyshyn, D.; Lemmens, M.; Glössl, J.; Adam, G. Toxin-dependent utilization of engineered ribosomal protein L3 limits trichothecene resistance in transgenic plants. Plant Biotechnol. J. 2004, 2, 329–340. [Google Scholar]
- Meskauskas, A.; Dinman, J.D. Ribosomal protein L3 functions as a “rocker switch” to aid in coordinating of large subunit-associated functions in eukaryotes and Archaea. Nucleic Acids Res. 2008, 36, 6175–6186. [Google Scholar]
- Meskauskas, A.; Petrov, A.N.; Dinman, J.D. Identification of functionally important amino acids of ribosomal protein L3 by saturation mutagenesis. Mol. Cell. Biol. 2005, 25, 10863–10874. [Google Scholar]
- Carter, C.J.; Cannon, M.; Smith, K.E. Inhibition of protein synthesis in reticulocyte lysates by trichodermin. Biochem. J. 1976, 154, 171–178. [Google Scholar]
- Cundliffe, E.; Cannon, M.; Davies, J. Mechanism of inhibition of eukaryotic protein synthesis by trichothecene fungal toxins. Proc. Natl. Acad. Sci. USA 1974, 71, 30–34. [Google Scholar]
- Shifrin, V.I.; Anderson, P. Trichothecene mycotoxins trigger a ribotoxic stress response that activates c-Jun N-terminal kinase and p38 mitogen-activated protein kinase and induces apoptosis. J. Biol. Chem. 1999, 274, 13985–13992. [Google Scholar] [CrossRef] [PubMed]
- Moon, Y.; Pestka, J.J. Vomitoxin-induced cyclooxygenase-2 gene expression in macrophages mediated by activation of ERK and p38 but not JNK mitogen-activated protein kinases. Toxicol. Sci. 2002, 69, 373–382. [Google Scholar]
- Yang, G.-H.; Jarvis, B.B.; Chung, Y.-J.; Pestka, J.J. Apoptosis induction by the satratoxins and other trichothecene mycotoxins: Relationship to ERK, p38 MAPK, and SAPK/JNK activation. Toxicol. Appl. Pharmacol. 2000, 164, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.-J.; Zhou, H.-R.; Pestka, J.J. Transcriptional and posttranscriptional roles for p38 mitogen-activated protein kinase in upregulation of TNF-alpha expression by deoxynivalenol (vomitoxin). Toxicol. Appl. Pharmacol. 2003, 193, 188–201. [Google Scholar]
- Zhou, H.-R.; Lau, A.S.; Pestka, J.J. Role of double-stranded RNA-activated protein kinase R (PKR) in deoxynivalenol-induced ribotoxic stress response. Toxicol. Sci. 2003, 74, 335–344. [Google Scholar]
- Packa, D. Cytogenetic changes in plant cells as influenced by mycotoxins. Mycotoxin Res. 1991, 7, 150–155. [Google Scholar]
- Bunner, D.L.; Morris, E.R. Alteration of multiple cell membrane functions in L-6 myoblasts by T-2 toxin: An important mechanism of action. Toxicol. Appl. Pharmacol. 1988, 92, 113–121. [Google Scholar]
- Pace, J.G.; Watts, M.R.; Canterbury, W.J. T-2 mycotoxin inhibits mitochondrial protein synthesis. Toxicon 1988, 26, 77–85. [Google Scholar]
- Pace, J.G. Effect of T-2 mycotoxin on rat liver mitochondria electron transport system. Toxicon 1983, 21, 675–680. [Google Scholar]
- Eudes, F.; Comeau, A.; Rioux, S.; Collin, J. Phytotoxicité de huit mycotoxines associées à la fusariose de l'épi chez le blé. Can. J. Plant Pathol. 2000, 22, 286–292. [Google Scholar]
- Desjardins, A.E.; McCormick, S.P.; Appell, M. Structure−activity relationships of trichothecene toxins in an Arabidopsis thaliana leaf assay. J. Agric. Food Chem. 2007, 55, 6487–6492. [Google Scholar] [CrossRef] [PubMed]
- Trusal, L.R.; O'Brien, J.C. Ultrastructural effects of T-2 mycotoxin on rat hepatocytes in vitro. Toxicon 1986, 24, 481–488. [Google Scholar]
- Shimada, T.; Otani, M. Effects of Fusarium mycotoxins on the growth of shoots and roots at germination in some Japanese wheat cultivars. Cereal Res. Commun. 1990, 18, 229–232. [Google Scholar]
- Ueno, Y. Trichothecenes: Chemical, Biological and Toxicological Aspects; Elsevier Scientific Publishers: Amsterdam, The Netherlands, 1983. [Google Scholar]
- Eudes, F.; Comeau, A.; Rioux, S.; Collin, J. Impact of trichothecenes on Fusarium head blight (Fusarium graminearum) development in spring wheat (Triticum aestivum). Can. J. Plant Pathol. 2001, 23, 318–322. [Google Scholar] [CrossRef]
- Steinmetz, W.E.; Rodarte, C.B.; Lin, A. 3D QSAR study of the toxicity of trichothecene mycotoxins. Eur. J. Med. Chem. 2009, 44, 4485–4489. [Google Scholar]
- Ueno, Y.; Ishii, K. Chemical and Biological Properties of Trichothecenes from Fusarium sporotrichioides. In Trichothecenes and Other Mycotoxins: Proceedings of the International Mycotoxin Symposium of Sydney, Australia,1984; John Wiley & Sons, Ltd.: New York, NY, USA, 1985; pp. 307–316. [Google Scholar]
- Minervini, F.; Fornelli, F.; Flynn, K.M. Toxicity and apoptosis induced by the mycotoxins nivalenol, deoxynivalenol and fumonisin B1 in a human erythroleukemia cell line. Toxicol. in Vitro 2004, 18, 21–28. [Google Scholar]
- Miller, J.D.; Greenhalgh, R.; Wang, Y.; Lu, M. Trichothecene chemotypes of three Fusarium species. Mycologia 1991, 83, 121–130. [Google Scholar] [CrossRef]
- Alexander, N.J.; McCormick, S.P.; Ziegenhorn, S.L. Phytotoxicity of selected trichothecenes using Chlamydomonas reinhardtii as a model system. Nat. Toxins 1999, 7, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Muhitch, M.J.; McCormick, S.P.; Alexander, N.J.; Hohn, T.M. Transgenic expression of the TRI101 or PDR5 gene increases resistance of tobacco to the phytotoxic effects of the trichothecene 4,15-diacetoxyscirpenol. Plant Sci. 2000, 157, 201–207. [Google Scholar]
- Ohsato, S.; Ochiai, F.T.; Nishiuchi, T.; Takahashi, A.N.; Koizumi, S.; Hamamoto, H.; Kudo, T.; Yamaguchi, I.; Kimura, M. Transgenic rice plants expressing trichothecene 3-O-acetyltransferase show resistance to the Fusarium phytotoxin deoxynivalenol. Plant Cell Rep. 2007, 26, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, A.E.; Hohn, T.M.; McCormick, S.P. Trichothecene biosynthesis in Fusarium species: Chemistry, genetics, and significance. Microbiol. Rev. 1993, 57, 595–604. [Google Scholar] [PubMed]
- Poppenberger, B.; Berthiller, F.; Lucyshyn, D.; Sieberer, T.; Schuhmacher, R.; Krska, R.; Kuchler, K.; Glossl, J.; Luschnig, C.; Adam, G. Detoxification of the Fusarium mycotoxin deoxynivalenol by a UDP-glucosyltransferase from Arabidopsis thaliana. J. Biol. Chem. 2003, 278, 47905–47914. [Google Scholar] [PubMed]
- Boutigny, A.L.; Richard, F.F.; Barreau, C. Natural mechanisms for cereal resistance to the accumulation of Fusarium trichothecenes. Eur. J. Plant Pathol. 2008, 121, 411–423. [Google Scholar] [CrossRef]
- Ueno, Y. The toxicology of mycotoxins. Crit. Rev. Toxicol. 1985, 14, 99–133. [Google Scholar]
- Anderson, D.W.; Black, R.M.; Lee, C.G.; Pottage, C.; Rickard, R.L.; Sandford, M.S.; Webber, T.D.; Williams, N.E. Structure-activity studies of trichothecenes: Cytotoxicity of analogs and reaction products derived from T-2 toxin and neosolaniol. J. Med. Chem. 1989, 32, 555–562. [Google Scholar]
- Khachatourians, G.G. Metabolic effects of trichothecene T-2 toxin. Can. J. Physiol. Pharmacol. 1990, 68, 1004–1008. [Google Scholar]
- Madhyastha, M.S.; Marquardt, R.R.; Abramson, D. Structure-activity relationships and interactions among trichothecene mycotoxins as assessed by yeast bioassay. Toxicon 1994, 32, 1147–1152. [Google Scholar]
- Dowd, P.F.; Miller, J.D.; Greenhalgh, R. Toxicity and interactions of some Fusarium graminearum metabolites to caterpillars. Mycologia 1989, 81, 646–650. [Google Scholar] [CrossRef]
- Grove, J.F.; Mortimer, P.H. The cytotoxicity of some transformation products of diacetoxyscirpenol. Biochem. Pharmacol. 1969, 18, 1473–1478. [Google Scholar]
- Chanh, T.C.; Hewetson, J.F. Structure/function studies of T-2 mycotoxin with a monoclonal antibody. Immunopharmacology 1991, 21, 83–89. [Google Scholar]
- Freeman, G.G.; Gill, J.E.; Waring, W.S. The structure of trichothecin and its hydrolysis products. J. Chem. Soc. 1959, 1105–1132. [Google Scholar] [CrossRef]
- Häubl, G.; Berthiller, F.; Hametner, C.; Rechthaler, J.; Jaunecker, G.; Freudenschuss, M.; Krska, R.; Schuhmacher, R. Characterization of (13C24) T-2 toxin and its use as an internal standard for the quantification of T-2 toxin in cereals with HPLC-MS/MS. Anal. Bioanal. Chem. 2007, 389, 931–940. [Google Scholar]
- Jarvis, B.B.; DeSilva, T.; McAlphine, J.B.; Swanson, S.J.; Whittern, D.N. New trichoverroids from Myrothecium verrucaria isolated by high speed countercurrent chromatography. J. Nat. Prod. 1992, 55, 1441–1446. [Google Scholar] [CrossRef] [PubMed]
- Krska, R.; Schothorst, R.C.; van Egmond, H.P.; Josephs, R.D.; Lepschy, J.; Pettersson, H.; Chan, D.; Berthiller, F.; Schuhmacher, R.; Kandler, W.; Parich, A.; Welzig, E. Processing and purity assessment of standards for the analysis of type-B trichothecene mycotoxins. Anal. Bioanal. Chem. 2005, 382, 1848–1858. [Google Scholar]
- Rhodes, G. Crystallography Made Crystal Clear: A Guide for Users of Macromolecular Model, 3rd ed; Academic Press: Waltham, MA, USA, 2006. [Google Scholar]
- McRee, D.E.; David, P.R. Pratical Protein Crystallography: Second Edition; Academic Press: San Diego, CA, USA, 1999. [Google Scholar]
- Harris, R.K.; Wasylishen, R.E.; Duer, M.J. NMR Crystallography; John Wiley & Sons, Ltd.: Chinchester, UK, 2009. [Google Scholar]
- McPhail, A.T.; Sim, G.A. Fungal metabolites. Part VI. The structure of verrucarin A: X-ray analysis of verrucarin A p-iodobenzenesulphonate. J. Chem. Soc. C 1966, 1394–1406. [Google Scholar] [CrossRef]
- Soriano-Garcia, M.; Ramos, F.M.; Chavez, G.T.; Ramirez-Medeles, M.D.; Hernandez, G.A.; Chicote, F.O. Crystal structure of Verrucarin A. Anal. Sci. 1999, 15, 403–404. [Google Scholar]
- Anderson, D.W.; Ashton, P.R.; Black, R.M.; Leigh, D.A.; Slawin, A.M.Z.; Stoddart, J.F.; Williams, D.J. The complexation properties of some unnatural and natural macrocyclic trichothecenes. J. Chem. Soc. Chem. Commun. 1988, 13, 904–908. [Google Scholar]
- Breitenstein, W.; Tamm, C.; Arnold, E.V.; Clardy, J. The absolute configuration of the fungal metabolite Verrucarin B. Biosynthetic consequences. Helv. Chim. Acta 1979, 62, 2699–2705. [Google Scholar] [CrossRef]
- Jarvis, B.B.; Midiwo, J.O.; Flippen-Anderson, J.L.; Mazzola, E.P. Stereochemistry of the roridins. J. Nat. Prod. 1982, 45, 440–448. [Google Scholar]
- Jarvis, B.B.; Wang, S. Stereochemistry of the roridins. Diastereomers of roridin E. J. Nat. Prod. 1999, 62, 1284–1289. [Google Scholar] [CrossRef] [PubMed]
- Flippen-Anderson, J.L.; Gilardi, R. Structure of isororidin E. Acta Crystallogr. Sect. C 1986, 42, 1184–1185. [Google Scholar]
- Gai, Y.; Zhao, L.-L.; Hu, C.-Q.; Zhang, H.-P. Roridin H. Acta Crystallogr. Sect. E 2007, 63, 2990–2991. [Google Scholar]
- Jarvis, B.B.; Çömezoğlu, F.T.; Lee, Y.-W.; Flippen-Anderson, J.L.; Gilardi, R.D.; George, C.F. Novel macrocyclic trichothecenes from Myrothecium roridum. Bull. Soc. Chim. Belg. 1986, 95, 681–697. [Google Scholar]
- Greenhalgh, R.; Hanson, A.W.; Miller, J.D.; Taylor, A. Production and X-ray crystal structure of 3-acetoxy-7,15-dihydroxy-12,13-epoxytrichothec-9-en-8-one. J. Agric. Food Chem. 1984, 32, 945–948. [Google Scholar]
- Greenhalgh, R.; Levandier, D.; Adams, W.; Miller, J.D.; Blackwell, B.A.; McAlees, A.J.; Taylor, A. Production and characterization of deoxynivalenol and other secondary metabolites of Fusarium culmorum (CMI 14764, HLX 1503). J. Agric. Food Chem. 1986, 34, 98–102. [Google Scholar] [CrossRef]
- Gilardi, R.; George, C.; Flippen-Anderson, J.L. The structure of T-2 toxin. Acta Crystallogr. Sect. C 1990, 46, 645–648. [Google Scholar]
- Mohr, P.; Tamm, C.; Zürcher, W.; Zehnder, M. Sambucinol and sambucoin, two new metabolites of Fusarium sambucinum possessing modified trichothecene structures. Helv. Chim. Acta 1984, 67, 406–412. [Google Scholar] [CrossRef]
- Levitt, M.H. Spin Dynamics: Basics of Nuclear Magnetic Resonance; John Wiley & Sons, Ltd.: Chinchester, UK, 2008. [Google Scholar]
- Vander Hart, D.L. Magnetic Susceptibility and High Resolution NMR of Liquids and Solids. In Encyclopedia of Nuclear Magnetic Resonance; Grant, D.M., Harris, R.K., Eds.; John Wiley & Sons, Ltd.: Chinchester, UK, 2009. [Google Scholar]
- Barfield, M. Indirect Coupling: Theory and Application in Organic Chemistry. In Encyclopedia of Nuclear Magnetic Resonance; Grant, D.M., Harris, R.K., Eds.; John Wiley & Sons, Ltd.: Chinchester, UK, 2009. [Google Scholar]
- Woessner, D.E. Brownian Motion and Correlation Times. In Encyclopedia of Nuclear Magnetic Resonance; Grant, D.M., Harris, R.K., Eds.; John Wiley & Sons, Ltd.: Chinchester, UK, 2009. [Google Scholar]
- Schaefer, T. Stereochemistry and Long Range Coupling Constants. In Encyclopedia of Nuclear Magnetic Resonance; Grant, D.M., Harris, R.K., Eds.; John Wiley & Sons, Ltd.: Chinchester, UK, 2009. [Google Scholar]
- Farrar, T.C.; Becker, E.D. Pulse and Fourier Transform NMR: Introduction to Theory and Methods; Academic Press: New York, NY, USA, 1971. [Google Scholar]
- Williamson, M.P. NOESY. In Encyclopedia of Nuclear Magnetic Resonance; Grant, D.M., Harris, R.K., Eds.; John Wiley & Sons, Ltd.: Chinchester, UK, 2009. [Google Scholar]
- Sakakibara, D.; Sasaki, A.; Ikeya, T.; Hamatsu, J.; Hanashima, T.; Mishima, M.; Yoshimasu, M.; Hayashi, N.; Mikawa, T.; Walchli, M.; Smith, B.O.; Shirakawa, M.; Guntert, P.; Ito, Y. Protein structure determination in living cells by in-cell NMR spectroscopy. Nature 2009, 458, 102–105. [Google Scholar]
- Inomata, K.; Ohno, A.; Tochio, H.; Isogai, S.; Tenno, T.; Nakase, I.; Takeuchi, T.; Futaki, S.; Ito, Y.; Hiroaki, H.; Shirakawa, M. High-resolution multi-dimensional NMR spectroscopy of proteins in human cells. Nature 2009, 458, 106–109. [Google Scholar]
- Ardenkjær-Larsen, J.H.; Fridlund, B.; Gram, A.; Hansson, G.; Hansson, L.; Lerche, M.H.; Servin, R.; Thaning, M.; Golman, K. Increase in signal-to-noise ratio of >10,000 times in liquid-state NMR. Proc. Natl.Acad. Sci. USA 2003, 100, 10158–10163. [Google Scholar]
- Hanson, J.R.; Marten, T.; Siverns, M. Studies in terpenoid biosynthesis. Part XII. Carbon-13 nuclear magnetic resonance spectra of the trichothecanes and the biosynthesis of trichothecolone from [2-13C]mevalonic acid. J. Chem. Soc. Perkin Trans. 1974, 1, 1033–1036. [Google Scholar]
- Blackwell, B.A.; Greenhalgh, R.; Bain, A.D. Carbon-13 and proton nuclear magnetic resonance spectral assignments of deoxynivalenol and other mycotoxins from Fusarium graminearum. J. Agric. Food Chem. 1984, 32, 1078–1083. [Google Scholar] [CrossRef]
- Doddrell, D.M.; Pegg, D.T.; Bendall, M.R. Distortionless enhancement of NMR signals by polarization transfer. J. Magn. Reson. 1982, 48, 323–327. [Google Scholar]
- Dang, J.; Bergdahl, B.M.; Separovic, F.; Brownlee, R.T.C.; Metzger, R.P. Difference in conformation of virginiamycin M1 in chloroform and bound form in the 50S ribosome or streptogramin acetyltransferase. Aust. J. Chem. 2004, 57, 415–418. [Google Scholar]
- Dang, J.; Bergdahl, M.; Separovic, F.; Brownlee, R.T.C.; Metzger, R.P. Solvent affects the conformation of virginiamycin M1 (pristinamycin IIA, streptogramin A). Org. Biomol. Chem. 2004, 2, 2919–2924. [Google Scholar]
- Duffy, M.J.; Reid, R.S. Measurement of the stability of T-2 toxin in aqueous solution. Chem. Res. Toxicol. 1993, 6, 524–529. [Google Scholar]
- Bamburg, J.R. Chemical and Biochemical Studies of the Trichothecene Mycotoxins. In Mycotoxins and Other Fungal Related Food Problems; American Chemical Society: Washington, DC, USA, 1976. [Google Scholar]
- Pace, J.G.; Matson, C.F. Stability of T-2, HT-2, and T-2 tetraol in biological fluid. J. Anal. Toxicol. 1988, 12, 48–50. [Google Scholar]
- Kay, L.E. Protein dynamics from NMR. Biochem. Cell Biol. 1998, 76, 145–152. [Google Scholar]
- Nagy, C.M.; Fejer, S.N.; Berek, L.; Molnar, J.; Viskolcz, B. Hydrogen bondings in deoxynivalenol (DON) conformations--a density functional study. J. Mol. Struct. 2005, 726, 55–59. [Google Scholar]
- Fragaki, G.; Stefanaki, I.; Dais, P.; Mikros, E. Conformational properties of the macrocyclic trichothecene mycotoxin verrucarin A in solution. Magn. Reson. Chem. 2008, 46, 1102–1111. [Google Scholar]
- Steinmetz, W.E.; Robustelli, P.; Edens, E.; Heineman, D. Structure and conformational dynamics of trichothecene mycotoxins. J. Nat. Prod. 2008, 71, 589–594. [Google Scholar]
- Taylor, R.E. Setting up 13C CP/MAS experiments. Concepts Magn. Reson. Part A 2004, 22A, 37–49. [Google Scholar]
- Slichter, C.P. Principles of Magnetic Resonance: Third Enlarged and Updated Edition; Springer-Verlag: Berlin, Germany, 1990. [Google Scholar]
- Madhu, P.K. High-resolution solid-state NMR spectroscopy of protons with homonuclear dipolar decoupling schemes under magic-angle spinning. Solid State Nucl. Magn. Reson. 2009, 35, 2–11. [Google Scholar]
- Geppi, M.; Mollica, G.; Borsacchi, S.; Veracini, C.A. Solid-state NMR studies of pharmaceutical systems. Appl. Spectrosc. Rev. 2008, 43, 202–302. [Google Scholar]
- Sewald, N.; von Gleissenthall, J.L.; Schuster, M.; Müller, G.; Aplin, R.T. Structure elucidation of a plant metabolite of 4-desoxynivalenol. Tetrahedron Asymmetry 1992, 3, 953–960. [Google Scholar]
- Otting, G. NMR studies of water bound to biological molecules. Prog. Nucl. Magn. Reson. Spectrosc. 1997, 31, 259–285. [Google Scholar]
- Rodnina, M.V.; Beringer, M.; Wintermeyer, W. How ribosomes make peptide bonds. TrendsBiochem. Sci. 2007, 32, 20–26. [Google Scholar]
- Pickard, C.J.; Mauri, F. All-electron magnetic response with pseudopotentials: NMR chemical shifts. Phys. Rev. B 2001, 63, 245101–245113. [Google Scholar]
- Vosegaard, T.; Hald, E.; Langer, V.; Skov, H.J.; Daugaard, P.; Bildsøe, H.; Jakobsen, H.J. Improved hardware and software for single-crystal NMR spectroscopy. J. Magn. Reson. 1998, 135, 126–132. [Google Scholar]
- Harris, R.K. Polymorphism and Related Phenomena. In Encyclopedia of Nuclear Magnetic Resonance; Grant, D.M., Harris, R.K., Eds.; John Wiley & Sons, Ltd.: Chinchester, UK, 2009. [Google Scholar]
- Christopher, E.A.; Harris, R.K.; Fletton, R.A. Assignments of solid-state 13C resonances for polymorphs of cortisone acetate using shielding tensor components. Solid State Nucl. Magn. Reson. 1992, 1, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Vega, S. NMR line shape analysis for two-site exchange in rotating solids. J. Chem. Phys. 1987, 87, 6895–6907. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Shank, R.A.; Foroud, N.A.; Hazendonk, P.; Eudes, F.; Blackwell, B.A. Current and Future Experimental Strategies for Structural Analysis of Trichothecene Mycotoxins—A Prospectus. Toxins 2011, 3, 1518-1553. https://doi.org/10.3390/toxins3121518
Shank RA, Foroud NA, Hazendonk P, Eudes F, Blackwell BA. Current and Future Experimental Strategies for Structural Analysis of Trichothecene Mycotoxins—A Prospectus. Toxins. 2011; 3(12):1518-1553. https://doi.org/10.3390/toxins3121518
Chicago/Turabian StyleShank, Roxanne A., Nora A. Foroud, Paul Hazendonk, François Eudes, and Barbara A. Blackwell. 2011. "Current and Future Experimental Strategies for Structural Analysis of Trichothecene Mycotoxins—A Prospectus" Toxins 3, no. 12: 1518-1553. https://doi.org/10.3390/toxins3121518
APA StyleShank, R. A., Foroud, N. A., Hazendonk, P., Eudes, F., & Blackwell, B. A. (2011). Current and Future Experimental Strategies for Structural Analysis of Trichothecene Mycotoxins—A Prospectus. Toxins, 3(12), 1518-1553. https://doi.org/10.3390/toxins3121518