Differential Retention and Loss of a Mycotoxin in Fungal Evolution


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
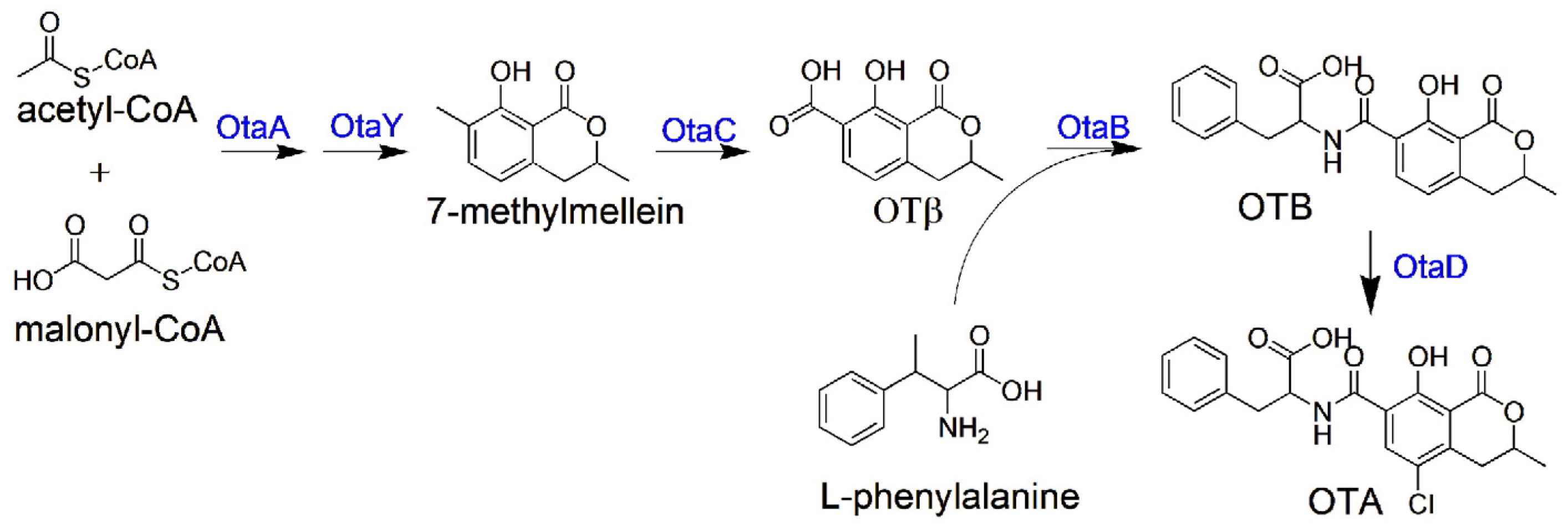
1. Introduction
2. Results
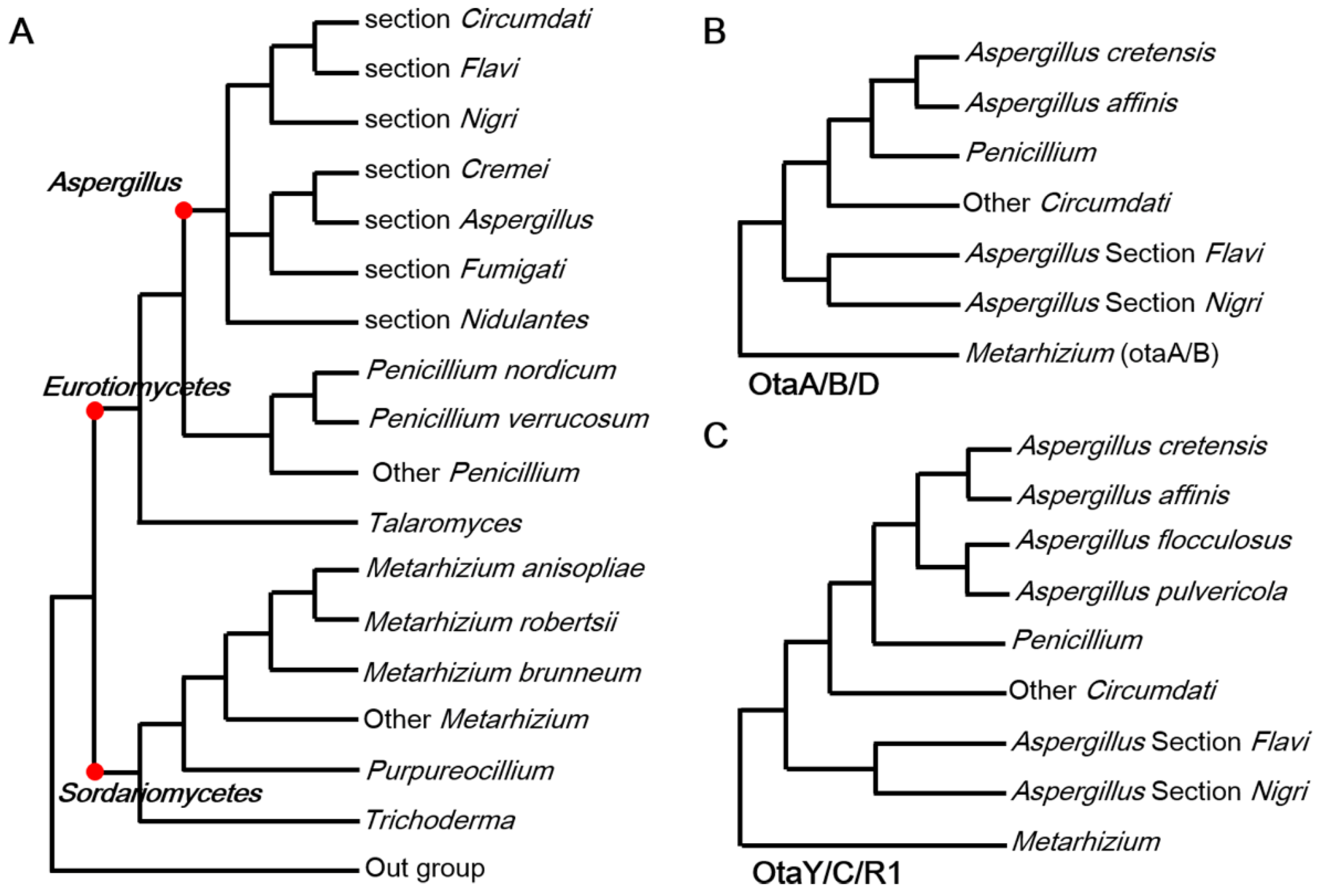
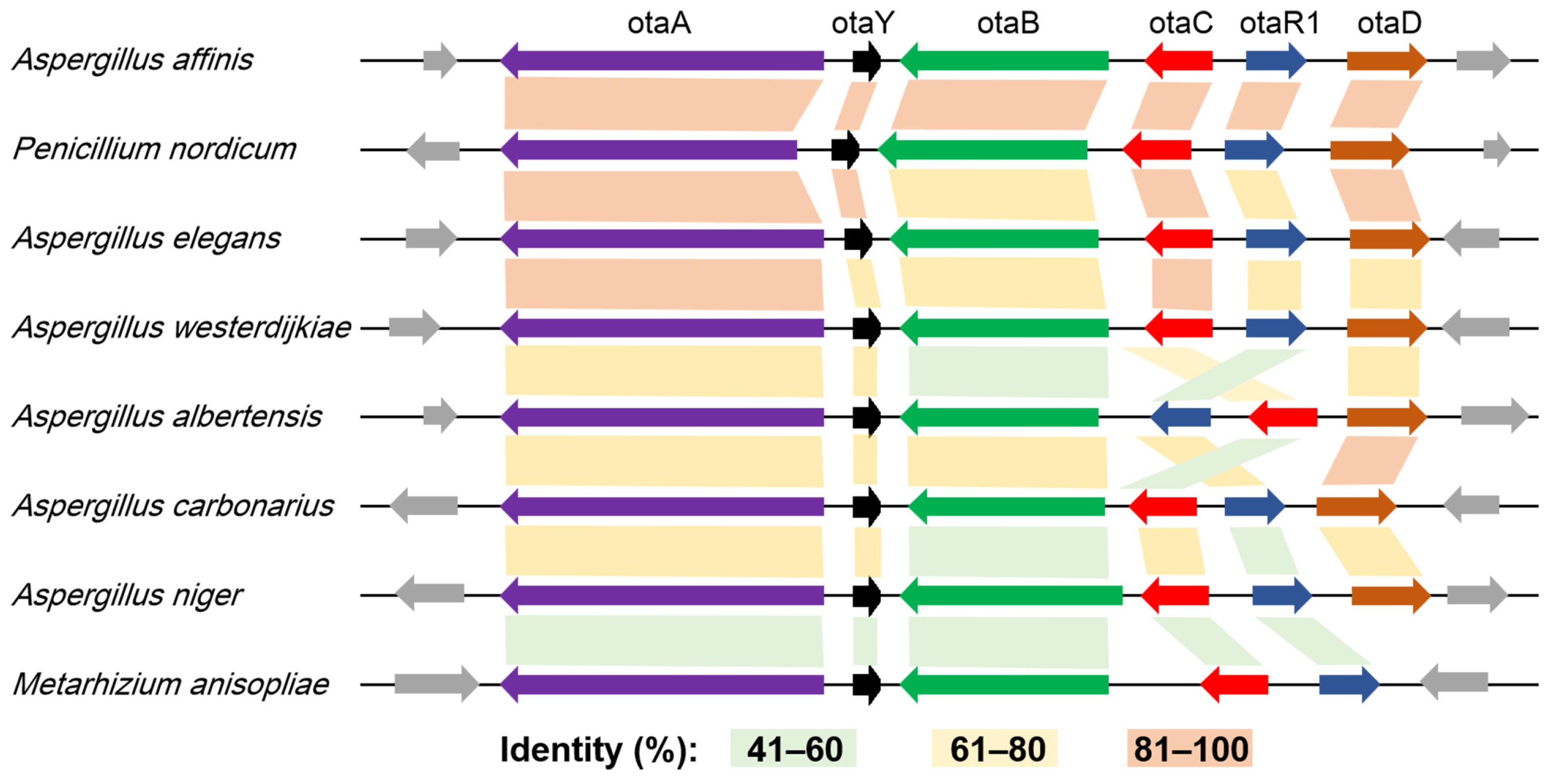
2.1. HGT Contributes to the Acquisition of OT Genes
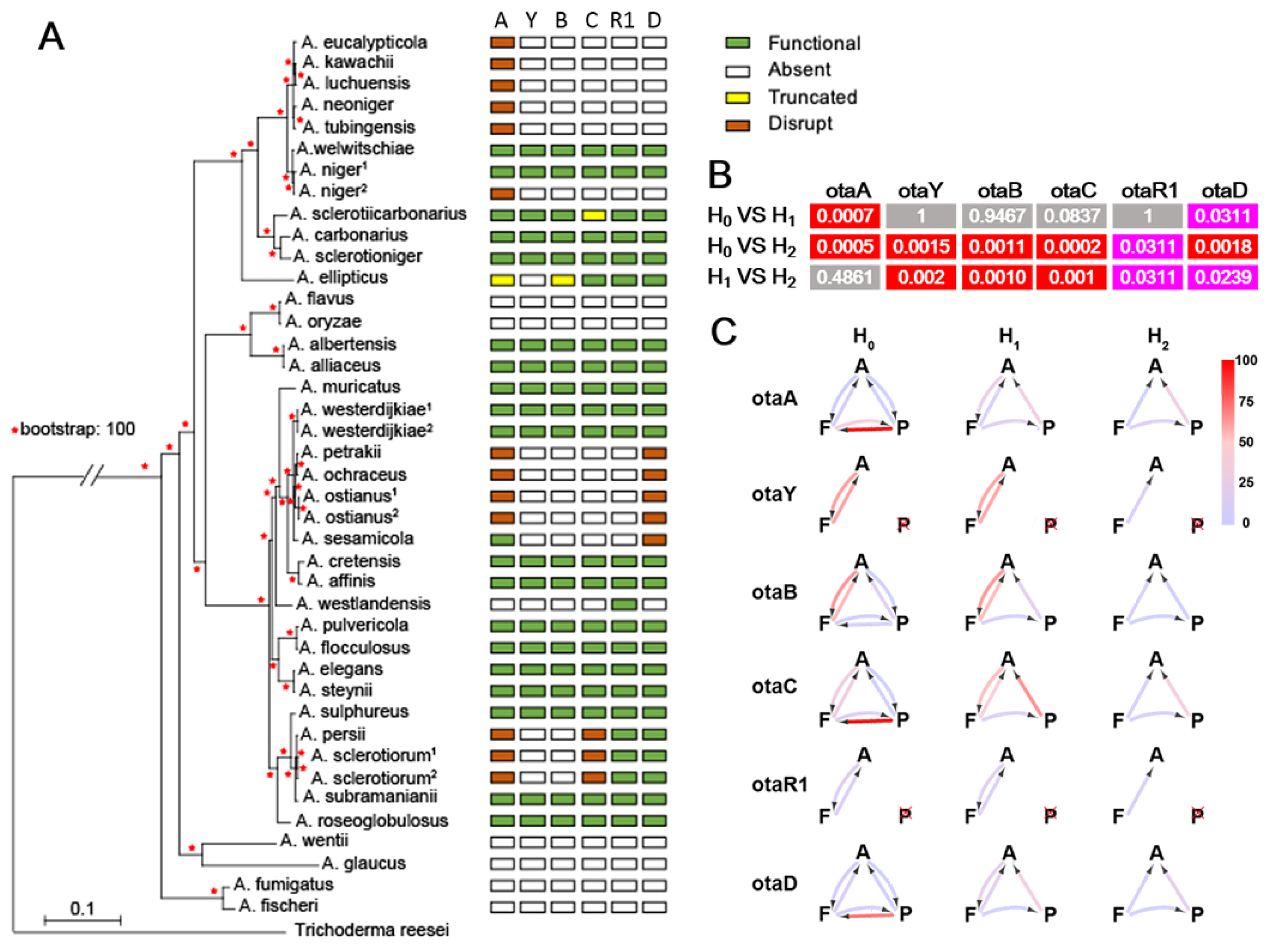
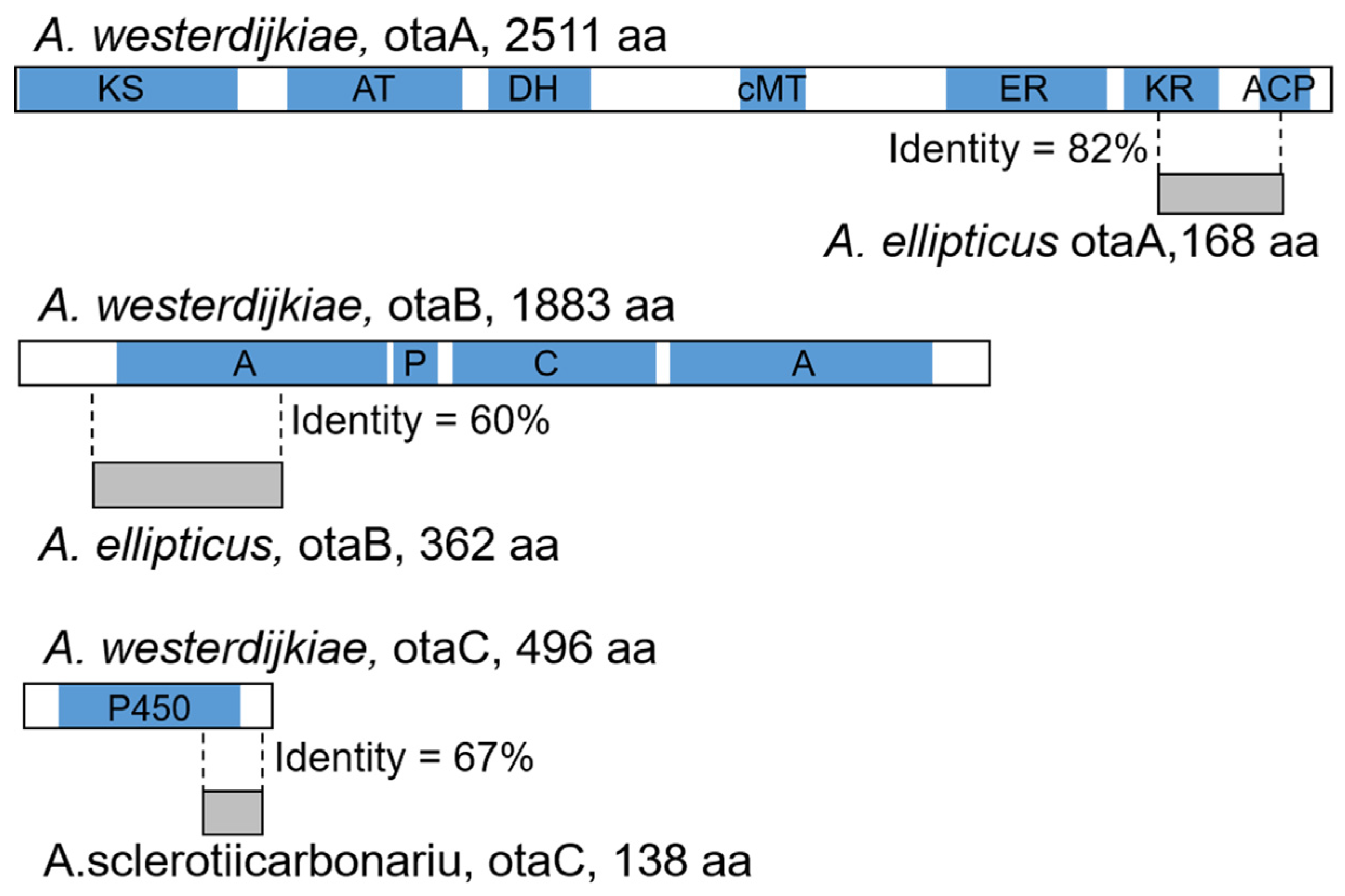
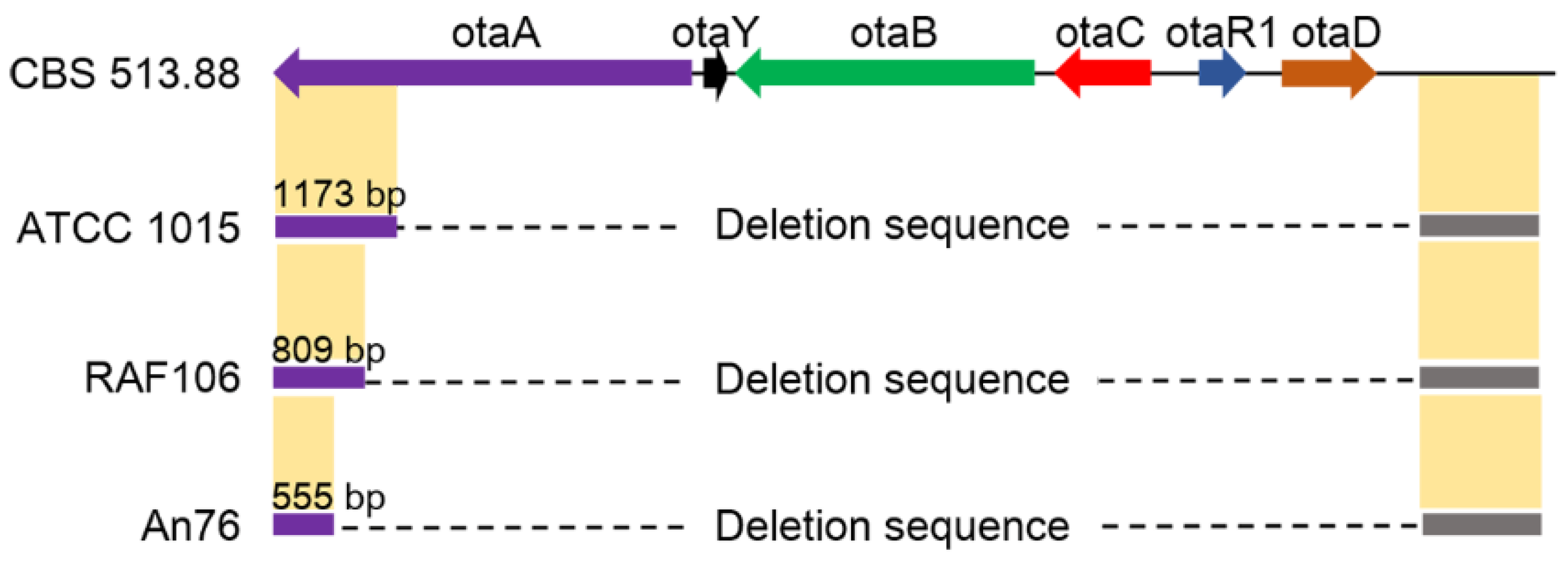
2.2. Degeneration of OT Cluster in Aspergillus
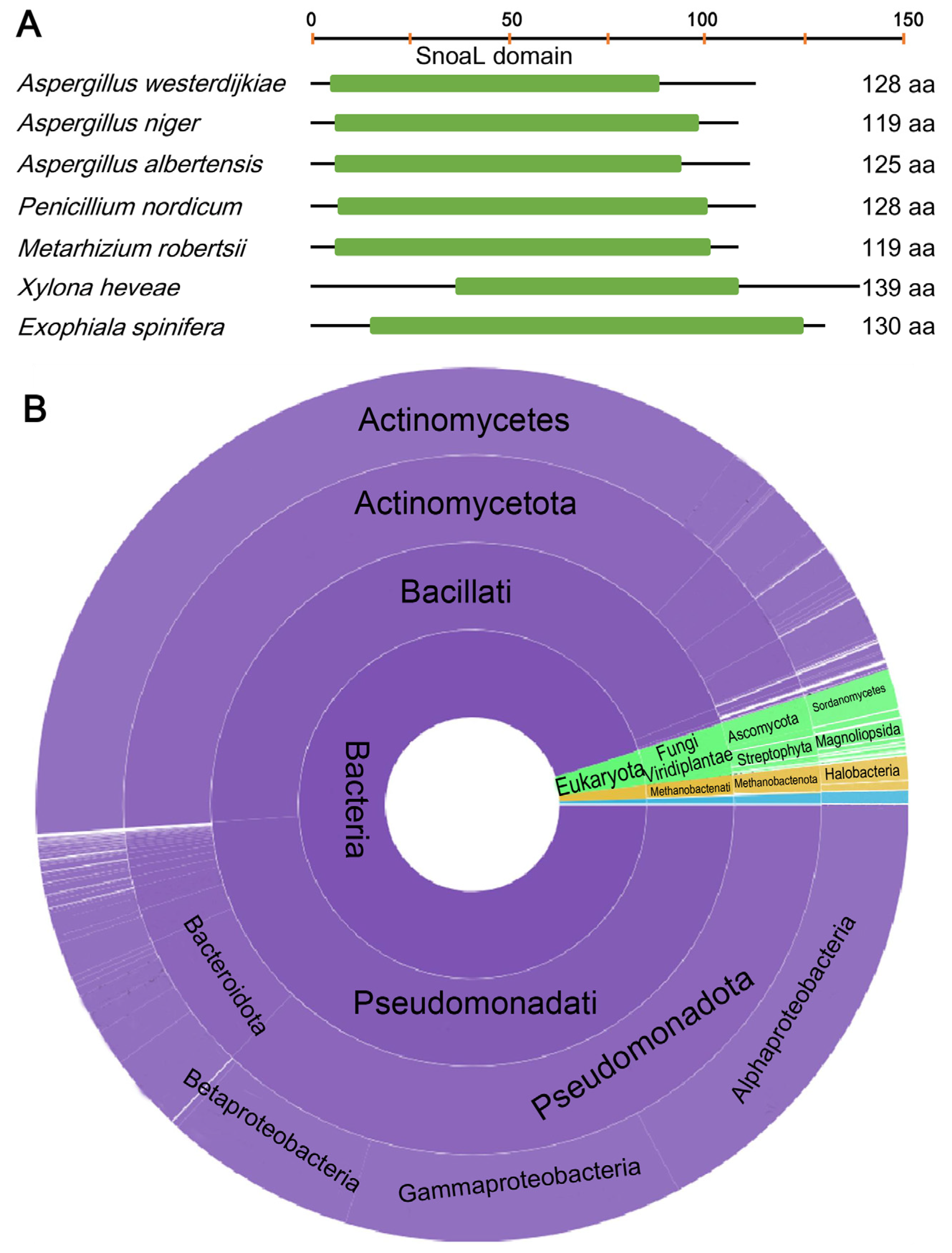
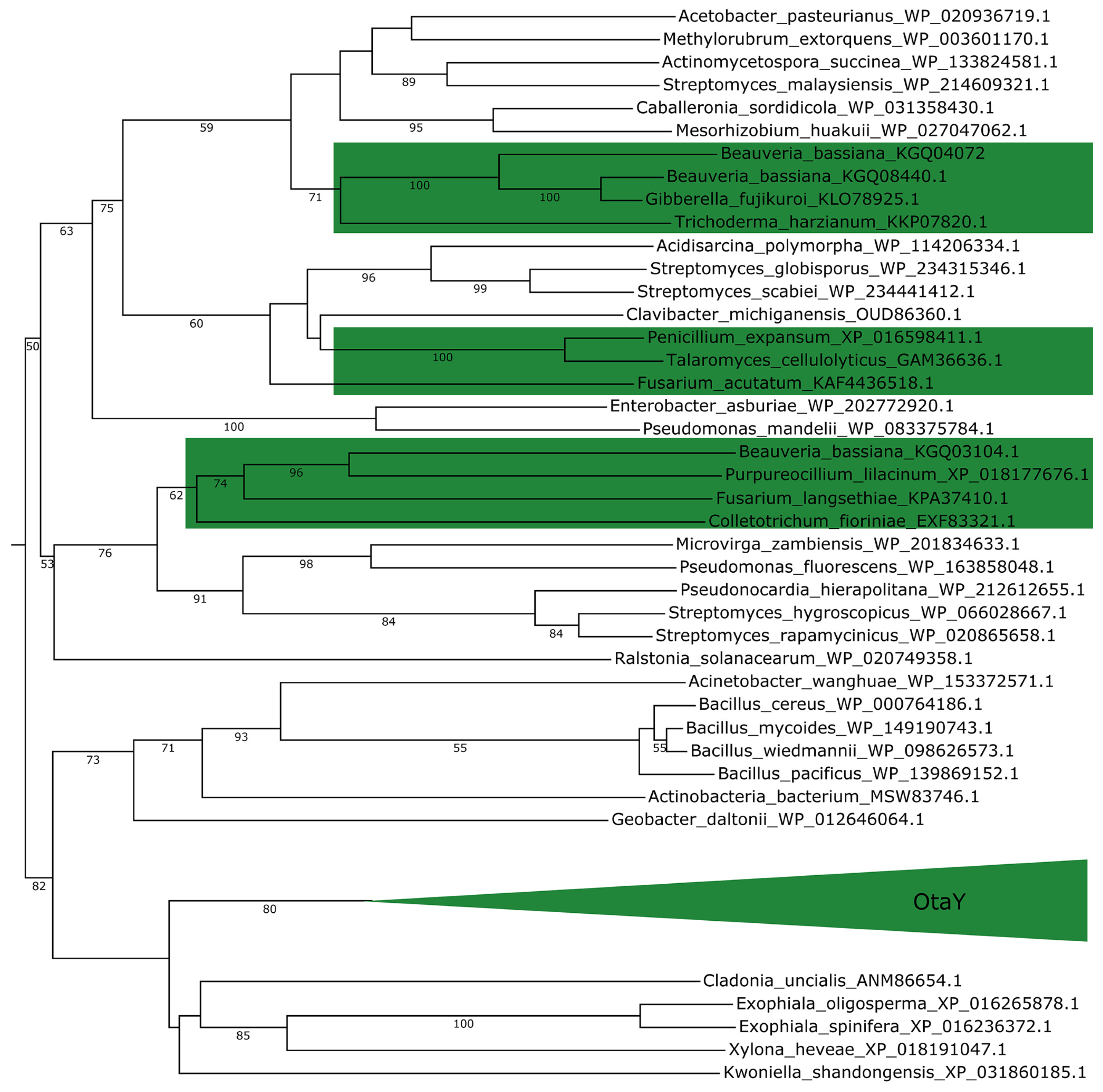
2.3. Fungi May Acquire otaY from Bacteria
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Strains and Culture Conditions
5.2. Bioinformatic Analysis
5.3. Phylogenetic Analysis
5.4. Estimation of Gene Decay Rates
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, G.; Li, E.; Gallo, A.; Perrone, G.; Varga, E.; Ma, J.; Yang, B.; Tai, B.; Xing, F. Impact of environmental factors on ochratoxin A: From natural occurrence to control strategy. Environ. Pollut. 2023, 317, 120767. [Google Scholar] [CrossRef] [PubMed]
- European Commission. Setting Maximum Levels for Certain Contaminants in Foodstuffs; European Commission Regulation 2006, EC No 1881/2006; European Commission: Brussels, Belgium, 2006. [Google Scholar]
- GB2761-2017; Maximum Limits of Mycotoxins in Food. National Criterion of China: Beijing, China, 2017.
- IARC. Ochratoxin A. Some Naturally Occurring Substances: Food Items and Constituents, Heterocyclic Aromatic Amines and Mycotoxins; IARC: Lyon, France, 1993. [Google Scholar]
- Wang, G.; Ran, H.; FAN, J.; Keller, N.; Liu, Z.; Wu, F.; Yin, W.-B. Fungal-fungal cocultivation leads to widespread secondary metabolite alteration requiring the partial loss-of-function VeA1 protein. Sci. Adv. 2022, 8, eabo6094. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, L.; Wu, F.; Liu, F.; Wang, Q.; Zhang, X.; Selvaraj, J.N.; Zhao, Y.; Xing, F.; Yin, W.-B.; et al. A consensus ochratoxin a biosynthetic pathway: Insights from the genome sequence of Aspergillus ochraceus and a comparative genomic analysis. Appl. Environ. Microbiol. 2018, 84, e01009-18. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, M.; Gallo, A.; Perrone, G.; Magistà, D.; Baker, S.E. Comparative genomic analysis of ochratoxin A biosynthetic cluster in producing fungi: New evidence of a cyclase gene involvement. Front. Microbiol. 2020, 11, 581309. [Google Scholar] [CrossRef]
- Ferrara, M.; Gallo, A.; Cervini, C.; Gambacorta, L.; Solfrizzo, M.; Baker, S.E.; Perrone, G. Evidence of the involvement of a cyclase gene in the biosynthesis of ochratoxin A in Aspergillus carbonarius. Toxins 2021, 13, 892. [Google Scholar] [CrossRef]
- Bachleitner, S.; Sulyok, M.; Sørensen, J.L.; Strauss, J.; Studt, L. The H4K20 methyltransferase Kmt5 is involved in secondary metabolism and stress response in phytopathogenic Fusarium species. Fungal Genet. Biol. 2021, 155, 103602. [Google Scholar] [CrossRef]
- Lim, S.; Bijlani, S.; Blachowicz, A.; Chiang, Y.-M.; Lee, M.-S.; Torok, T.; Venkateswaran, K.; Wang, C.C.C. Identification of the pigment and its role in UV resistance in Paecilomyces variotii, a Chernobyl isolate, using genetic manipulation strategies. Fungal Genet. Biol. 2021, 152, 103567. [Google Scholar] [CrossRef]
- Chase, A.; Sweeney, D.; Muskat, M.; Guillén-Matus, D.; Jensen, P. Vertical Inheritance Facilitates Interspecies Diversification in Biosynthetic Gene Clusters and Specialized Metabolites. mBio 2021, 12, e0270021. [Google Scholar] [CrossRef]
- Lawrence, J. Selfish operons: The evolutionary impact of gene clustering in prokaryotes and eukaryotes. Curr. Opin. Genet. Dev. 1999, 9, 642–648. [Google Scholar] [CrossRef]
- Lawrence, J.G.; Roth, J.R. Selfish operons: Horizontal transfer may drive the evolution of gene clusters. Genetics 1996, 143, 1843–1860. [Google Scholar] [CrossRef]
- Philippe, H.; Douady, C. Horizontal gene transfer and phylogenetics. Curr. Opin. Microbiol. 2003, 6, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Khaldi, N.; Collemare, J.; Lebrun, M.-H.; Wolfe, K.H. Evidence for horizontal transfer of a secondary metabolite gene cluster between fungi. Genome Biol. 2008, 9, R18. [Google Scholar] [CrossRef]
- Reynolds, H.T.; Slot, J.C.; Divon, H.H.; Lysøe, E.; Proctor, R.H.; Brown, D.W. Differential retention of gene functions in a secondary metabolite cluster. Mol. Biol. Evol. 2017, 34, 2002–2015. [Google Scholar] [CrossRef] [PubMed]
- Slot, J.C.; Rokas, A. Horizontal transfer of a large and highly toxic secondary metabolic gene cluster between fungi. Curr. Biol. 2011, 21, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Gil-Serna, J.; García-Díaz, M.; González-Jaén, M.; Vázquez, C.; Patiño, B. Description of an orthologous cluster of ochratoxin A biosynthetic genes in Aspergillus and Penicillium species. A comparative analysis. Int. J. Food Microbiol. 2018, 268, 35–43. [Google Scholar] [CrossRef]
- Wang, G.; Wu, W.; Keller, N.P.; Guo, X.; Li, E.; Ma, J.; Xing, F. Metarhizium spp. encode an ochratoxin cluster and a high efficiency ochratoxin-degrading amidohydrolase revealed by genomic analysis. J. Adv. Res. 2025, 72, 85–95. [Google Scholar] [CrossRef]
- Arnold, B.J.; Huang, I.; Hanage, W.P. Horizontal gene transfer and adaptive evolution in bacteria. Nat. Rev. Microbiol. 2021, 20, 206–218. [Google Scholar] [CrossRef]
- Wang, W.; Drott, M.; Greco, C.; Luciano-Rosario, D.; Wang, P.; Keller, N.P. Transcription factor repurposing offers insights into evolution of biosynthetic gene cluster regulation. mBio 2021, 12, e0139921. [Google Scholar] [CrossRef]
- Campbell, M.A.; Rokas, A.; Slot, J.C. Horizontal transfer and death of a fungal secondary metabolic gene cluster. Genome Biol. Evol. 2012, 4, 289–293. [Google Scholar] [CrossRef]
- Liu, A.; Juan Chen, A.; Liu, B.; Wei, Q.; Bai, J.; Hu, Y. Investigation of citrinin and monacolin K gene clusters variation among pigment producer Monascus species. Fungal Genet. Biol. 2022, 160, 103687. [Google Scholar] [CrossRef]
- Sultana, A.; Kallio, P.; Jansson, A.; Wang, J.-S.; Niemi, J.; Mäntsälä, P.; Schneider, G. Structure of the polyketide cyclase SnoaL reveals a novel mechanism for enzymatic aldol condensation. EMBO J. 2004, 23, 1911–1921. [Google Scholar] [CrossRef] [PubMed]
- He, B.-B.; Zhou, T.; Bu, X.-L.; Weng, J.-Y.; Xu, J.; Lin, S.; Zheng, J.-T.; Zhao, Y.-L.; Xu, M.-J. Enzymatic pyran formation involved in xiamenmycin biosynthesis. ACS Catal. 2019, 9, 5391–5399. [Google Scholar] [CrossRef]
- Tatum, E.L.; Lederberg, J. Gene recombination in the bacterium Escherichia coli. J. Bacteriol. 1947, 53, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Guo, Z.; Yang, Z.; Han, H.; Wang, S.; Xu, H.; Yang, X.; Yang, F.; Wu, Q.; Xie, W.; et al. Whitefly hijacks a plant detoxification gene that neutralizes plant toxins. Cell 2021, 184, 1693–1705.e17. [Google Scholar] [CrossRef]
- Park, C.; Zhang, J. High expression hampers horizontal gene transfer. Genome Biol. Evol. 2012, 4, 523–532. [Google Scholar] [CrossRef]
- Soucy, S.M.; Huang, J.; Gogarten, J.P. Horizontal gene transfer: Building the web of life. Nat. Rev. Genet. 2015, 16, 472–482. [Google Scholar] [CrossRef]
- Schmidt-Heydt, M.; Graf, E.; Batzler, J.; Geisen, R. The application of transcriptomics to understand the ecological reasons of ochratoxin A biosynthesis by Penicillium nordicum on sodium chloride rich dry cured foods. Trends Food Sci. Technol. 2011, 22, S39–S48. [Google Scholar] [CrossRef]
- Schmidt-Heydt, M.; Graf, E.; Stoll, D.; Geisen, R. The biosynthesis of ochratoxin A by Penicillium as one mechanism for adaptation to NaCl rich foods. Food Microbiol. 2012, 29, 233–241. [Google Scholar] [CrossRef]
- Chen, A.J.; Tang, D.; Zhou, Y.Q.; Sun, B.D.; Li, X.J.; Wang, L.Z.; Gao, W.W. Identification of ochratoxin A producing fungi associated with fresh and dry liquorice. PLoS ONE 2013, 8, e78285. [Google Scholar] [CrossRef]
- Mao, X.-M.; Zhan, Z.-J.; Grayson, M.N.; Tang, M.-C.; Xu, W.; Li, Y.-Q.; Yin, W.-B.; Lin, H.-C.; Chooi, Y.-H.; Houk, K.N.; et al. Efficient biosynthesis of fungal polyketides containing the dioxabicyclo-octane ring system. J. Am. Chem. Soc. 2015, 137, 11904–11907. [Google Scholar] [CrossRef]
- Zhou, H.; Li, Y.; Tang, Y. Cyclization of aromatic polyketides from bacteria and fungi. Nat. Prod. Rep. 2010, 27, 839–868. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.F.; Chiang, Y.-M.; Szewczyk, E.; Davidson, A.D.; Ahuja, M.; Elizabeth Oakley, C.; Woo Bok, J.; Keller, N.; Oakley, B.R.; Wang, C.C.C. Molecular genetic analysis of the orsellinic acid/F9775 gene cluster of Aspergillus nidulans. Mol. Biosyst. 2010, 6, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.J. Polyketides, proteins and genes in fungi: Programmed nano-machines begin to reveal their secrets. Org. Biomol. Chem. 2007, 5, 2010–2026. [Google Scholar] [CrossRef] [PubMed]
- Herbst, D.A.; Townsend, C.A.; Maier, T. The architectures of iterative type I PKS and FAS. Nat. Prod. Rep. 2018, 35, 1046–1069. [Google Scholar] [CrossRef]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Zdobnov, E.M. BUSCO: Assessing genomic data quality and beyond. Curr. Protoc. 2021, 12, e323. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef]
- Wang, G.; Liu, Z.; Lin, R.; Li, E.; Mao, Z.; Ling, J.; Yang, Y.; Yin, W.-B.; Xie, B. Biosynthesis of antibiotic leucinostatins in bio-control fungus Purpureocillium lilacinum and their inhibition on Phytophthora revealed by genome mining. PLoS Pathog. 2016, 12, e1005685. [Google Scholar] [CrossRef] [PubMed]
- Pagel, M.; Meade, A.; Barker, D. Bayesian estimation of ancestral character states on phylogenies. Syst. Biol. 2004, 53, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.; Meade, A.; Pagel, M. Constrained models of evolution lead to improved prediction of functional linkage from correlated gain and loss of genes. Bioinformatics 2006, 23, 14–20. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.; Yan, Z.; Yang, B.; Tai, B.; Li, W.; Li, E.; Wang, G.; Xing, F. Differential Retention and Loss of a Mycotoxin in Fungal Evolution. Toxins 2025, 17, 311. https://doi.org/10.3390/toxins17060311
Chen L, Yan Z, Yang B, Tai B, Li W, Li E, Wang G, Xing F. Differential Retention and Loss of a Mycotoxin in Fungal Evolution. Toxins. 2025; 17(6):311. https://doi.org/10.3390/toxins17060311
Chicago/Turabian StyleChen, Lin, Ziying Yan, Bolei Yang, Bowen Tai, Weizhao Li, Erfeng Li, Gang Wang, and Fuguo Xing. 2025. "Differential Retention and Loss of a Mycotoxin in Fungal Evolution" Toxins 17, no. 6: 311. https://doi.org/10.3390/toxins17060311
APA StyleChen, L., Yan, Z., Yang, B., Tai, B., Li, W., Li, E., Wang, G., & Xing, F. (2025). Differential Retention and Loss of a Mycotoxin in Fungal Evolution. Toxins, 17(6), 311. https://doi.org/10.3390/toxins17060311