1. Introduction
Sepsis constitutes life-threatening organ dysfunction and is characterized by a complicated host response to invading pathogenic microorganisms. Due to life-threatening hemodynamic instability, sepsis is a major contributor to mortality in the hospital [
1,
2]. In China, the incidence of sepsis in intensive care unit (ICU) patients is approximately 25.5% [
3]. Sepsis accounts for 13% of hospital costs and over 24 billion clinical burdens in the USA [
4]. It is a fact that acute systemic inflammation caused by sepsis will first spread to the respiratory system, where the lungs tend to become the first affected organs, and it is generally believed that approximately half of patients with sepsis develop acute lung injury (ALI) [
5]. Notably, ALI and its most severe form, acute respiratory distress syndrome (ARDS), constitute devastating clinical complications arising from sepsis. They are characterized by inflammatory cell infiltration, refractory hypoxemia, pulmonary edema, and serious respiratory failure [
5]. Mortality caused by ARDS is over 40%, and it leads to approximately 75,000 deaths annually in the USA [
6]. Despite abundant advances, current therapeutic intervention has not been convincingly effective [
7].
As a proverbial inflammation-related disease, sepsis-evoked ALI/ARDS chiefly manifests as immune cell infiltration and produces inflammatory mediators, such as TNF-α. Inflammatory cytokines and cells will enter blood circulation and accumulate in the lungs, leading to the release of abundant inflammatory cytokines and oxygen free radicals to further aggravate inflammatory response in the lungs [
8,
9]. An excessive inflammatory response in ALI will result in pulmonary structural cell injury, especially alveolar epithelial cells [
10]. It is known that alveolar epithelial cell damage leads to the dysfunction of the alveolar epithelial barrier, allowing inflammatory cells into the alveolar space from the blood, further exacerbating lung tissue hypoxia and aggravating lung damage in sepsis [
5,
11]. Oxidative stress is also known as a major pathological mechanism for ALI. During the progression of ALI, the systemic inflammatory response also induces abundant production of ROS to evoke excessive oxidative stress [
12]. Oxidative stress accumulation can lead to fluid leakage, alveolar epithelial cell apoptosis, and the penetration of inflammatory cells, exacerbating the progression of ALI [
12]. Currently, the targeting of the inflammatory response and oxidative stress represents a promising potential therapeutic strategy in ALI/ADRS [
12,
13].
It is known that sepsis concerns life-threatening systemic inflammation caused by pathogens, especially bacteria. Antimicrobial peptides constitute the first natural defense barrier blocking invading pathogens in multiple mammalian species, playing important roles in inflammation-related diseases, including sepsis. LL-37, a 37-residue helical peptide, represents the only proverbial human antimicrobial peptide. It is believed that LL-37 is majorly released from the neutrophils and epithelial cells of the testis, gastrointestinal tissue, skin, and respiratory tract [
14]. Compared with traditional antibiotics, LL-37 possesses broad antibacterial activity and is not susceptible to drug resistance or immune rejection. Intriguingly, increasing attention has revealed that LL-37 plays important roles in diverse biological processes, including immune regulation, inflammatory response, re-epithelialization, and wound healing [
14]. Notably, a previous study confirmed the anti-apoptotic efficacy of LL-37 in LPS-treated human microvascular blood vessel endothelial cells [
15]. Also notably, a recent study highlighted that LL-37 attenuates bacterial burden and improved the survival of septic mice [
16]. Nevertheless, the roles and underlying mechanism of LL-37 in sepsis-evoked ALI remain undefined.
In the present research, we assessed LL-37’s therapeutic potential using preclinical models. Moreover, the underlying mechanism involved in the above processes was further elucidated.
3. Discussion
Sepsis-induced ALI usually leads to organ failure and constitutes a heavy burden on families and society due to high mortality and morbidity. A previous study revealed the therapeutic roles of LL-37 in septic mice [
16]. Nevertheless, its roles in sepsis-evoked ALI are still unclear. LPS, a major component in Gram-negative bacteria, can induce systemic inflammation and microvascular lung injury. Therefore, LPS-induced injury has been recognized as a useful experimental model due to its similarities with the characteristics of ALI in humans [
11,
19,
20]. In this research, LL-37 attenuated LPS-induced oxidative stress injury and inflammatory response in pulmonary epithelial cells. Moreover, LL-37 enhanced autophagy by inhibiting ZBP1 expression, which accounted for LL-37-mediated anti-oxidative injury and anti-inflammation in LPS-stimulated pulmonary epithelial cells. Importantly, in vivo, LL-37 alleviated CLP-induced ALI by blocking ZBP1 via ameliorating pathological changes, lung edema, and lung injury scores. Moreover, LL-37 inhibited oxidative stress and inflammation in the ALI mouse model, concomitant with autophagy activation. Thus, the current study highlights that LL-37 may represent a feasible therapeutic agent against sepsis-induced ALI by regulating ZBP-mediated autophagy activation.
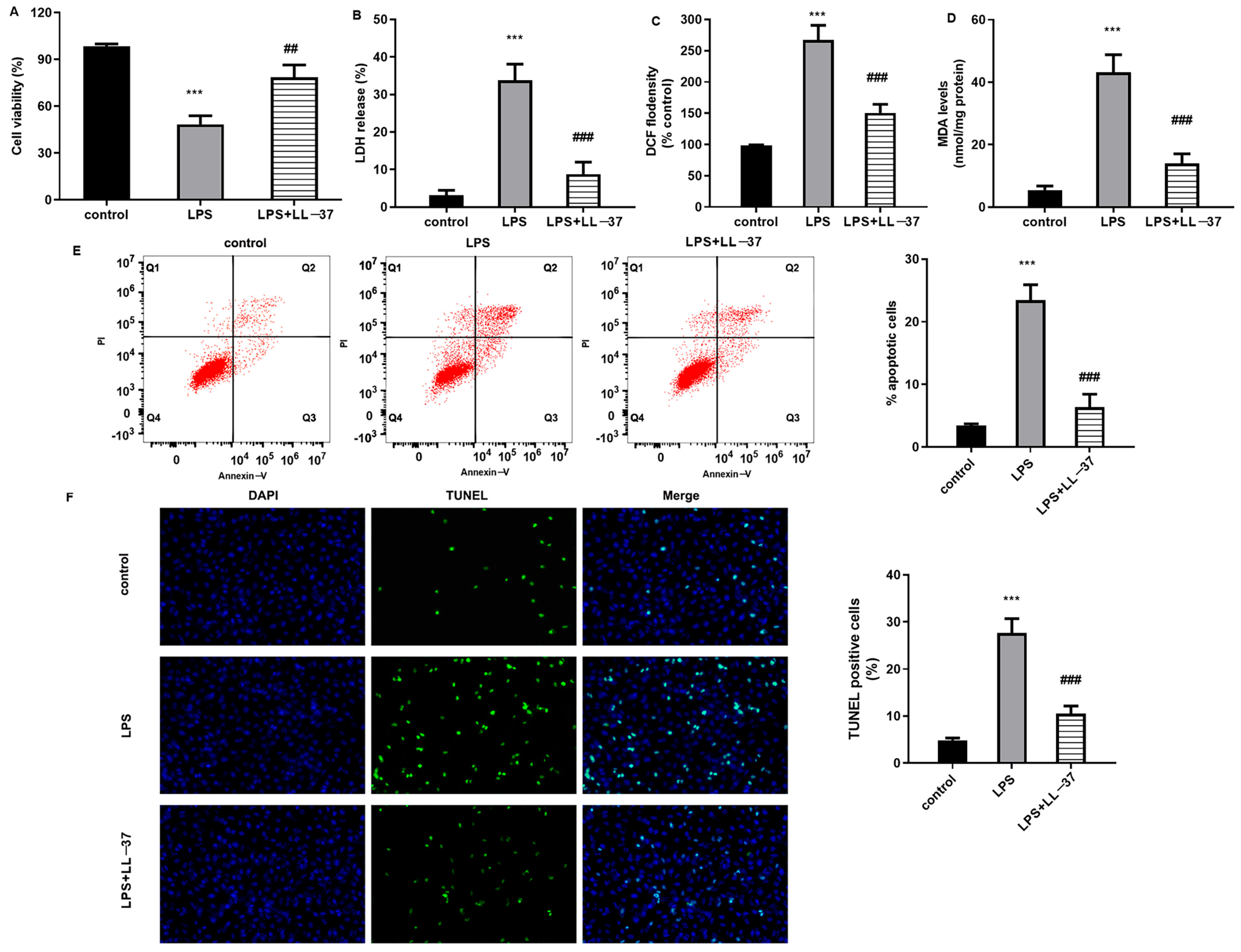
Alveolar epithelial cells exert a critical function in antagonizing lung tissue injury and can establish a capillary alveolar epithelial barrier to prevent alveolar collapse and extra-vascular inflamed cell entrance into the alveolar space. It is generally believed that oxidative stress injury represents a known pathological mechanism in sepsis-induced ALI [
12]. Under a sepsis-induced systemic inflammatory response, excessive inflammation leads to the overproduction of free radicals and induction of alveolar epithelial cell oxidative damage, which will further aggravate cell apoptosis and necrosis [
12,
20]. LPS, as a major component of Gram-negative bacteria, is a main cause of ALI and can be applied to mimic sepsis-induced ALI models in vitro [
13,
20]. Thus, we stimulated human alveolar pulmonary epithelial cells with LPS and confirmed oxidative stress injury after LPS stimulation. Interestingly, the LL-37 treatment alleviated LPS-evoked oxidative injury in alveolar pulmonary epithelial cells. Similarly, an emerging study also confirms that LL-37 attenuates heat-stroke-induced cytotoxicity in intestinal goblet cells by exerting its antioxidant function [
21]. Several reports have revealed anti-oxidative stress as a potential approach for sepsis-induced ALI therapy [
12,
20]. In this study, LL-37 also attenuated oxidative stress levels in lung tissues from ALI mice. Thus, these findings reveal that LL-37 may attenuate the development of sepsis-induced ALI by suppressing oxidative injury.
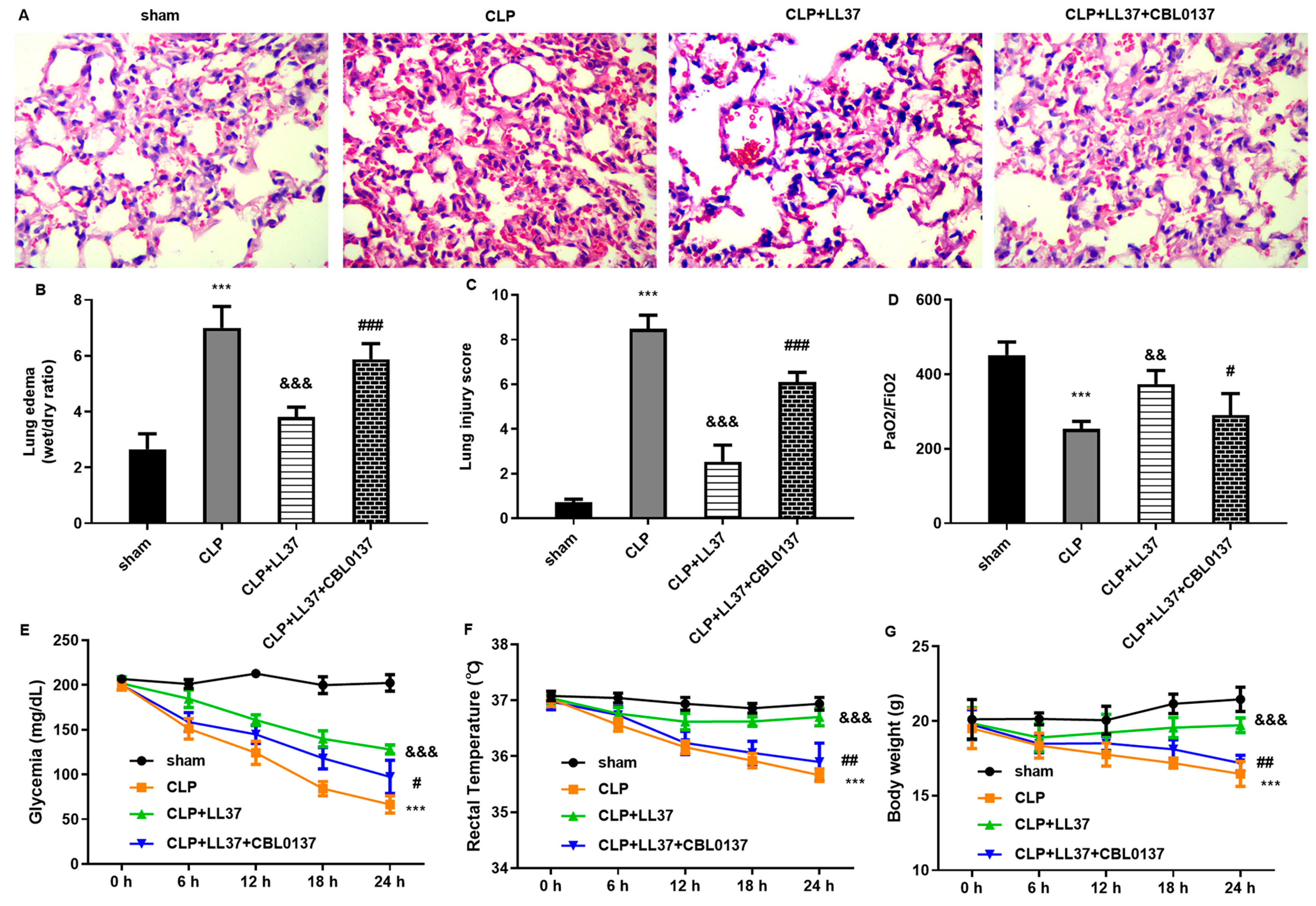
Pathologically, the lung ranks as the most vulnerable organ after sepsis. Abundant inflammatory mediators caused by infection will induce alveolar epithelial cell injury and disrupt the alveolar microvascular barrier, which allows for inflammatory cell infiltration and evokes lung dysfunction manifested as ALI [
10,
12,
20]. Currently, targeting the inflammatory response represents a promising therapeutic strategy for ALI caused by sepsis [
9,
20,
22]. Therefore, before the investigation of LL-37 in ALI in vivo, we first constructed LPS-evoked inflammation in pulmonary epithelial cells. Consistent with a previous study [
19,
22], LPS stimulation evoked excessive inflammatory responses in alveolar pulmonary epithelial cells. Importantly, LL-37 restrained the above LPS-evoked inflammation. Moreover, in the ALI mouse model, LL-37 also suppressed CLP-increased inflammatory cytokines levels. Intriguingly, a previous study revealed that LL-37 could stimulate neutrophils to produce antimicrobial microvesicles to ameliorate pathological changes in sepsis [
16]. Furthermore, LL-37 also inhibits contents of pro-inflammatory cytokines (TNF-α, IL-1β, and IL-6) in peritoneal fluids and sera in septic mice [
23]. Importantly, the current findings substantiate that LL-37 alleviated CLP-evoked hemorrhage, inflammatory cells infiltration, diffuse alveolar damage, lung tissue edema, and injury score. Thus, LL-37 may attenuate the progression of ALI triggered by sepsis by inhibiting inflammation.
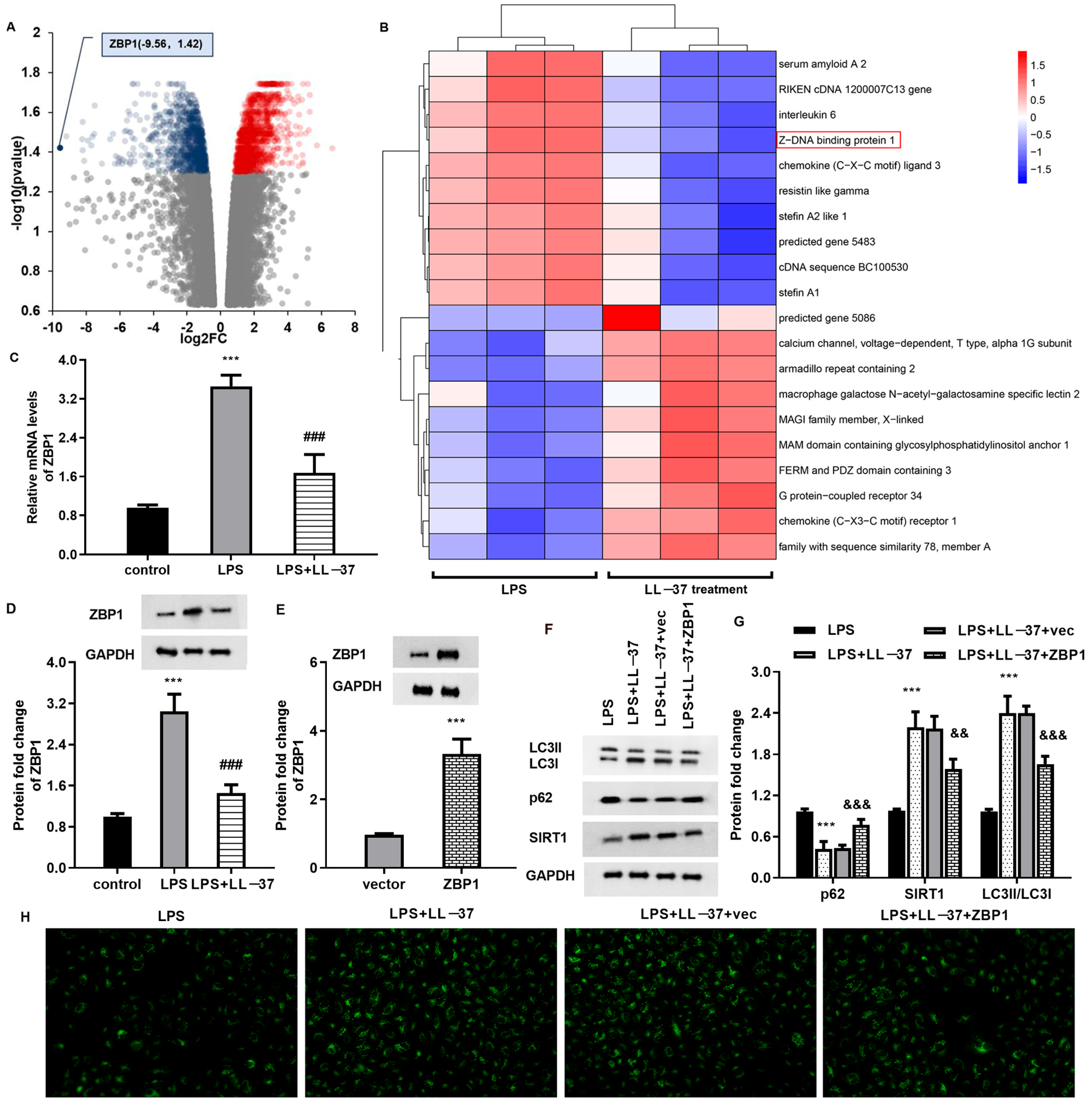
Accumulating evidence reveals that impaired autophagic–lysosomal degradation is responsible for lung tissue injury caused by sepsis [
18,
24]. Autophagy is an evolutionary conserved process that can exert a protective mechanism to degrade and recycle the damaged organelles. Under systemic inflammation after sepsis, autolysosomes are reduced in lung tissue and lead to autophagy dysfunction in ALI. Several studies confirm the dysregulation of autophagy in the lung tissues of patients with sepsis and in animal models [
18,
25]. Autophagy impairment can result in the accumulation of damaged mitochondria to induce ROS overproduction, which further aggravates oxidative injury and ALI progression [
26]. Moreover, autophagy is implicated in pathophysiological process of sepsis-evoked ALI, including cell apoptosis and inflammatory response [
27]. Intriguingly, LC3II-related autophagy was increased over 24 h in a CLP-induced ALI model; however, the impaired autophagosome–lysosome fusion gradually reduced within 24 h in sepsis-induced ALI [
27]. Thus, increasing research supports that enhancing autophagy activation may attenuate the progression of ALI caused by sepsis [
18,
28]. LC3II is a standard marker for autophagosomes, and its high expression and down-regulation of autophagic substrate p62 indicate the activation of autophagy. In this study, a decreased expression of LC3II and increased expression of p62 were observed in LPS-treated pulmonary epithelial cells and lung tissues from the ALI model, indicating the impaired autophagy in the ALI model. Notably, LL-37 restored autophagy impairment in ALI cells and in the mice model, indicating that LL-37 may ameliorate the progression of ALI caused by sepsis by enhancing autophagy. Intriguingly, LL-37 can induce autophagy activation to eliminate live Porphyromonas gingivalis in keratinocytes [
29]. Furthermore, RNA-seq sequencing analysis substantiated ZBP1 as a down-regulated gene with the largest fold change. Moreover, LL-37 treatment suppressed ZBP1 expression, whilst ZBP1 overexpression inhibited autophagy and reversed LL-37-mediated protection against LPS-induced oxidative injury and inflammation. Importantly, ZBP1 elevation reversed the protective function of LL-37 in lung injury in sepsis-caused ALI mice. Similarly, a previous study also revealed that ZBP2 might attenuate H
2O
2-induced cell autophagy in endothelial cells [
30]. However, how does ZBP1 affect autophagy? It is unknown whether ZBP1 regulates autophagy through an autophagy-related signaling pathway (such as PI3K/AKT) or its domain. This is a limitation of our work and will be explored in the future.
5. Materials and Methods
5.1. Cell Culture and Sepsis-Induced Lung Injury Model In Vitro
Human A549 pulmonary epithelial cells were bought from Shanghai Cell Collection (Shanghai, China). All cells were maintained in DMEM/F12 medium (Gibco, Waltham, MA, USA) containing 10% fetal bovine serum, 100 μg/mL streptomycin, and penicillin in a 5% CO2 incubator at 37 °C. To mimic sepsis-evoked lung injury, A549 cells were stimulated with lipopolysaccharide (LPS; 2 μg/mL; Sigma-Aldrich, St Louis, MO, USA) or LL-37 (50 μg/mL; Sangon Biotech, Shanghai, China) for 24 h in vitro.
5.2. Recombinant Plasmid Construction and Transfection
Before the construction of the ZBP recombinant vector, RNA was extracted from the A549 cells using commercial TRIzol Reagent (Invitrogen, Carlsbad, CA, USA). Then, first-strand cDNA was synthesized according to the protocols of a SuperScript™ IV First-Strand Synthesis System (Invitrogen). Subsequently, ZBP1 cDNA was obtained via PCR amplification and inserted into a pcDNA3.1(+) plasmid (Invitrogen) to prepare the ZBP1 recombinant vector pcDNA-ZBP1. After that, the cells were placed in six-well plates and transfected with empty or pcDNA-ZBP1 vector when cells had grown to 70–80% confluence. Forty-eight hours later, the efficacy of ZBP1 vector transfection was evaluated using Western blotting.
5.3. Cell Viability Detection
A549 cells, whether transfected with ZBP1 plasmids or not, were treated with LL-37 under LPS exposure (2 μg/mL) for 24 h. After that, all the specimens were collected and incubated with CCK-8 solution (10 μL; Nanjing Jiancheng Bioengineering Institute, Nanjing, China). Subsequently, the cell viability was measured at 4 h post-reaction when all specimens were analyzed using a spectrophotometer (Bio-Rad, Hercules, CA, USA) to measure the absorbance at 450 nm.
5.4. Detection of Lactate Dehydrogenase (LDH) Release
Following the treatment with plasmid or LL-37 under the LPS conditions, the A549 cell specimens were centrifuged for 15 min. Then, an LDH detection solution (Beyotime Biotechnology, Shanghai, China) was added into the prepared cell supernatant samples, shielding them from light for 30 min. The release of LDH was assessed by detecting OD490 nm.
5.5. Cell Apoptosis Analysis Using Flow Cytometer
After treatments under various conditions, the A549 cells were collected. Following rinsing with cold PBS (Sangon Biotech, Shanghai, China), 500 μL of binding buffer was added to re-suspend the collected samples (Beyotime, Shanghai, China). Then, annexin V-FITC (10 μL) and PI (5 μL) were added to the specimens at room temperature according to the instructions of a FITC-Annexin V Apoptosis Detection Kit. All specimens were then analyzed using a FACScan flow cytometer (BD Biosciences; San Jose, CA, USA).
5.6. TUNEL Staining
The treated A549 cells were fixed with 4% paraformaldehyde at room temperature. A total of 20 min later, the samples were rinsed with PBS three times, then re-suspended in PBS solution containing 0.3% of Triton X-100 (Sangon Biotech, Shanghai, China) and incubated for 5 min. Subsequently, all the specimens were reacted with TUNEL detection solution (50 μL) for 1 h at 37 °C in the dark. Finally, cells were analyzed under a fluorescence microscope (excitation wavelength of 450 nm and tracking emission wavelength of 520 nm).
5.7. qRT-PCR
After the synthesis of first-strand cDNA, real-time PCR was carried out on an ABI PRISM 7000 sequence detection system (Applied Biosystems, Foster City, CA, USA) to quantify the transcriptional expression of TNF-α, IL-18, IL-1β, p62, and SIRT1. All the protocols were constructed in reference to the instructions of a SYBR Premix Ex TaqTM II Kit (Takara, Dalian, China). All primers were prepared by Sangon Biotech (Shanghai, China). The transcriptional levels of targeted genes were quantified via 2−ΔΔCt and an internal control GAPDH.
5.8. Fluorescence Detection of Autophagy Level
To analyze the autophagy levels in A549 cells under various treatments, cell samples were infected with Ad-GFP-LC3B (Beyotime) at an MOI (Multiplicity of Infection) of 40. All the protocols were performed according to a commercial Ad-GFP-LC3B Kit, and all the samples were observed under a fluorescence microscope (Olympus, Tokyo, Japan).
5.9. RNA Transcriptome Sequencing Assay
A549 cells were stimulated with LPS and LL-37 for 24 h. The total RNA from the cells was extracted, purified using oligo dT magnetic beads and fragmented, and then applied to synthesize cDNA. Subsequently, a sequencing library was constructed using a DNA library construction kit (Illumina, San Diego, CA, USA), and the Illumina hiseq 2000 platform was applied for mRNA sequencing. After the data were taken off the plane, the FastQC software (version 0.11.8) was used for data quality control. Then, transcriptome data were compared using the HISAT2 software (version 2.2.1), and the gene expression was calculated using the Stringtie software (version 2.1.2) and Deseq2 (version 1.14.1). A heatmap and a volcano plot were used to analyze differentially expressed genes (DEGs) between both groups. DEGs are shown as a log2 fold change >1 and adjusted p-value < 0.05.
5.10. Experimental Animal and Ethics Statement
Male C57BL/6 mice (8 weeks, 20–25 g) were obtained from the Laboratory Animals Center, the Fourth Military Medical University. All mice were maintained in specific pathogen-free (SPF) conditions (12 h light/dark cycle at 22 ± 2 °C). All animals were fed with standard chow and autoclaved water for one week. All reasonable efforts were carried out to minimize suffering. All the experimental procedures were conducted following the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals, and all the experiments were approved via the Institutional Animal Care and Use Committee of West China Second University Hospital, Sichuan University.
5.11. Construction of Animal Model
Sepsis-associated ALI mice were constructed via CLP as previously described [
19]. Mice were randomly divided into five groups (
n = 5 in each group): the sham group, CLP group, CLP and LL37 group, and CLP + LL37 + CBL0137 (ZBP1 activator) group. For the CLP groups, mice were anesthetized intraperitoneally with pentobarbital sodium (60 mg/kg). Then, a 1 cm midline incision was generated in the lower abdomen to allow for cecum exposure. Subsequently, the cecum was ligated using 3-0 silk and then perforated with a 18-G needle to gently squeeze a droplet of feces into the peritoneal cavity to induce infection. The cecum was then repositioned and closed. After surgery, mice were resuscitated immediately through the subcutaneous injection of prewarmed normal saline (50 mL/kg). Mice in the sham groups underwent the same procedure without ligature and puncture. In the CLP and LL37 group, CLP model mice were intravenously injected with LL-37 (10 mg/kg). In the CLP + LL37 + CBL0137 (ZBP1 activator) group, CLP mice were intravenously injected with LL-37 (10 mg/kg) and CBL0137 (30 mg/kg). The weight, rectal temperature, and blood glucose were tested at 6 h intervals.
5.12. Detection of Oxygenation Index (PaO2/FiO2)
At 24 h after the operation, mice were anesthetized, and a 0.5 cm long incision was generated along the central axis of the anterior chest. Chest wall tissue was carefully separated to expose the chest cavity. Then, a 0.2 mL blood sample was collected from the heart using an arterial needle for blood oxygen analysis. Then, the oxygenation index (PaO2/FiO2) was calculated.
5.13. Hematoxylin and Eosin (HE) Staining and Lung Injury Score
Lung samples were collected and fixed with 4% paraformaldehyde. Then, all the specimens were embedded in paraffin. The tissue samples were cut into 5 μm thick serial sections. A histopathological evaluation was performed via HE staining, and lung injury scores were then assessed by two blinded pathologists, as previously reported [
17].
5.14. Lung Edema Evaluation
To evaluate lung edema, the wet/dry weight (W/D) ratio was used. Briefly, the fresh upper portion of left lungs was excised and then rinsed with PBS. The weight of a wet lung was recorded. Then, the lung tissues were dried for 24 h at 80 °C in an oven, and the dry weight of the tissues was subsequently weighed.
5.15. Immunohistochemical Assay
For a histochemical analysis of ZBP1 expression in the lung tissues, lung sections were incubated with normal goat serum. One hour later, all samples were treated with anti-ZBP1 antibody (1:2000) at 4 °C overnight. Then, all section specimens were treated with biotin-labeled goat anti-rabbit secondary antibody at room temperature. Approximately 1 h later, slides were then developed with diaminobenzidine reagent (Zhongshan Company, Beijing, China) and counterstained with hematoxylin. The final images were observed using an Olympus BX53 microscope (Olympus, Tokyo, Japan).
5.16. Analysis of ROS and Malondialdehyde (MDA) Levels
To measure intracellular ROS levels in cells and tissue, an ROS Detection Kit (Nanjing Jiancheng Bioengineering Institute) was used. In brief, cells under various treatments were maintained in serum-free medium containing 10 µM 2′,7′-dichlorodihydrofluorescin diacetate (DCFH-DA). Approximately 0.5 h later, a microplate reader was used to measure the fluorescence intensity (with an excitation wavelength of 490 nm and tracking emission wavelength of 525 nm, respectively).
The contents of MDA in cells and lung tissues were determined using MDA Detection Kits (Nanjing Jiancheng Bioengineering Institute), following the manufacturer’s instructions. The absorbance at 450 nm was measured to quantify the levels of MDA.
5.17. Western Blotting Assay
The total protein from cells and tissue specimens was treated with RIPA lysis buffer. Subsequently, a BCA protein assay kit (Beyotime) was used to measure the prepared protein concentration. Then, approximately 30 μg of protein was subjected to 10% SDS-PAGE and transferred to a PVDF membrane. Following interdiction with non-fat dry milk (5%), the membrane was incubated with the primary antibody against LC3 (1:2000), p62 (1:20,000), SIRT1 (1:1000), and ZBP1 (1:1000) (all from Abcam, Cambridge, MA, USA) at 4 °C overnight. Subsequently, samples were hatched with horseradish-peroxidase-conjugated secondary antibodies. One hour later, binding bands were incubated with a chemiluminescent detection system and quantified using the Image J software (version 1.8.0).
5.18. Inflammatory Cytokine Detection via ELISA
The contents of inflammatory cytokines (TNF-α, IL-18, IL-1β) in the serum of mice and cell supernatant were detected using commercial ELISA kits (eBioscience, San Diego, CA, USA). All the procedures were conducted with reference to the manufacturer’s instructions.
5.19. Statistical Analysis
All experiments were carried out at least three times, and all data are shown as the mean ± standard deviation (SD). All statistical assays were analyzed using the SPSS19.0 software. A significant difference was determined using Student’s t-test for two groups and ANOVA with Tukey’s test for multiple groups. p < 0.05 was defined as statistically significant.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}