In Vivo Genotoxicity and Toxicity Assessment of Sterigmatocystin Individually and in Mixture with Aflatoxin B1

 , ,
, ,  , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Organ Weight, Clinical Biochemistry and Histopathology
2.2. Standard and Fpg-Modified Alkaline Comet Assay
2.3. Micronucleus Assay
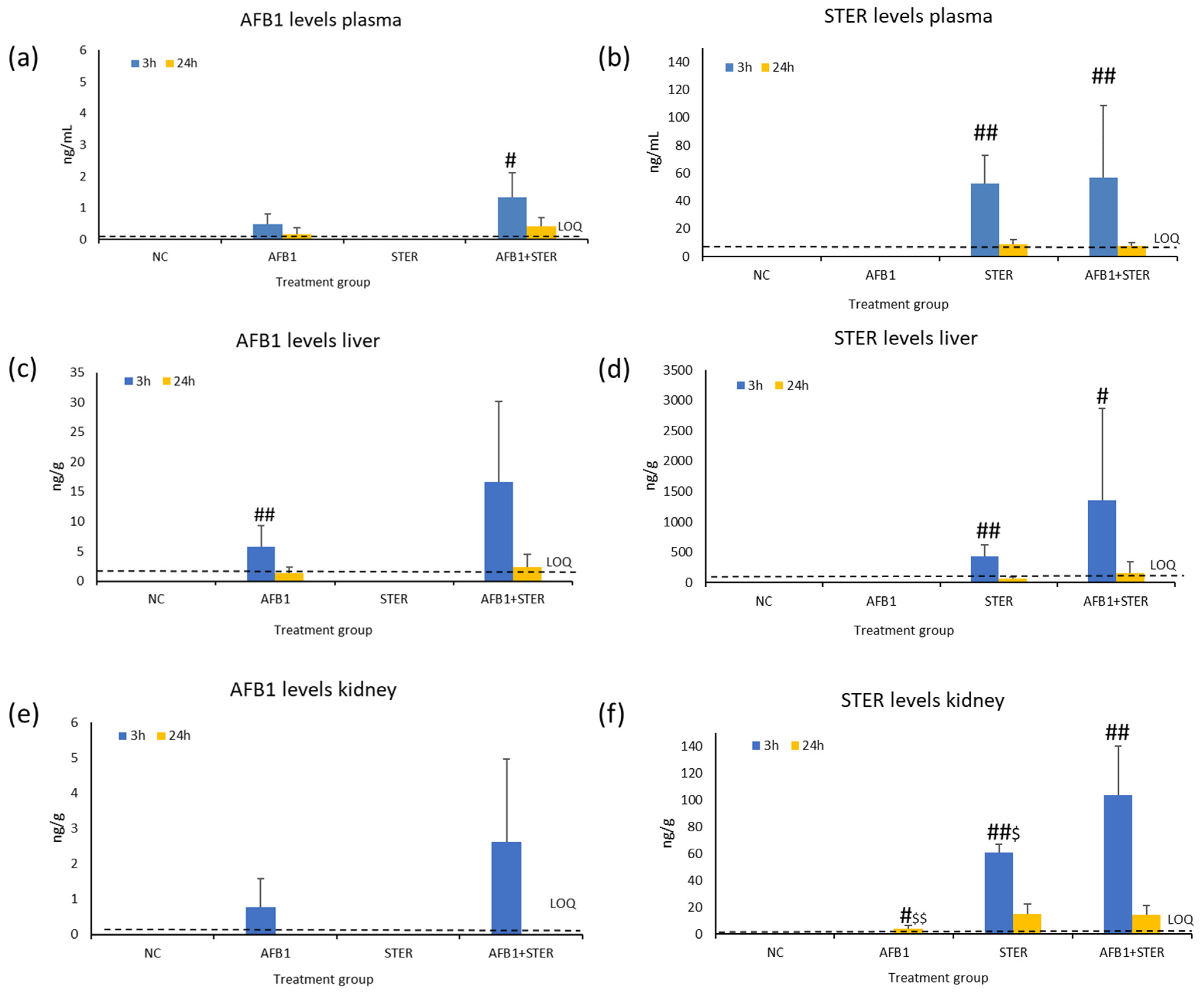
2.4. Mycotoxin Determination in Plasma, Liver, and Kidney
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Chemicals and Reagents
5.2. Mycotoxins Safety Precautions
5.3. Animals
5.4. Study Design and Treatments
5.4.1. Dose Selection
5.4.2. In Vivo Study Design
5.5. Sample Collection
5.6. Body Weight, Organ/Body Weight Index, Clinical Biochemistry, and Histopathology
Results Evaluation
5.7. Standard and Fpg-Modified Comet Assay
Results Evaluation
5.8. Micronucleus Assay
Results Evaluation
5.9. Determination of Mycotoxins in Plasma, Kidney, and Liver
5.9.1. Sample Preparation
Plasma Samples
Liver and Kidney Samples
5.9.2. Preparation of Calibration Samples
5.9.3. Results Evaluation
5.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bennett, J.W.; Klich, M. Mycotoxins. Clin. Microbiol. Rev. 2003, 16, 497–516. [Google Scholar] [CrossRef] [PubMed]
- Alassane-Kpembi, I.; Schatzmayr, G.; Taranu, I.; Marin, D.; Puel, O.; Oswald, I.P. Mycotoxins co-contamination: Methodological aspects and biological relevance of combined toxicity studies. Crit. Rev. Food Sci. Nutr. 2017, 57, 3489–3507. [Google Scholar] [CrossRef] [PubMed]
- Klarić, M.; Cvetnić, Z.; Pepeljnjak, S.; Kosalec, I. Co-occurrence of Aflatoxins, Ochratoxin A, Fumonisins, and Zearalenone in Cereals and Feed, Determined by Competitive Direct Enzyme-Linked Immunosorbent Assay and Thin-Layer Chromatography. Arch. Ind. Hyg. Toxicol. 2009, 60, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Kagot, V.; De Boevre, M.; Landschoot, S.; Obiero, G.; Okoth, S.; De Saeger, S. Comprehensive analysis of multiple mycotoxins and Aspergillus flavus metabolites in maize from Kenyan households. Int. J. Food Microbiol. 2022, 363, 109502. [Google Scholar] [CrossRef] [PubMed]
- EFSA (European Food Safety Authority). Risk assessment of aflatoxins in food. EFSA J. 2020, 18, e06040. [Google Scholar] [CrossRef]
- EFSA (European Food Safety Authority). Scientific Opinion on the risk for public and animal health related to the presence of sterigmatocystin in food and feed. EFSA J. 2013, 11, 3254. [Google Scholar] [CrossRef]
- Theumer, M.G.; Henneb, Y.; Khoury, L.; Snini, S.P.; Tadrist, S.; Canlet, C.; Puel, O.; Oswald, I.P.; Audebert, M. Genotoxicity of aflatoxins and their precursors in human cells. Toxicol. Lett. 2018, 287, 100–107. [Google Scholar] [CrossRef]
- Yogendrarajah, P.; Jacxsens, L.; De Saeger, S.; De Meulenaer, B. Co-occurrence of multiple mycotoxins in dry chilli (Capsicum annum L.) samples from the markets of Sri Lanka and Belgium. Food Control 2014, 46, 26–34. [Google Scholar] [CrossRef]
- Ingenbleek, L.; Sulyok, M.; Adegboye, A.; Hossou, S.E.; Koné, A.Z.; Oyedele, A.D.; Kisito, C.S.K.J.; Dembélé, Y.K.; Eyangoh, S.; Verger, P.; et al. Regional Sub-Saharan Africa Total Diet Study in Benin, Cameroon, Mali and Nigeria Reveals the Presence of 164 Mycotoxins and Other Secondary Metabolites in Foods. Toxins 2019, 11, 54. [Google Scholar] [CrossRef]
- Arce-López, B.; Lizarraga, E.; Vettorazzi, A.; González-Peñas, E. Human Biomonitoring of Mycotoxins in Blood, Plasma and Serum in Recent Years: A Review. Toxins 2020, 12, 147. [Google Scholar] [CrossRef]
- Joint Expert Committee on Food Additives (JECFA). Safety Evaluation of Certain Contaminants in Food: Prepared by the Eighty-Third Meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA); World Health Organization Food Agriculture Organization United Nations: Geneva, Switzerland, 2018; Volume 992, pp. 1–992.
- Arce-López, B.; Lizarraga, E.; Irigoyen, Á.; González-Peñas, E. Presence of 19 Mycotoxins in Human Plasma in a Region of Northern Spain. Toxins 2020, 12, 750. [Google Scholar] [CrossRef]
- De Ruyck, K.; Huybrechts, I.; Yang, S.; Arcella, D.; Claeys, L.; Abbeddou, S.; De Keyzer, W.; De Vries, J.; Ocke, M.; Ruprich, J.; et al. Mycotoxin exposure assessments in a multi-center European validation study by 24-h dietary recall and biological fluid sampling. Environ. Int. 2020, 137, 105539. [Google Scholar] [CrossRef]
- Ezekiel, C.N.; Abia, W.A.; Braun, D.; Šarkanj, B.; Ayeni, K.I.; Oyedele, O.A.; Michael-Chikezie, E.C.; Ezekiel, V.C.; Mark, B.N.; Ahuchaogu, C.P.; et al. Mycotoxin exposure biomonitoring in breastfed and non-exclusively breastfed Nigerian children. Environ. Int. 2022, 158, 106996. [Google Scholar] [CrossRef]
- Cao, X.; Li, X.; Li, J.; Niu, Y.; Shi, L.; Fang, Z.; Zhang, T.; Ding, H. Quantitative determination of carcinogenic mycotoxins in human and animal biological matrices and animal-derived foods using multi-mycotoxin and analyte-specific high performance liquid chromatography-tandem mass spectrometric methods. J. Chromatogr. B 2018, 1073, 191–200. [Google Scholar] [CrossRef]
- IARC (International Agency for Research on Cancer). Monographs on the Evaluation of Carcinogenic Risks to Humans. Chem. Agents Relat. Occup. 2012, 100F, 225–249. [Google Scholar]
- EFSA (European Food Safety Authority). Opinion of the scientific panel on contaminants in the food chain [CONTAM] related to the potential increase of consumer health risk by a possible increase of the existing maximum levels for aflatoxins in almonds, hazelnuts and pistachios and derived prod. EFSA J. 2007, 5, 446. [Google Scholar] [CrossRef]
- IARC (International Agency for Research on Cancer). Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Man: Some Naturally Occurring Substances. Am. Ind. Hyg. Assoc. J. 1976, 10, 1–331. Available online: https://publications.iarc.fr/Book-And-Report-Series/Iarc-Monographs-On-The-Identification-Of-Carcinogenic-Hazards-To-Humans/Some-Naturally-Occurring-Substances-1976 (accessed on 6 February 2023).
- McCann, J.; Spingarn, N.E.; Kobori, J.; Ames, B.N. Detection of carcinogens as mutagens: Bacterial tester strains with R factor plasmids. Proc. Natl. Acad. Sci. USA 1975, 72, 979–983. [Google Scholar] [CrossRef]
- Kuczuk, M.H.; Benson, P.M.; Heath, H.; Wallace Hayes, A. Evaluation of the mutagenic potential of mycotoxins using Salmonella typhimurium and Saccharomyces cerevisiae. Mutat. Res. Mutagen. Relat. Subj. 1978, 53, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Wehner, F.C.; Thiel, P.G.; van Rensburg, S.J.; Demasius, I.P.C. Mutagenicity to Salmonella typhimurium of some Aspergillus and Penicillium mycotoxins. Mutat. Res. Toxicol. 1978, 58, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Ellard, S.; Parry, J.M. A comparative study of the use of primary Chinese hamster liver cultures and genetically engineered immortal V79 Chinese hamster cell lines expressing rat liver CYP1A1, 1A2 and 2B1 cDNAs in micronucleus assays. Toxicology 1993, 82, 131–149. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Jiang, L.; Ge, L.; Chen, M.; Geng, C.; Yang, G.; Li, Q.; Ji, F.; Yan, Q.; Zou, Y.; et al. Sterigmatocystin-induced oxidative DNA damage in human liver-derived cell line through lysosomal damage. Toxicol. In Vitro 2015, 29, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Curry, P.T.; Reed, R.N.; Martino, R.M.; Kitchin, R.M. Induction of sister-chromatid exchanges in vivo in mice by the mycotoxins sterigmatocystin and griseofulvin. Mutat. Res. Toxicol. 1984, 137, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Ueda, N.; Fujie, K.; Gotoh-Mimura, K.; Chattopadhyay, S.C.; Sugiyama, T. Acute cytogenetic effect of sterigmatocystin on rat bone-marrow cells in vivo. Mutat. Res. Lett. 1984, 139, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Dubravka, R.; Daniela, J.; Andrea, H.T.; Domagoj, K.; Nevenka, K.; Lada, R.; Davor, Ž.; Maja, P.; Maja, Š.K. Sterigmatocystin moderately induces oxidative stress in male Wistar rats after short-term oral treatment. Mycotoxin Res. 2020, 36, 181–191. [Google Scholar] [CrossRef]
- Jakšić, D.; Ćurtović, I.; Kifer, D.; Rašić, D.; Kopjar, N.; Micek, V.; Peraica, M.; Klarić, M.Š. Single-Dose Toxicity of Individual and Combined Sterigmatocystin and 5-Methoxysterigmatocistin in Rat Lungs. Toxins 2020, 12, 734. [Google Scholar] [CrossRef]
- Essigmann, J.M.; Barker, L.J.; Fowler, K.W.; Francisco, M.A.; Reinhold, V.N.; Wogan, G.N. Sterigmatocystin-DNA interactions: Identification of a major adduct formed after metabolic activation in vitro. Proc. Natl. Acad. Sci. USA 1979, 76, 179–183. [Google Scholar] [CrossRef]
- Pfeiffer, E.; Fleck, S.C.; Metzler, M. Catechol Formation: A Novel Pathway in the Metabolism of Sterigmatocystin and 11-Methoxysterigmatocystin. Chem. Res. Toxicol. 2014, 27, 2093–2099. [Google Scholar] [CrossRef]
- Alonso-Jauregui, M.; Font, M.; González-Peñas, E.; López de Cerain, A.; Vettorazzi, A. Prioritization of Mycotoxins Based on Their Genotoxic Potential with an In Silico-In Vitro Strategy. Toxins 2021, 13, 734. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Jauregui, M.; González-Peñas, E.; López de Cerain, A.; Vettorazzi, A. Genotoxicity of 12 Mycotoxins by the SOS/umu Test: Comparison of Liver and Kidney S9 Fraction. Toxins 2022, 14, 400. [Google Scholar] [CrossRef]
- Richard, J.L.; Thurston, J.R.; Lillehoj, E.B.; Cysewski, S.J.; Booth, G.D. Complement activity, serum protein, and hepatic changes in guinea pigs given sterigmatocystin or aflatoxin, alone or in combination. Am. J. Vet. Res. 1978, 39, 163–166. [Google Scholar]
- Abdelhamid, A.M.; Dorra, T.M. Effect of feed-borne pollution with some mycotoxin combinations on broiler chicks. Arch. Für Tierernaehrung 1993, 44, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Du, M.; Zhang, G. Proapoptotic activity of aflatoxin B 1 and sterigmatocystin in HepG2 cells. Toxicol. Rep. 2014, 1, 1076–1086. [Google Scholar] [CrossRef] [PubMed]
- Kövesi, B.; Kulcsár, S.; Ancsin, Z.; Zándoki, E.; Erdélyi, M.; Mézes, M.; Balogh, K. Individual and Combined Effects of Aflatoxin B1 and Sterigmatocystin on Lipid Peroxidation and Glutathione Redox System of Common Carp Liver. Toxins 2021, 13, 109. [Google Scholar] [CrossRef] [PubMed]
- OECD (Organization for Economic Cooperation and Development). Test No. 489: In Vivo Mammalian Alkaline Comet Assay. In OECD Guidelines for the Testing of Chemicals, Section 4; OECD: Paris, France, 2016; pp. 1–27. ISBN 9789264264885. [Google Scholar]
- OECD (Organization for Economic Cooperation and Development). Test No. 474: Mammalian Erythrocyte Micronucleus Test. In OECD Guidelines for the Testing of Chemicals, Section 4; OECD: Paris, France, 2016; pp. 1–21. ISBN 9789264264762. [Google Scholar]
- International Council on Harmonization (ICH). Guideline S2 (R1) on Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended for Human Use; International Counci on Harmonization: Amsterdam, The Netherlands, 2012; pp. 1–28. [Google Scholar]
- Hubrecht, C. The 3Rs and Humane Experimental Technique: Implementing Change. Animals 2019, 9, 754. [Google Scholar] [CrossRef] [PubMed]
- Bowen, D.E.; Whitwell, J.H.; Lillford, L.; Henderson, D.; Kidd, D.; Mc Garry, S.; Pearce, G.; Beevers, C.; Kirkland, D.J. Evaluation of a multi-endpoint assay in rats, combining the bone-marrow micronucleus test, the Comet assay and the flow-cytometric peripheral blood micronucleus test. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2011, 722, 7–19. [Google Scholar] [CrossRef]
- Corcuera, L.-A.; Vettorazzi, A.; Arbillaga, L.; Pérez, N.; Gil, A.G.; Azqueta, A.; González-Peñas, E.; García-Jalón, J.A.; López de Cerain, A. Genotoxicity of Aflatoxin B1 and Ochratoxin A after simultaneous application of the in vivo micronucleus and comet assay. Food Chem. Toxicol. 2015, 76, 116–124. [Google Scholar] [CrossRef]
- Vasquez, M.Z. Combining the in vivo comet and micronucleus assays: A practical approach to genotoxicity testing and data interpretation. Mutagenesis 2010, 25, 187–199. [Google Scholar] [CrossRef]
- Collins, A.R. Measuring oxidative damage to DNA and its repair with the comet assay. Biochim. Biophys. Acta-Gen. Subj. 2014, 1840, 794–800. [Google Scholar] [CrossRef]
- Anwar, W.A.; Khalil, M.M.; Wild, C.P. Micronuclei, chromosomal aberrations and aflatoxin-albumin adducts in experimental animals after exposure to aflatoxin B1. Mutat. Res. Toxicol. 1994, 322, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Trzos, R.J.; Petzold, G.L.; Brunden, M.N.; Swenberg, J.A. The evaluation of sixteen carcinogens in the rat using the micronucleus test. Mutat. Res. Toxicol. 1978, 58, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Madle, E.; Korte, A.; Beek, B. Species differences in mutagenicity testing: I. Micronucleus and SCE tests in rats, mice, and Chinese hamsters with aflatoxin B1. Teratog. Carcinog. Mutagen. 1986, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Raj, H.G.; Kohli, E.; Rohil, V.; Dwarakanath, B.; Parmar, V.S.; Malik, S.; Adhikari, J.; Tyagi, Y.K.; Goel, S.; Gupta, K.; et al. Acetoxy-4-methylcoumarins confer differential protection from aflatoxin B1-induced micronuclei and apoptosis in lung and bone marrow cells. Mutat. Res. Toxicol. Environ. Mutagen. 2001, 494, 31–40. [Google Scholar] [CrossRef]
- Miyata, M. Grapefruit juice intake does not enhance but rather protects against aflatoxin B1-induced liver DNA damage through a reduction in hepatic CYP3A activity. Carcinogenesis 2003, 25, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Belloir, C.; Siess, M.-H.; Le Bon, A.-M. Inhibition of Carcinogen-Induced DNA Damage in Rat Liver and Colon by Garlic Powders With Varying Alliin Content. Nutr. Cancer 2006, 55, 178–184. [Google Scholar] [CrossRef]
- Ferk, F.; Huber, W.W.; Grasl-Kraupp, B.; Speer, K.; Buchmann, S.; Bohacek, R.; Mišík, M.; Edelbauer, L.; Knasmüller, S. Protective effects of coffee against induction of DNA damage and pre-neoplastic foci by aflatoxin B 1. Mol. Nutr. Food Res. 2014, 58, 229–238. [Google Scholar] [CrossRef]
- Bakheet, S.A.; Alhuraishi, A.M.; Al-Harbi, N.O.; Al-Hosaini, K.A.; Al-Sharary, S.D.; Attia, M.M.; Alhoshani, A.R.; Al-Shabanah, O.A.; Al-Harbi, M.M.; Imam, F.; et al. Alleviation of Aflatoxin B1-Induced Genomic Damage by Proanthocyanidins via Modulation of DNA Repair. J. Biochem. Mol. Toxicol. 2016, 30, 559–566. [Google Scholar] [CrossRef]
- Corcuera, L.A.; Arbillaga, L.; Vettorazzi, A.; Azqueta, A.; López de Cerain, A. Ochratoxin A reduces aflatoxin B1 induced DNA damage detected by the comet assay in Hep G2 cells. Food Chem. Toxicol. 2011, 49, 2883–2889. [Google Scholar] [CrossRef]
- Muruzabal, D.; Langie, S.A.S.; Pourrut, B.; Azqueta, A. The enzyme-modified comet assay: Enzyme incubation step in 2 vs 12-gels/slide systems. Mutat. Res. Toxicol. Environ. Mutagen. 2019, 845, 402981. [Google Scholar] [CrossRef]
- Arce-López, B.; Lizarraga, E.; Flores-Flores, M.; Irigoyen, Á.; González-Peñas, E. Development and validation of a methodology based on Captiva EMR-lipid clean-up and LC-MS/MS analysis for the simultaneous determination of mycotoxins in human plasma. Talanta 2020, 206, 120193. [Google Scholar] [CrossRef]
- Food and Drug Aministration (FDA). Bionalytical Method Validation; Food and Drug Administration: Silver Spring, MD, USA, 2018; Volume 1043, pp. 1–44.
- EC (European Comission). Commission Decision of 12 August 2002 Implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and the Interpretation of Results (Notified under Document Number C (2002) 3044) (Text with EEA Relevance) (2002/657/EC). Off. J. Eur. Communities 2002, 1–29. Available online: http://data.europa.eu/eli/dec/2002/657/oj (accessed on 6 February 2023).


{kind=link}
| Group | Organs Absolute Weight (g) | Organ/Body Weight Index | ||
|---|---|---|---|---|
| Liver | Kidney | Liver | Kidney | |
| IC 95% (inf-sup) | 7.6–13.5 | 0.7–1.5 | - | - |
| NC 3 h | 8.4 ± 0.6 | 0.9 ± 0.07 | 3.14 ± 0.2 | 0.4 ± 0.03 |
| AFB1 3 h | 7.8 ± 0.7 | 0.9 ± 0.06 | 3 ± 0.1 | 0.3 ± 0.03 |
| STER 3 h | 8.9 ± 0.4 | 0.9 ± 0.07 | 3.3 ± 0.3 | 0.3 ± 0.01 |
| AFB1+STER 3 h | 7.8 ± 1.2 | 0.9 ± 0.09 | 2.9 ± 0.4 | 0.4 ± 0.02 |
| NC 24 h | 11.7 ± 1.1 | 0.8 ± 0.06 | 4.1 ± 0.2 | 0.3 ± 0.03 |
| AFB1 24 h | 11.9 ± 2 | 1.03 ± 0.08 | 4.1 ± 0.6 | 0.3 ± 0.02 |
| STER 24 h | 12.8 ± 1.1 | 1.1 ± 0.1 | 4.2 ± 0.4 | 0.4 ± 0.03 |
| AFB1+STER 24 h | 10.7 ± 2.1 | 0.9 ± 0.1 | 3.6 ± 0.6 | 0.3 ± 0.04 |
| Group | Total Protein (g/dL) | Albumin (g/dL) | Urea (mg/dL) | AST (U/L) | ALT (U/L) | Creatinine (mg/dL) |
|---|---|---|---|---|---|---|
| IC 95% (inf-sup) | 5.1–6.7 | 3.7–4.7 | 21–58 | 52–138 | 14–63 | 0.17–0.5 |
| NC 3 h (n = 4) | 5.2 ± 0.2 | 3.5 ± 0.2 | 27.5 ± 2.4 | 144 ± 43.6 | 41 ± 5.2 | 0.3 ± 0.03 |
| NC 24 h (n = 5) | 4.8 ± 0.7 | 3.2 ± 0.5 | 20.8 ± 2.2 | 124.8 ± 50.1 | 43.8 ± 11.2 | 0.2 ± 0.03 |
| AFB1 3 h (n = 3) | 5.1 ± 0.3 | 3.5 ± 0.1 | 26.3 ± 6.6 | 98 ± 19.7 | 35 ± 3.6 | 0.3 ± 0.06 |
| AFB1 24 h (n = 5) | 4.9 ± 0.2 | 3.4 ± 0.2 | 23.6 ± 2.5 | 96.4 ± 20.1 | 41.8 ± 12 | 0.3 ± 0.03 |
| STER 3 h (n = 4) | 5.3 ± 0.6 | 3.5 ± 0.3 | 29.7 ± 11.1 | 131.7 ± 38.4 | 43.7 ± 5.7 | 0.2 ± 0.06 |
| STER 24 h (n = 5) | 5 ± 0.4 | 3.3 ± 0.4 | 20.8 ± 2.3 | 123 ± 68.4 | 38.4 ± 8.4 | 0.2 ± 0.04 |
| AFB1+STER 3 h (n = 4) | 5.6 ± 0.4 | 3.7 ± 0.2 | 32.7 ± 7.6 | 121.2 ± 18.6 | 42.5 ± 7.8 | 0.3 ± 0.04 |
| AFB1+STER 24 h (n = 3) | 4.8 ± 0.5 | 3.3 ± 0.1 | 23.3 ± 3.2 | 107 ± 25.9 | 48.7 ± 4.7 | 0.2 ± 0.06 |
| Group | Standard (% DNA in Tail) | Fpg-Modified (Fpg-Sensitive Sites) | ||
|---|---|---|---|---|
| Liver | Kidney | Liver | Kidney | |
| NC | 0.4 ± 0.9 | 0.4 ± 0.3 | 0.5 ± 0.8 | 0.5 ± 0.8 |
| AFB1 | 1.8 ± 1.06 * | 1.5 ± 0.9 | 0.18 ± 0.4 | 4.4 ± 7.6 |
| STER | 0.8 ± 1.2 | 1.9 ± 1.3 | 1.5 ± 2 | 1.2 ± 1.4 |
| AFB1+STER | 1.9 ± 1.7 | 0.3 ± 0.4 | 2.2 ± 1.6 * | 2.03 ± 1.7 |
| PC | 17.3 ± 6.3 ** | 36.4 ± 14.3 * | - | - |
| AC | - | - | 59.4 ± 9.4 ** | 59.4 ± 9.4 ** |
| Group ID | PCE 1 | NCE | PCE (%) | PCE 2 | MN | MN (%) |
|---|---|---|---|---|---|---|
| NC | 255.2 ± 39.6 | 266.6 ± 42.4 | 49 ± 7.8 | 4003.4 ± 2.9 | 14 ± 5.3 | 0.3 ± 0.2 |
| AFB1 | 242.8 ± 100.4 | 274.2 ± 108.1 | 47.2 ± 20.1 | 4002.4 ± 2.6 | 20 ± 10.9 | 0.5 ± 0.3 |
| STER | 247.2 ± 34.1 | 277.2 ± 37.5 | 47.2 ± 6.7 | 4003 ± 2 | 10 ± 2.9 | 0.2 ± 0.1 |
| AFB1+STER | 221.4 ± 83.7 | 282 ± 89.8 | 44.7 ± 17.2 | 4001.8 ± 1.3 | 11 ± 2.3 | 0.3 ± 0.04 |
| PC | 194.2 ± 86.1 | 332.2 ± 101 | 37.3 ± 17.6 | 4004.8 ± 1.3 | 36 ± 20.3 * | 0.9 ± 0.5 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alonso-Jauregui, M.; López de Cerain, A.; Azqueta, A.; Rodriguez-Garraus, A.; Gil, A.G.; González-Peñas, E.; Vettorazzi, A. In Vivo Genotoxicity and Toxicity Assessment of Sterigmatocystin Individually and in Mixture with Aflatoxin B1. Toxins 2023, 15, 491. https://doi.org/10.3390/toxins15080491
Alonso-Jauregui M, López de Cerain A, Azqueta A, Rodriguez-Garraus A, Gil AG, González-Peñas E, Vettorazzi A. In Vivo Genotoxicity and Toxicity Assessment of Sterigmatocystin Individually and in Mixture with Aflatoxin B1. Toxins. 2023; 15(8):491. https://doi.org/10.3390/toxins15080491
Chicago/Turabian StyleAlonso-Jauregui, Maria, Adela López de Cerain, Amaya Azqueta, Adriana Rodriguez-Garraus, Ana Gloria Gil, Elena González-Peñas, and Ariane Vettorazzi. 2023. "In Vivo Genotoxicity and Toxicity Assessment of Sterigmatocystin Individually and in Mixture with Aflatoxin B1" Toxins 15, no. 8: 491. https://doi.org/10.3390/toxins15080491
APA StyleAlonso-Jauregui, M., López de Cerain, A., Azqueta, A., Rodriguez-Garraus, A., Gil, A. G., González-Peñas, E., & Vettorazzi, A. (2023). In Vivo Genotoxicity and Toxicity Assessment of Sterigmatocystin Individually and in Mixture with Aflatoxin B1. Toxins, 15(8), 491. https://doi.org/10.3390/toxins15080491