Mycolactone A vs. B: Multiscale Simulations Reveal the Roles of Localization and Association in Isomer-Specific Toxicity


Abstract
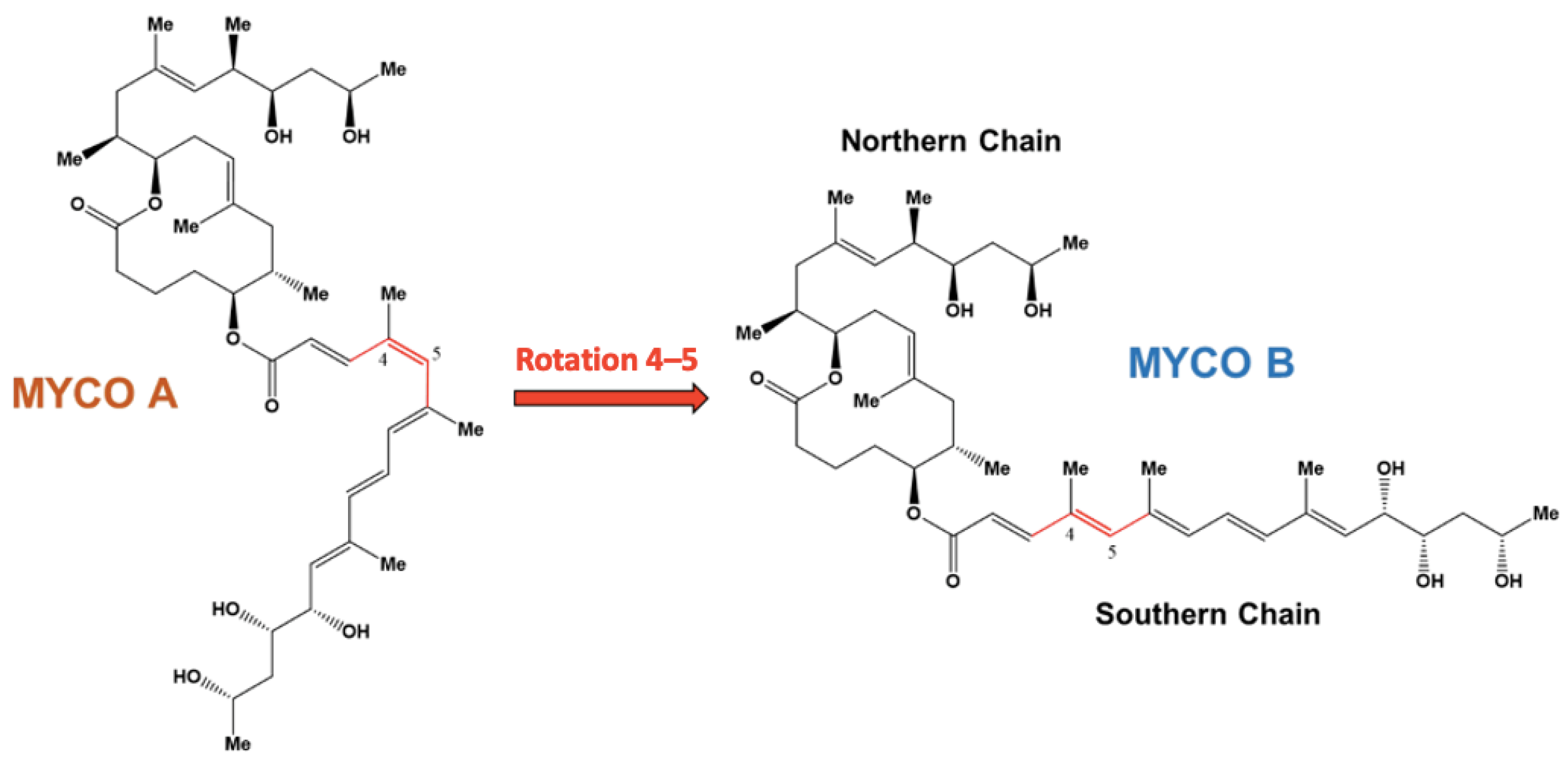
1. Introduction
2. Results
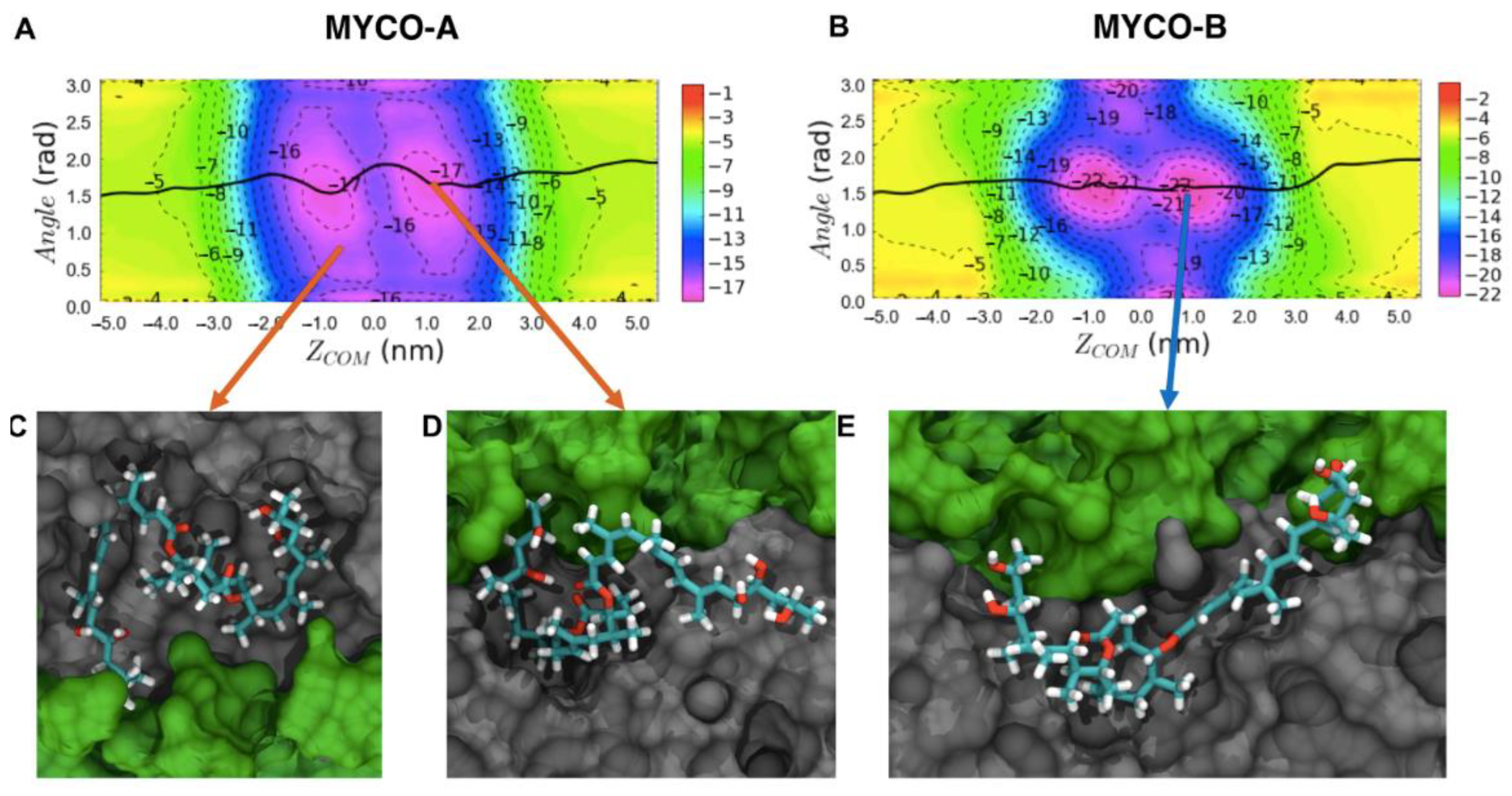
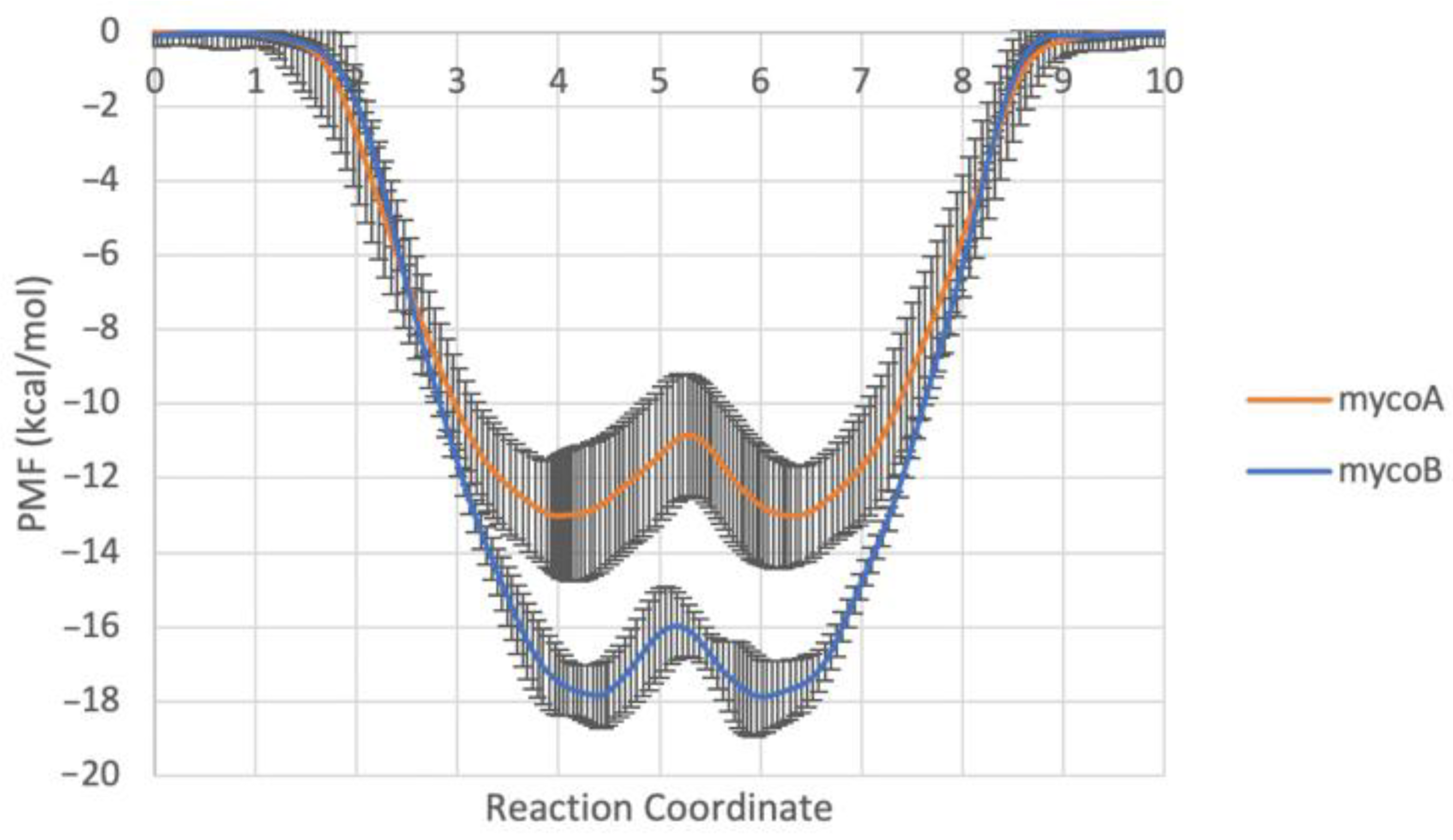
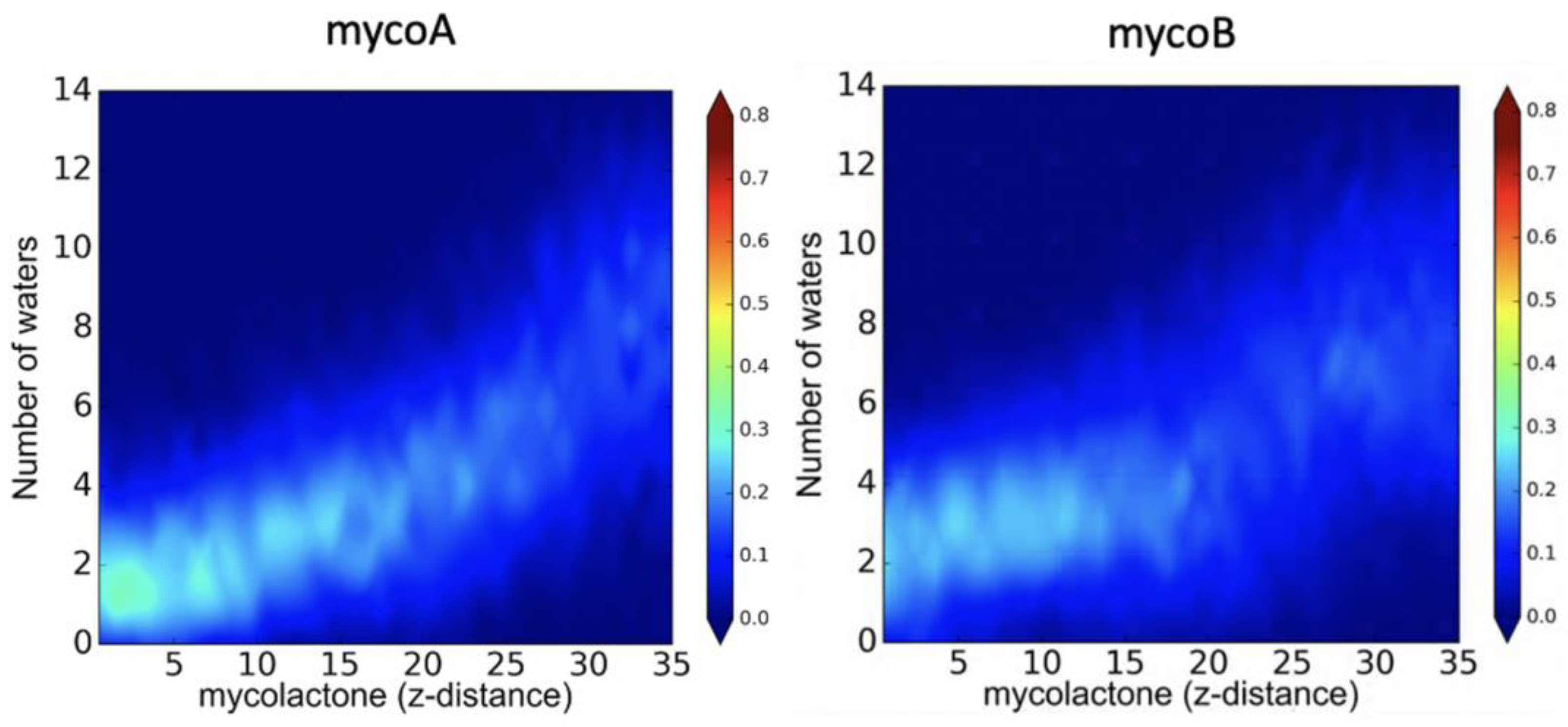
2.1. Endoplasmic Reticulum Membrane Association
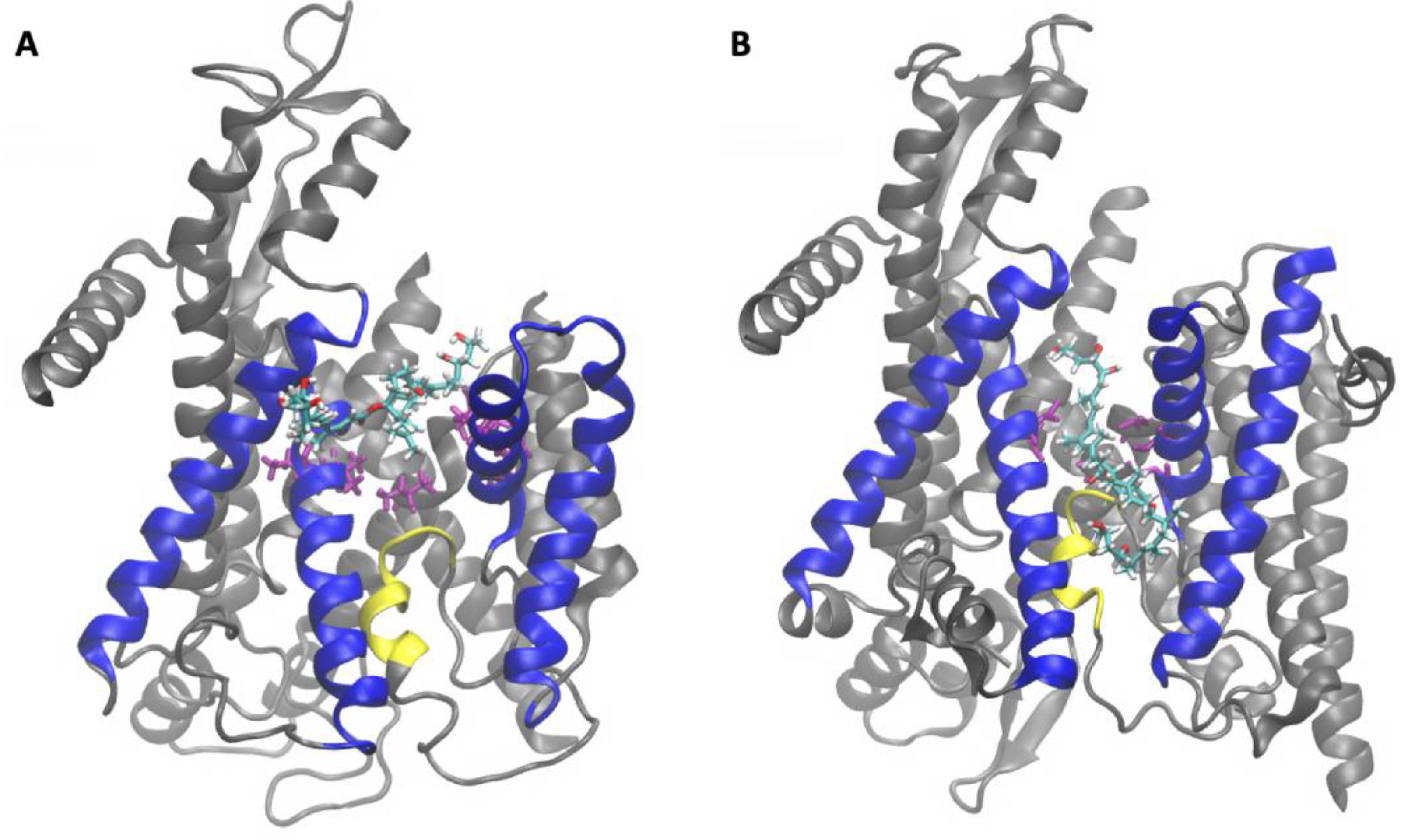
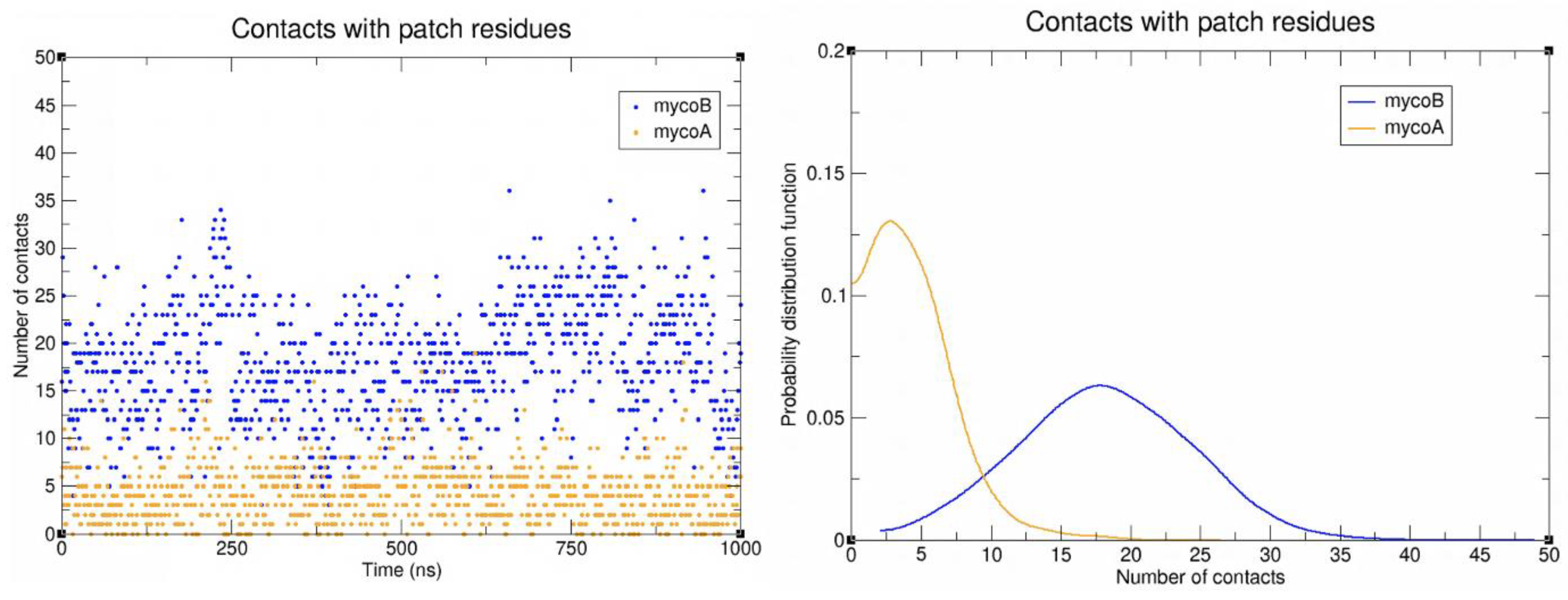
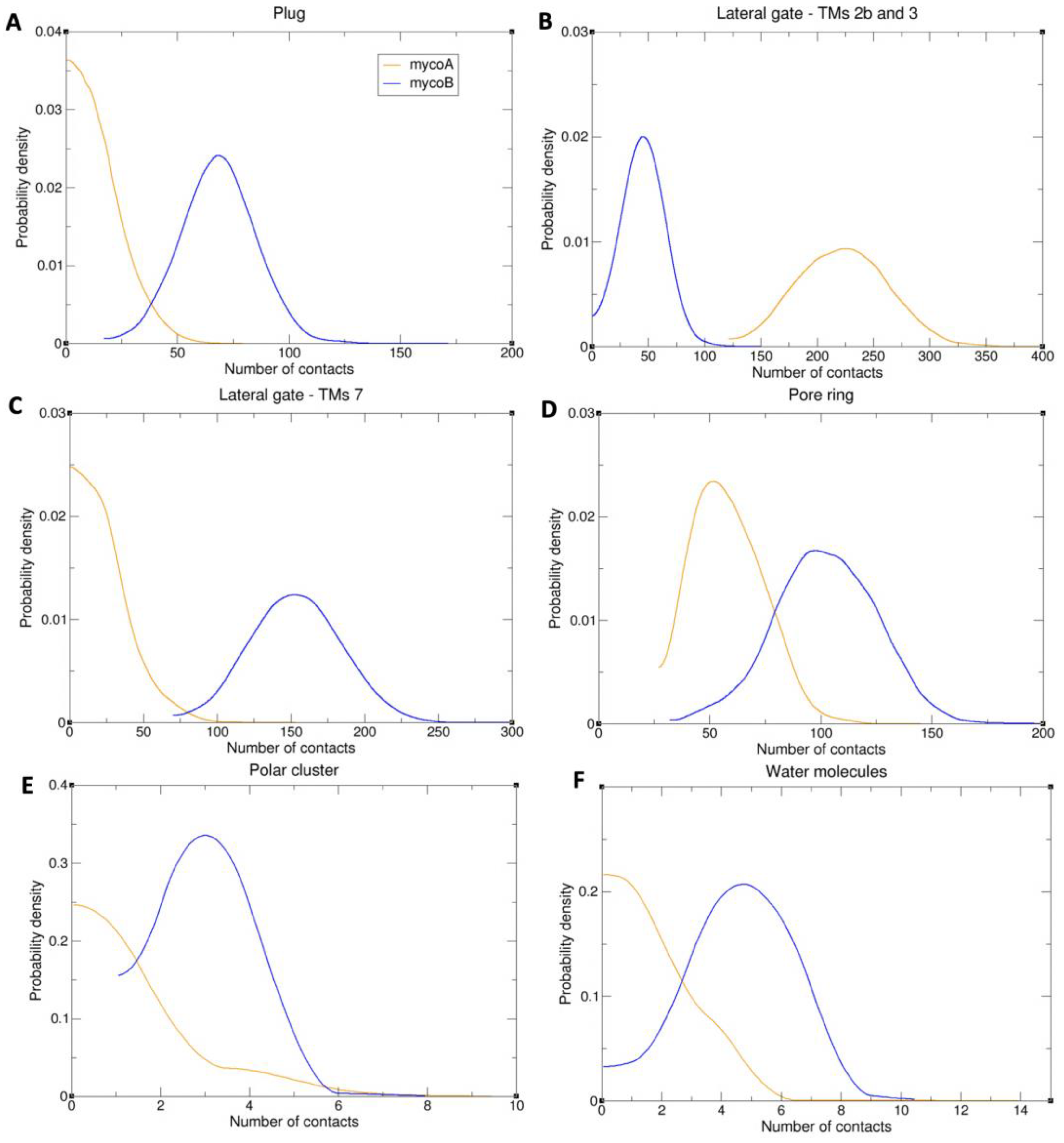
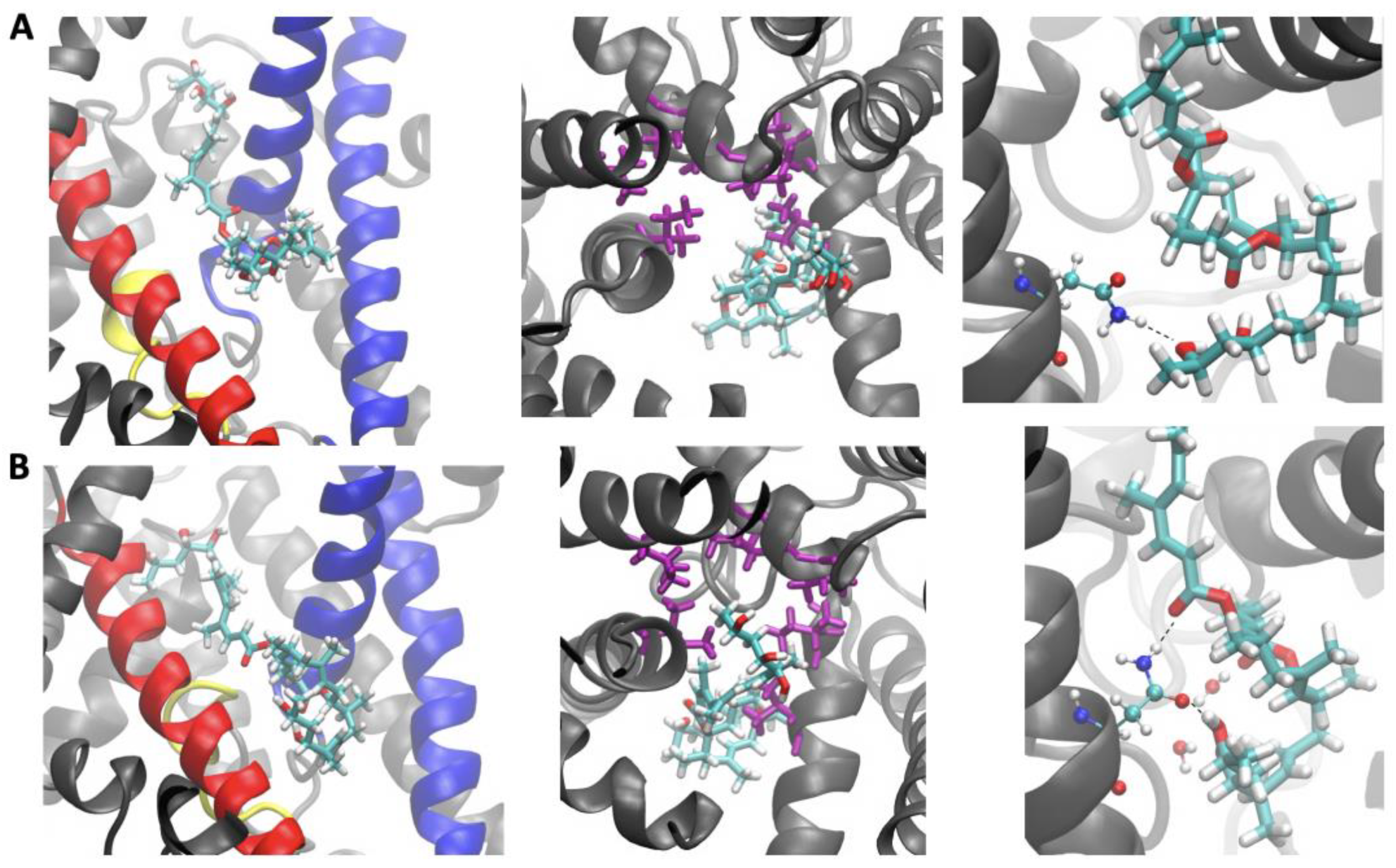
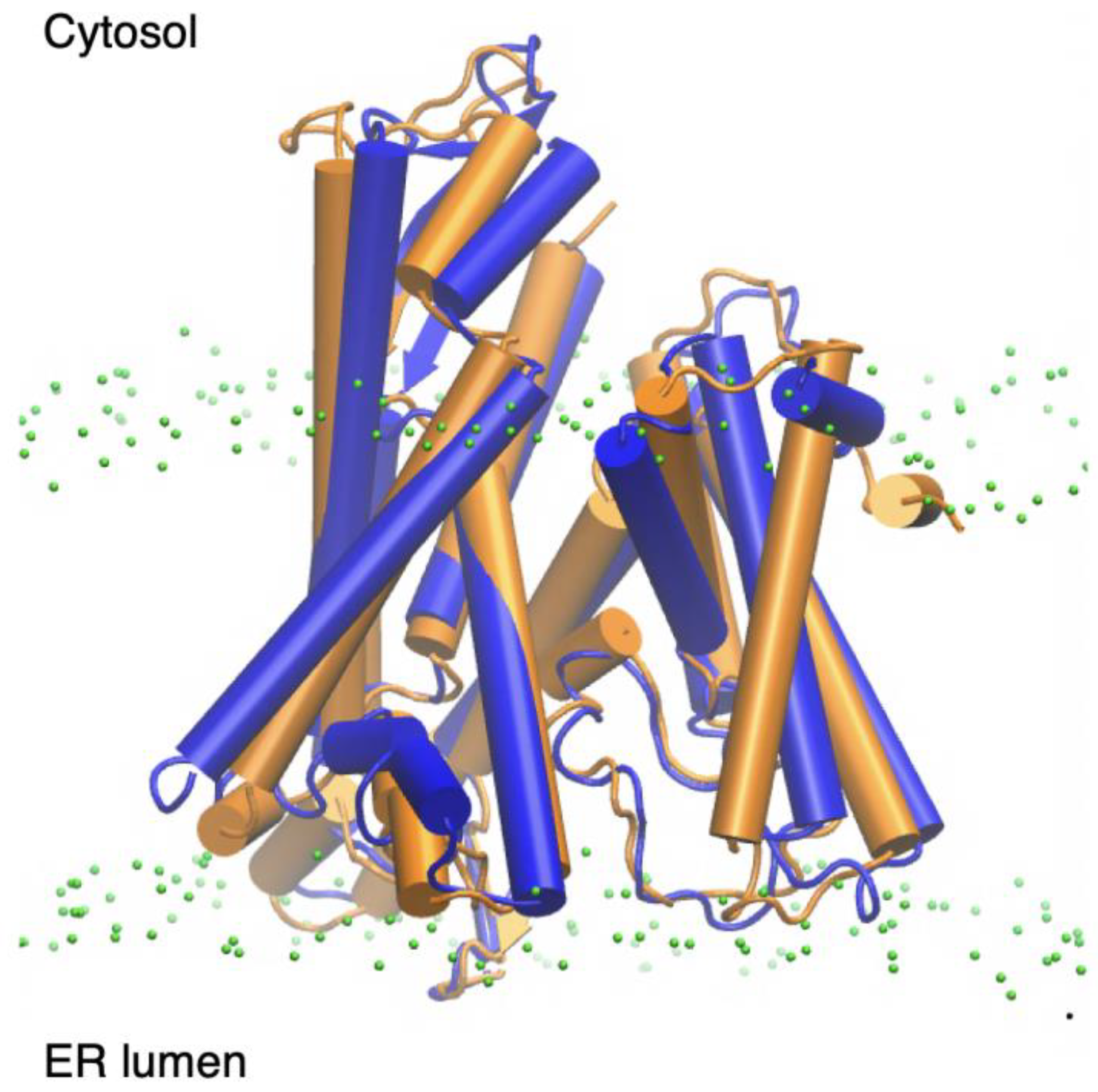
2.2. Sec61-Toxin Complex 1
2.3. Sec61-Toxin Complex 2
3. Discussion
4. Conclusions
5. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hall, B.S.; Hill, K.; McKenna, M.; Ogbechi, J.; High, S.; Willis, A.E.; Simmonds, R.E. The pathogenic mechanism of the Mycobacterium ulcerans virulence factor, mycolactone, depends on blockade of protein translocation into the ER. PLoS Pathog. 2014, 10, e1004061. [Google Scholar] [CrossRef] [PubMed]
- Sarfo, F.S.; Phillips, R.; Wansbrough-Jones, M.; Simmonds, R.E. Recent advances: Role of mycolactone in the pathogenesis and monitoring of Mycobacterium ulcerans infection/Buruli ulcer disease. Cell Microbiol. 2016, 18, 17–29. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Buruli Ulcer (Mycobacterium ulcerans Infection); WHO: Geneva, Switzerland, 2023. [Google Scholar]
- Marsollier, L.; Brodin, P.; Jackson, M.; Kordulakova, J.; Tafelmeyer, P.; Carbonnelle, E.; Aubry, J.; Milon, G.; Legras, P.; Andre, J.P.; et al. Impact of Mycobacterium ulcerans biofilm on transmissibility to ecological niches and Buruli ulcer pathogenesis. PLoS Pathog. 2007, 3, e62. [Google Scholar] [CrossRef]
- Hall, B.; Simmonds, R. Pleiotropic molecular effects of the Mycobacterium ulcerans virulence factor mycolactone underlying the cell death and immunosuppression seen in Buruli ulcer. Biochem. Soc. Trans. 2014, 42, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Baron, L.; Paatero, A.O.; Morel, J.D.; Impens, F.; Guenin-Mace, L.; Saint-Auret, S.; Blanchard, N.; Dillmann, R.; Niang, F.; Pellegrini, S.; et al. Mycolactone subverts immunity by selectively blocking the Sec61 translocon. J. Exp. Med. 2016, 213, 2885–2896. [Google Scholar] [CrossRef]
- Ogbechi, J.; Hall, B.S.; Sbarrato, T.; Taunton, J.; Willis, A.E.; Wek, R.C.; Simmonds, R.E. Inhibition of Sec61-dependent translocation by mycolactone uncouples the integrated stress response from ER stress, driving cytotoxicity via translational activation of ATF4. Cell Death Dis. 2018, 9, 397. [Google Scholar] [CrossRef]
- Guenin-Mace, L.; Baron, L.; Chany, A.C.; Tresse, C.; Saint-Auret, S.; Jonsson, F.; Le Chevalier, F.; Bruhns, P.; Bismuth, G.; Hidalgo-Lucas, S.; et al. Shaping mycolactone for therapeutic use against inflammatory disorders. Sci. Transl. Med. 2015, 7, 289ra285. [Google Scholar] [CrossRef]
- Gunawardana, G.; Chatterjee, D.; George, K.M.; Brennan, P.; Whittern, D.; Small, P.L. Characterization of Novel Macrolide Toxins, Mycolactones A and B, from a Human Pathogen, Mycobacterium ulcerans. J. Am. Chem. Soc. 1999, 121, 6092–6093. [Google Scholar] [CrossRef]
- Gehringer, M.; Mader, P.; Gersbach, P.; Pfeiffer, B.; Scherr, N.; Dangy, J.P.; Pluschke, G.; Altmann, K.H. Configurationally Stabilized Analogs of M. ulcerans Exotoxins Mycolactones A and B Reveal the Importance of Side Chain Geometry for Mycolactone Virulence. Org. Lett. 2019, 21, 5853–5857. [Google Scholar] [CrossRef]
- Snyder, D.S.; Small, P.L. Uptake and cellular actions of mycolactone, a virulence determinant for Mycobacterium ulcerans. Microb. Pathog. 2003, 34, 91–101. [Google Scholar] [CrossRef]
- Lopez, C.A.; Unkefer, C.J.; Swanson, B.I.; Swanson, J.M.J.; Gnanakaran, S. Membrane perturbing properties of toxin mycolactone from Mycobacterium ulcerans. PLoS Comput. Biol. 2018, 14, e1005972. [Google Scholar] [CrossRef] [PubMed]
- Nitenberg, M.; Benarouche, A.; Maniti, O.; Marion, E.; Marsollier, L.; Gean, J.; Dufourc, E.J.; Cavalier, J.F.; Canaan, S.; Girard-Egrot, A.P. The potent effect of mycolactone on lipid membranes. PLoS Pathog. 2018, 14, e1006814. [Google Scholar] [CrossRef] [PubMed]
- Aydin, F.; Sun, R.; Swanson, J.M.J. Mycolactone Toxin Membrane Permeation: Atomistic versus Coarse-Grained MARTINI Simulations. Biophys. J. 2019, 117, 87–98. [Google Scholar] [CrossRef] [PubMed]
- da Hora, G.C.A.; Nguyen, J.D.M.; Swanson, J.M.J. Can membrane composition traffic toxins? Mycolactone and preferential membrane interactions. Biophys. J. 2022, 121, 4260–4270. [Google Scholar] [CrossRef]
- Castello-Serrano, I.; Heberle, F.A.; Diaz-Rohrer, B.; Ippolito, R.; Shurer, C.R.; Lujan, P.; Campelo, F.; Levental, K.R.; Levental, I. Partitioning to ordered membrane domains regulates the kinetics of secretory traffic. bioRxiv 2023. [Google Scholar] [CrossRef]
- Marion, E.; Song, O.R.; Christophe, T.; Babonneau, J.; Fenistein, D.; Eyer, J.; Letournel, F.; Henrion, D.; Clere, N.; Paille, V.; et al. Mycobacterial toxin induces analgesia in buruli ulcer by targeting the angiotensin pathways. Cell 2014, 157, 1565–1576. [Google Scholar] [CrossRef]
- Song, O.R.; Kim, H.B.; Jouny, S.; Ricard, I.; Vandeputte, A.; Deboosere, N.; Marion, E.; Queval, C.J.; Lesport, P.; Bourinet, E.; et al. A Bacterial Toxin with Analgesic Properties: Hyperpolarization of DRG Neurons by Mycolactone. Toxins 2017, 9, 227. [Google Scholar] [CrossRef]
- Thrasher, A.J.; Burns, S.O. WASP: A key immunological multitasker. Nat. Rev. Immunol. 2010, 10, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Hayward, R.D.; Leong, J.M.; Koronakis, V.; Campellone, K.G. Exploiting pathogenic Escherichia coli to model transmembrane receptor signalling. Nat. Rev. Microbiol. 2006, 4, 358–370. [Google Scholar] [CrossRef]
- Guenin-Mace, L.; Veyron-Churlet, R.; Thoulouze, M.I.; Romet-Lemonne, G.; Hong, H.; Leadlay, P.F.; Danckaert, A.; Ruf, M.T.; Mostowy, S.; Zurzolo, C.; et al. Mycolactone activation of Wiskott-Aldrich syndrome proteins underpins Buruli ulcer formation. J. Clin. Investig. 2013, 123, 1501–1512. [Google Scholar] [CrossRef]
- Chany, A.C.; Veyron-Churlet, R.; Tresse, C.; Mayau, V.; Casarotto, V.; Le Chevalier, F.; Guenin-Mace, L.; Demangel, C.; Blanchard, N. Synthetic variants of mycolactone bind and activate Wiskott-Aldrich syndrome proteins. J. Med. Chem. 2014, 57, 7382–7395. [Google Scholar] [CrossRef]
- Hsieh, L.T.-H.; Hall, B.S.; Newcombe, J.; Mendum, T.A.; Umrania, Y.; Deery, M.J.; Shi, W.Q.; Salguero, F.J.; Simmonds, R.E. Mycolactone causes catastrophic Sec61-dependent loss of the endothelial glycocalyx and basement membrane: A new indirect mechanism driving tissue necrosis in Mycobacterium ulcerans infection. bioRxiv 2023. [Google Scholar] [CrossRef]
- Demangel, C.; High, S. Sec61 blockade by mycolactone: A central mechanism in Buruli ulcer disease. Biol. Cell 2018, 110, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Gérard, S.F.; Hall, B.S.; Zaki, A.M.; Corfield, K.A.; Mayerhofer, P.U.; Costa, C.; Whelligan, D.K.; Biggin, P.C.; Simmonds, R.E.; Higgins, M.K. Structure of the Inhibited State of the Sec Translocon. Mol. Cell 2020, 79, 406–415.e7. [Google Scholar] [CrossRef] [PubMed]
- Itskanov, S.; Wang, L.; Junne, T.; Sherriff, R.; Xiao, L.; Blanchard, N.; Shi, W.Q.; Forsyth, C.; Hoepfner, D.; Spiess, M.; et al. A common mechanism of Sec61 translocon inhibition by small molecules. Nat. Chem. Biol. 2023, 1–9. [Google Scholar] [CrossRef]
- Van den Berg, B.; Clemons, W.M., Jr.; Collinson, I.; Modis, Y.; Hartmann, E.; Harrison, S.C.; Rapoport, T.A. X-ray structure of a protein-conducting channel. Nature 2004, 427, 36–44. [Google Scholar] [CrossRef]
- Pauwels, E.; Shewakramani, N.R.; De Wijngaert, B.; Camps, A.; Provinciael, B.; Stroobants, J.; Kalies, K.U.; Hartmann, E.; Maes, P.; Vermeire, K.; et al. Structural insights into TRAP association with ribosome-Sec61 complex and translocon inhibition by a CADA derivative. Sci. Adv. 2023, 9, eadf0797. [Google Scholar] [CrossRef]
- Rehan, S.; Tranter, D.; Sharp, P.P.; Craven, G.B.; Lowe, E.; Anderl, J.L.; Muchamuel, T.; Abrishami, V.; Kuivanen, S.; Wenzell, N.A.; et al. Signal peptide mimicry primes Sec61 for client-selective inhibition. Nat. Chem. Biol. 2023, 1–9. [Google Scholar] [CrossRef]
- Voorhees, R.M.; Hegde, R.S. Structure of the Sec61 channel opened by a signal sequence. Science 2016, 351, 88–91. [Google Scholar] [CrossRef]
- Harant, H.; Lettner, N.; Hofer, L.; Oberhauser, B.; de Vries, J.E.; Lindley, I.J. The translocation inhibitor CAM741 interferes with vascular cell adhesion molecule 1 signal peptide insertion at the translocon. J. Biol. Chem. 2006, 281, 30492–30502. [Google Scholar] [CrossRef]
- Harant, H.; Wolff, B.; Schreiner, E.P.; Oberhauser, B.; Hofer, L.; Lettner, N.; Maier, S.; de Vries, J.E.; Lindley, I.J. Inhibition of vascular endothelial growth factor cotranslational translocation by the cyclopeptolide CAM741. Mol. Pharmacol. 2007, 71, 1657–1665. [Google Scholar] [CrossRef] [PubMed]
- Klein, W.; Westendorf, C.; Schmidt, A.; Conill-Cortes, M.; Rutz, C.; Blohs, M.; Beyermann, M.; Protze, J.; Krause, G.; Krause, E.; et al. Defining a conformational consensus motif in cotransin-sensitive signal sequences: A proteomic and site-directed mutagenesis study. PLoS ONE 2015, 10, e0120886. [Google Scholar] [CrossRef]
- Gehringer, M.; Altmann, K.H. The chemistry and biology of mycolactones. Beilstein J. Org. Chem. 2017, 13, 1596–1660. [Google Scholar] [CrossRef]
- Wang, G.; Yin, N.; Negishi, E. Highly stereoselective total synthesis of fully hydroxy-protected mycolactones A and B and their stereoisomerization upon deprotection. Chemistry 2011, 17, 4118–4130. [Google Scholar] [CrossRef] [PubMed]
- Kubicek-Sutherland, J.Z.; Vu, D.M.; Anderson, A.S.; Sanchez, T.C.; Converse, P.J.; Marti-Arbona, R.; Nuermberger, E.L.; Swanson, B.I.; Mukundan, H. Understanding the Significance of Biochemistry in the Storage, Handling, Purification, and Sampling of Amphiphilic Mycolactone. Toxins 2019, 11, 202. [Google Scholar] [CrossRef]
- Gérard, S.F.; Simmonds, R.E.; Higgins, M.K. Evaluation/Discussion of This Paper. Available online: https://www.biorxiv.org/content/10.1101/2022.08.11.503542v1#comment-5960826580 (accessed on 7 June 2023).
- Nyathi, Y.; Wilkinson, B.M.; Pool, M.R. Co-translational targeting and translocation of proteins to the endoplasmic reticulum. Biochim. Biophys. Acta 2013, 1833, 2392–2402. [Google Scholar] [CrossRef]
- Hegde, R.S.; Keenan, R.J. Tail-anchored membrane protein insertion into the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 2011, 12, 787–798. [Google Scholar] [CrossRef]
- Johnson, N.; Powis, K.; High, S. Post-translational translocation into the endoplasmic reticulum. Biochim. Biophys. Acta 2013, 1833, 2403–2409. [Google Scholar] [CrossRef]
- McKenna, M.; Simmonds, R.E.; High, S. Mechanistic insights into the inhibition of Sec61-dependent co- and post-translational translocation by mycolactone. J. Cell Sci. 2016, 129, 1404–1415. [Google Scholar] [CrossRef]
- Fidanze, S.; Song, F.; Szlosek-Pinaud, M.; Small, P.L.; Kishi, Y. Complete structure of the mycolactones. J. Am. Chem. Soc. 2001, 123, 10117–10118. [Google Scholar] [CrossRef]
- George, K.M.; Chatterjee, D.; Gunawardana, G.; Welty, D.; Hayman, J.; Lee, R.; Small, P.L. Mycolactone: A polyketide toxin from Mycobacterium ulcerans required for virulence. Science 1999, 283, 854–857. [Google Scholar] [CrossRef]
- Judd, T.C.; Bischoff, A.; Kishi, Y.; Adusumilli, S.; Small, P.L. Structure determination of mycolactone C via total synthesis. Org. Lett. 2004, 6, 4901–4904. [Google Scholar] [CrossRef]
- Kim, H.J.; Kishi, Y. Total synthesis and stereochemistry of mycolactone F. J. Am. Chem. Soc. 2008, 130, 1842–1844. [Google Scholar] [CrossRef] [PubMed]
- Mve-Obiang, A.; Lee, R.E.; Portaels, F.; Small, P.L. Heterogeneity of mycolactones produced by clinical isolates of Mycobacterium ulcerans: Implications for virulence. Infect. Immun. 2003, 71, 774–783. [Google Scholar] [CrossRef]
- Mve-Obiang, A.; Lee, R.E.; Umstot, E.S.; Trott, K.A.; Grammer, T.C.; Parker, J.M.; Ranger, B.S.; Grainger, R.; Mahrous, E.A.; Small, P.L. A newly discovered mycobacterial pathogen isolated from laboratory colonies of Xenopus species with lethal infections produces a novel form of mycolactone, the Mycobacterium ulcerans macrolide toxin. Infect. Immun. 2005, 73, 3307–3312. [Google Scholar] [CrossRef] [PubMed]
- Ranger, B.S.; Mahrous, E.A.; Mosi, L.; Adusumilli, S.; Lee, R.E.; Colorni, A.; Rhodes, M.; Small, P.L. Globally distributed mycobacterial fish pathogens produce a novel plasmid-encoded toxic macrolide, mycolactone F. Infect. Immun. 2006, 74, 6037–6045. [Google Scholar] [CrossRef]
- Song, F.; Fidanze, S.; Benowitz, A.B.; Kishi, Y. Total synthesis of the mycolactones. Org. Lett. 2002, 4, 647–650. [Google Scholar] [CrossRef]
- Pauwels, E.; Schulein, R.; Vermeire, K. Inhibitors of the Sec61 Complex and Novel High Throughput Screening Strategies to Target the Protein Translocation Pathway. Int. J. Mol. Sci. 2021, 22, 12007. [Google Scholar] [CrossRef]
- Linxweiler, M.; Schick, B.; Zimmermann, R. Let’s talk about Secs: Sec61, Sec62 and Sec63 in signal transduction, oncology and personalized medicine. Signal Transduct. Target. Ther. 2017, 2, 17002. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Jiang, X.; Wang, J.; Sang, Z.; Guo, L.; Yin, G.; Wang, Y. SEC61G is upregulated and required for tumor progression in human kidney cancer. Mol. Med. Rep. 2021, 23, 427. [Google Scholar] [CrossRef]
- Ravindran, M.S.; Bagchi, P.; Cunningham, C.N.; Tsai, B. Opportunistic intruders: How viruses orchestrate ER functions to infect cells. Nat. Rev. Microbiol. 2016, 14, 407–420. [Google Scholar] [CrossRef]
- Perez-Hernandez, G.; Paul, F.; Giorgino, T.; De Fabritiis, G.; Noe, F. Identification of slow molecular order parameters for Markov model construction. J. Chem. Phys. 2013, 139, 015102. [Google Scholar] [CrossRef]
- Jo, S.; Lim, J.B.; Klauda, J.B.; Im, W. CHARMM-GUI Membrane Builder for mixed bilayers and its application to yeast membranes. Biophys. J. 2009, 97, 50–58. [Google Scholar] [CrossRef]
- Doktorova, M.; Heberle, F.A.; Eicher, B.; Standaert, R.F.; Katsaras, J.; London, E.; Pabst, G.; Marquardt, D. Preparation of asymmetric phospholipid vesicles for use as cell membrane models. Nat. Protoc. 2018, 13, 2086–2101. [Google Scholar] [CrossRef] [PubMed]
- Monje-Galvan, V.; Klauda, J.B. Modeling Yeast Organelle Membranes and How Lipid Diversity Influences Bilayer Properties. Biochemistry 2015, 54, 6852–6861. [Google Scholar] [CrossRef]
- Horvath, S.E.; Wagner, A.; Steyrer, E.; Daum, G. Metabolic link between phosphatidylethanolamine and triacylglycerol metabolism in the yeast Saccharomyces cerevisiae. Biochim. Et Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2011, 1811, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Zinser, E.; Sperka-Gottlieb, C.D.; Fasch, E.V.; Kohlwein, S.D.; Paltauf, F.; Daum, G. Phospholipid synthesis and lipid composition of subcellular membranes in the unicellular eukaryote Saccharomyces cerevisiae. J. Bacteriol. 1991, 173, 2026–2034. [Google Scholar] [CrossRef]
- Klug, L.; Daum, G. Yeast lipid metabolism at a glance. FEMS Yeast Res. 2014, 14, 369–388. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham III, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Jämbeck, J.P.M.; Lyubartsev, A.P. Derivation and Systematic Validation of a Refined All-Atom Force Field for Phosphatidylcholine Lipids. J. Phys. Chem. B 2012, 116, 3164–3179. [Google Scholar] [CrossRef]
- Jämbeck, J.P.M.; Lyubartsev, A.P. Another Piece of the Membrane Puzzle: Extending Slipids Further. J. Chem. Theory Comput. 2013, 9, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Olson, A.J. Using AutoDock for Ligand-Receptor Docking. Curr. Protoc. Bioinform. 2008, 24, 8.14.11–18.14.40. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Jämbeck, J.P.M.; Lyubartsev, A.P. An Extension and Further Validation of an All-Atomistic Force Field for Biological Membranes. J. Chem. Theory Comput. 2012, 8, 2938–2948. [Google Scholar] [CrossRef]
- Grote, F.; Lyubartsev, A.P. Optimization of Slipids Force Field Parameters Describing Headgroups of Phospholipids. J. Phys. Chem. B 2020, 124, 8784–8793. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Gowers, R.; Linke, M.; Barnoud, J.; Reddy, T.; Melo, M.; Seyler, S.; Domański, J.; Dotson, D.; Buchoux, S.; Kenney, I.; et al. MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations. In Proceedings of the 15th Python in Science Conference, Austin, TX, USA, 11–17 July 2016; pp. 98–105. [Google Scholar]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph 1996, 14, 33–38, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [PubMed]
- Laio, A.; Gervasio, F.L. Metadynamics: A method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Rep. Prog. Phys. 2008, 71, 126601. [Google Scholar] [CrossRef]
- Dama, J.F.; Rotskoff, G.; Parrinello, M.; Voth, G.A. Transition-tempered metadynamics: Robust, convergent metadynamics via on-the-fly transition barrier estimation. J. Chem. Theory Comput. 2014, 10, 3626–3633. [Google Scholar] [CrossRef]
- Sun, R.; Dama, J.F.; Tan, J.S.; Rose, J.P.; Voth, G.A. Transition-Tempered Metadynamics Is a Promising Tool for Studying the Permeation of Drug-like Molecules through Membranes. J. Chem. Theory Comput. 2016, 12, 5157–5169. [Google Scholar] [CrossRef]
- Aydin, F.; Durumeric, A.E.P.; da Hora, G.C.A.; Nguyen, J.D.M.; Oh, M.I.; Swanson, J.M.J. Improving the accuracy and convergence of drug permeation simulations via machine-learned collective variables. J. Chem. Phys. 2021, 155, 045101. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Vanden-Eijnden, E. Simplified and improved string method for computing the minimum energy paths in barrier-crossing events. J. Chem. Phys. 2007, 126, 164103. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mycolactone Interaction Energies (kcal mol−1) | ||||
|---|---|---|---|---|
| Lipids | Water | Intramolecular | Total | |
| mycoA | −85.89 ± 0.32 | −28.33 ± 0.36 | −24.89 ± 0.16 | −139.11 |
| mycoB | −103.44 ± 0.43 | −31.54 ± 0.36 | −9.99 ± 0.19 | −144.97 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, J.D.M.; da Hora, G.C.A.; Swanson, J.M.J. Mycolactone A vs. B: Multiscale Simulations Reveal the Roles of Localization and Association in Isomer-Specific Toxicity. Toxins 2023, 15, 486. https://doi.org/10.3390/toxins15080486
Nguyen JDM, da Hora GCA, Swanson JMJ. Mycolactone A vs. B: Multiscale Simulations Reveal the Roles of Localization and Association in Isomer-Specific Toxicity. Toxins. 2023; 15(8):486. https://doi.org/10.3390/toxins15080486
Chicago/Turabian StyleNguyen, John D. M., Gabriel C. A. da Hora, and Jessica M. J. Swanson. 2023. "Mycolactone A vs. B: Multiscale Simulations Reveal the Roles of Localization and Association in Isomer-Specific Toxicity" Toxins 15, no. 8: 486. https://doi.org/10.3390/toxins15080486
APA StyleNguyen, J. D. M., da Hora, G. C. A., & Swanson, J. M. J. (2023). Mycolactone A vs. B: Multiscale Simulations Reveal the Roles of Localization and Association in Isomer-Specific Toxicity. Toxins, 15(8), 486. https://doi.org/10.3390/toxins15080486