Next-Generation Sequencing for Venomics: Application of Multi-Enzymatic Limited Digestion for Inventorying the Snake Venom Arsenal

 ,
,  , , , , and
, , , , and
Abstract
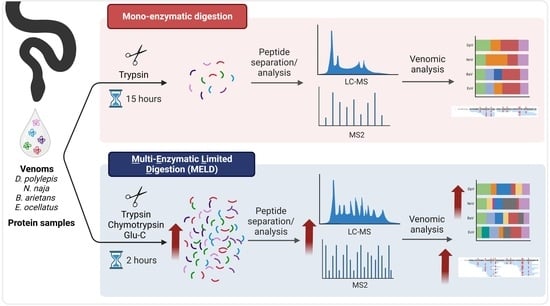
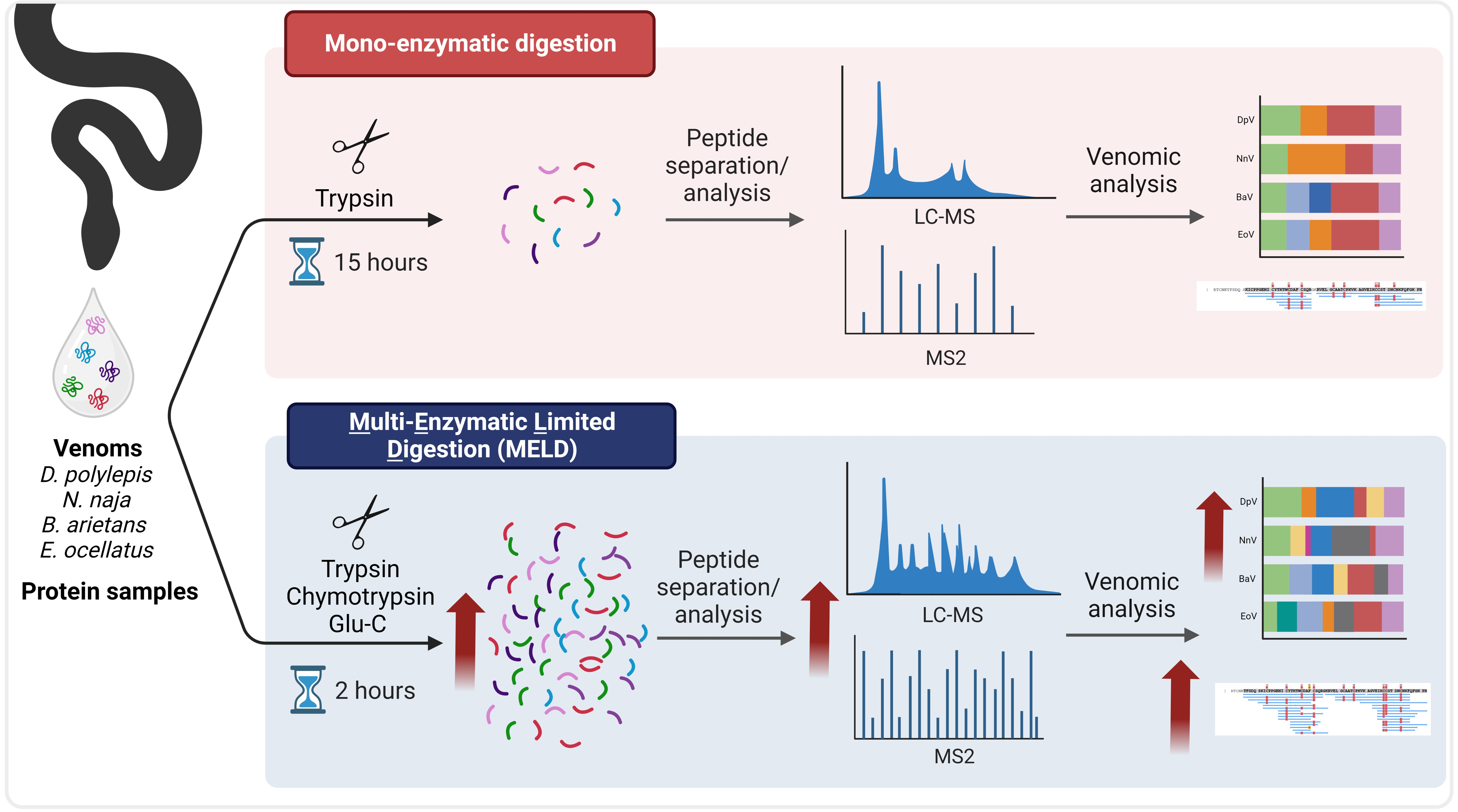
1. Introduction
2. Results and Discussion
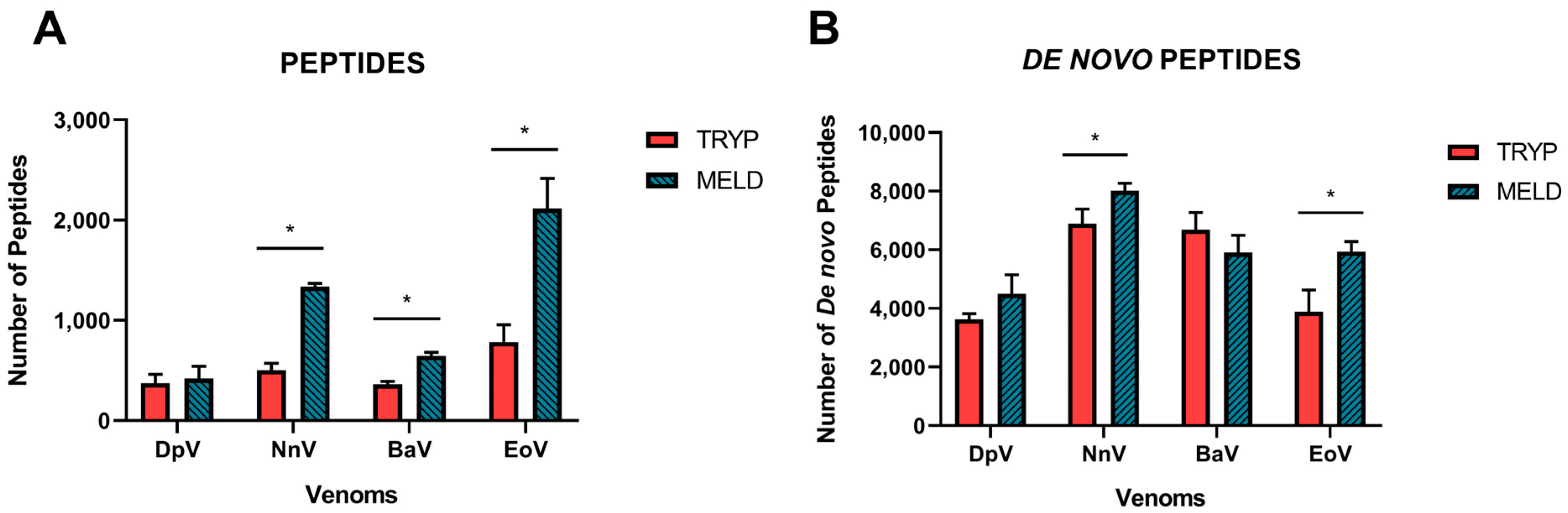
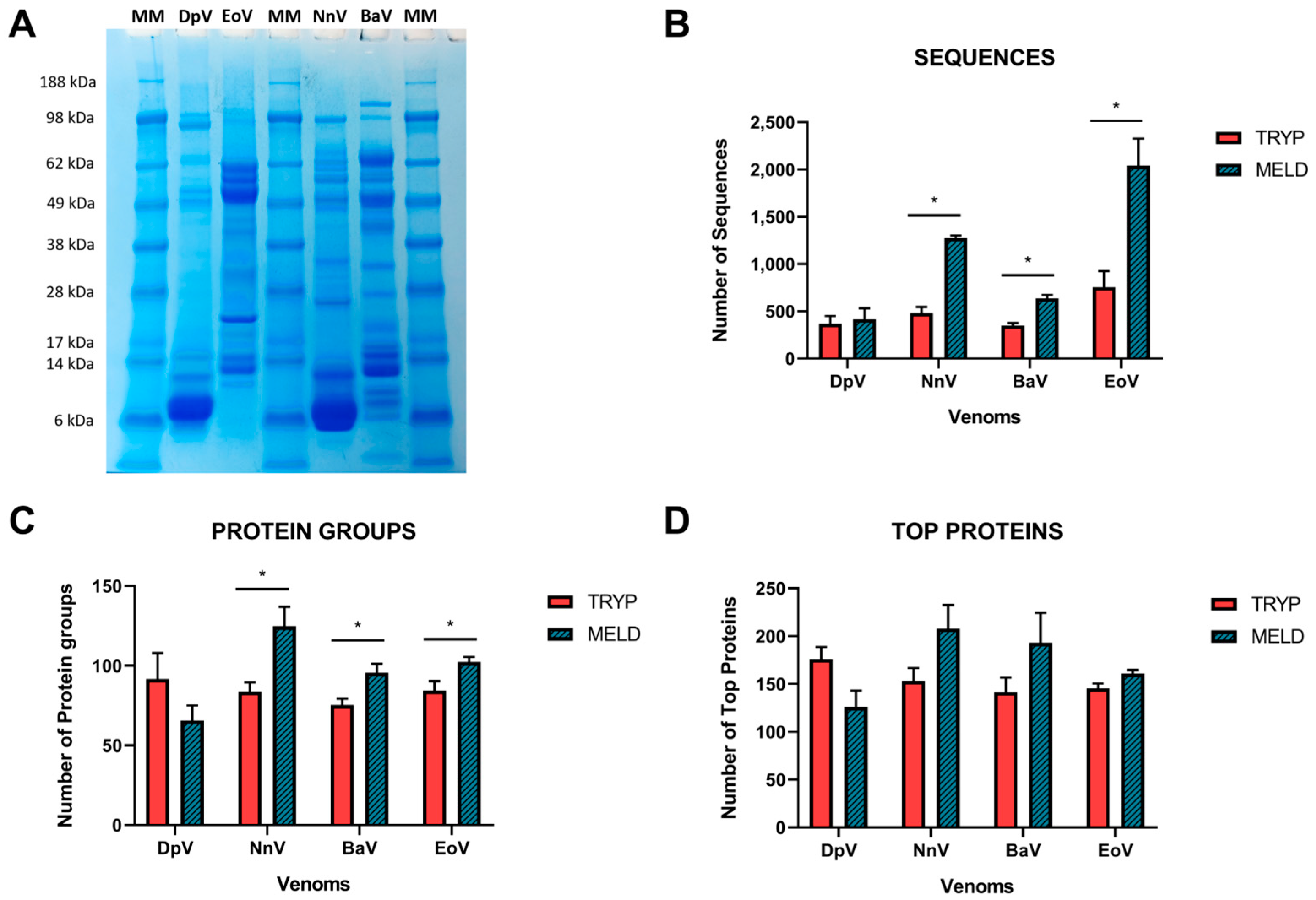
2.1. Data Acquisition
2.2. Database Search
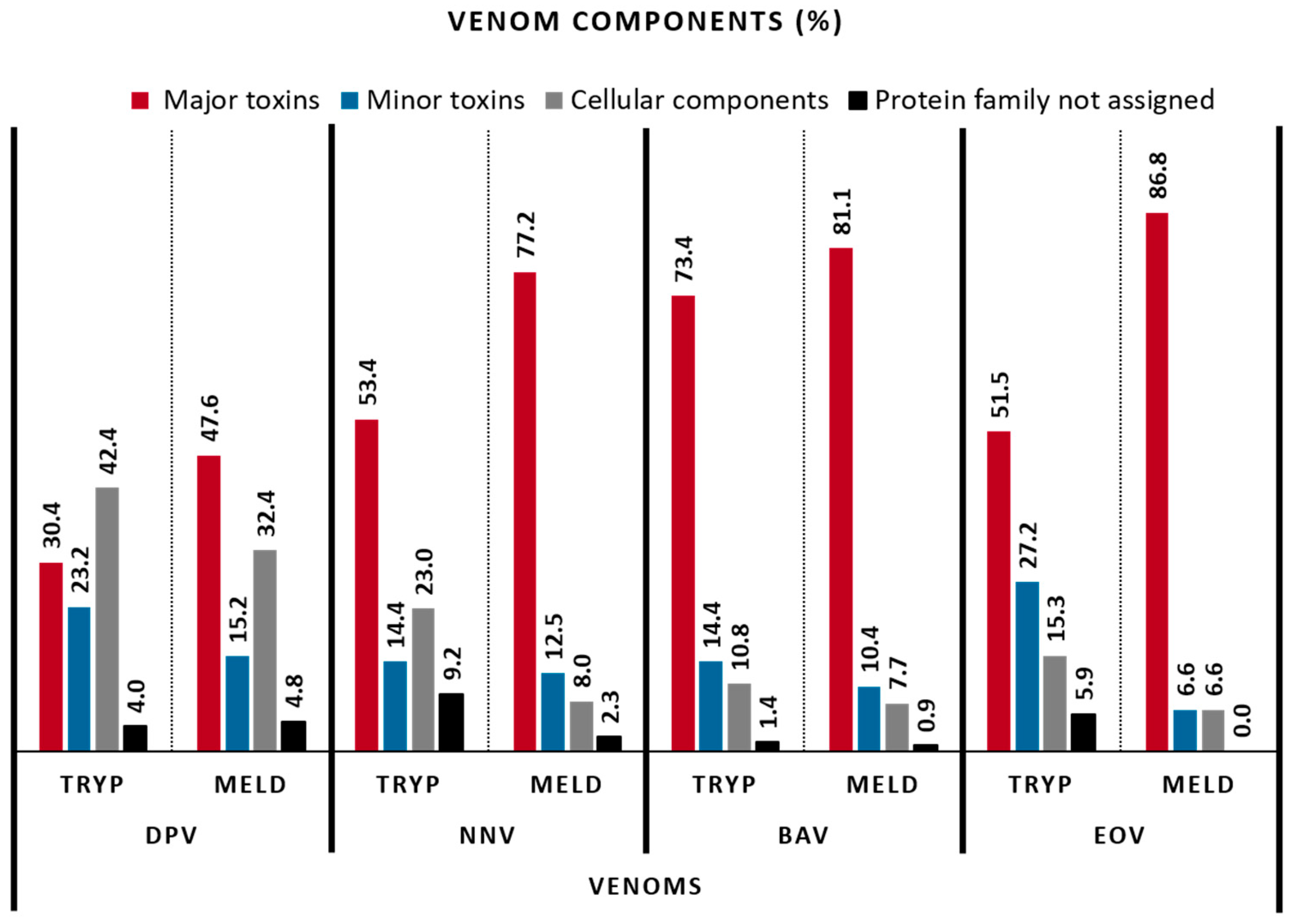
2.3. Venom Composition
2.4. Sequence Coverage of Major Toxins
3. Conclusions
4. Materials and Methods
4.1. Venoms Selection
4.2. Polyacrylamide Gel Electrophoresis (SDS-PAGE)
4.3. Digestion Protocols
4.3.1. Mono-Enzymatic Protocol
4.3.2. Synergic Multi-Enzymatic and Limited Digestion (MELD)
4.4. MALDI-TOF/MS
4.5. Liquid Chromatography—Mass Spectrometry Analysis
4.6. Data Analysis
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- D’adamo, G.L.; Widdop, J.T.; Giles, E.M. The future is now? Clinical and translational aspects of “Omics” technologies. Immunol. Cell Biol. 2020, 99, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.G.; Sokolowska, I.; Wetie, A.G.N.; Wormwood, K.; Aslebagh, R.; Patel, S.; Darie, C.C. Mass Spectrometry for Proteomics-Based Investigation. Adv. Exp. Med. Biol. 2014, 806, 1–32. [Google Scholar] [CrossRef]
- Dupree, E.J.; Jayathirtha, M.; Yorkey, H.; Mihasan, M.; Petre, B.A.; Darie, C.C. A Critical Review of Bottom-Up Proteomics: The Good, the Bad, and the Future of This Field. Proteomes 2020, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Morsa, D.; Baiwir, D.; La Rocca, R.; Zimmerman, T.A.; Hanozin, E.; Grifnée, E.; Longuespée, R.; Meuwis, M.-A.; Smargiasso, N.; De Pauw, E.; et al. Multi-Enzymatic Limited Digestion: The Next-Generation Sequencing for Proteomics? J. Proteome Res. 2019, 18, 2501–2513. [Google Scholar] [CrossRef] [PubMed]
- Karpievitch, Y.V.; Polpitiya, A.D.; Anderson, G.A.; Smith, R.D.; Dabney, A.R. Liquid chromatography mass spectrometry-based proteomics: Biological and technological aspects. Ann. Appl. Stat. 2010, 4, 1797–1823. [Google Scholar] [CrossRef]
- Zhang, Y.; Fonslow, B.R.; Shan, B.; Baek, M.-C.; Yates, J.R., 3rd. Protein Analysis by Shotgun/Bottom-up Proteomics. Chem. Rev. 2013, 113, 2343–2394. [Google Scholar] [CrossRef]
- Yates, J.R.; Ruse, C.I.; Nakorchevsky, A. Proteomics by Mass Spectrometry: Approaches, Advances, and Applications. Annu. Rev. Biomed. Eng. 2009, 11, 49–79. [Google Scholar] [CrossRef]
- Cristobal, A.; Marino, F.; Post, H.; Toorn, H.W.P.v.D.; Mohammed, S.; Heck, A.J.R. Toward an Optimized Workflow for Middle-Down Proteomics. Anal. Chem. 2017, 89, 3318–3325. [Google Scholar] [CrossRef]
- Wilson, D.; Daly, N.L. Venomics: A Mini-Review. Biotech 2018, 7, 19. [Google Scholar] [CrossRef]
- Casewell, N.R.; Jackson, T.N.W.; Laustsen, A.H.; Sunagar, K. Causes and Consequences of Snake Venom Variation. Trends Pharmacol. Sci. 2020, 41, 570–581. [Google Scholar] [CrossRef]
- WHO. Snakebite Envenoming. World Health Organization (WHO). Available online: https://www.who.int/health-topics/snakebite#tab=tab_1 (accessed on 4 April 2023).
- Calvete, J.J.; Sanz, L.; Angulo, Y.; Lomonte, B.; Gutiérrez, J.M. Venoms, venomics, antivenomics. FEBS Lett. 2009, 583, 1736–1743.13. [Google Scholar] [CrossRef] [PubMed]
- Morjen, M.; Abdelkafi-Koubaa, Z.; Luis, J.; Othman, H.; Srairi-Abid, N.; El Ayeb, M.; Marrakchi, N. Snake venom Kunitz/BPTI family: Structure, classification and pharmacological potential. Arch. L’institut Pasteur Tunis 2014, 91, 3–13. [Google Scholar]
- Slagboom, J.; Kaal, C.; Arrahman, A.; Vonk, F.J.; Somsen, G.W.; Calvete, J.J.; Wüster, W.; Kool, J. Analytical strategies in venomics. Microchem. J. 2022, 175, 107187. [Google Scholar] [CrossRef]
- Zhang, J.; Xin, L.; Shan, B.; Chen, W.; Xie, M.; Yuen, D.; Zhang, W.; Zhang, Z.; Lajoie, G.A.; Ma, B. PEAKS DB: De novo Sequencing Assisted Database Search for Sensitive and Accurate Peptide Identification. Mol. Cell. Proteom. 2012, 11, M111.010587. [Google Scholar] [CrossRef]
- Dau, T.; Bartolomucci, G.; Rappsilber, J. Proteomics Using Protease Alternatives to Trypsin Benefits from Sequential Digestion with Trypsin. Anal. Chem. 2020, 92, 9523–9527. [Google Scholar] [CrossRef]
- Ma, B.; Zhang, K.; Hendrie, C.; Liang, C.; Li, M.; Doherty-Kirby, A.; Lajoie, G. PEAKS: Powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun. Mass Spectrom 2003, 17, 2337–2342. [Google Scholar] [CrossRef]
- Damm, M.; Hempel, B.-F.; Süssmuth, R.D. Old World vipers—A review about snake venom proteomics of viperinae and their variations. Toxins 2021, 13, 427. [Google Scholar] [CrossRef]
- Boldrini-França, J.; Cologna, C.T.; Pucca, M.B.; Bordon, K.D.C.F.; Amorim, F.G.; Anjolette, F.A.P.; Cordeiro, F.A.; Wiezel, G.A.; Cerni, F.A.; Pinheiro-Junior, E.L.; et al. Minor snake venom proteins: Structure, function and potential applications. Biochim. Et Biophys. Acta (BBA)-Gen. Subj. 2017, 1861, 824–838. [Google Scholar] [CrossRef]
- Tasoulis, T.; Pukala, T.L.; Isbister, G.K. Investigating Toxin Diversity and Abundance in Snake Venom Proteomes. Front. Pharmacol. 2022, 12, 3869. [Google Scholar] [CrossRef]
- Petras, D.; Hempel, B.-F.; Göçmen, B.; Karis, M.; Whiteley, G.; Wagstaff, S.; Heiss, P.; Casewell, N.; Nalbantsoy, A.; Süssmuth, R.D. Intact protein mass spectrometry reveals intraspecies variations in venom composition of a local population of Vipera kaznakovi in Northeastern Turkey. J. Proteom. 2019, 199, 31–50. [Google Scholar] [CrossRef]
- Ghezellou, P.; Albuquerque, W.; Garikapati, V.; Casewell, N.R.; Kazemi, S.M.; Ghassempour, A.; Spengler, B. Integrating Top-Down and Bottom-Up Mass Spectrometric Strategies for Proteomic Profiling of Iranian Saw-Scaled Viper, Echis carinatus sochureki, Venom. J. Proteome Res. 2020, 20, 895–908. [Google Scholar] [CrossRef] [PubMed]
- Tsiatsiani, L.; Heck, A.J.R. Proteomics beyond trypsin. FEBS J. 2015, 282, 2612–2626. [Google Scholar] [CrossRef]
- Morrison, H. Enzyme Active Sites and Their Reaction Mechanisms; Academic Press: Elsevier, Cambridge, MA, USA, 2021. [Google Scholar]
- Drapeau, G.R. [21] Cleavage at glutamic acid with staphylococcal protease. Methods Enzymol. 1977, 47, 189–191. [Google Scholar] [CrossRef]
- Walker, J.M. The Protein Protocols Handbook; Springer Science & Business Media: Berlin, Germany, 1996. [Google Scholar]
- Olsen, J.; Ong, S.-E.; Mann, M. Trypsin Cleaves Exclusively C-terminal to Arginine and Lysine Residues. Mol. Cell. Proteom. 2004, 3, 608–614. [Google Scholar] [CrossRef]
- Swaney, D.L.; Wenger, C.D.; Coon, J.J. Value of Using Multiple Proteases for Large-Scale Mass Spectrometry-Based Proteomics. J. Proteome Res. 2010, 9, 1323–1329. [Google Scholar] [CrossRef]
- Choudhury, M.; McCleary, R.J.; Kesherwani, M.; Kini, R.M.; Velmurugan, D. Comparison of proteomic profiles of the venoms of two of the ‘Big Four’snakes of India, the Indian cobra (Naja naja) and the common krait (Bungarus caeruleus), and analyses of their toxins. Toxicon 2017, 135, 33–42. [Google Scholar] [CrossRef]
- Chanda, A.; Mukherjee, A.K. Quantitative proteomics to reveal the composition of Southern India spectacled cobra (Naja naja) venom and its immunological cross-reactivity towards commercial antivenom. Int. J. Biol. Macromol. 2020, 160, 224–232. [Google Scholar] [CrossRef]
- Modahl, C.; Frietze, S.E.; Mackessy, S.P. Transcriptome-facilitated proteomic characterization of rear-fanged snake venoms reveal abundant metalloproteinases with enhanced activity. J. Proteom. 2018, 187, 223–234. [Google Scholar] [CrossRef]
- Possani, L.D.; de la Vega, R. R: Scorpion venom peptides. In Handbook of Biologically Active Peptides; Academic Press; Elsevier: Cambridge, MA, USA, 2006; Volume 1, pp. 339–354. [Google Scholar]
- Trevisan-Silva, D.; Bednaski, A.V.; Fischer, J.S.; Veiga, S.S.; Bandeira, N.; Guthals, A.; Marchini, F.K.; Leprevost, F.V.; Barbosa, V.C.; Senff-Ribeiro, A.; et al. A multi-protease, multi-dissociation, bottom-up-to-top-down proteomic view of the Loxosceles intermedia venom. Sci. Data 2017, 4, 170090. [Google Scholar] [CrossRef]
- Tan, C.H.; Tan, K.Y.; Tan, N.H. A protein decomplexation strategy in snake venom proteomics. In Functional Proteomics; Springer: Berlin/Heidelberg, Germany, 2019; pp. 83–92. [Google Scholar]
- El-Aziz, T.M.A.; Soares, A.G.; Stockand, J.D. Advances in venomics: Modern separation techniques and mass spectrometry. J. Chromatogr. B 2020, 1160, 122352. [Google Scholar] [CrossRef]
- Zainal Abidin, S.A.; Rajadurai, P.; Chowdhury ME, H.; Ahmad Rusmili, M.R.; Othman, I.; Naidu, R. Proteomic Characterization and Comparison of malaysian Tropidolaemus wagleri and Cryptelytrops purpureomaculatus venom using shotgun-proteomics. Toxins 2016, 8, 299. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VENOM CLASSES (%) | VENOMS | |||||||
|---|---|---|---|---|---|---|---|---|
| DpV | NnV | BaV | EoV | |||||
| TRYP | MELD | TRYP | MELD | TRYP | MELD | TRYP | MELD | |
| Major toxins | ||||||||
| 3-finger toxins | 15.2 | 21.9 | 19.54 | 39.2 | 1.4 | 13.5 | 0 | 0 |
| Snake venom metalloproteinases | 6.3 | 13.3 | 10.34 | 11.8 | 27.3 | 18.9 | 23.8 | 48.2 |
| Snake venom serine proteinases | 6.3 | 1.0 | 5.75 | 5.7 | 12.9 | 14.9 | 9.4 | 14.2 |
| Venom Kunitz-type family | 1.3 | 6.7 | 3.45 | 2.3 | 0.7 | 1.4 | 0.5 | 1 |
| Disintegrins | 0.4 | 0.0 | 0.00 | 0.0 | 5.8 | 8.6 | 2 | 0.5 |
| Natriuretic peptides | 0.4 | 4.8 | 0.00 | 0.0 | 0.0 | 0.0 | 0 | 0 |
| Phospholipases A2 | 0.4 | 0.0 | 5.75 | 14.8 | 8.6 | 4.5 | 1.5 | 2.5 |
| Bradykinin-potentiating peptides | 0.0 | 0.0 | 0.00 | 0.0 | 0.0 | 0.5 | 0 | 0 |
| C-type lectin-like | 0.0 | 0.0 | 4.60 | 0.4 | 9.4 | 14.0 | 9.9 | 17.8 |
| Cysteine-rich venom proteins | 0.0 | 0.0 | 2.87 | 2.3 | 5.0 | 3.6 | 3.5 | 1.5 |
| L-amino-acid oxidases | 0.0 | 0.0 | 1.15 | 0.8 | 2.2 | 1.4 | 1 | 1 |
| Minor toxins | ||||||||
| Ohanin/vespryn family | 5.4 | 0.0 | 4.02 | 0.8 | 0.0 | 0.0 | 14.4 | 0.0 |
| Phosphodiesterases | 4.9 | 1.0 | 1.15 | 1.1 | 2.2 | 1.8 | 2.0 | 1.5 |
| Nerve growth factors | 3.6 | 4.8 | 0.57 | 0.8 | 0.7 | 0.5 | 2.5 | 0.0 |
| Hyaluronidases | 2.7 | 4.8 | 0.00 | 0.0 | 0.7 | 0.0 | 0.0 | 0.0 |
| Aminopeptidases | 1.8 | 1.9 | 1.15 | 1.1 | 2.2 | 1.4 | 5.0 | 1.0 |
| Venom endothelial growth factors | 1.8 | 0.0 | 0.00 | 3.0 | 1.4 | 0.9 | 2.5 | 2.5 |
| Cathepsins | 1.3 | 0.0 | 1.15 | 0.0 | 0.0 | 0.5 | 0.0 | 0.0 |
| Cystatins | 0.9 | 0.0 | 0.57 | 0.4 | 0.7 | 1.4 | 0.0 | 0.0 |
| 5′-nucleotidase family | 0.4 | 1.9 | 2.30 | 1.9 | 4.3 | 2.7 | 0.5 | 1.0 |
| Prokineticins | 0.4 | 1.0 | 0.00 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Waprins | 0.0 | 0.0 | 0.57 | 0.4 | 0.0 | 0.0 | 0.0 | 0.0 |
| Phospholipases B | 0.0 | 0.0 | 0.00 | 0.8 | 2.2 | 1.4 | 0.5 | 0.5 |
| Venom complement C3-likes | 0.0 | 0.0 | 2.87 | 2.3 | 0.0 | 0.0 | 0.0 | 0.0 |
| Others | ||||||||
| Cellular components | 42.4 | 32.4 | 22.99 | 8.0 | 10.8 | 7.7 | 15.3 | 6.6 |
| Protein family not assigned | 4.0 | 4.8 | 9.20 | 2.3 | 1.4 | 0.9 | 5.9 | 0.0 |
| Major Toxins Coverage | VENOMS | |||||||
|---|---|---|---|---|---|---|---|---|
| DpV | NnV | BaV | EoV | |||||
| Alpha-Elapitoxin-Dpp2d (C0HJD7) | Cytotoxin 10 (P86541) | Bitiscetin-3 Subunit Alpha (A0A5A4WNG2) | Group III Snake Venom Metalloproteinase (E9KJY6) | |||||
| TRYP | MELD | TRYP | MELD | TRYP | MELD | TRYP | MELD | |
| Protein length | 72 amino acids | 60 amino acids | 156 amino acids | 515 amino acids | ||||
| Peptide coverage | 59/72 = 82% | 64/72 = 89% | 60/60 = 100% | 60/60 = 100% | 104/156 = 67% | 117/156 = 75% | 290/515 = 56% | 372/515 = 72% |
| #peptides | 11 | 23 | 19 | 92 | 27 | 76 | 98 | 320 |
| #denovo tags | 784 | 1128 | 2596 | 3067 | 892 | 748 | 843 | 988 |
| −10lgP | 193.23 | 218.36 | 264.71 | 305.99 | 308.08 | 242.11 | 409.2 | 418.29 |
| #spectras | 125 | 77 | 387 | 886 | 645 | 403 | 442 | 1086 |
| #unique peptides | 7 | 13 | 1 | 23 | 23 | 63 | 7 | 20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amorim, F.G.; Redureau, D.; Crasset, T.; Freuville, L.; Baiwir, D.; Mazzucchelli, G.; Menzies, S.K.; Casewell, N.R.; Quinton, L. Next-Generation Sequencing for Venomics: Application of Multi-Enzymatic Limited Digestion for Inventorying the Snake Venom Arsenal. Toxins 2023, 15, 357. https://doi.org/10.3390/toxins15060357
Amorim FG, Redureau D, Crasset T, Freuville L, Baiwir D, Mazzucchelli G, Menzies SK, Casewell NR, Quinton L. Next-Generation Sequencing for Venomics: Application of Multi-Enzymatic Limited Digestion for Inventorying the Snake Venom Arsenal. Toxins. 2023; 15(6):357. https://doi.org/10.3390/toxins15060357
Chicago/Turabian StyleAmorim, Fernanda Gobbi, Damien Redureau, Thomas Crasset, Lou Freuville, Dominique Baiwir, Gabriel Mazzucchelli, Stefanie K. Menzies, Nicholas R. Casewell, and Loïc Quinton. 2023. "Next-Generation Sequencing for Venomics: Application of Multi-Enzymatic Limited Digestion for Inventorying the Snake Venom Arsenal" Toxins 15, no. 6: 357. https://doi.org/10.3390/toxins15060357
APA StyleAmorim, F. G., Redureau, D., Crasset, T., Freuville, L., Baiwir, D., Mazzucchelli, G., Menzies, S. K., Casewell, N. R., & Quinton, L. (2023). Next-Generation Sequencing for Venomics: Application of Multi-Enzymatic Limited Digestion for Inventorying the Snake Venom Arsenal. Toxins, 15(6), 357. https://doi.org/10.3390/toxins15060357