Antimicrobial Peptide Arsenal Predicted from the Venom Gland Transcriptome of the Tropical Trap-Jaw Ant Odontomachus chelifer

 , , , ,
, , , ,
Abstract
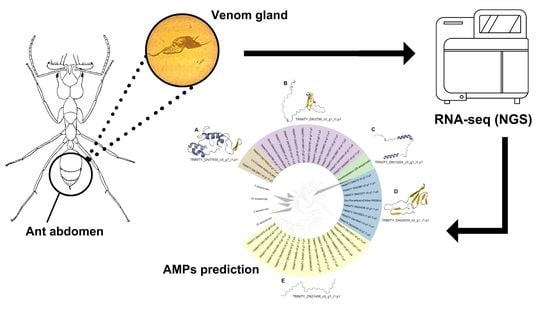
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. O. chelifer Body Preprocessing and Assembly
2.2. Potentially Secreted Peptides Found in O. chelifer
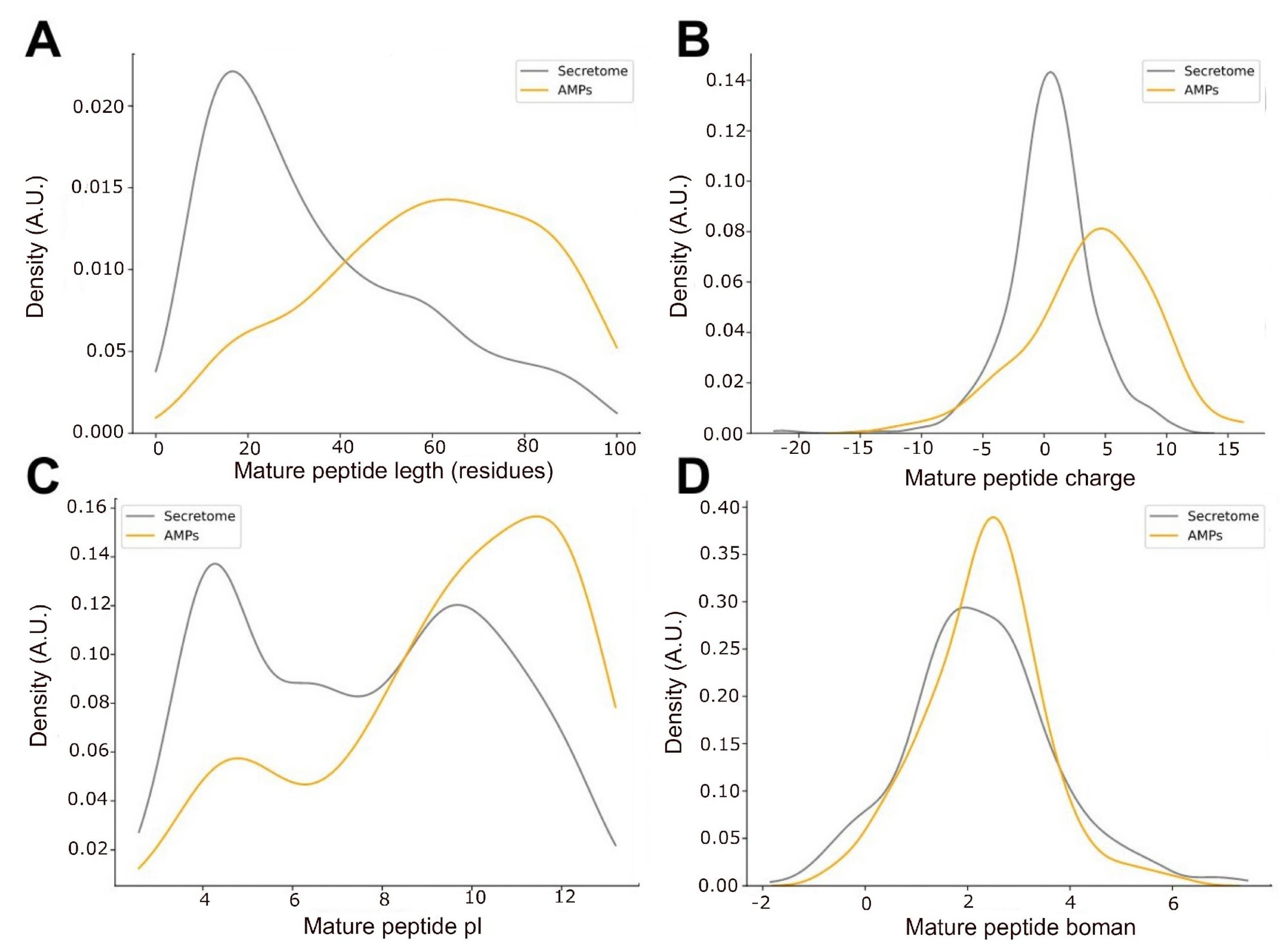
2.3. Predicted Antimicrobial Activity of Peptides That May Perform as Toxins in the Venom Gland from O. chelifer
2.4. Phylogeny of AMP Candidates
2.5. AMPs May Also Compose the Immune System of O. chelifer
2.6. Conservation of Candidate AMP Transcription across the Genus Odontomachus
3. Conclusions
4. Materials and Methods
4.1. O. chelifer Venom Gland Transcriptome
4.2. O. chelifer Body RNA Extraction and Sequencing
4.3. O. chelifer Body Quality Control, Preprocessing, and De Novo Assembly
4.4. Obtaining Coding Protein Regions from O. chelifer Transcriptome
4.5. Prediction and Annotation of the Peptides Composing the O. chelifer Venom Gland Secretome
4.6. Prediction of Potentially Secreted Antimicrobial Peptides
4.7. Cross-Mapping Experiments
4.8. Statistical Testing
4.9. Predicted AMP Clustering and Phylogeny
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Palma, M.S. Insect Venom Peptides. In Handbook of Biologically Active Peptides, 1st ed.; Kastin, A.J., Ed.; Academic Press: Burlington, NJ, USA, 2006; pp. 389–396. ISBN 9780123694423. [Google Scholar]
- Aili, S.R.; Touchard, A.; Escoubas, P.; Padula, M.P.; Orivel, J.; Dejean, A.; Nicholson, G.M. Diversity of peptide toxins from stinging ant venoms. Toxicon 2014, 92, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Touchard, A.; Aili, S.R.; Fox, E.G.P.; Escoubas, P.; Orivel, J.; Nicholson, G.M.; Dejean, A. The Biochemical Toxin Arsenal from Ant Venoms. Toxins 2016, 8, 30. [Google Scholar] [CrossRef]
- Agarwal, S.; Sharma, G.; Verma, K.; Latha, N.; Mathur, V. Pharmacological potential of ants and their symbionts—A review. Entomol. Exp. Appl. 2022, 170, 1032–1048. [Google Scholar] [CrossRef]
- Tonk, M.; Vilcinskas, A. The Medical Potential of Antimicrobial Peptides from Insects. Curr. Top. Med. Chem. 2016, 17, 554–575. [Google Scholar] [CrossRef]
- Li, Y.; Xiang, Q.; Zhang, Q.; Huang, Y.; Su, Z. Overview on the recent study of antimicrobial peptides: Origins, functions, relative mechanisms and application. Peptides 2012, 37, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Amorim-Carmo, B.; Parente, A.M.S.; Souza, E.S.; Silva-Junior, A.A.; Araújo, R.M.; Fernandes-Pedrosa, M.F. Antimicrobial Peptide Analogs From Scorpions: Modifications and Structure-Activity. Front. Mol. Biosci. 2022, 9, 887763. [Google Scholar] [CrossRef]
- Guzman, J.; Téné, N.; Touchard, A.; Castillo, D.; Belkhelfa, H.; Haddioui-Hbabi, L.; Treilhou, M.; Sauvain, M. Anti-Helicobacter pylori Properties of the Ant-Venom Peptide Bicarinalin. Toxins 2017, 10, 21. [Google Scholar] [CrossRef]
- Inagaki, H.; Akagi, M.; Imai, H.T.; Taylor, R.W.; Kubo, T. Molecular cloning and biological characterization of novel antimicrobial peptides, pilosulin 3 and pilosulin 4, from a species of the Australian ant genus Myrmecia. Arch. Biochem. Biophys. 2004, 428, 170–178. [Google Scholar] [CrossRef]
- Wiese, M.D.; Brown, S.G.A.; Chataway, T.K.; Davies, N.W.; Milne, R.W.; Aulfrey, S.J.; Heddle, R.J. Original article: Myrmecia pilosula (Jack Jumper) ant venom: Identification of allergens and revised nomenclature: Allergens in Myrmecia pilosula (Jack Jumper) ant venom. Allergy 2007, 62, 437–443. [Google Scholar] [CrossRef]
- Wanandy, T.; Gueven, N.; Davies, N.W.; Brown, S.G.A.; Wiese, M.D. Pilosulins: A review of the structure and mode of action of venom peptides from an Australian ant Myrmecia pilosula. Toxicon 2015, 98, 54–61. [Google Scholar] [CrossRef]
- Donovan, G.; Baldo, B. Method of Cell Inhibition Using Polypeptides Derived from the Venom of the Austrialian Jumper Ant Myrmecia pilosula. U.S. Patent No. 6,294,649 B1, 25 September 2001. [Google Scholar]
- King, M.A.; Wu, Q.-X.; Donovan, G.R.; Baldo, B.A. Flow cytometric analysis of cell killing by the jumper ant venom peptide pilosulin 1. Cytometry 1998, 32, 268–273. [Google Scholar] [CrossRef]
- Wu, Q.-X.; King, M.A.; Donovan, G.R.; Alewood, D.; Alewood, P.; Sawyer, W.H.; Baldo, B.A. Cytotoxicity of pilosulin 1, a peptide from the venom of the jumper ant Myrmecia pilosula. Biochim. Biophys. Acta (BBA) Gen. Subj. 1998, 1425, 74–80. [Google Scholar] [CrossRef]
- Zelezetsky, I.; Pag, U.; Antcheva, N.; Sahl, H.-G.; Tossi, A. Identification and optimization of an antimicrobial peptide from the ant venom toxin pilosulin. Arch. Biochem. Biophys. 2005, 434, 358–364. [Google Scholar] [CrossRef]
- Bolton, B. A taxonomic and zoogeographical census of the extant ant taxas (Hymenoptera: Formicidae). J. Nat. Hist. 1995, 29, 1037–1056. [Google Scholar] [CrossRef]
- Raimundo, R.L.G.; Freitas, A.V.L.; Oliveira, P.S. Seasonal Patterns in Activity Rhythm and Foraging Ecology in the Neotropical Forest-Dwelling Ant, Odontomachus chelifer (Formicidae: Ponerinae). Ann. Entomol. Soc. Am. 2009, 102, 1151–1157. [Google Scholar] [CrossRef]
- Eberhardt, R.Y.; Haft, D.H.; Punta, M.; Martin, M.; O’Donovan, C.; Bateman, A. AntiFam: A tool to help identify spurious ORFs in protein annotation. Database 2012, 2012, bas003. [Google Scholar] [CrossRef] [PubMed]
- Necci, M.; Piovesan, D.; Tosatto, S.C.E. Large-scale analysis of intrinsic disorder flavors and associated functions in the protein sequence universe: Large-Scale Analysis of Disorder in Protein Sequences. Protein Sci. 2016, 25, 2164–2174. [Google Scholar] [CrossRef]
- Erdős, G.; Pajkos, M.; Dosztányi, Z. IUPred3: Prediction of protein disorder enhanced with unambiguous experimental annotation and visualization of evolutionary conservation. Nucleic Acids Res. 2021, 49, W297–W303. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef]
- Li, S.; Wang, Y.; Xue, Z.; Jia, Y.; Li, R.; He, C.; Chen, H. The structure-mechanism relationship and mode of actions of antimicrobial peptides: A review. Trends Food Sci. Technol. 2021, 109, 103–115. [Google Scholar] [CrossRef]
- Hansen, I.K.Ø.; Lövdahl, T.; Simonovic, D.; Hansen, K.Ø.; Andersen, A.J.; Devold, H.; Richard, C.S.M.; Andersen, J.H.; Strøm, M.B.; Haug, T. Antimicrobial activity of small synthetic peptides based on the marine peptide turgencin A: Prediction of antimicrobial peptide sequences in a natural peptide and strategy for optimization of potency. Int. J. Mol. Sci. 2020, 21, 5460. [Google Scholar] [CrossRef]
- Schendel, V.; Rash, L.D.; Jenner, R.A.; Undheim, E.A.B. The Diversity of Venom: The Importance of Behavior and Venom System Morphology in Understanding Its Ecology and Evolution. Toxins 2019, 11, 666. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhu, S. Comparative genomics analysis of five families of antimicrobial peptide-like genes in seven ant species. Dev. Comp. Immunol. 2012, 38, 262–274. [Google Scholar] [CrossRef] [PubMed]
- Smith, V.J.; Fernandes, J.M.O.; Kemp, G.D.; Hauton, C. Crustins: Enigmatic WAP domain-containing antibacterial proteins from crustaceans. Dev. Comp. Immunol. 2008, 32, 758–772. [Google Scholar] [CrossRef] [PubMed]
- Koehbach, J. Structure-Activity Relationships of Insect Defensins. Front. Chem. 2017, 5, 45. [Google Scholar] [CrossRef]
- Landon, C.; Barbault, F.; Legrain, M.; Guenneugues, M.; Vovelle, F. Rational design of peptides active against the gram positive bacteria Staphylococcus aureus: Rational Design of Peptides. Proteins 2008, 72, 229–239. [Google Scholar] [CrossRef]
- Yi, H.Y.; Chowdhury, M.; Huang, Y.D.; Yu, X.Q. Insect antimicrobial peptides and their applications. Appl. Microbiol. Biotechnol. 2014, 98, 5807–5822. [Google Scholar] [CrossRef]
- Koehbach, J.; Craik, D.J. The Vast Structural Diversity of Antimicrobial Peptides. Trends Pharmacol. Sci. 2019, 40, 517–528. [Google Scholar] [CrossRef]
- Craik, D.J.; Daly, N.L.; Waine, C. The cystine knot motif in toxins and implications for drug design. Toxicon 2001, 39, 43–60. [Google Scholar] [CrossRef]
- Graf, M.; Mardirossian, M.; Nguyen, F.; Seefeldt, A.C.; Guichard, G.; Scocchi, M.; Innis, C.A.; Wilson, D.N. Proline-rich antimicrobial peptides targeting protein synthesis. Nat. Prod. Rep. 2014, 34, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Bulet, P.; Hetru, C.; Dimarcq, J.L.; Hoffmann, D. Antimicrobial peptides in insects; structure and function. Dev. Comp. Immunol. 1999, 23, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, M.H.; Meneguetti, B.T.; Costa, B.O.; Buccini, D.F.; Oshiro, K.G.N.; Preza, S.L.E.; Carvalho, C.M.; Migliolo, L.; Franco, O.L. Non-lytic antibacterial peptides that translocate through bacterial membranes to act on intracellular targets. Int. J. Mol. Sci. 2019, 20, 4877. [Google Scholar] [CrossRef]
- Yang, S.T.; Shin, S.Y.; Shin, S.H. The central PXXP motif is crucial for PMAP-23 translocation across the lipid bilayer. Int. J. Mol. Sci. 2021, 22, 9752. [Google Scholar] [CrossRef]
- Quinet, Y.; Vieira, R.H.S.F.; Sousa, M.R.; Evangelista-Barreto, N.S.; Carvalho, F.C.T.; Guedes, M.I.F.; Alves, C.R.; de Biseau, J.C.; Heredia, A. Antibacterial properties of contact defensive secretions in neotropical Crematogaster ants. J. Venom. Anim. Toxins Incl. Trop. Dis. 2012, 18, 441–445. [Google Scholar] [CrossRef]
- Tani, N.; Kazuma, K.; Ohtsuka, Y.; Shigeri, Y.; Masuko, K.; Konno, K.; Inagaki, H. Mass Spectrometry Analysis and Biological Characterization of the Predatory Ant Odontomachus monticola Venom and Venom Sac Components. Toxins 2019, 11, 50. [Google Scholar] [CrossRef]
- Kim, M.-S.; Zhong, J.; Pandey, A. Common errors in mass spectrometry-based analysis of post-translational modifications. Proteomics 2016, 16, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Westbrook, A.; Ramsdell, J.; Schuelke, T.; Normington, L.; Bergeron, R.D.; Thomas, W.K.; MacManes, M.D. PALADIN: Protein alignment for functional profiling whole metagenome shotgun data. Bioinformatics 2017, 33, 1473–1478. [Google Scholar] [CrossRef]
- Guimarães, D.O.; Ferro, M.; Santos, T.S.; Costa, T.R.; Yoneyama, K.A.G.; Rodrigues, V.M.; Henrique-Silva, F.; Rodrigues, R.S. Transcriptomic and biochemical analysis from the venom gland of the neotropical ant Odontomachus chelifer. Toxicon 2023, 223, 107006. [Google Scholar] [CrossRef]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef]
- Hansen, K.D.; Brenner, S.E.; Dudoit, S. Biases in Illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Res. 2010, 38, e131. [Google Scholar] [CrossRef] [PubMed]
- Zhbannikov, I.Y.; Arbeev, K.G.; Yashin, A.I. rqt: An R package for gene-level meta-analysis. Bioinformatics 2017, 33, 3129–3130. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Götz, S.; Garcia-Gomez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talon, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, B.K.; Gardner, P.P.; Lim, C.S. Razor: Annotation of signal peptides from toxins. bioRxiv, 2021; Preprint. [Google Scholar] [CrossRef]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef]
- Santos-Júnior, C.D.; Pan, S.; Zhao, X.M.; Coelho, L.P. Macrel: Antimicrobial peptide screening in genomes and metagenomes. PeerJ 2020, 8, e10555. [Google Scholar] [CrossRef] [PubMed]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Lin, T.-T.; Yang, L.-Y.; Lu, I.-H.; Cheng, W.-C.; Hsu, Z.-R.; Chen, S.-H.; Lin, C.-Y. AI4AMP: An Antimicrobial Peptide Predictor Using Physicochemical Property-Based Encoding Method and Deep Learning. mSystems 2021, 6, e00299-21. [Google Scholar] [CrossRef]
- Li, C.; Sutherland, D.; Hammond, S.A.; Yang, C.; Taho, F.; Bergman, L.; Houston, S.; Warren, R.L.; Wong, T.; Hoang, L.M.N.; et al. AMPlify: Attentive deep learning model for discovery of novel antimicrobial peptides effective against WHO priority pathogens. BMC Genom. 2022, 23, 77. [Google Scholar] [CrossRef]
- Veltri, D.; Kamath, U.; Shehu, A. Deep learning improves antimicrobial peptide recognition. Bioinformatics 2018, 34, 2740–2747. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.J.; Carper, D.L.; Spangler, M.K.; Carrell, A.A.; Rush, T.A.; Minter, S.J.; Weston, D.J.; Labbé, J.L. amPEPpy 1.0: A portable and accurate antimicrobial peptide prediction tool. Bioinformatics 2021, 37, 2058–2060. [Google Scholar] [CrossRef]
- Fingerhut, L.C.H.W.; Miller, D.J.; Strugnell, J.M.; Daly, N.L.; Cooke, I.R. ampir: An R package for fast genome-wide prediction of antimicrobial peptides. Bioinformatics 2020, 36, 5256–5263. [Google Scholar] [CrossRef]
- UniProt Consortium. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- Waghu, F.H.; Barai, R.S.; Gurung, P.; Idicula-Thomas, S. CAMP R3: A database on sequences, structures and signatures of antimicrobial peptides: Table 1. Nucleic Acids Res. 2016, 44, D1094–D1097. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Farrell, C.M.; Feldgarden, M.; Fine, A.M.; Funk, K.; et al. Database resources of the National Center for Biotechnology Information in 2023. Nucleic Acids Res. 2023, 51, D29–D38. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0 Contributors. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Waskom, M. seaborn: Statistical data visualization. J. Open Source Softw. 2021, 6, 3021. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Steenwyk, J.L.; Buida, T.J.; Li, Y.; Shen, X.-X.; Rokas, A. ClipKIT: A multiple sequence alignment trimming software for accurate phylogenomic inference. PLoS Biol. 2020, 18, e3001007. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Le, S.Q.; Lartillot, N.; Gascuel, O. Phylogenetic mixture models for proteins. Philos. Trans. R. Soc. B Biol. Sci. 2008, 363, 3965–3976. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef]
- Buchan, D.W.A.; Jones, D.T. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. 2019, 47, W402–W407. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menk, J.J.; Matuhara, Y.E.; Sebestyen-França, H.; Henrique-Silva, F.; Ferro, M.; Rodrigues, R.S.; Santos-Júnior, C.D. Antimicrobial Peptide Arsenal Predicted from the Venom Gland Transcriptome of the Tropical Trap-Jaw Ant Odontomachus chelifer. Toxins 2023, 15, 345. https://doi.org/10.3390/toxins15050345
Menk JJ, Matuhara YE, Sebestyen-França H, Henrique-Silva F, Ferro M, Rodrigues RS, Santos-Júnior CD. Antimicrobial Peptide Arsenal Predicted from the Venom Gland Transcriptome of the Tropical Trap-Jaw Ant Odontomachus chelifer. Toxins. 2023; 15(5):345. https://doi.org/10.3390/toxins15050345
Chicago/Turabian StyleMenk, Josilene J., Yan E. Matuhara, Henrique Sebestyen-França, Flávio Henrique-Silva, Milene Ferro, Renata S. Rodrigues, and Célio D. Santos-Júnior. 2023. "Antimicrobial Peptide Arsenal Predicted from the Venom Gland Transcriptome of the Tropical Trap-Jaw Ant Odontomachus chelifer" Toxins 15, no. 5: 345. https://doi.org/10.3390/toxins15050345
APA StyleMenk, J. J., Matuhara, Y. E., Sebestyen-França, H., Henrique-Silva, F., Ferro, M., Rodrigues, R. S., & Santos-Júnior, C. D. (2023). Antimicrobial Peptide Arsenal Predicted from the Venom Gland Transcriptome of the Tropical Trap-Jaw Ant Odontomachus chelifer. Toxins, 15(5), 345. https://doi.org/10.3390/toxins15050345