Hosts and Heterologous Expression Strategies of Recombinant Toxins for Therapeutic Purposes


Abstract
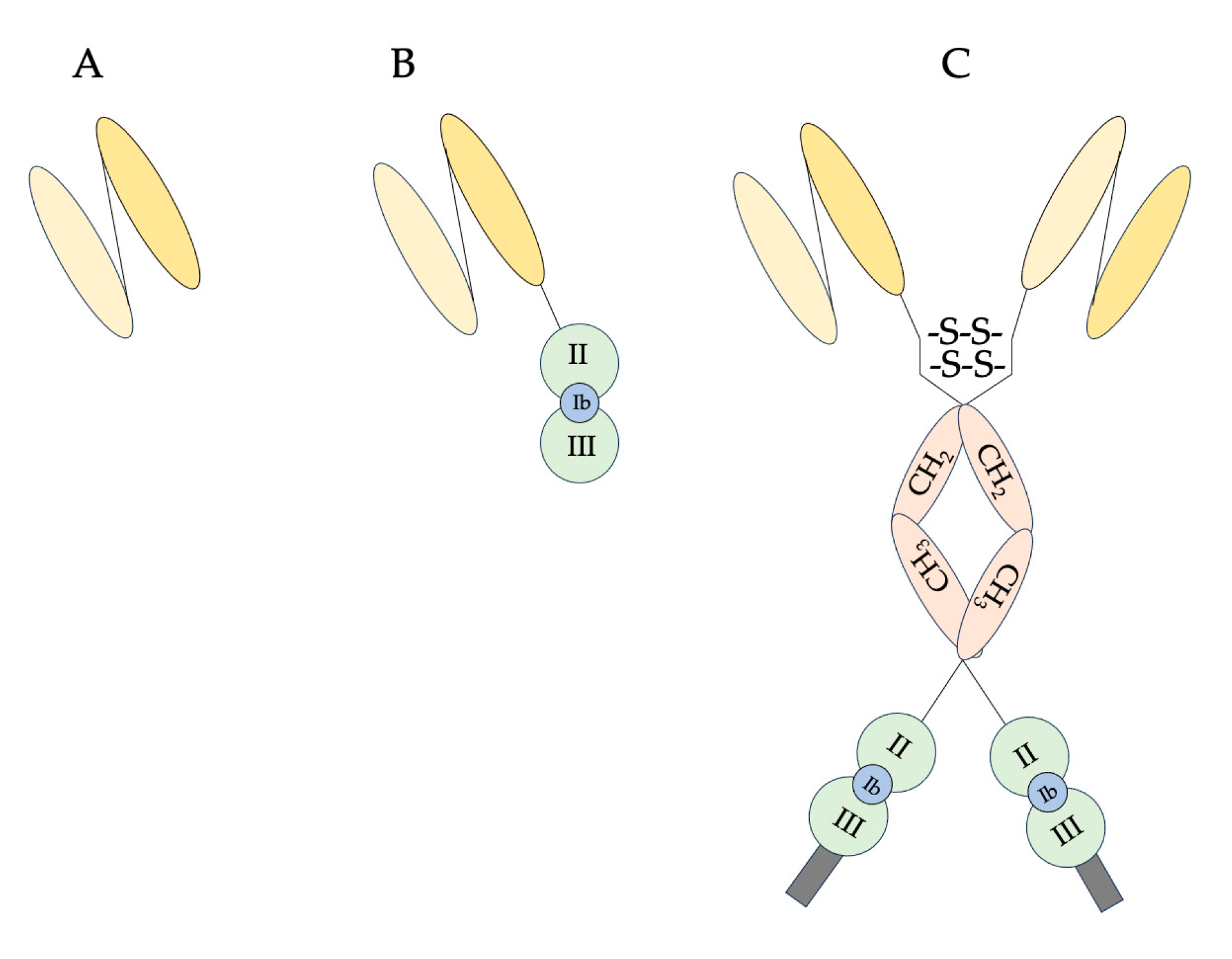
1. Introduction
2. Bacterial Toxins
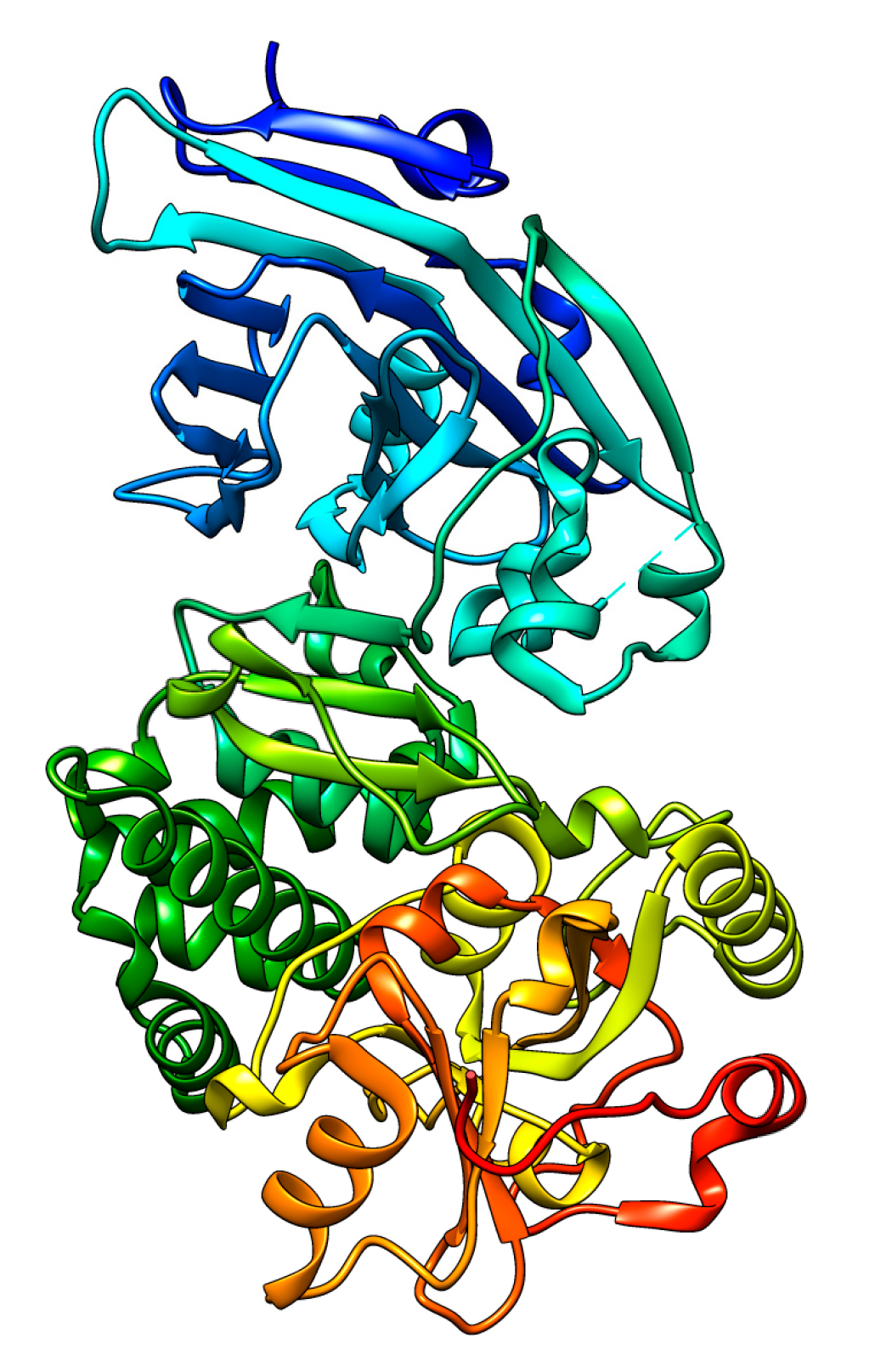
2.1. Diphtheria Toxin
Expression Hosts for DT or Its Mutants
- DT expression in Bacteria
- DT Yeast Expression in Biological Research
- Adenovirus and Lentivirus in Gene Therapy with DT Gene
- DT expression in tobacco
- DT expression in Mammalian cells
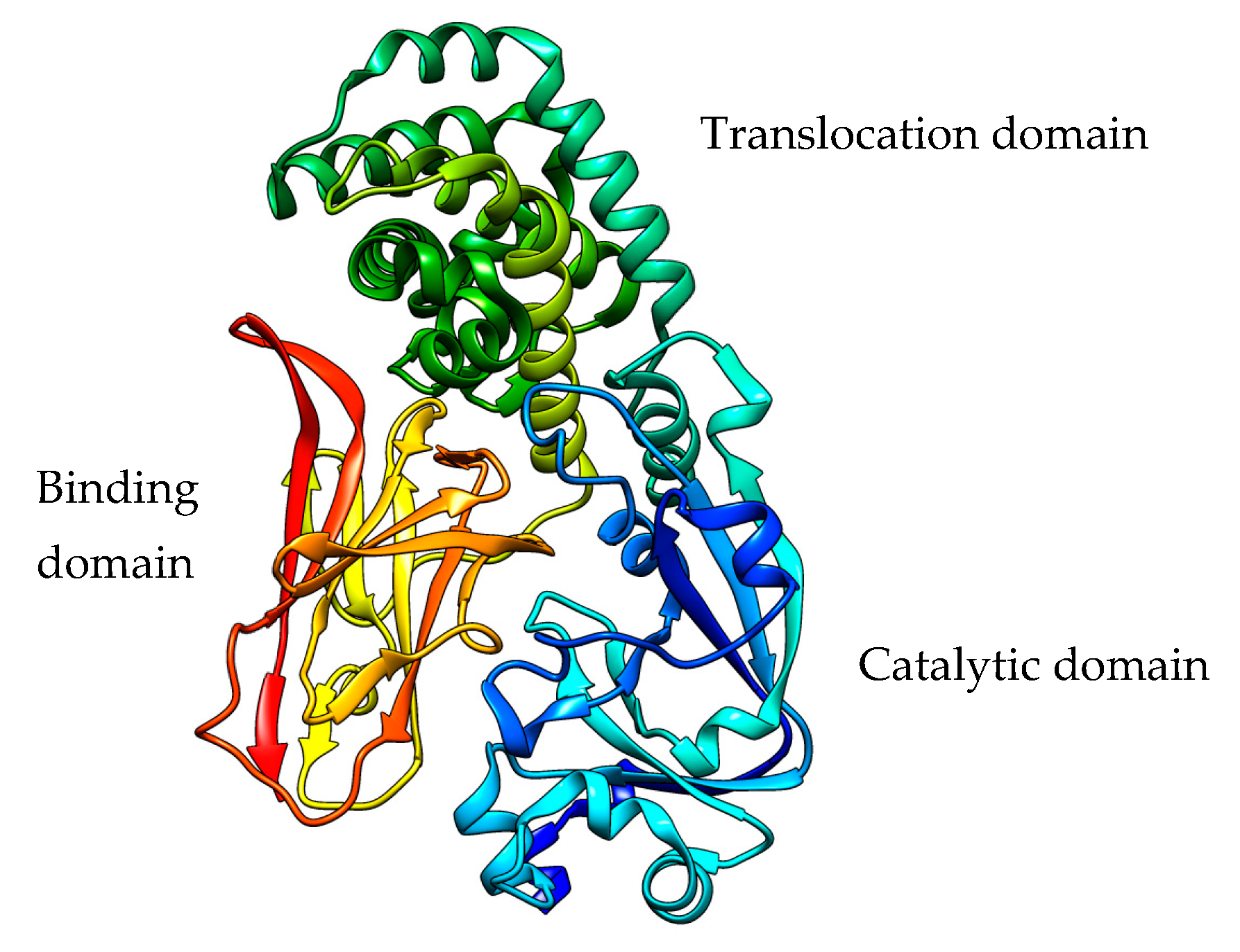
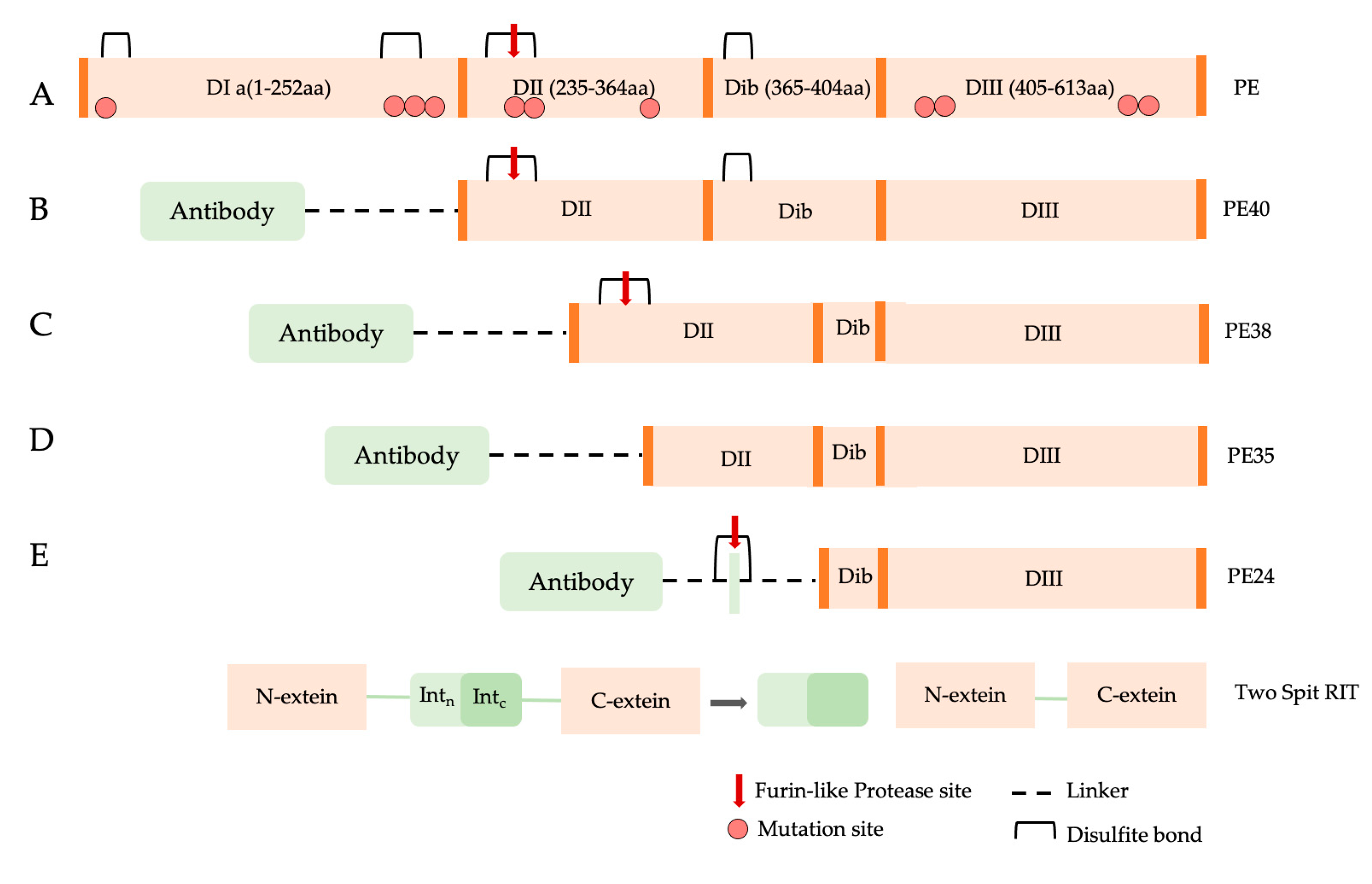
2.2. Pseudomonas aeruginosa Exotoxin A
PE Expression Hosts
- PE Bacterial expression systems
- PE Yeasts expression systems
- PE expression in Algal chloroplasts
3. Plant Toxins
3.1. Saporin SO6
Saporin Expression Hosts
- Saporin Bacterial expression systems
- Saporin Yeasts expression systems
- Saporin expression in Tobacco protoplasts
- Gene therapy with saporin gene
3.2. Ricin A Chain (RTA)
RTA Expression Hosts
- RTA Bacterial expression systems
3.3. Other Plant Toxins
3.3.1. Bouganin
- Bacterial expression systems
3.3.2. Pulchellin
4. Animal Toxins
Melittin
Melittin Expression Hosts
- Melittin Bacterial expression systems
- Melittin Yeasts expression systems
- Melittin Adenovirus expression systems
- Gene therapy with melittin non-viral vectors
5. Conclusions
- Prokaryotic hosts
- Eukaryotic hosts
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gomes, A.R.; Byregowda, S.M.; Veeregowda, B.M.; Balamurugan, V. An Overview of Heterologous Expression Host Systems for the Production of Recombinant Proteins. Adv. Anim. Vet. Sci. 2016, 4, 346–356. [Google Scholar] [CrossRef]
- Petrosini, L.; De Bartolo, P.; Cutuli, D. Neurotoxic Effects, Mechanisms, and Outcome of 192-IgG Saporin. In Handbook of Neurotoxicity; Kostrzewa, R., Ed.; Springer: New York, NY, USA, 2014. [Google Scholar] [CrossRef]
- Abbas, M.S.T. Genetically engineered (modified) crops (Bacillus thuringiensis crops) and the world controversy on their safety. Egypt. J. Biol. Pest. Control 2018, 28, 52. [Google Scholar] [CrossRef]
- Abulmagd, S.; Emara, M.; Aziz, S.; El-Domany, R. Evaluation and characterisation of A and B fragments of Corynebacterium diphtheriae toxin towards recombinant diphtheria vaccine. Indian J. Med. Microbiol. 2013, 31, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Oppenheimer, N.J.; Bodley, J.W. Diphtheria toxin. Site and configuration of ADP-ribosylation of diphthamide in elongation factor 2. J. Biol. Chem. 1981, 256, 8579–8581. [Google Scholar] [CrossRef] [PubMed]
- Uchida, T.; Pappenheimer, A.M., Jr.; Greany, R. Diphtheria toxin and related proteins. I. Isolation and properties of mutant proteins serologically related to diphtheria toxin. J. Biol. Chem. 1973, 248, 3838–3844. [Google Scholar] [CrossRef] [PubMed]
- Holmes R., K. Biology and molecular epidemiology of diphtheria toxin and the tox gene. J. Infect. Dis. 2000, 181 (Suppl. S1), S156–S167. [Google Scholar] [CrossRef]
- Broker, M.; Costantino, P.; De Tora, L.; McIntosh, E.D. Biochemical and biological characteristics of cross-reacting material 197 CRM197, a non-toxic mutant of diphtheria toxin: Use as a conjugation protein in vaccines and other potential clinical applications. Biologicals 2011, 39, 195–204. [Google Scholar] [CrossRef]
- Uchida, T.; Gill, D.M.; Pappenheimer, A.M., Jr. Mutation in the structural gene for diphtheria toxin carried by temperate phage. Nat. New Biol. 1971, 233, 8–11. [Google Scholar] [CrossRef]
- Khodak, Y.A.; Ryazanova, A.Y.; Vorobiev, I.I.; Kovalchuk, A.L.; Ovechko, N.N.; Aparin, P.G. High-Level Production of Soluble Cross-Reacting Material 197 in Escherichia coli Cytoplasm Due to Fine Tuning of the Target Gene’s mRNA Structure. BioTech 2023, 12, 9. [Google Scholar] [CrossRef]
- Kageyama, T.; Ohishi, M.; Miyamoto, S.; Mizushima, H.; Iwamoto, R.; Mekada, E. Diphtheria toxin mutant CRM197 possesses weak EF2 ADP-ribosyl activity that potentiates its anti-tumorigenic activity. J. Biochem. 2007, 142, 95–104. [Google Scholar] [CrossRef]
- Shinefield, H.R. Overview of the development and current use of CRM(197) conjugate vaccines for pediatric use. Vaccine 2010, 28, 4335–4339. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, P.C.; Sharyan, A.; Sheikhi Moghaddam, L. Meningococcal vaccines: Current status and emerging strategies. Vaccines 2018, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Yotsumoto, F.; Fukagawa, S.; Miyata, K.; Nam, S.O.; Katsuda, T.; Miyahara, D. HB-EGF Is a Promising Therapeutic Target for Lung Cancer with Secondary Mutation of EGFRT790M. Anticancer Res. 2017, 37, 3825–3831. [Google Scholar] [CrossRef] [PubMed]
- Mitamura, T.; Higashiyama, S.; Taniguchi, N.; Klagsbrun, M.; Mekada, E. Diphtheria toxin binds to the epidermal growth factor (EGF)-like domain of human heparin-binding EGF-like growth factor/diphtheria toxin receptor and inhibits specifically its mitogenic activity. J. Biol. Chem. 1995, 270, 1015–1019. [Google Scholar] [CrossRef] [PubMed]
- Rappuoli, R. Isolation and characterization of Corynebacterium diphtheriae nontandem double lysogens hyperproducing CRM197. Appl. Environ. Microbiol. 1983, 46, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Oram, D.M.; Jacobson, A.D.; Holmes, R.K. Transcription of the contiguous sigB, dtxR, and galE genes in Corynebacterium diphtheriae: Evidence for multiple transcripts and regulation by environmental factors. J. Bacteriol. 2006, 188, 2959–2973. [Google Scholar] [CrossRef] [PubMed]
- Retallack, D.M.; Chew, L. High Level Expression of Recombinant Toxin Proteins. U.S. Patent 896636B22014, 9 December 2014. [Google Scholar]
- Murakami, S.; Bodley, J.W.; Livingston, D.M. Saccharomyces cerevisiae spheroplasts are sensitive to the action of diphtheria toxin. Mol. Cell Biol. 1982, 2, 588–592. [Google Scholar] [CrossRef]
- Tweten, R.K.; Collier, R.J. Molecular cloning and expression of gene fragments from corynebacteriophage beta encoding enzymatically active peptides of diphtheria toxin. J. Bacteriol. 1983, 156, 680–685. [Google Scholar] [CrossRef]
- Abulmagd, S.; Khattab, A.E.A.; Zedan, H. Expression of full and fragment-B of diphtheria toxin genes in Escherichia coli for generating of recombinant diphtheria vaccines. Clin. Exp. Vaccine Res. 2022, 11, 12–29. [Google Scholar] [CrossRef]
- Barbieri, J.T.; Collier, R.J. Expression of a mutant, full-length form of diphtheria toxin in Escherichia coli. Infect. Immun. 1987, 55, 1647–1651. [Google Scholar] [CrossRef]
- Gilliland, D.G.; Steplewski, Z.; Collier, R.J.; Mitchell, K.F.; Chang, T.H.; Koprowski, H. Anti-directed cytotoxic agents: Use of monoclonal antibody to direct the action of toxin A chains to colorectal carcinoma cells. Proc. Natl. Acad. Sci. USA 1980, 77, 4539–4543. [Google Scholar] [CrossRef]
- Shapira, A.; Benhar, I. Toxin-based therapeutic approaches. Toxins 2010, 2, 2519–2583. [Google Scholar] [CrossRef]
- Beilhartz, G.L.; Sugiman-Marangos, S.N.; Melnyk, R.A. Repurposing bacterial toxins for intracellular delivery of therapeutic proteins. Biochem. Pharmacol. 2017, 142, 13–20. [Google Scholar] [CrossRef]
- Williams, D.P.; Parker, K.; Bacha, P.; Bishai, W.; Borowski, M.; Genbauffe, F.; Strom, T.B.; Murphy, J.R. Diphtheria toxin receptor binding domain substitution with interleukin-2: Genetic construction and properties of a diphtheria toxin-related interleukin-2 fusion protein. Protein Eng. Des. Sel. 1987, 1, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Zarkar, N.; Nasiri Khalili, M.A.; Khodadadi, S.; Zeinoddini, M.; Ahmadpour, F. Expression and purification of soluble and functional fusion protein DAB389 IL-2 into the E. coli strain Rosetta-gami (DE3). Biotechnol. Appl. Biochem. 2020, 67, 206–212. [Google Scholar] [CrossRef]
- Peraino, J.S.; Zhang, H.; Rajasekera, P.V.; Wei, M.; Madsen, J.C.; Sachs, D.H.; Huang, C.A.; Wang, Z. Diphtheria toxin-based bivalent human IL-2 fusion toxin with improved efficacy for targeting human CD25(+) cells. J. Immunol. Methods 2014, 405, 57–66. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cheung, L.S.; Fu, J.; Kumar, P.; Kumar, A.; Urbanowski, M.E.; Ihms, E.A. Second-generation IL-2 receptor-targeted diphtheria fusion toxin exhibits antitumor activity and synergy with anti-PD-1 in melanoma. Proc. Natl. Acad. Sci. USA 2019, 116, 3100–3105. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.E.; Ramage, J.; Latimer, A.; Feely, T.; Delatte, S.; Hall, P.; Tagge, E.; Kreitman, R.; Willingham, M. High-level expression and purification of the recombinant diphtheria fusion toxin DTGM for PHASE I clinical trials. Protein Expr. Purif. 1999, 16, 190–201. [Google Scholar] [CrossRef]
- Urieto, J.O.; Liu, T.F.; Black, J.H.; Cohen, K.A.; Hall, P.D.; Willingham, M.C.; Pennell, L.K.; Hogge, D.E.; Kreitman, R.J.; Frankel, A.E. Expression and purification of the recombinant diphtheria fusion toxin DT388IL3 for phase I clinical trials. Protein Expr. Purif. 2004, 33, 123–133. [Google Scholar] [CrossRef]
- Liu, T.F.; Cohen, K.A.; Ramage, J.G.; Willingham, M.C.; Thorburn, A.M.; Frankel, A.E. A diphtheria toxin-epidermal growth factor fusion protein is cytotoxic to human glioblastoma multiforme cells. Cancer Res. 2003, 63, 1834–1837. [Google Scholar]
- Hashemi, Y.H.; Heiat, M.; Kieliszek, M.; Alavian, S.M.; Rezaie, E. DT389-YP7, a Recombinant Immunotoxin against Glypican-3 That Inhibits Hepatocellular Cancer Cells: An In Vitro Study. Toxins 2021, 13, 749. [Google Scholar] [CrossRef]
- Woo, J.H.; Liu, Y.Y.; Stavrou, S.; Neville, D.M., Jr. Increasing secretion of a bivalent anti-T-cell immunotoxin by Pichia pastoris. Appl. Environ. Microbiol. 2004, 70, 3370–3376. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vallera, D.A.; Todhunter, D.A.; Kuroki, D.W.; Shu, Y.; Sicheneder, A.; Chen, H. A Bispecific Recombinant Immunotoxin, DT2219, Targeting Human CD19 and CD22 Receptors in a Mouse Xenograft Model of B-Cell Leukemia/Lymphoma. Clin. Cancer Res. 2005, 11, 3879–3888. [Google Scholar] [CrossRef] [PubMed]
- Vallera, D.A.; Li, C.; Jin, N.; Panoskaltsis-Mortari, A.; Hall, W.A. Targeting Urokinase-Type Plasminogen Activator Receptor on Human Glioblastoma Tumors With Diphtheria Toxin Fusion Protein DTAT. JNCI J. Natl. Cancer Inst. 2002, 94, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Stish, B.J.; Vickers, S.M.; Buchsbaum, D.J.; Saluja, A.K.; Vallera, D.A. A new drug delivery method of bispecific ligand-directed toxins, which reduces toxicity and promotes efficacy in a model of orthotopic pancreatic cancer. Pancreas 2010, 39, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wei, M.; Zhang, H.; Chen, H.; Germana, S.; Huang, C.A.; Madsen, J.C.; Sachs, D.H.; Wang, Z. Diphtheria-toxin based anti-human CCR4 immunotoxin for targeting human CCR4(+) cells in vivo. Mol. Oncol. 2015, 9, 1458–1470. [Google Scholar] [CrossRef] [PubMed]
- Shafiee, F.; Rabbani, M.; Jahanian-Najafabadi, A. Optimization of the Expression of DT386-BR2 Fusion Protein in Escherichia coli using Response Surface Methodology. Adv. Biomed. Res. 2017, 6, 22. [Google Scholar] [CrossRef]
- Babavalian, E.; Zeinoddini, M.; Saeedinia, A.R.; Mohammadi, R.; Xodadadi, N. Design of a recombinant immunotoxin against the human granulocyte-colony stimulating factor receptor. Mol. Biol. Rep. 2019, 46, 1093–1097. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, S.; Zhuang, H.; Lu, F. Cytotoxicity of a novel fibroblast growth factor receptor targeted immunotoxin on a human ovarian teratocarcinoma cell line. Cancer Biother. Radiopharm. 2006, 21, 321–332. [Google Scholar] [CrossRef]
- Vanderspek, J.C.; Sutherland, J.A.; Zeng, H.; Battey, J.F.; Jensen, R.T.; Murphy, J.R. Inhibition of protein synthesis in small cell lung cancer cells induced by the diphtheria toxin-related fusion protein DAB389 GRP. Cancer Res. 1997, 57, 290–294. [Google Scholar]
- Shulga-Morskoy, S.; Rich, B. Bioactive IL7-diphtheria fusion toxin secreted by mammalian cells. Protein Eng. Des. Sel. PEDS 2005, 18, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Lv, P.; Ye, S.; Zhang, Y.; Li, S.; Kan, C.; Fan, L.; Liu, R.; Luo, D.; Wang, A.; et al. DT390-triTMTP1, a novel fusion protein of diphtheria toxin with tandem repeat TMTP1 peptide, preferentially targets metastatic tumors. Mol. Pharm. 2013, 10, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Potala, S.; Verma, R.S. A Novel Fusion Protein Diphtheria Toxin–Stem Cell Factor (DT-SCF)—Purification and Characterization. Appl. Biochem. Biotechnol. 2010, 162, 1258–1269. [Google Scholar] [CrossRef] [PubMed]
- Rustamzadeh, E.; Hall, W.A.; Todhunter, D.A.; Low, W.C.; Liu, H.; Panoskaltsis-Mortari, A.; Vallera, D.A. Intracranial therapy of glioblastoma with the fusion protein DTIL13 in immunodeficient mice. Int. J. Cancer 2006, 118, 2594–2601. [Google Scholar] [CrossRef]
- Citius Pharmaceuticals, Inc. Citius Pharmaceuticals Completes Enrollment in the Pivotal Phase 3 Study of Its Cancer Immunotherapy I/ONTAK for the Treatment of Cutaneous t-cell Lymphoma. News Release Citius Pharm, 6 December 2021. Available online: https://bit.ly/3DQNZnm (accessed on 6 December 2021).
- Tao, X.; Boyd, J.; Murphy, J.R. Specific binding of the diphtheria tox regulatory element DtxR to the tox operator requires divalent heavy metal ions and a 9-base-pair interrupted palindromic sequence. Proc. Natl. Acad. Sci. USA 1992, 89, 5897–5901. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Upadhyay, V.; Upadhyay, A.K.; Singh, S.M.; Panda, A.K. Protein recovery from inclusion bodies of Escherichia coli using mild solubilization process. Microb. Cell Fact. 2015, 14, 41. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, L.; Yu, J.; Mei, J.; Yi, Y.; Chen, J.; Ying, G. Preparation of Recombinant Human Thymic Stromal Lymphopoietin Protein. Prep. Biochem. Biotechnol. 2017, 47, 934–938. [Google Scholar] [CrossRef]
- Oganesyan, N.; Lees, A.; Fina Biosolutions, Llc. Expression and Purification of crm197 and Related. Proteins. Patent WO2015117093 A1, 2 February 2015. [Google Scholar]
- Peraino, J.S.; Schenk, M.; Li, G.; Zhang, H.; Farkash, E.A.; Sachs, D.H.; Huang, C.A.; Duran-Struuck, R.; Wang, Z. Development of a diphtheria toxin-based recombinant porcine IL-2 fusion toxin for depleting porcine CD25+ cells. J. Immunol. Methods 2013, 398–399, 33–43. [Google Scholar] [CrossRef][Green Version]
- Rudolph, R.; Lilie, H. In vitro folding of inclusion body proteins. FASEB J. 1996, 10, 49–56. [Google Scholar] [CrossRef]
- Park, A.-R.; Jang, S.-W.; Kim, J.-S.; Park, Y.-G.; Koo, B.-S.; Lee, H.-C. Efficient recovery of recombinant CRM197 expressed as inclusion bodies in E. coli. PLoS ONE 2018, 13, e0201060. [Google Scholar] [CrossRef]
- Messens, J.; Collet, J.F. Pathways of disulfide bond formation in Escherichia coli. Int. J. Biochem. Cell Biol. 2006, 38, 1050–1062. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Lee, R.; Lee, S.H.; Yim, S.S.; Jeong, K.J. Enhanced secretion of recombinant proteins via signal recognition particle (SRP)-dependent secretion pathway by deletion of rrsE in Escherichia coli. Biotechnol. Bioeng. 2016, 113, 2453–2461. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.H.; Kim, S.K.; Kim, J.F. Secretory production of recombinant proteins in Escherichia coli. Recent Pat. Biotechnol. 2010, 4, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Goffin, P.; Dewerchin, M.; De Rop, P.; Blais, N.; Dehottay, P. High-yield production of recombinant CRM197, a non-toxic mutant of diphtheria toxin, in the periplasm of Escherichia coli. Biotechnol. J. 2017, 12, 1700168. [Google Scholar] [CrossRef] [PubMed]
- Stefan, A.; Conti, M.; Rubboli, D.; Ravagli, L.; Presta, E.; Hochkoeppler, A. Overexpression and purification of the recombinant diphtheria toxin variant CRM197 in Escherichia coli. J. Biotechnol. 2011, 156, 245–252. [Google Scholar] [CrossRef]
- Mahamad, P.; Boonchird, C.; Panbangred, W. High level accumulation of soluble diphtheria toxin mutant (CRM197) with co-expression of chaperones in recombinant Escherichia coli. Appl. Microbiol. Biotechnol. 2016, 100, 6319–6330. [Google Scholar] [CrossRef]
- Bessette, P.H.; Åslund, F.; Beckwith, J.; Georgiou, G. Efficient Folding of Proteins with Multiple Disulfide Bonds in the Escherichia coli Cytoplasm. Proc. Natl. Acad. Sci. USA 1999, 96, 13703–13708. [Google Scholar] [CrossRef]
- Roth, R.; Van Zyl, P.; Tsekoa, T.; Stoychev, S.; Mamputha, S.; Buthelezi, S.; Crampton, M. Co-expression of sulphydryl oxidase and protein disulphide isomerase in Escherichia coli allows for production of soluble CRM197. J. Appl. Microbiol. 2017, 122, 1402–1411. [Google Scholar] [CrossRef]
- Woo, J.H.; Liu, Y.Y.; Mathias, A.; Stavrou, S.; Wang, Z.; Thompson, J.; Neville, D.M. Gene optimization is necessary to express a bivalent anti-human anti-T cell immunotoxin in Pichia pastoris. Protein Expr. Purif. 2002, 25, 270–282. [Google Scholar] [CrossRef]
- Woo, J.H.; Liu, Y.Y.; Neville, D.M., Jr. Minimization of aggregation of secreted bivalent anti-human T cell immunotoxin in Pichia pastoris bioreactor culture by optimizing culture conditions for protein secretion. J. Biotechnol. 2006, 121, 75–85. [Google Scholar] [CrossRef]
- Aw, R.; Ashik, M.R.; Islam, A.A.Z.M.; Khan, I.; Mainuddin, M.; Islam, M.A.; Ahasan, M.M.; Polizzi, K.M. Production and purification of an active CRM197 in Pichia pastoris and its immunological characterization using a Vi-typhoid antigen vaccine. Vaccine 2021, 39, 7379–7386. [Google Scholar] [CrossRef]
- Zhou, J.; Petracca, R. Secretory expression of recombinant diphtheria toxin mutants in B. subtilis. J. Tongji Med. Univ. 1999, 19, 253–256. [Google Scholar] [CrossRef]
- Wang, Z.; Duran, R.; Struuck, R.; Crepeau, A.; Matar, I.; Hanekamp, S.; Srinivasan, D.M.; Neville, D.H.; Sachs, C.A.; Huang, C.A. Development of a diphtheria toxin based antiporcine CD3 recombinant immunotoxin. Bioconjugate Chem. 2011, 22, 2014. [Google Scholar] [CrossRef] [PubMed]
- Delhove, J.; Osenk, I.; Prichard, I.; Donnelley, M. Public acceptability of gene therapy and gene editing for human use: A systematic review. Hum. Gene Ther. 2020, 31, 20–46. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Tuhin, I.J.; Monty, M.A.; Luo, S.; Shao, J.; Yan, Z.; Yu, L. Gene therapy for hepatocellular carcinoma using adenoviral vectors delivering a gene encoding IL-17A-neutralizing antibody fragments. Hum. Gene Ther. 2020, 31, 1074–1085. [Google Scholar] [CrossRef]
- Gahéry-Ségard, H.; Molinier-Frenkel, V.; Le Boulaire, C.; Saulnier, P.; Opolon, P.; Lengagne, R.; Gautier, E.; Le Cesne, A.; Zitvogel, L.; Venet, A.; et al. Phase I trial of recombinant adenovirus gene transfer in lung cancer. Longitudinal study of the immune responses to transgene and viral products. J. Clin. Investig. 1997, 100, 2218–2226. [Google Scholar] [CrossRef]
- Dinney, C.P.; Fisher, M.B.; Navai, N.; O’Donnell, M.A.; Cutler, D.; Abraham, A.; Young, S.; Hutchins, B.; Caceres, M.; Kishnani, N.; et al. Phase I trial of intravesical recombinant adenovirus mediated interferon-α2b formulated in Syn3 for Bacillus Calmette-Guérin failures in nonmuscle invasive bladder cancer. J. Urol. 2013, 190, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; McCadden, J.; Ferrer, F.; Kruszewski, M.; Carducci, M.; Simons, J.; Rodriguez, R. Prostate-specific expression of the diphtheria toxin A chain (DT-A): Studies of inducibility and specificity of expression of prostate-specific antigen promoter-driven DT-A adenoviral-mediated gene transfer. Cancer Res. 2002, 62, 2576–2582. [Google Scholar]
- Dai, L.; Yu, X.; Huang, S.; Peng, Y.; Liu, J.; Chen, T.; Wang, X.; Liu, Q.; Zhu, Y.; Chen, D.; et al. The therapeutic potential of attenuated diphtheria toxin delivered by an adenovirus vector with survivin promoter on human lung cancer cells. Cancer Biol. Ther. 2021, 22, 79–87. [Google Scholar] [CrossRef]
- Fogar, P.; Navaglia, F.; Basso, D.; Zambon, C.F.; Moserle, L.; Indraccolo, S.; Stranges, A.; Greco, E.; Fadi, E.; Padoan, A.; et al. Heat-induced transcription of diphtheria toxin A or its variants, CRM176 and CRM197: Implications for pancreatic cancer gene therapy. Cancer Gene Ther. 2010, 17, 58–68. [Google Scholar] [CrossRef]
- Wang, Z.; Tang, Z.; Zheng, Y.; Yu, D.; Spear, M.; Iyer, S.R.; Bishop, B.; Wu, Y. Development of a nonintegrating Rev-dependent lentiviral vector carrying diphtheria toxin A chain and human TRAF6 to target HIV reservoirs. Gene Ther. 2010, 17, 1063–1076. [Google Scholar] [CrossRef]
- Lange, M.J.; Lyddon, T.D.; Johnson, M.C. Diphtheria Toxin A-Resistant Cell Lines Enable Robust Production and Evaluation of DTA-Encoding Lentiviruses. Sci. Rep. 2019, 9, 8985. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H. Production of biopharmaceuticals and vaccines in plants via the chloroplast genome. Biotechnol. J. 2006, 1, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.L.; Liang, B.G.; Jin, Y.S.; Zhang, W.J.; Wang, T. Oral immunization with pBsVP6 transgenic alfalfa protects mice against rotavirus infection. Virology 2005, 339, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Mason, H.S.; Ball, J.M.; Shi, J.J.; Jiang, X.; Estes, M.K.; Arntzen, C.J. Expression of Norwalk virus capsid protein in transgenic tobacco and protein and its oral immunogenicity in mice. Proc. Natl. Acad. Sci. USA 1996, 93, 5335–5340. [Google Scholar] [CrossRef] [PubMed]
- Marquet-Blouin, E.; Bouche, F.B.; Steinmetz, A.; Muller, C.P. Neutralizing immunogenicity of transgenic carrot (Daucus carota L.)-derived measles virus hemagglutinin. Plant Mol. Biol. 2003, 51, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Rosales-Mendoza, S.; Soria-Guerra, R.E.; Olivera-Flores, M.J.; López-Revilla, R.; Argüello-Astorga, G.; Jiménez-Bremont, J.F.; García-de la Cruz, R.F.; Loyola-Rodríguez, J.P.; Alpuche-Solís, A.G. Expression of Escherichia coli heat-labile enterotoxin b subunit (LTB) in carrot (Daucus carota L.). Plant Cell Rep. 2007, 26, 969–976. [Google Scholar] [CrossRef]
- Sandhu, J.S.; Krasnyanski, S.F.; Domier, L.; Korban, S.S.; Osadjan, M.D.; Buetow, D.E. Oral immunization of mice with transgenic tomato fruit expressing respiratory syncytial virus-F protein induces a systemic immune response. Transgenic Res. 2000, 9, 127–135. [Google Scholar] [CrossRef]
- Liu, H.L.; Li, W.S.; Lei, T.; Zheng, J.; Zhang, Z.; Yan, X.F.; Wang, Z.Z.; Wang, Y.L.; Si, L.S. Expression of human papillomavirus type 16 L1 protein in transgenic tobacco plants. Act. Biochem. Biophys. Sin. 2005, 37, 153–158. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, X.; Liu, M.; Zhang, J.; Li, Y.; Zheng, C.C. Expression and characterization of Helicobacter pylori heat-shock protein A (HspA) protein in transgenic tobacco (Nicotiana tabacum) plants. Biotechnol. Appl. Biochem. 2006, 43, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Soria-Guerra, R.E.; Alpuche-Solís, A.G.; Rosales-Mendoza, S.; Moreno-Fierros, L.; Bendik, E.M.; Martínez-González, L.; Korban, S.S. Expression of a multi-epitope DPT fusion protein in transplastomic tobacco plants retains both antigenicity and immunogenicity of all three components of the functional oligomer. Planta 2009, 229, 1293–1302. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.S.; Siminovitch, L. Diphtheria-toxin-resistant mutants of CHO cells affected in protein synthesis: A novel phenotype. Somat. Cell Mol. Genet. 1978, 4, 553–571. [Google Scholar] [CrossRef] [PubMed]
- Kohno, K.; Uchida, T. Highly frequent single amino acid substitution in mammalian elongation factor 2 (EF-2) results in expression of resistance to EF-2-ADP-ribosylating toxins. J. Biol. Chem. 1987, 262, 12298–12305. [Google Scholar] [CrossRef]
- Foley, B.T.; Moehring, J.M.; Moehring, T.J. A mutation in codon 717 of the CHO-K1 elongation factor 2 gene prevents the first step in the biosynthesis of diphthamide. Somat. Cell Mol. Genet. 1992, 18, 227–231. [Google Scholar] [CrossRef]
- Foley, B.T.; Moehring, J.M.; Moehring, T.J. Mutations in the elongation factor 2 gene which confer resistance to diphtheria toxin and Pseudomonas exotoxin A. Genetic and biochemical analyses. J. Biol. Chem. 1995, 270, 23218–23225. [Google Scholar] [CrossRef] [PubMed]
- DuBridge, R.B.; Tang, P.; Hsia, H.C.; Leong, P.M.; Miller, J.H.; Calos, M.P. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol. Cell. Biol. 1987, 7, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Hedstrom, R.C.; Funk, C.R.; Kaper, J.B.; Pavlovskis, O.R.; Galloway, D.R. Cloning of a gene involved in regulation of exotoxin A expression in Pseudomonas aeruginosa. Infect. Immun. 1986, 51, 37–42. [Google Scholar] [CrossRef]
- Allured, V.S.; Collier, R.J.; Carroll, S.F.; McKay, D.B. Structure of exotoxin A of Pseudomonas aeruginosa at 3.0-Angstrom resolution. Proc. Natl. Acad. Sci. USA 1986, 83, 1320–1324. [Google Scholar] [CrossRef]
- Weldon, J.E.; Pastan, I. A guide to taming a toxin–recombinant immunotoxins constructed from Pseudomonas exotoxin A for the treatment of cancer. FEBS J. 2011, 278, 4683–4700. [Google Scholar] [CrossRef]
- Havaei, S.M.; Aucoin, M.G.; Jahanian-Najafabadi, A. Pseudomonas Exotoxin-Based Immunotoxins: Over Three Decades of Efforts on Targeting Cancer Cells with the Toxin. Front. Oncol. 2021, 11, 781800. [Google Scholar] [CrossRef]
- Amoozadeh, S.; Hemmati, M.; Farajollahi, M.M.; Akbari, N.; Tarighi, P. Preparation of Diphtheria and Pseudomonas Exotoxin A Immunotoxins and Evaluation of Their Cytotoxicity Effect on SK-BR-3, BT-474, and MDA-MB-231 Breast Cancer Cell Lines. Cancer Investig. 2019, 37, 546–557. [Google Scholar] [CrossRef]
- Chang, J.; Liu, X.; Ren, H.; Lu, S.; Li, M.; Zhang, S.; Zhao, K.; Li, H.; Zhou, X.; Peng, L.; et al. Pseudomonas Exotoxin A-Based Immunotherapy Targeting CCK2R-Expressing Colorectal Malignancies: An In Vitro and In Vivo Evaluation. Mol. Pharm. 2021, 18, 2285–2297. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Zhu, J. Recent development and optimization of pseudomonas aeruginosa exotoxin immunotoxins in cancer therapeutic applications. Int. Immunopharmacol. 2021, 96, 107759. [Google Scholar] [CrossRef]
- Allahyari, H.; Heidari, S.; Ghamgosha, M.; Saffarian, P.; Amani, J. Immunotoxin: A new tool for cancer therapy. Tumour. Biol. 2017, 39, 1010428317692226. [Google Scholar] [CrossRef] [PubMed]
- Mazor, R.; Crown, D.; Addissie, S.; Jang, Y.; Kaplan, G.; Pastan, I. Elimination of murine and human T-cell epitopes in recombinant immunotoxin eliminates neutralizing and anti-drug antibodies in vivo. Cell Mol. Immunol. 2017, 14, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Weldon, J.E.; Xiang, L.; Chertov, O.; Margulies, I.; Kreitman, R.J.; FitzGerald, D.J.; Pastan, I. A protease-resistant immunotoxin against CD22 with greatly increased activity against CLL and diminished animal toxicity. Blood 2009, 113, 3792–3800. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Han, L.; Chen, J.; Xie, Y.; Jiang, H.; Zhu, J. Reduction of non-specific toxicity of immunotoxin by intein mediated reconstitution on target cells. Int. Immunopharmacol. 2019, 66, 288–295. [Google Scholar] [CrossRef]
- Pirzer, T.; Becher, K.S.; Rieker, M.; Meckel, T.; Mootz, H.D.; Kolmar, H. Generation of Potent Anti-HER1/2 Immunotoxins by Protein Ligation Using Split Inteins. ACS Chem. Biol. 2018, 13, 2058–2066. [Google Scholar] [CrossRef]
- Onda, M.; Beers, R.; Xiang, L.; Lee, B.; Weldon, J.E.; Kreitman, R.J.; Pastan, I. Recombinant immunotoxin against B-cell malignancies with no immunogenicity in mice by removal of B-cell epitopes. Proc. Natl. Acad. Sci. USA 2011, 108, 5742–5747. [Google Scholar] [CrossRef]
- Mazor, R.; Vassall, A.N.; Eberle, J.A.; Beers, R.; Weldon, J.E.; Venzon, D.J.; Tsang, K.Y.; Benhar, I.; Pastan, I. Identification and elimination of an immunodominant T-cell epitope in recombinant immunotoxins based on Pseudomonas exotoxin A. Proc. Natl. Acad. Sci. USA 2012, 109, E3597–E3603. [Google Scholar] [CrossRef]
- Mazor, R.; Eberle, J.A.; Hu, X.; Vassall, A.N.; Onda, M.; Beers, R.; Lee, E.C.; Kreitman, R.J.; Lee, B.; Baker, D.; et al. Recombinant immunotoxin for cancer treatment with low immunogenicity by identification and silencing of human T-cell epitopes. Proc. Natl. Acad. Sci. USA 2014, 111, 8571–8576. [Google Scholar] [CrossRef] [PubMed]
- Buchner, J.; Pastan, I.; Brinkmann, U. A method for increasing the yield of properly folded recombinant fusion proteins: Single-chain immunotoxins from renaturation of bacterial inclusion bodies. Anal. Biochem. 1992, 205, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Qaiser, H.; Aslam, F.; Iftikhar, S.; Farooq, A. Construction and recombinant expression of Pseudomonas aeruginosa truncated exotoxin A in Escherichia coli. Cell. Mol. Biol. 2018, 64, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Zanjani, L.S.; Shapouri, R.; Dezfulian, M.; Mahdavi, M.; Ardestani, M.S. Exotoxin A-PLGA nanoconjugate vaccine against Pseudomonas aeruginosa infection: Protectivity in murine model. World J. Microbiol. Biotechnol. 2019, 35, 94. [Google Scholar] [CrossRef]
- Seetharam, S.; Chaudhary, V.K.; FitzGerald, D.; Pastan, I. Increased cytotoxic activity of Pseudomonas exotoxin and two chimeric toxins ending in KDEL. J. Biol. Chem. 1991, 266, 17376–17381. [Google Scholar] [CrossRef] [PubMed]
- Chandramohan, V.; Pegram, C.N.; Piao, H.; Szafranski, S.E.; Kuan, C.T.; Pastan, I.H.; Bigner, D.D. Production and quality control assessment of a GLP-grade immunotoxin, D2C7-(scdsFv)-PE38KDEL, for a phase I/II clinical trial. Appl. Microbiol. Biotechnol. 2017, 101, 2747–2766. [Google Scholar] [CrossRef] [PubMed]
- Della Cristina, P.; Castagna, M.; Lombardi, A.; Barison, E.; Tagliabue, G.; Ceriotti, A.; Koutris, I.; Di Leandro, L.; Giansanti, F.; Vago, R.; et al. Systematic comparison of single-chain Fv antibody-fusion toxin constructs containing Pseudomonas Exotoxin A or saporin produced in different microbial expression systems. Microb. Cell Fact. 2015, 14, 19. [Google Scholar] [CrossRef]
- Tran, M.; Van, C.; Barrera, D.J.; Pettersson, P.L.; Peinado, C.D.; Bui, J.D.; Mayfield, S.P. Production of unique immunotoxin cancer therapeutics in algal chloroplasts. Proc. Natl. Acad. Sci. USA 2012, 110, E15–E22. [Google Scholar] [CrossRef]
- Lappi, D.A.; Esch, F.S.; Barbieri, L.; Stirpe, F.; Soria, M. Characterization of a Saponaria officinalis seed ribosome-inactivating protein: Immunoreactivity and sequence homologies. Biochem. Biophys. Res. Commun. 1985, 129, 934–942. [Google Scholar] [CrossRef]
- Pizzo, E.; Di Maro, A. A new age for biomedical applications of Ribosome Inactivating Proteins (RIPs): From bioconjugate to nanoconstructs. J. Biomed. Sci. 2016, 23, 54. [Google Scholar] [CrossRef]
- Santanché, S.; Bellelli, A.; Brunori, M. The Unusual Stability of Saporin, a Candidate for the Synthesis of Immunotoxins. Biochem. Biophys. Res. Commun. 1997, 234, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, A.; Tazzari, P.L.; Tassi, C.; Gromo, G.; Gobbi, M.; Stirpe, F. A comparison of anti-lymphocyte immunotoxins containing different ribosoma-inactivating proteins and antibodies. Clin. Exp. Immunol. 1992, 89, 341–346. [Google Scholar] [CrossRef]
- Maras, B.; Ippoliti, R.; De Luca, E.; Lendaro, E.; Bellelli, A.; Barra, D.; Bossa, F.; Brunori, M. The amino acid sequence of a ribosome-inactivating protein from Saponaria officinalis seeds. Biochem. Int. 1990, 21, 831–838. [Google Scholar]
- Endo, Y.; Tsurugi, K. The RNA N-glycosidase activity of ricin A-chain. The characteristics of the enzymatic activity of ricin A-chain with ribosomes and with rRNA. J. Biol. Chem. 1988, 263, 8735–8739. [Google Scholar] [CrossRef]
- Barbieri, L.; Battelli, M.G.; Stirpe, F. Ribosome-inactivating proteins from plants. Biochim. Biophys. Acta 1993, 1154, 237–282. [Google Scholar] [CrossRef]
- Bagga, S.; Seth, D.; Batra, J.K. The cytotoxic activity of ribosome-inactivating protein saporin-6 is attributed to its rRNA N-glycosidase and internucleosomal DNA fragmentation activities. J. Biol. Chem. 2003, 278, 4813–4820. [Google Scholar] [CrossRef]
- Stirpe, F. Ribosome-inactivating proteins: From toxins to useful proteins. Toxicon 2013, 67, 12–16. [Google Scholar] [CrossRef]
- Polito, L.; Bortolotti, M.; Farini, V.; Battelli, M.G.; Barbieri, L.; Bolognesi, A. Saporin induces multiple death pathways in lymphoma cells with different intensity and timing as compared to ricin. Int. J. Biochem. Cell Biol. 2009, 41, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, P.E.; Brown, A.N.; Bremner, J.A.G., Jr.; Foxwell, B.M.; Stirpe, F. An immunotoxin composed of monoclonal anti-Thy 1.1 antibody and a ribosome-inactivating protein from Saponaria officinalis: Potent antitumor effects in vitro and in vivo. J. Natl. Cancer Inst. 1985, 75, 151–159. [Google Scholar]
- Glennie, M.J.; McBride, H.M.; Stirpe, F.; Thorpe, P.E.; Worth, A.T.; Stevenson, G.T. Emergence of immunoglobulin variants following treatment of a B cell leukemia with an immunotoxin composed of antiidiotypic antibody and saporin. J. Exp. Med. 1987, 166, 43–62. [Google Scholar] [CrossRef] [PubMed]
- Siena, S.; Lappi, D.A.; Bregni, M.; Formosa, A.; Villa, S.; Soria, M.; Bonadonna, G.; Gianni, A.M. Synthesis and characterization of an antihuman T-lymphocyte saporin immunotoxin (OKT1-SAP) with in vivo stability into nonhuman primates. Blood 1988, 72, 756–765. [Google Scholar] [CrossRef]
- Tazzari, P.L.; Bolognesi, A.; De Totero, D.; Lemoli, R.M.; Fortuna, A.; Conte, R.; Crumpton, M.J.; Stirpe, F. Immunotoxins containing saporin linked to different CD2 monoclonal antibodies: In vitro evaluation. Br. J. Haematol. 1994, 86, 97–105. [Google Scholar] [CrossRef]
- Morland, B.J.; Barley, J.; Boehm, D.; Flavell, S.U.; Ghaleb, N.; Kohler, J.A.; Okayama, K.; Wilkins, B.; Flavell, D.J. Effectiveness of HB2 (anti-CD7)-Saporin immunotoxin in an in vivo model of human T-cell leukaemia developed in severe combined immunodeficient mice. Br. J. Cancer 1994, 69, 279–285. [Google Scholar] [CrossRef]
- Flavell, D.J.; Boehm, D.A.; Okayama, K.; Kohler, J.A.; Flavell, S.U. Therapy of human T-cell acute lymphoblastic leukaemia in severe combined immunodeficient mice with two different anti-CD7-saporin immunotoxins containing hindered or non-hindered disulphide cross-linkers. Int. J. Cancer 1994, 58, 407–414. [Google Scholar] [CrossRef]
- Flavell, D.J.; Flavell, S.U.; Boehm, D.; Emery, L.; Noss, A.; Ling, N.R.; Richardson, P.R.; Hardie, D.; Wright, D.H. Preclinical studies with the anti-CD19-saporin immunotoxin BU12-SAPORIN for the treatment of human-B-cell tumours. Br. J. Cancer 1995, 72, 1373–1379. [Google Scholar] [CrossRef]
- Flavell, D.J.; Boehm, D.A.; Noss, A.M.; Flavell, S.U. Comparison of the potency and therapeutic efficacy of the anti-CD7 immunotoxin HB2-saporin constructed with one or two saporin moieties per immunotoxin molecule. Br. J. Cancer 1997, 75, 1035–1043. [Google Scholar] [CrossRef]
- Flavell, D.J.; Warnes, S.; Noss, A.; Flavell, S.U. Host-mediated antibody-dependent cellular cytotoxicity contributes to the in vivo therapeutic efficacy of an anti-CD7-saporin immunotoxin in a severe combined immunodeficient mouse model of human T-cell acute lymphoblastic leukemia. Cancer Res. 1998, 58, 5787–5794. [Google Scholar]
- Flavell, D.J.; Warnes, S.L.; Noss, A.L.; Flavell, S.U. Anti-CD7 antibody and immunotoxin treatment of human CD7(+)T-cell leukaemia is significantly less effective in NOD/LtSz-scid mice than in CB.17 scid mice. Br. J. Cancer 2000, 83, 1755–1761. [Google Scholar] [CrossRef]
- Polito, L.; Bortolotti, M.; Mercatelli, D.; Battelli, M.G.; Bolognesi, A. Saporin-S6: A useful tool in cancer therapy. Toxins 2013, 5, 1698–1722. [Google Scholar] [CrossRef] [PubMed]
- Puri, M.; Kaur, I.; Perugini, M.A.; Gupta, R.C. Ribosome-inactivating proteins: Current status and biomedical applications. Drug Discov. Today 2012, 17, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, L.; Bolognesi, A.; Valbonesi, P.; Polito, L.; Olivieri, F.; Stirpe, F. Polynucleotide: Adenosine glycosidase activity of immunotoxins containing ribosome-inactivating proteins. J. Drug Target. 2000, 8, 281–288. [Google Scholar] [CrossRef]
- Qi, L.; Nett, T.M.; Allen, M.C.; Sha, X.; Harrison, G.S.; Frederick, B.A.; Crawford, E.D.; Glode, L.M. Binding and cytotoxicity of conjugated and recombinant fusion proteins targeted to the gonadotropin-releasing hormone receptor. Cancer Res. 2004, 64, 2090–2095. [Google Scholar] [CrossRef]
- Frankel, A.; Welsh, P.; Richardson, J.; Robertus, J.D. Role of arginine 180 and glutamic acid 177 of ricin toxin A chain in enzymatic inactivation of ribosomes. Mol. Cell. Biol. 1990, 10, 6257–6263. [Google Scholar] [CrossRef]
- Robertus, J.D.; Piatak, M.; Ferris, R.; Houston, L.L. Crystallization of ricin A chain obtained from a cloned gene expressed in Escherichia coli. J. Biol. Chem. 1987, 262, 19–20. [Google Scholar] [CrossRef]
- Habuka, N.; Akiyama, K.; Tsuge, H.; Miyano, M.; Matsumoto, T.; Noma, M. Expression and secretion of Mirabilis antiviral protein in Escherichia coli and its inhibition of in vitro eukaryotic and prokaryotic protein synthesis. J. Biol. Chem. 1990, 265, 10988–10992. [Google Scholar] [CrossRef]
- Kataoka, J.; Habuka, N.; Furuno, M.; Miyano, M.; Takanami, Y.; Koiwai, A. Expression of a pokeweed antiviral protein in Escherichia coli and its characterization. J. Biol. Chem. 1991, 266, 8426–8430. [Google Scholar] [CrossRef]
- Legname, G.; Fossati, G.; Monzini, N.; Gromo, G.; Marcucci, F.; Mascagni, P.; Modena, D. Heterologous expression, purification, activity and conformational studies of different forms of dianthin 30. Biomed. Pept. Proteins Nucleic Acids 1995, 1, 61–68. [Google Scholar]
- Shaw, P.C.; Yun, M.H.; Zhu, R.H.; Ho, W.K.K.; Ng, T.B.; Yeung, H.W. Cloning of trichosanthin cDNA and its expression in Escherichia coli. Gene 1991, 97, 267–272. [Google Scholar] [CrossRef]
- Bass, H.W.; Krawetz, J.E.; OBrian, G.R.; Zinselmeier, C.; Habben, J.E.; Boston, R.S. Maize ribosome-inactivating proteins (RIPs) with distinct expression patterns have similar requirements for proenzyme activation. J. Exp. Bot. 2004, 55, 2219–2233. [Google Scholar] [CrossRef]
- Ding, G.B.; Wu, G.; Li, B.; Yang, P.; Li, Z. High-yield expression in Escherichia coli, biophysical characterization, and biological evaluation of plant toxin gelonin. 3 Biotech 2019, 9, 19. [Google Scholar] [CrossRef]
- Hartley, M.R.; Legname, G.; Osborn, R.; Chen, Z.; Lord, J.M. Single-chain ribosome inactivating proteins from plants depurinate Escherichia coli 23S ribosomal RNA. FEBS Lett. 1991, 290, 65–68. [Google Scholar] [CrossRef]
- Barthelemy, I.; Martineau, D.; Ong, M.; Matsunami, R.; Ling, N.; Benatti, L.; Cavallaro, U.; Soria, M.; Lappi, D.A. The expression of saporin, a ribosome-inactivating protein from the plant Saponaria officinalis, in Escherichia coli. J. Biol. Chem. 1993, 268, 6541–6548. [Google Scholar] [CrossRef]
- Fabbrini, M.S.; Rappocciolo, E.; Carpani, D.; Solinas, M.; Valsasina, B.; Breme, U.; Cavallaro, U.; Nykjaer, A.; Rovida, E.; Legname, G.; et al. Characterization of a saporin isoform with lower ribosome-inhibiting activity. Biochem. J. 1997, 322, 719–727. [Google Scholar] [CrossRef]
- Benatti, L.; Saccardo, M.B.; Dani, M.; Nitti, G.P.; Sassano, M.; Lorenzetti, R.; Lappi, D.A.; Soria, M. Nucleotide sequence of cDNA coding for saporin-6, a type-1 ribosome-inactivating protein from Saponaria officinalis. Eur. J. Biochem. 1989, 183, 465–470. [Google Scholar] [CrossRef]
- Benatti, L.; Nitti, G.; Solinas, M.; Valsasina, B.; Vitale, A.; Ceriotti, A.; Soria, M.R. A Saporin-6 cDNA containing a precursor sequence coding for a carboxyl-terminal extension. FEBS Lett. 1991, 291, 285–288. [Google Scholar] [CrossRef]
- Pittaluga, E.; Poma, A.; Tucci, A.; Spanò, L. Expression and characterisation in E. coli of mutant forms of saporin. J. Biotechnol. 2005, 117, 263–266. [Google Scholar] [CrossRef]
- Günhan, E.; Swe, M.; Palazoglu, M.; Voss, J.C.; Chalupa, L.M. Expression and purification of cysteine introduced recombinant saporin. Protein Expr. Purif. 2008, 58, 203–209. [Google Scholar] [CrossRef][Green Version]
- Giansanti, F.; di Leandro, L.; Koutris, I.; Pitari, G.; Fabbrini, M.S.; Lombardi, A.; Flavel, D.J.; Flavell, S.U.; Gianni, S.; Ippoliti, R. Engineering a switchable toxin: The potential use of PDZ domains in the expression, targeting and activation of modified saporin variants. Protein Eng. Des. Sel. 2010, 23, 61–68. [Google Scholar] [CrossRef]
- Giansanti, F.; Sabatini, D.; Pennacchio, M.R.; Scotti, S.; Angelucci, F.; Dhez, A.C.; Antonosante, A.; Cimini, A.; Giordano, A.; Ippoliti, R. PDZ Domain in the Engineering and Production of a Saporin Chimeric Toxin as a Tool for targeting Cancer Cells. J. Cell. Biochem. 2015, 116, 1256–1266. [Google Scholar] [CrossRef]
- Weng, A.; Thakur, M.; von Mallinckrodt, B.; Beceren-Braun, F.; Gilabert-Oriol, R.; Wiesner, B.; Eichhorst, J.; Böttger, S.; Melzig, M.F.; Fuchs, H. Saponins modulate the intracellular trafficking of protein toxins. J. Control Release 2012, 164, 74–86. [Google Scholar] [CrossRef]
- Zuppone, S.; Assalini, C.; Minici, C.; Botrugno, O.A.; Curnis, F.; Degano, M.; Corti, A.; Montorsi, F.; Salonia, A.; Vago, R.A. Novel RGD-4C-Saporin Conjugate Inhibits Tumor Growth in Mouse Models of Bladder Cancer. Front. Oncol. 2022, 12, 846958. [Google Scholar] [CrossRef]
- Lombardi, A.; Bursomanno, S.; Lopardo, T.; Traini, R.; Colombatti, M.; Ippoliti, R.; Flavell, D.J.; Flavell, S.U.; Ceriotti, A.; Fabbrini, M.S. Pichia pastoris as a host for secretion of toxic saporin chimeras. FASEB J. 2010, 24, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Mattanovich, D.; Branduardi, P.; Dato, L.; Gasser, B.; Sauer, M.; Porro, D. Recombinant protein production in yeasts. In Methods in Molecular Biology (Clifton, N.J.); Humana Press: Totowa, NJ, USA, 2012; Volume 824, pp. 329–358. [Google Scholar]
- Heisler, I.; Keller, J.; Tauber, R.; Sutherland, M.; Fuchs, H. A cleavableadapter to reduce nonspecific cytotoxicity of recombinant immuno-toxins. Int. J. Cancer 2003, 103, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Vitetta, E.S. Immunotoxins and vascular leak syndrome. Cancer J. 2000, 6, 218–224. [Google Scholar]
- Chaudhary, V.K.; Gallo, M.G.; FitzGerald, D.J.; Pastan, I. A recombinant single-chain immunotoxin composed of anti-Tac variable regions and a truncated diphtheria toxin. Proc. Natl. Acad. Sci. USA 1990, 87, 9491–9494. [Google Scholar] [CrossRef]
- Madhumathi, J.; Verma, R.S. Therapeutic targets and recent advances in protein immunotoxins. Curr. Opin. Microbiol. 2012, 15, 300–309. [Google Scholar] [CrossRef]
- Fabbrini, M.S.; Flavell, D.J.; Ippoliti, R. Plant protein toxins: Structure, function and biotechnological applications. In Bacterial, Plant and Animal Toxins; Ascenzi, P.P.F., Visca, P., Eds.; Research Signpost: Kerala, India, 2003; pp. 66–69. [Google Scholar]
- Fabbrini, M.S.; Carpani, D.; Bello-Rivero, I.; Soria, M.R. The amino-terminal fragment of human urokinase directs a recombinant chimeric toxin to target cells: Internalization is toxin mediated. FASEB J. 1997, 11, 1169–1176. [Google Scholar] [CrossRef]
- Errico Provenzano, A.; Posteri, R.; Giansanti, F.; Angelucci, F.; Flavell, S.U.; Flavell, D.J.; Fabbrini, M.S.; Porro, D.; Ippoliti, R.; Ceriotti, A.; et al. Optimization of construct design and fermentation strategy for the production of bioactive ATF-SAP, a saporin based anti-tumoral uPAR-targeted chimera. Microb. Cell Fact. 2016, 15, 194. [Google Scholar] [CrossRef]
- Marshall, R.S.; D’Avila, F.; Di Cola, A.; Traini, R.; Spano, L.; Fabbrini, M.S.; Ceriotti, A. Signal peptide-regulated toxicity of a plant ribosome-inactivating protein during cell stress. Plant J. 2011, 65, 218–229. [Google Scholar] [CrossRef]
- Frigerio, L.; Vitale, A.; Lord, J.M.; Ceriotti, A.; Roberts, L.M. Free ricin A chain, proricin, and native toxin have different cellular fates when expressed in tobacco protoplasts. J. Biol. Chem. 1998, 273, 14194–14199. [Google Scholar] [CrossRef]
- Krishnan, R.; McDonald, K.A.; Dandekar, A.M.; Jackman, A.P.; Falk, B. Expression of recombinant trichosanthin, a ribosome-inactivating protein, in transgenic tobacco. J. Biotechnol. 2002, 97, 69–88. [Google Scholar] [CrossRef]
- Zarovni, N.; Vago, R.; Soldà, T.; Monaco, L.; Fabbrini, M.S. Saporin as a novel suicide gene in anticancer gene therapy. Cancer Gene Ther. 2007, 14, 165–173. [Google Scholar] [CrossRef]
- Zarovni, N.; Vago, R.; Fabbrini, M.S. Saporin suicide gene therapy. Methods Mol. Biol. 2009, 542, 261–283. [Google Scholar] [CrossRef] [PubMed]
- Min, K.A.; He, H.; Yang, V.C.; Shin, M.C. Construction and characterization of gelonin and saporin plasmids for toxic gene-based cancer therapy. Arch. Pharm. Res. 2016, 39, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Salvioni, L.; Zuppone, S.; Andreata, F.; Monieri, M.; Mazzucchelli, S.; Di Carlo, C.; Morelli, L.; Cordiglieri, C.; Donnici, L.; De Francesco, R.; et al. Nanoparticle-Mediated Suicide Gene Therapy for Triple Negative Breast Cancer Treatment. Adv. Therap. 2020, 3, 2000007. [Google Scholar] [CrossRef]
- di Leandro, L.; Giansanti, F.; Mei, S.; Ponziani, S.; Colasante, M.; Ardini, M.; Angelucci, F.; Pitari, G.; d’Angelo, M.; Cimini, A.; et al. Aptamer-Driven Toxin Gene Delivery in U87 Model Glioblastoma Cells. Front. Pharmacol. 2021, 12, 588306. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, X.-P.; Kahn, J.N.; Tumer, N.E. Functional Assays for Measuring the Catalytic Activity of Ribosome Inactivating Proteins. Toxins 2018, 10, 240. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.W.; Tang, Y.S.; Sze, S.Y.; Zhu, Z.N.; Wong, K.B.; Shaw, P.C. Crystal Structure of Ribosome-Inactivating Protein Ricin A Chain in Complex with the C-Terminal Peptide of the Ribosomal Stalk Protein P2. Toxins 2016, 8, 296. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Kawasaki, T.; Sako, N.; Funatsu, G. Actions of pokeweed antiviral protein on virus-infected protoplasts. Biosci. Biotechnol. Biochem. 1997, 61, 994–997. [Google Scholar] [CrossRef]
- Gilabert-Oriol, R.; Weng, A.; Mallinckrodt, B.; Melzig, M.F.; Fuchs, H.; Thakur, M. Immunotoxins constructed with ribosome-inactivating proteins and their enhancers: A lethal cocktail with tumor specific efficacy. Curr. Pharm. Des. 2014, 20, 6584–6643. [Google Scholar] [CrossRef]
- Bortolotti, M.; Polito, L.; Bolognesi, A. Toxin and Immunotoxin Based Therapeutic Approaches. Toxins 2022, 14, 63. [Google Scholar] [CrossRef] [PubMed]
- Polito, L.; Djemil, A.; Bortolotti, M. Plant Toxin-Based Immunotoxins for Cancer Therapy: A Short Overview. Biomedicines 2016, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Słomińska-Wojewódzka, M.; Sandvig, K. Ricin and Ricin-Containing Immunotoxins: Insights into Intracellular Transport and Mechanism of action in Vitro. Antibodies 2013, 2, 236–269. [Google Scholar] [CrossRef]
- Schnell, R.; Borchmann, P.; Staak, J.O.; Schindler, J.; Ghetie, V.; Vitetta, E.S.; Engert, A. Clinical evaluation of ricin A-chain immunotoxins in patients with Hodgkin’s lymphoma. Ann. Oncol. 2003, 14, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Messmann, R.A.; Vitetta, E.S.; Headlee, D.; Senderowicz, A.M.; Figg, W.D.; Schindler, J.; Michiel, D.F.; Creekmore, S.; Steinberg, S.M.; Kohler, D.; et al. A phase I study of combination therapy with immunotoxins IgG-HD37-deglycosylated ricin A chain (dgA) and IgG-RFB4-dgA (Combotox) in patients with refractory CD19(+), CD22(+) B cell lymphoma. Clin. Cancer Res. 2000, 6, 1302–1313. [Google Scholar] [PubMed]
- LoRusso, P.M.; Lomen, P.L.; Redman, B.G.; Poplin, E.; Bander, J.J.; Valdivieso, M. Phase I study of monoclonal antibody-ricin A chain immunoconjugate Xomazyme-791 in patients with metastatic colon cancer. Am. J. Clin. Oncol. 1995, 18, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Engert, A.; Diehl, V.; Schnell, R.; Radszuhn, A.; Hatwig, M.T.; Drillich, S.; Schön, G.; Bohlen, H.; Tesch, H.; Hansmann, M.L.; et al. A phase-I study of an anti-CD25 ricin A-chain immunotoxin (RFT5-SMPT-dgA) in patients with refractory Hodgkin’s lymphoma. Blood 1997, 89, 403–410. [Google Scholar] [CrossRef]
- de Virgilio, M.; Degryse, B. Harnessing the Destructive Power of Ricin to Fight Human Cancer. In Ricin Toxin; John, W.C., Ed.; Bentham Science Publishers: Soest, The Netherlands, 2014; pp. 208–237. [Google Scholar] [CrossRef]
- Olsnes, S.; Pihl, A. Molecular Action of Toxins and Viruses; Cohen, P., Van Heyningen, S., Eds.; Elsevier Scientific Publishing Co., Inc.: New York, NY, USA, 1982; pp. 51–105. [Google Scholar]
- Naemi, A.A.; Salmanian, A.H.; Noormohammadi, Z.; Amani, J. A novel EGFR-specific recombinant ricin-panitumumab (scFv) immunotoxin against breast and colorectal cancer cell lines; in silico and in vitro analyses. Eur. J. Pharmacol. 2023, 955, 175894. [Google Scholar] [CrossRef]
- Park, S.G.; Kim, H.; Jun, H.; Choi, S.Y.; Kim, E.; Kang, S. Directing ricin-based immunotoxins with targeting affibodies and KDEL signal peptide to cancer cells effectively induces apoptosis and tumor suppression. J. Nanobiotechnology 2022, 20, 387. [Google Scholar] [CrossRef]
- Mlsna, D.; Monzingo, A.F.; Katzin, B.J.; Ernst, S.; Robertus, J.D. Structure of recombinant ricin A chain at 2.3 Å. Protein Sci. 1993, 2, 429–435. [Google Scholar] [CrossRef]
- O’Hare, M.; Roberts, L.M.; Thorpe, P.E.; Watson, G.J.; Prior, B.; Lord, J.M. Expression of ricin a chain in Escherichia coli. FEBS Lett. 1987, 216, 73–78. [Google Scholar] [CrossRef]
- FitzGerald, D.J.; Bjorn, M.J.; Ferris, R.J.; Winkelhake, J.L.; Frankel, A.E.; Hamilton, T.C.; Ozols, R.F.; Willingham, M.C.; Pastan, I. Antitumor activity of an immunotoxin in a nude mouse model of human ovarian cancer. Cancer Res. 1987, 47, 1407–1410. [Google Scholar] [PubMed]
- Li, B.Y.; Ramakrishnan, S. Recombinant hybrid toxin with dual enzymatic activities. Potential use in preparing highly effective immunotoxins. J. Biol. Chem. 1994, 269, 2652–2658. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.; Ge, L.; Shen, J.; Wang, K.; Zheng, S. A trans-Golgi network retention signal YQRL fused to ricin A chain significantly enhances its cytotoxicity. Biochem. Biophys. Res. Commun. 2004, 313, 1053–1057. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.; Stayton, P.; Press, O.W. Modification of ricin A chain, by addition of endoplasmic reticulum (KDEL) or Golgi (YQRL) retention sequences, enhances its cytotoxicity and translocation. Cancer Immunol. Immunother. 1998, 46, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Tagge, E.; Chandler, J.; Tang, B.L.; Hong, W.; Willingham, M.C.; Frankel, A. Cytotoxicity of KDEL-terminated ricin toxins correlates with distribution of the KDEL receptor in the Golgi. J. Histochem. Cytochem. 1996, 44, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, R.; Dianat-Moghadam, H.; Poorebrahim, M.; Siapoush, S.; Poortahmasebi, V.; Salahlou, R.; Rahmati, M. Recombinant immunotoxins development for HER2-based targeted cancer therapies. Cancer Cell Int. 2021, 21, 470. [Google Scholar] [CrossRef]
- Westby, M.; Argent, R.H.; Pitcher, C.; Lord, J.M.; Roberts, L.M. Preparation and characterization of recombinant proricin containing an alternative protease-sensitive linker sequence. Bioconjugate Chem. 1992, 3, 375–381. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Pastan, I. Recombinant toxins. Adv. Pharmacol. 1994, 28, 193–219. [Google Scholar]
- Benhar, I.; Pastan, I. Cloning, expression and characterization of the Fv fragments of the anti-carbohydrate mAbs B1 and B5 as single-chain immunotoxins. Protein Eng. 1994, 7, 1509–1515. [Google Scholar] [CrossRef]
- Sørensen, H.P.; Mortensen, K.K. Soluble expression of recombinant proteins in the cytoplasm of Escherichia coli. Microb. Cell Fact. 2005, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Hassan, Y.; Ogg, S. Gene cloning and construction of prokaryotic and plant expression vectors of RICIN-A-Chain/PAP-S1 fusion protein and its inhibition of protein synthesis. bioRxiv 2016. [Google Scholar] [CrossRef]
- Hassan, Y.; Ogg, S. Expression of Pokeweed Antiviral Protein Isoform S1 (PAP-S1) And of Ricin-A-Chain/PAP-S1 Novel Fusion Protein (RTA/PAP-S1) In Escherichia coli And Their Comparative Inhibition of Protein Synthesis In Vitro. bioRxiv 2017. [Google Scholar] [CrossRef]
- Hassan, Y.; Ogg, S.; Ge, H. Expression of novel fusion antiviral proteins ricin a chain-pokeweed antiviral proteins (RTA-PAPs) in Escherichia coli and their inhibition of protein synthesis and of hepatitis B virus in vitro. BMC Biotechnol. 2018, 18, 47. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.P.; Savage, P.M.; Lord, J.M.; Roberts, L.M. Biologically active interleukin 2-ricin A chain fusion proteins may require intracellular proteolytic cleavage to exhibit a cytotoxic effect. Bioconjugate Chem. 1993, 4, 440–447. [Google Scholar] [CrossRef]
- Asrorov, A.M.; Gu, Z.; Min, K.A.; Shin, M.C.; Huang, Y. Advances on Tumor-Targeting Delivery of Cytotoxic Proteins. ACS Pharmacol. Transl. Sci. 2019, 3, 107–118. [Google Scholar] [CrossRef]
- Bortolotti, M.; Bolognesi, A.; Polito, L. Bouganin, an Attractive Weapon for Immunotoxins. Toxins 2018, 10, 323. [Google Scholar] [CrossRef]
- Bolognesi, A.; Polito, L.; Olivieri, F.; Valbonesi, P.; Barbieri, L.; Battelli, M.G.; Carusi, M.V.; Benvenuto, E.; Del Vecchio Blanco, F.; Di Maro, A.; et al. New Ribosome-Inactivating Proteins with Polynucleotide: Adenosine Glycosidase and Antiviral Activities from Basella Rubra, L. and Bougainvillea Spectabilis Willd. Planta 1997, 203, 422–429. [Google Scholar] [CrossRef]
- Den Hartog, M.T.; Lubelli, C.; Boon, L.; Heerkens, S.; Ortiz Buijsse, A.P.; de Boer, M.; Stirpe, F. Cloning and Expression of cDNA Coding for Bouganin. Eur. J. Biochem. 2002, 269, 1772–1779. [Google Scholar] [CrossRef]
- Fermani, S.; Tosi, G.; Farini, V.; Polito, L.; Falini, G.; Ripamonti, A.; Barbieri, L.; Chambery, A.; Bolognesi, A. Structure/Function Studies on Two Type 1 Ribosome Inactivating Proteins: Bouganin and Lychnin. J. Struct. Biol. 2009, 168, 278–287. [Google Scholar] [CrossRef]
- Cizeau, J.; Grenkow, D.M.; Brown, J.G.; Entwistle, J.; MacDonald, G.C. Engineering and Biological Characterization of VB6-845, an Anti-EpCAM Immunotoxin Containing a T-Cell Epitope-Depleted Variant of the Plant Toxin Bouganin. J. Immunother. 2009, 32, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Dillon, R.L.; Chooniedass, S.; Premsukh, A.; Adams, G.P.; Entwistle, J.; MacDonald, G.C.; Cizeau, J. Trastuzumab-deBouganin Conjugate Overcomes Multiple Mechanisms of T-DM1 Drug Resistance. J. Immunother. 2016, 39, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Chooniedass, S.; Dillon, R.L.; Premsukh, A.; Adams, G.P.; MacDonald, G.C.; Cizeau, J. Abstract 79: Trastuzumab and C6.5 Diabody Armed with deBouganin Overcome Drug Resistance to ADCs Comprised of Anti-Microtubule Agents. Cancer Res. 2017, 77, 79. [Google Scholar] [CrossRef]
- Kowalski, M.; Brazas, L.; Zaretsky, R.; Rasamoelisolo, M.; MacDonald, G.; Cuthbert, W.; Glover, N. A Phase I Study of VB6–845, an Anti-EpCAM Fusion Protein Targeting Advanced Solid Tumours of Epithelial Origin: Preliminary Results. J. Clin. Oncol. 2008, 26, 14663. [Google Scholar] [CrossRef]
- Silva, A.L.; Goto, L.S.; Dinarte, A.R.; Hansen, D.; Moreira, R.A.; Beltramini, L.M.; Araújo, A.P. Pulchellin, a Highly Toxic Type 2 Ribosome-inactivating Protein from Abrus Pulchellus: Cloning, Heterologous Expression of A-chain and Structural Studies. FEBS J. 2005, 272, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Castilho, P.V.; Goto, L.S.; Roberts, L.M.; Araújo, A.P.U. Isolation and Characterization of Four Type 2 Ribosome Inactivating Pulchellin Isoforms from Abrus Pulchellus Seeds. FEBS J. 2008, 275, 948–959. [Google Scholar] [CrossRef]
- Sdraeian, M.; Guimarães, F.E.G.; Araújo, A.P.U.; Worthylake, D.K.; LeCour, L.J.; Pincus, S.H. Selective Cytotoxicity of a Novel Immunotoxin Based on Pulchellin A Chain for Cells Expressing HIV Envelope. Sci. Rep. 2017, 7, 7579. [Google Scholar] [CrossRef]
- Amorim, F.G.; Cordeiro, F.A.; Pinheiro-Júnior, E.L.; Boldrini-França, J.; Arantes, E.C. Microbial production of toxins from the scorpion venom: Properties and applications. Appl. Microbiol. Biotechnol. 2018, 102, 6319–6331. [Google Scholar] [CrossRef]
- Chen, N.; Xu, S.; Zhang, Y.; Wang, F. Animal protein toxins: Origins and therapeutic applications. Biophys. Rep. 2018, 4, 233–242. [Google Scholar] [CrossRef]
- Rivera-de-Torre, E.; Rimbault, C.; Jenkins, T.P.; Sørensen, C.V.; Damsbo, A.; Saez, N.J.; Duhoo, Y.; Hackney, C.M.; Ellgaard, L.; Laustsen, A.H. Strategies for Heterologous Expression, Synthesis, and Purification of Animal Venom Toxins. Front. Bioeng. Biotechnol. 2022, 9, 811905. [Google Scholar] [CrossRef]
- Qian, C.Y.; Wang, K.L.; Fang, F.F.; Gu, W.; Huang, F.; Wang, F.Z. Triple-controlled oncolytic adenovirus expressing melittin to exert inhibitory efficacy on hepatocellular carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 10403–10411. [Google Scholar]
- Su, M.; Chang, W.; Cui, M.; Lin, Y.; Wu, S.; Xu, T. Expression and anticancer activity analysis of recombinant human uPA1-43-melittin. Int. J. Oncol. 2015, 46, 619–626. [Google Scholar] [CrossRef]
- Su, M.; Chang, W.; Zhang, K.; Cui, M.; Wu, S.; Xu, T. Expression and purification of recombinant ATF-mellitin, a new type fusion protein targeting ovarian cancer cells, in P. pastoris. Oncol. Rep. 2016, 35, 1179–1185. [Google Scholar] [CrossRef]
- Shin, M.C.; Minm, K.A.; Cheong, H.; Moon, C.; Huang, Y.; He, H.; Yang, V.C. Preparation and characterization of gelonin-melittin fusion biotoxin for synergistically enhanced anti-tumor activity. Pharm. Res. 2016, 33, 2218–2228. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zong, J.; Liu, Z.; Li, L.; Zheng, X.; Wang, B.; Sun, G. A novel melittin-MhIL-2 fusion protein inhibits the growth of human ovarian cancer SKOV3 cells in vitro and in vivo tumor growth. Cancer Immunol. Immunother. 2013, 62, 889–895. [Google Scholar] [CrossRef]
- Liu, H.; Han, Y.; Fu, H.; Liu, M.; Wu, J.; Chen, X.; Zhang, S.; Chen, Y. Construction and expression of sTRAIL-melittin combining enhanced anticancer activity with antibacterial activity in Escherichia coli. Appl. Microbiol. Biotechnol. 2013, 97, 2877–2884. [Google Scholar] [CrossRef] [PubMed]
- Holle, L.; Song, W.; Holle, E.; Wei, Y.; Li, J.; Wagner, T.E.; Yu, X. In vitro- and in vivo-targeted tumor lysis by an MMP2 cleavable melittin-LAP fusion protein. Int. J. Oncol. 2009, 35, 829–835. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhao, X.; Yu, Z.; Dai, W.; Yao, Z.; Zhou, W.; Zhou, W.; Zhou, J.; Yang, Y.; Zhu, Y.; Chen, S.; et al. Construction and characterization of an anti-asialoglycoprotein receptor single-chain variable-fragment-targeted melittin. Biotechnol. Appl. Biochem. 2011, 58, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.Q.; Li, B.; Zhang, C.; Zhu, D.Z.; Huang, X.Q.; Gu, W.; Li, S.X. Inhibitory effect of recombinant adenovirus carrying melittin gene on hepatocellular carcinoma. Ann. Oncol. 2005, 16, 109–115. [Google Scholar] [CrossRef]
- Qu, L.; Jiang, M.; Li, Z.; Pu, F.; Gong, L.; Sun, L.; Gong, R.; Ji, G.; Si, J. Inhibitory Effect of Biosynthetic Nanoscale Peptide Melittin on Hepatocellular Carcinoma, Driven by Survivin Promoter. J. Biomed. Nanotechnol. 2014, 10, 695–706. [Google Scholar] [CrossRef]
- Jin, H.; Li, C.; Li, D.; Cai, M.; Li, Z.; Wang, S.; Hong, X.; Shi, B. Construction and characterization of a CTLA-4-targeted scFv-melittin fusion protein as a potential immunosuppressive agent for organ transplant. Cell Biochem. Biophys. 2013, 67, 1067–1074. [Google Scholar] [CrossRef]
- Dunn, R.D.; Weston, K.M.; Longhurst, T.J.; Lilley, G.G.; Rivett, D.E.; Hudson, P.J.; Raison, R.L. Antigen binding and cytotoxic properties of a recombinant immunotoxin incorporating the lytic peptide, melittin. Immunotechnology 1996, 3, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Choo, A.B.; Dunn, R.D.; Broady, K.W.; Raison, R.L. Soluble expression of a functional recombinant cytolytic immunotoxin in insect cells. Protein Expr. Purif. 2002, 3, 338–347. [Google Scholar] [CrossRef]
- Kong, G.M.; Zhang, X.R.; Wu, K.Y.; Liao, Y.X.; Bo, P. Anti-proliferative activity of recombinant melittin expressed in Escherichia coli toward U937 cells. Afr. J. Biotechnol. 2012, 11, 3026–3030. [Google Scholar] [CrossRef]
- Wang, D.; Hu, L.; Su, M.; Wang, J.; Xu, T. Preparation and functional characterization of human vascular endothelial growth factor-melittin fusion protein with analysis of the antitumor activity in vitro and in vivo. Int. J. Oncol. 2015, 47, 1160–1168. [Google Scholar] [CrossRef]
- Wang, W.; Luo, J.; Xu, L.; Zeng, J.; Cao, L.; Dong, J.; Cai, S. Expression of scFv-Mel-Gal4 triple fusion protein as a targeted DNA-carrier in Escherichia coli. Cell Biochem. Funct. 2013, 31, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Qian, D.; Shao, G.; Yan, Z.; Li, R.; Hua, X.; Song, X.; Wang, B. High-level expression, purification and study of bioactivity of fusion protein M-IL-2 (88)Arg, (125)Ala in Pichia pastoris. Protein Expr. Purif. 2014, 101, 99–105. [Google Scholar] [CrossRef]
- Klocek, G.; Schulthess, T.; Shai, Y.; Seelig, J. Thermodynamics of melittin binding to lipid bilayers: Aggregation and pore formation. Biochemistry 2009, 48, 2586–2596. [Google Scholar] [CrossRef]
- Wimley, W.C. How does melittin permeabilize membranes? Biophys. J. 2018, 114, 251–253. [Google Scholar] [CrossRef]
- Yang, Z.; Choi, H.; Weisshaar, J.C. Melittin-induced permeabilization, re-sealing, and re-permeabilization of E. coli membranes. Biophys. J. 2017, 114, 368–379. [Google Scholar] [CrossRef]
- Krauson, A.J.; He, J.; Wimley, W.C. Gain-of-function analogues of the pore-forming peptide melittin selected by orthogonal high-throughput screening. J. Am. Chem. Soc. 2012, 134, 12732–12741. [Google Scholar] [CrossRef] [PubMed]
- Hristova, K.; Dempsey, C.E.; White, S.H. Structure, location, and lipid perturbations of melittin at the membrane interface. Biophys. J. 2001, 80, 801–881. [Google Scholar] [CrossRef]
- Son, D.J.; Lee, J.W.; Lee, Y.H.; Song, H.S.; Lee, C.K.; Hong, J.T. Therapeutic application of anti-arthritis, pain-releasing, and anti-cancer effects of bee venom and its constituent compounds. Pharmacol. Ther. 2007, 115, 246–270. [Google Scholar] [CrossRef]
- Russell, P.J.; Hewish, D.; Carter, T.; Sterling-Levis, K.; Ow, K.; Hattarki, M.; Doughty, L.; Guthrie, R.; Shapira, D.; Molloy, P.L.; et al. Cytotoxic properties of immunoconjugates containing melittin-like peptide 101 against prostate cancer: In vitro and in vivo studies. Cancer Immunol. Immunother. 2004, 53, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Badawi, J.K. Bee Venom Components as Therapeutic Tools against Prostate Cancer. Toxins 2021, 13, 337. [Google Scholar] [CrossRef] [PubMed]
- Rady, I.; Siddiqui, I.A.; Rady, M.; Mukhtar, H. Melittin, a major peptide component of bee venom, and its conjugates in cancer therapy. Cancer Lett. 2017, 402, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Soman, N.R.; Schlesinger, P.H.; Lanza, G.M.; Wickline, S.A. Cytolytic peptide nanoparticles (‘NanoBees’) for cancer therapy. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2011, 3, 318–327. [Google Scholar] [CrossRef]
- DeGrado, W.F.; Musso, G.F.; Lieber, M.; Kaiser, E.T.; Kezdy, F.J. Kinetics and mechanism of hemolysis induced by melittin and by a synthetic melittin analogue. Biophys. J. 1982, 37, 329–338. [Google Scholar] [CrossRef]
- Soman, N.R.; Lanza, G.M.; Heuser, J.M.; Schlesinger, P.H.; Wickline, S.A. Synthesis and characterization of stable fluorocarbon nanostructures as drug delivery vehicles for cytolytic peptides. Nano Lett. 2008, 8, 1131–1136. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, Z.; Xu, G.; Li, L.; Xuan, K.; Xu, Y.; Zhang, R. Expression of Melittin in Fusion with GST in Escherichia coli and Its Purification as a Pure Peptide with Good Bacteriostatic Efficacy. ACS Omega 2020, 5, 9251–9258. [Google Scholar] [CrossRef]
- Rayahin, J.E.; Buhrman, J.S.; Gemeinhart, R.A. Melittin-glutathione S-transferase fusion protein exhibits anti-inflammatory properties and minimal toxicity. Eur. J. Pharm. Sci. 2014, 65, 112–121. [Google Scholar] [CrossRef]
- Shi, W.J.; Xu, H.J.; Cheng, J.A.; Zhang, C.X. Expression of the melittin gene of Apis cerana cerana in Escherichia coli. Protein Expr. Purif. 2004, 37, 213–219. [Google Scholar] [CrossRef]
- Chen, Q.; Liu, L.; Yu, T.; Tang, L.; Yi, M.; Zhu, W.; Jiang, X.; Wang, H. High-Level Expression and Purification of Melittin in Escherichia coli Using SUMO Fusion Partner. Int. J. Pept. Res. Ther. 2021, 27, 9–15. [Google Scholar] [CrossRef]
- Liu, M.; Wang, H.; Liu, L.; Wang, B.; Sun, G. Melittin-MIL-2 fusion protein as a candidate for cancer immunotherapy. J. Transl. Med. 2016, 14, 155. [Google Scholar] [CrossRef] [PubMed]
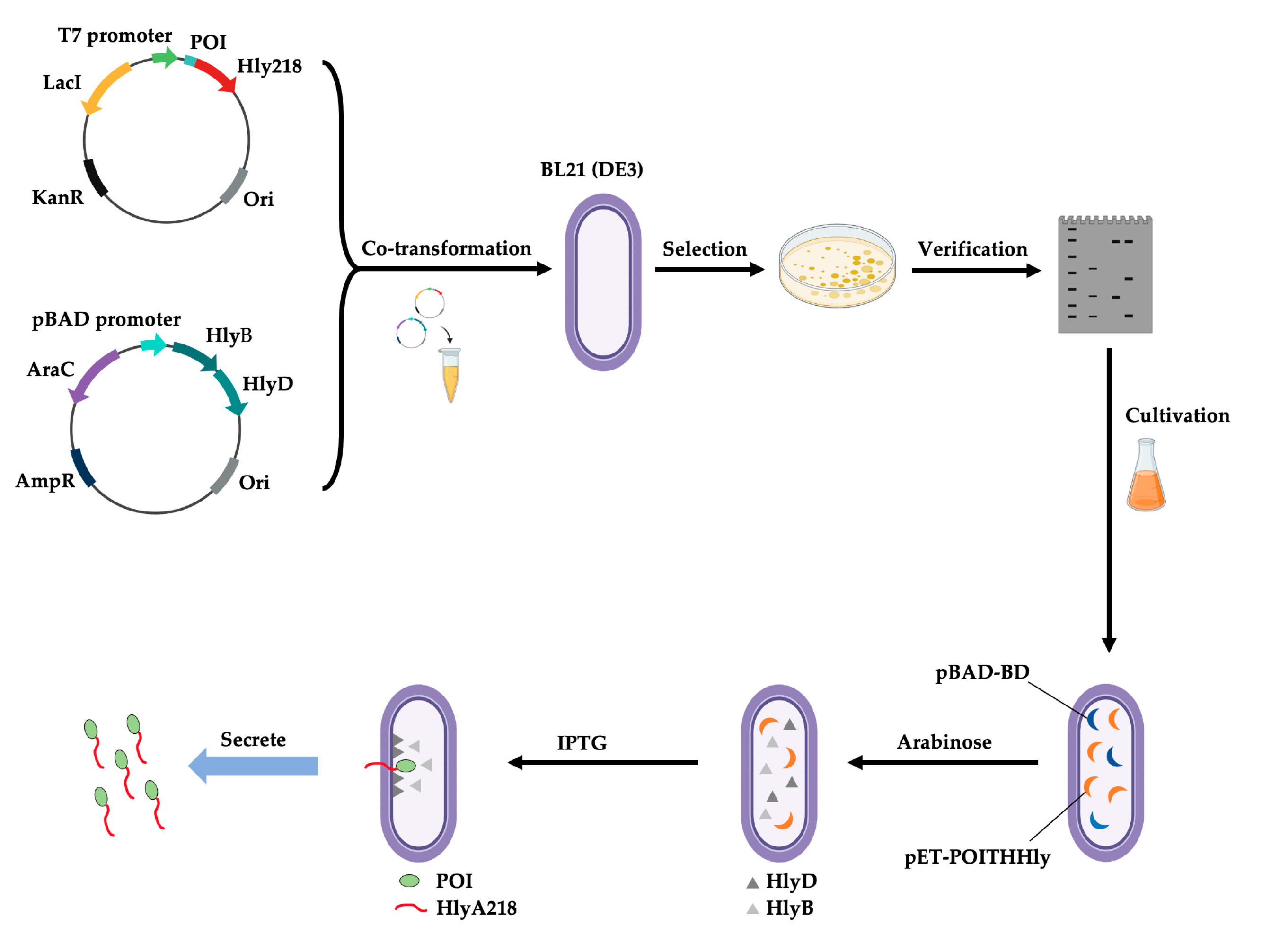
- Zhu, W.; Wang, Y.; Lv, L.; Wang, H.; Shi, W.; Liu, Z.; Yang, W.; Zhu, J.; Lu, H. SHTXTHHly, an extracellular secretion platform for the preparation of bioactive peptides and proteins in Escherichia coli. Microb. Cell Fact. 2022, 21, 128. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Li, W.; Baloch, A.R.; Wang, M.; Li, H.; Cao, B.; Zhang, H. Efficient biosynthesis of a cecropin A-melittin mutant in Bacillus subtilis WB700. Sci. Rep. 2017, 7, 40587. [Google Scholar] [CrossRef]
- Kim, H.; Park, S.Y.; Lee, G. Potential therapeutic applications of bee venom on skin disease and its mechanisms: A literature review. Toxins 2019, 11, 374. [Google Scholar] [CrossRef] [PubMed]
- Moridi, K.; Hemmaty, M.; Eidgahi, M.R.A.; Najafi, M.F.; Zare, H.; Ghazvini, K.; Neshani, A. Construction, cloning, and expression of Melittin antimicrobial peptide using Pichia pastoris expression system. Gene Rep. 2020, 21, 100900. [Google Scholar] [CrossRef]
- Cregg, J.M.; Tolstorukov, I.; Kusari, A.; Sunga, J.; Madden, K.; Chappell, T. Chapter 13 expression in the yeast Pichia pastoris. Methods Enzymol. 2009, 463, 169–189. [Google Scholar]
- Neshani, A.; Tanhaeian, A.; Zare, H.; Eidgahi, M.R.A.; Ghazvini, K. Preparation and evaluation of a new biopesticide solution candidate for plant disease control using pexiganan gene and Pichia pastoris expression system. Gene Rep. 2019, 17, 100509. [Google Scholar] [CrossRef]
- Lopes, M.; Oliveira, C.; Domingues, L.; Mota, M.; Belo, I. Enhanced heterologous protein production in Pichia pastoris under increased air pressure. Biotechnol. Prog. 2014, 30, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Tarahomjoo, S.; Bandehpour, M.; Aghaebrahimian, M.; Ahangaran, S. Soluble Diphtheria Toxin Variant, CRM 197 was Obtained in Escherichia coli at High Productivity Using SUMO Fusion and an Adjusted Expression Strategy. Protein Pept. Lett. 2022, 29, 350–359. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Host | DT Fusion Protein/Immunotoxin | Target | References |
|---|---|---|---|
| E. coli | DAB486IL-2 | Tumor cells | [26] |
| E. coli Rosetta Gami (DE3) | DAB389IL-2 (Denileukin DiftitoxTM) | Cutaneous form of T-cell lymphoma | [27] |
| P. pastoris | DAB389-IL2IL2 | Human CD25(+) cells (regulatory T cells) | [28] |
| C. diphteria | s-DAB-IL2(V6A) | PD-1 in melanoma cells | [29] |
| E. coli BL21 (DE3) | DT388-GM-CSF | Acute myeloid leukemia blasts | [30] |
| E. coli BLR (DE3) | DT388-IL3 | Acute myeloid leukemia blasts | [31] |
| E. coli | DT389-EGF | Human glioblastoma multiforme cells | [32] |
| E. coli | DT389-YP7 | Hepatocellular cancer cells | [33] |
| P. pastoris | A-dmDT390-bisFV | T-cell leukemia and autoimmune diseases | [34] |
| E. coli | DT2219 | Human CD19 and CD22 receptors in a mouse xenograft model of B-cell leukemia/lymphoma | [35] |
| E. coli | DTAT/DTAT13/DTATEG | Urokinase-type plasminogen activator receptor on human glioblastoma tumors | [36] |
| E. coli | DTEGF13 | Pancreatic cancer cells | [37] |
| P. pastoris | DT-antiCCR4 | Human CCR4(+) cells | [38] |
| E. coli | DT386-BR2 | Tumor cells | [39] |
| E. coli | DT389GCSF | Granulocyte colony-stimulating factor (G-CSF) receptor | [40] |
| E. coli | DL9F and DL2F | Human ovarian teratocarcinoma | [41] |
| E. coli | DT389-GRP | Small cell lung cancer cells | [42] |
| 293T cells | DAB389-IL7 | IL-7(+) tumor cells | [43] |
| E. coli | DT390-biTMTP1/DT390-triTMTP1 | Highly metastatic tumors | [44] |
| E. coli | DT-SCF | c-kit(+) tumor cells | [45] |
| E. coli BL21 (DER) | DTIL13 | Human glioblastoma multiforme cells | [46] |
| Host | Recombinant MEL Constructs | Target | References |
|---|---|---|---|
| Adenovirus | QG511-HA–melittin | - | [219] |
| P. pastoris | RhuPA1-43–melittin | Hepatocellular carcinoma cells | [220] |
| P. pastoris | rATF–melittin | Ovarian cancer cells | [221] |
| E.coli | Gelonin–melittin | Ovarian cancer cells | [222] |
| E. coli | Melittin–MhIL-2 | Tumor cells | [223] |
| E. coli | sTRAIL–melittin | Human ovarian cancer SKOV cells | [224] |
| Adenovirus | MEL-MMP2-LAP | Leukemia cells and liver carcinoma cells | [225] |
| E. coli | C1–melittin immunotoxin | Ovarian cancer cells | [226] |
| Adenovirus | Ad-rAFP-Mel | Hepatocellular carcinoma | [227] |
| Adenovirus | pSURV–Mel (non-viral vector) | Hepatocellular carcinoma | [228] |
| E. coli | anti-CTLA-4-scFv-melittin | Hepatocellular carcinoma | [229] |
| E. coli | scFvK121-melittin | Potential immunosuppressive agent for organ transplant | [230] |
| Baculovirus | scFv-mel-FLAG | KMA(+) tumor cells | [231] |
| E. coli | GST-melittin | Human lymphoblastoid cells | [232] |
| P. pastoris | VEGF165-melittin | - | [233] |
| E. coli | scFv-Mel-Gal4 | Hepatocellular carcinoma | [234] |
| P. pastoris | M-IL-2((88)Arg, (125)Ala) | Liver diseases | [235] |
| Toxin | Recombinant Toxins | Expression Sistem | Compartmentalization | Yield | Reference | Year |
|---|---|---|---|---|---|---|
| Diphtheria Toxin | Full protein | Bacteria | ||||
| E. coli BLR (DE3) | Cytoplasmatic, inclusion bodies | 1.84 mg/mL | [31] | 2004 | ||
| E. coli BL21 (DE3) PlysS | - | 1.2 mg/mL | [21] | 2022 | ||
| CRM197 | Corynebacterium diphtheriae | Secreted | 175–250 mg/mL | [16] | 1983 | |
| Bacillus subtilis | Secreted | 7.1 mg/mL | [66] | 1999 | ||
| E.coli BL21AI | Cytoplasmatic, inclusion bodies | 7.1 mg/mL | [59] | 2011 | ||
| E. coli | Cytoplasmatic | 154 mg/mL | [60] | 2016 | ||
| E. coli B843 (DE3) | Periplasmic | >3 mg/mL | [58] | 2017 | ||
| E. coli | Cytoplasmatic, soluble | 106 mg/L | [62] | 2017 | ||
| E. coli ClearColi BL21 (DE3) | Cytoplasmatic, inclusion bodies | 196 mg/mL | [54] | 2018 | ||
| E. coli | Cytoplasmatic, soluble | 130 mg/mL | [260] | 2022 | ||
| E. coli Shuffle T7 | Cytoplasmatic, soluble | 150–270 mg/L | [10] | 2023 | ||
| Yeast | ||||||
| Pichia pastoris | Secreted | >100 mg/L | [7] | 2021 | ||
| Bacteria | ||||||
| Exotoxin A | Full protein | Pseudomonas aeruginosa, PAO1 | Secreted | - | [107] | 2018 |
| PE38KDEL | E. coli BL21 (XDEB) | Cytoplasmatic, inclusion bodies | 1 mg/mL | [109] | 1991 | |
| PE40-antiCD22 | E. coli | Cytoplasmatic, inclusion bodies | 3 mg/L | [111] | 2015 | |
| D2C7-(scdsFv)-PE38KDEL | E.coli BLR DE3 | Cytoplasmatic, inclusion bodies | 30 mg/L | [110] | 2017 | |
| Bacteria | ||||||
| Saporin S6 | Full protein | E. coli strain JA221 | Periplasmic | 4 ug/L | [146] | 1993 |
| E. coli BL21 (DE3) PlysS | Soluble fraction | 1–3 mg/L | [163] | 1997 | ||
| pET-E176K | pET-K234stop | E. coli BL21 (DE3) | Soluble fraction | 0.1–0.3 mg/L | [150] | 2005 | |
| SAP-Ser255Cys | E. coli BL21 (DE3) PlysS | Soluble fraction | 2.7 mg/L | [151] | 2008 | |
| SapVSAV | E. coli BL21 (DE3) PlysS | Cytosolic fraction | - | [152] | 2010 | |
| hCASK- SAP | (hCASK)2-SAP | E. coli Rosetta GamiTM B pLysS(DE3) | BL21 (DE3) | Soluble fraction | 0.5 mg/L | [153] | 2015 | |
| CYS- SAP | RGD-SAP | E. coli BL21 (DE3) | Soluble fraction | 0.6–1.2 mg/L | [155] | 2022 | |
| Yeast | ||||||
| ATF-Saporin | Pichia pastoris | Secreted | 3.5 mg/L | [156] | 2010 | |
| SAP-antiCD22 | Pichia pastoris GS115 | Secreted | 1–2 mg/mL | [111] | 2015 | |
| ATF-SAP chimera | Pichia pastoris GS115 | Secreted | 1–5 mg/L * | [164] | 2016 | |
| Secreted | 3–7 mg/L * | [164] | 2016 | |||
| Bacteria | ||||||
| Ricin A chain | Full protein | E. coli 7118 | Secreted | 2–3 mg/L | [189] | 1987 |
| RTA–KDEL | RTA-YQRL | E. coli JM 109 | Cytoplasmatic, soluble | 10 mg/L | [193] | 1998 | |
| rRTA | rRTA–YQRL | E. coli JM 109 | Cytoplasmatic, soluble | 10 mg/L | [192] | 2004 | |
| RTA-PAP1 | E. coli | Cytoplasmatic, inclusion bodies | 0.22 mg/mL | [202] | 2018 | |
| RTAM-PAP1 | E.coli | Soluble fraction | 0.1 mg/mL | [202] | 2018 | |
| Ricin-panitumumab | E. coli BL21 (DE3) | Cytoplasmatic, inclusion bodies | 0.18 mg/mL | [186] | 2023 | |
| Bacteria | ||||||
| Bouganin | Full protein | E. coli BL21 (DE3) | Periplasmic | 1 mg/L | [207] | 2002 |
| Trastuzumab-deBouganin Conjugate | E. coli | - | 0.15–0.5 mg/L | [210] | 2016 | |
| Bacteria | ||||||
| Pulchellin | rPAC | E. coli | Soluble fraction | 3 mg/L | [213] | 2005 |
| Melittin | sTRAIL–melittin | E. coli BL21 (DE3) | Cytoplasmatic, inclusion bodies | - | [224] | 2013 |
| rGel-Mel | E. coli BL21 star (DE3) | Cytoplasmatic, soluble | 3 mg/L | [222] | 2016 | |
| Cecropin A-melittin mutant | Bacillus subtilis WB700 | Secreted | 159 mg/L | [254] | 2017 | |
| GST-MET | E. coli BL21 (DE3) | Cytoplasmatic, soluble | 3.5 mg/L | [248] | 2020 | |
| Full protein | E. coli BL21 (DE3) | Cytoplasmatic, soluble | 25 mg/L | [251] | 2021 | |
| Yeast | ||||||
| rhuPA1-43-melittin | Pichia pastoris | Secreted | 128 mg/L | [220] | 2015 | |
| rATF-mellitin | Pichia pastoris | Secreted | 312 mg/L | [221] | 2016 | |
| Cecropin A-melittin mutant | Pichia pastoris | Secreted | 125 mg/L | [254] | 2017 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
di Leandro, L.; Colasante, M.; Pitari, G.; Ippoliti, R. Hosts and Heterologous Expression Strategies of Recombinant Toxins for Therapeutic Purposes. Toxins 2023, 15, 699. https://doi.org/10.3390/toxins15120699
di Leandro L, Colasante M, Pitari G, Ippoliti R. Hosts and Heterologous Expression Strategies of Recombinant Toxins for Therapeutic Purposes. Toxins. 2023; 15(12):699. https://doi.org/10.3390/toxins15120699
Chicago/Turabian Styledi Leandro, Luana, Martina Colasante, Giuseppina Pitari, and Rodolfo Ippoliti. 2023. "Hosts and Heterologous Expression Strategies of Recombinant Toxins for Therapeutic Purposes" Toxins 15, no. 12: 699. https://doi.org/10.3390/toxins15120699
APA Styledi Leandro, L., Colasante, M., Pitari, G., & Ippoliti, R. (2023). Hosts and Heterologous Expression Strategies of Recombinant Toxins for Therapeutic Purposes. Toxins, 15(12), 699. https://doi.org/10.3390/toxins15120699