A Colonic Organoid Model Challenged with the Large Toxins of Clostridioides difficile TcdA and TcdB Exhibit Deregulated Tight Junction Proteins

 , , ,
, , ,  , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
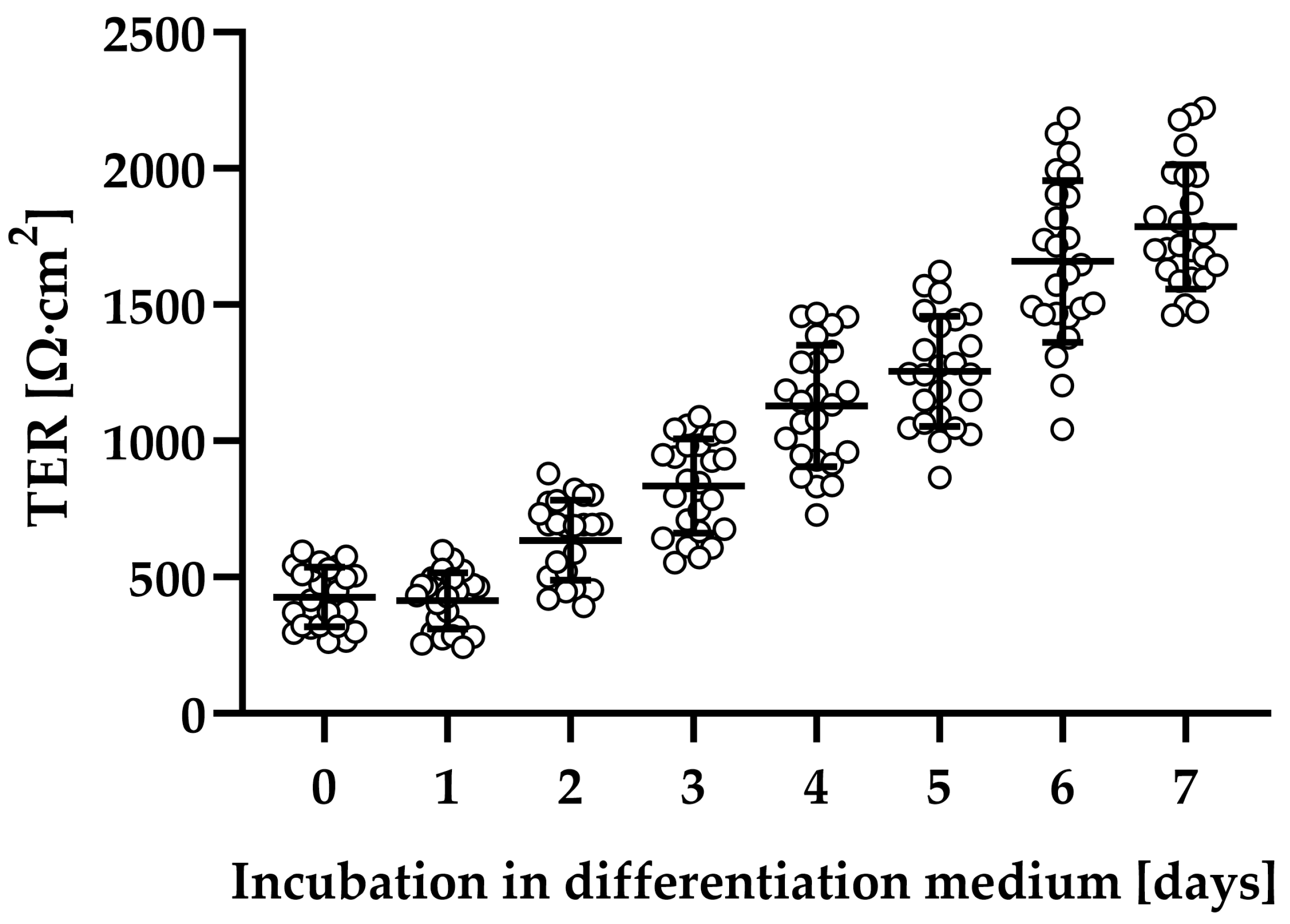
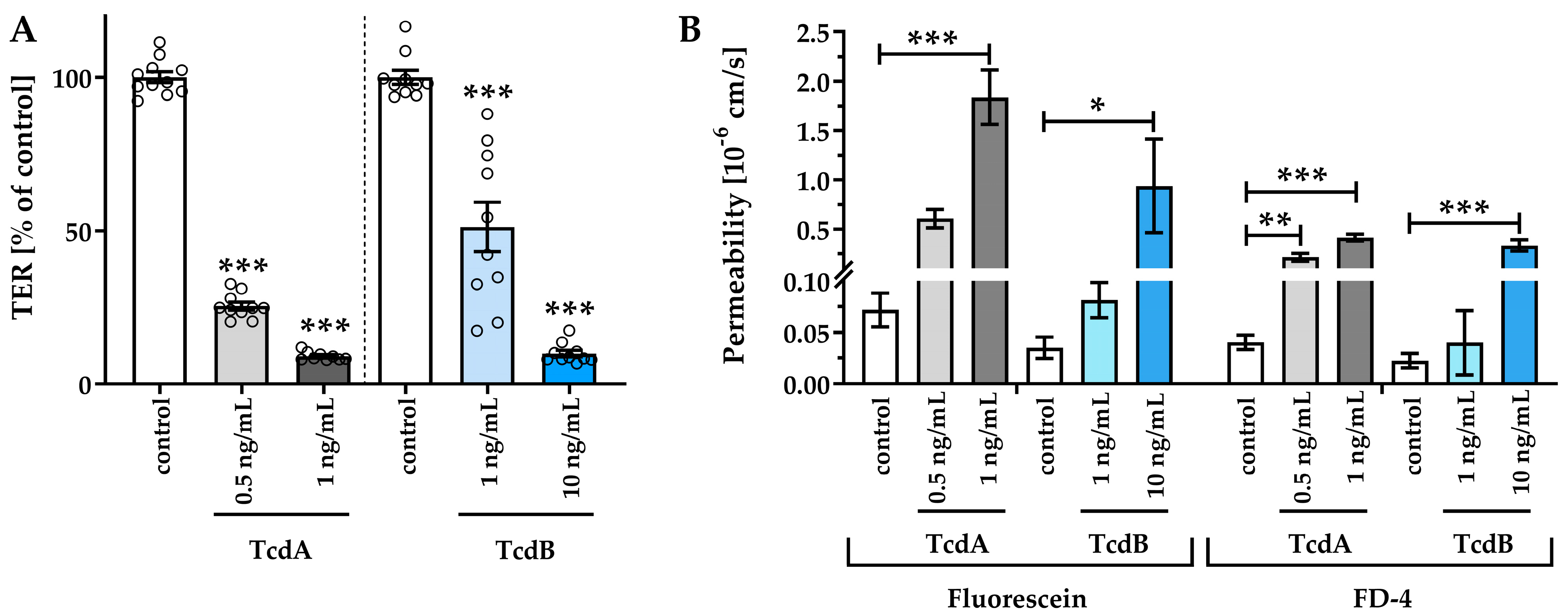
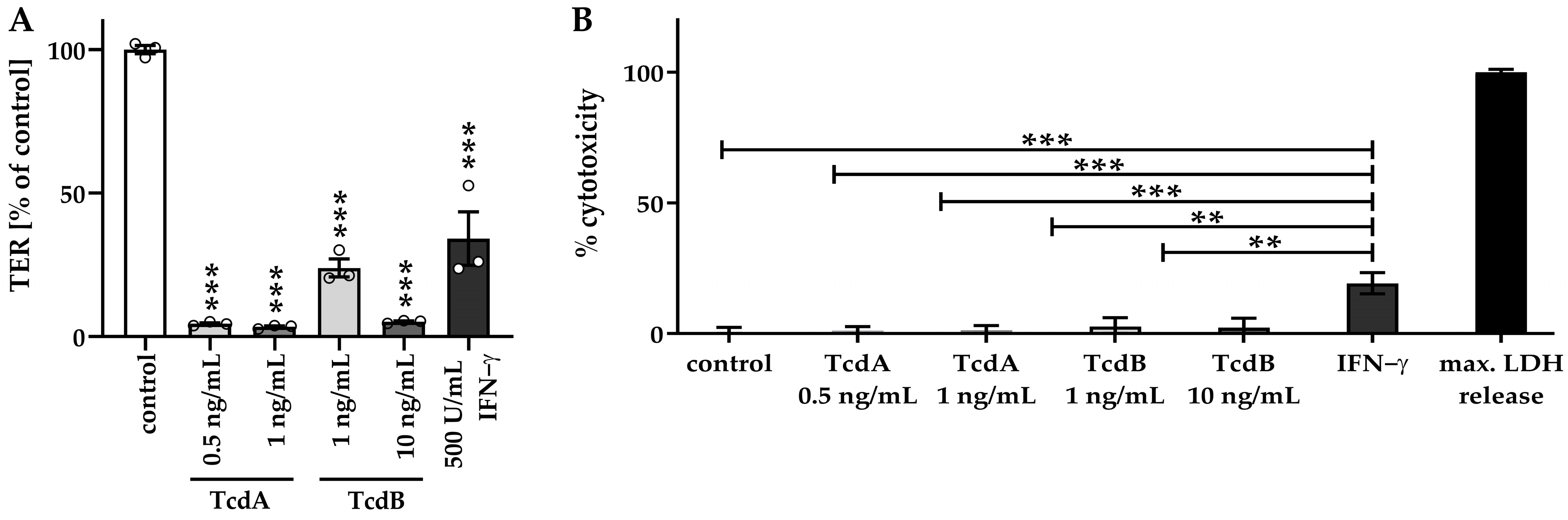
2.1. Clostridial Toxins TcdA and TcdB Caused Barrier Dysfunction in Adult Stem Cell-Derived Human Colonic Organoid Monolayers
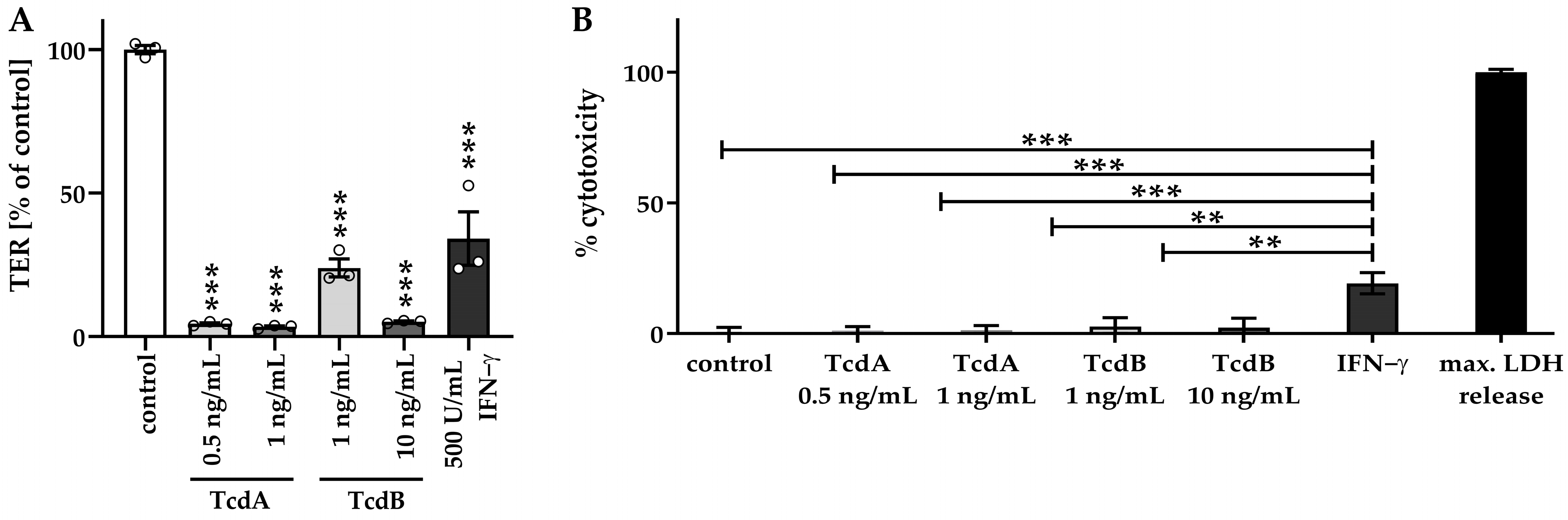
2.2. Impaired Barrier Function Emerges before Induction of Necrosis
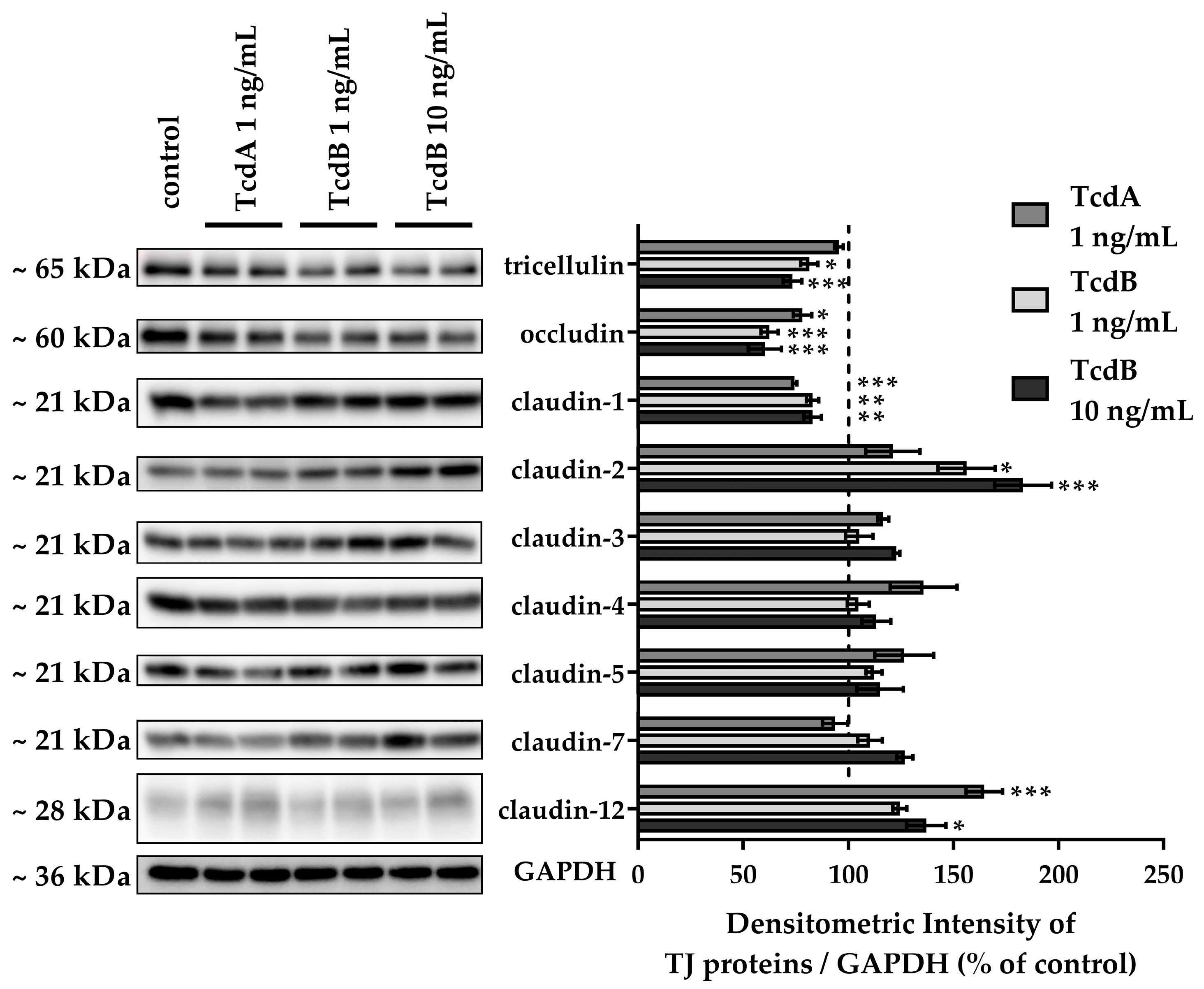
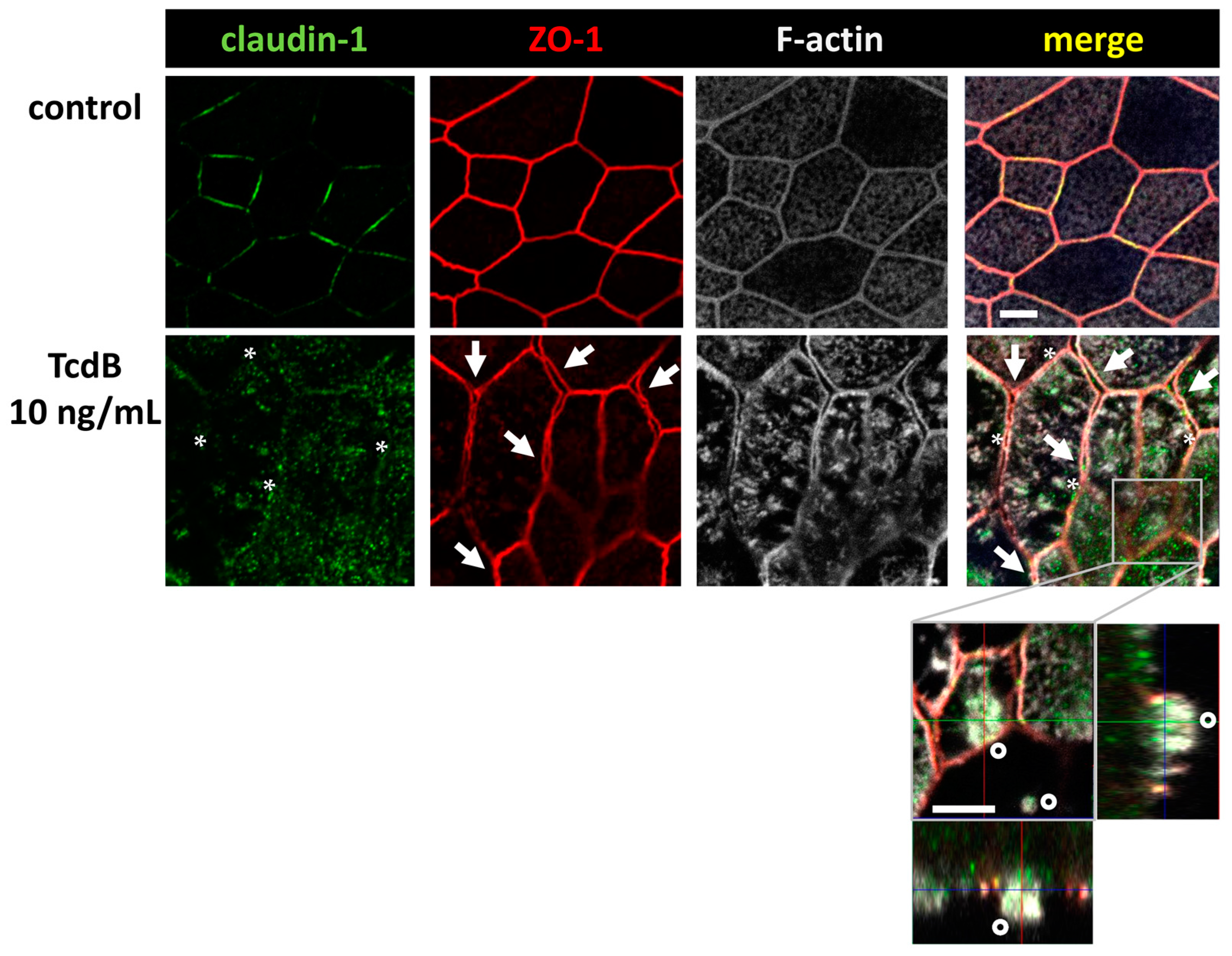
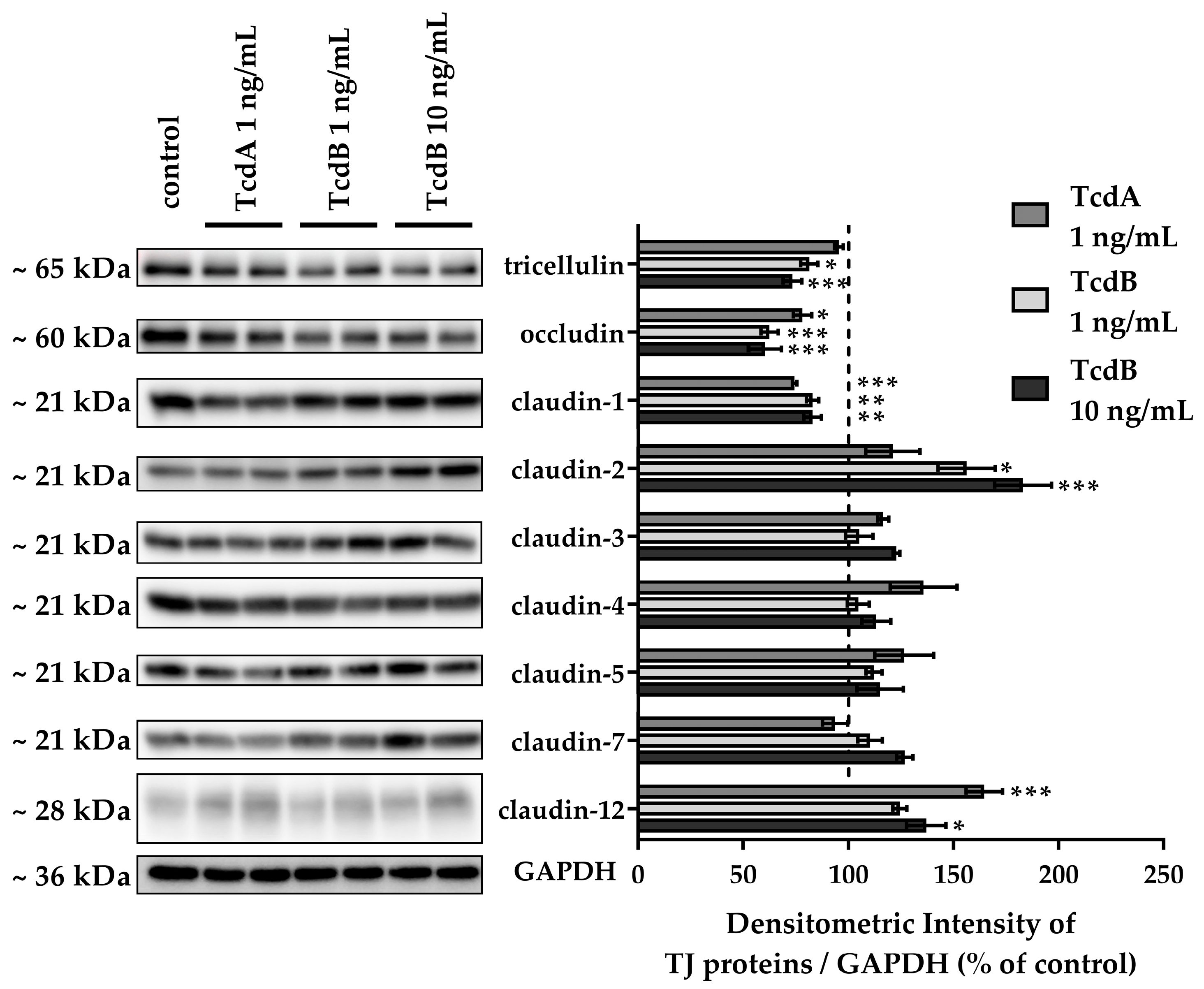
2.3. Down-Regulation of Tight Junction Proteins Claudin-1, Occludin, and Tricellulin via TcdB Caused Disruption of the Epithelial Barrier
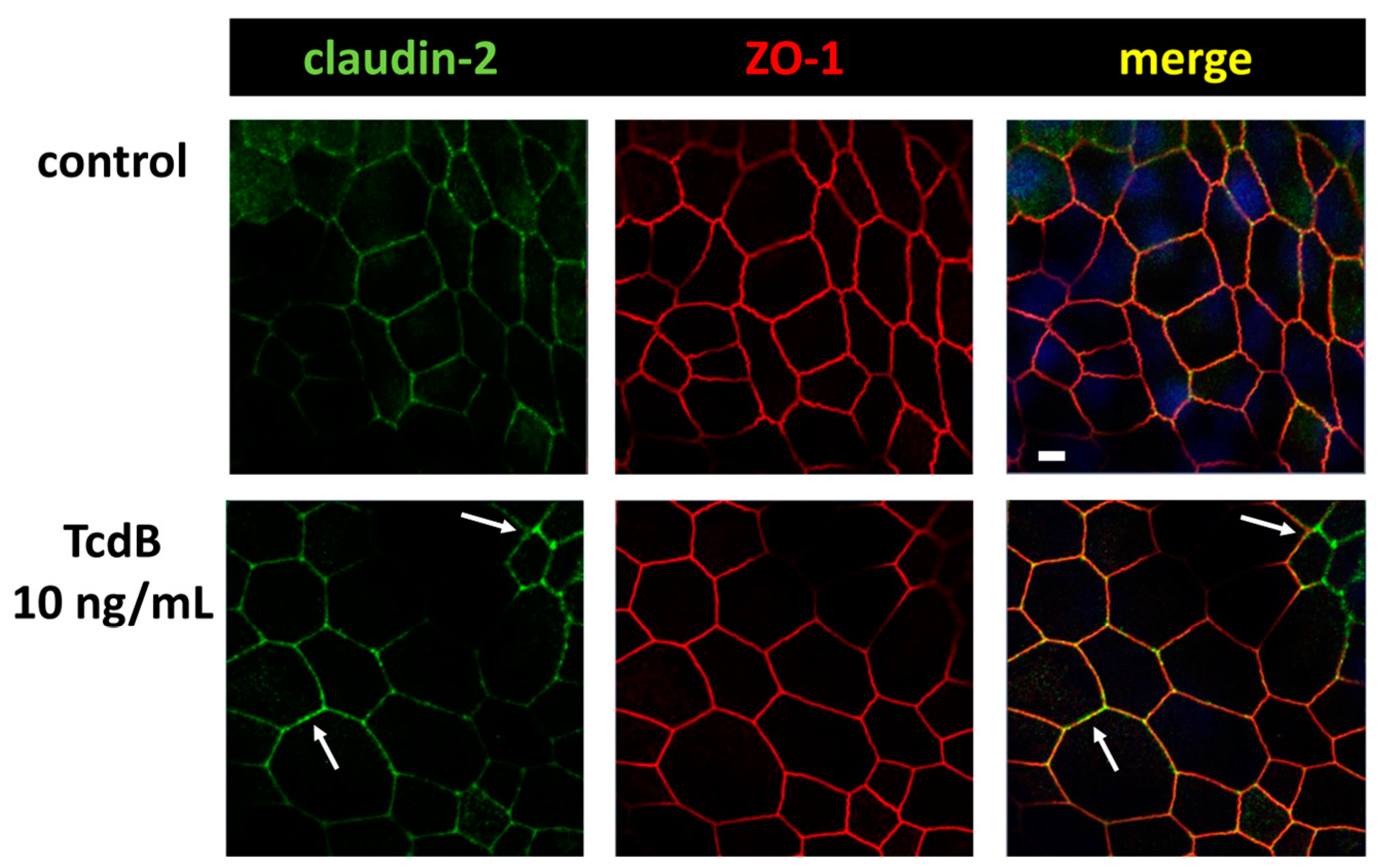
2.4. In TJ Domain Integrated Claudin-2 as Defense Reaction towards TcdB
2.5. Cytokine Measurements in Toxin-Exposed Monolayers
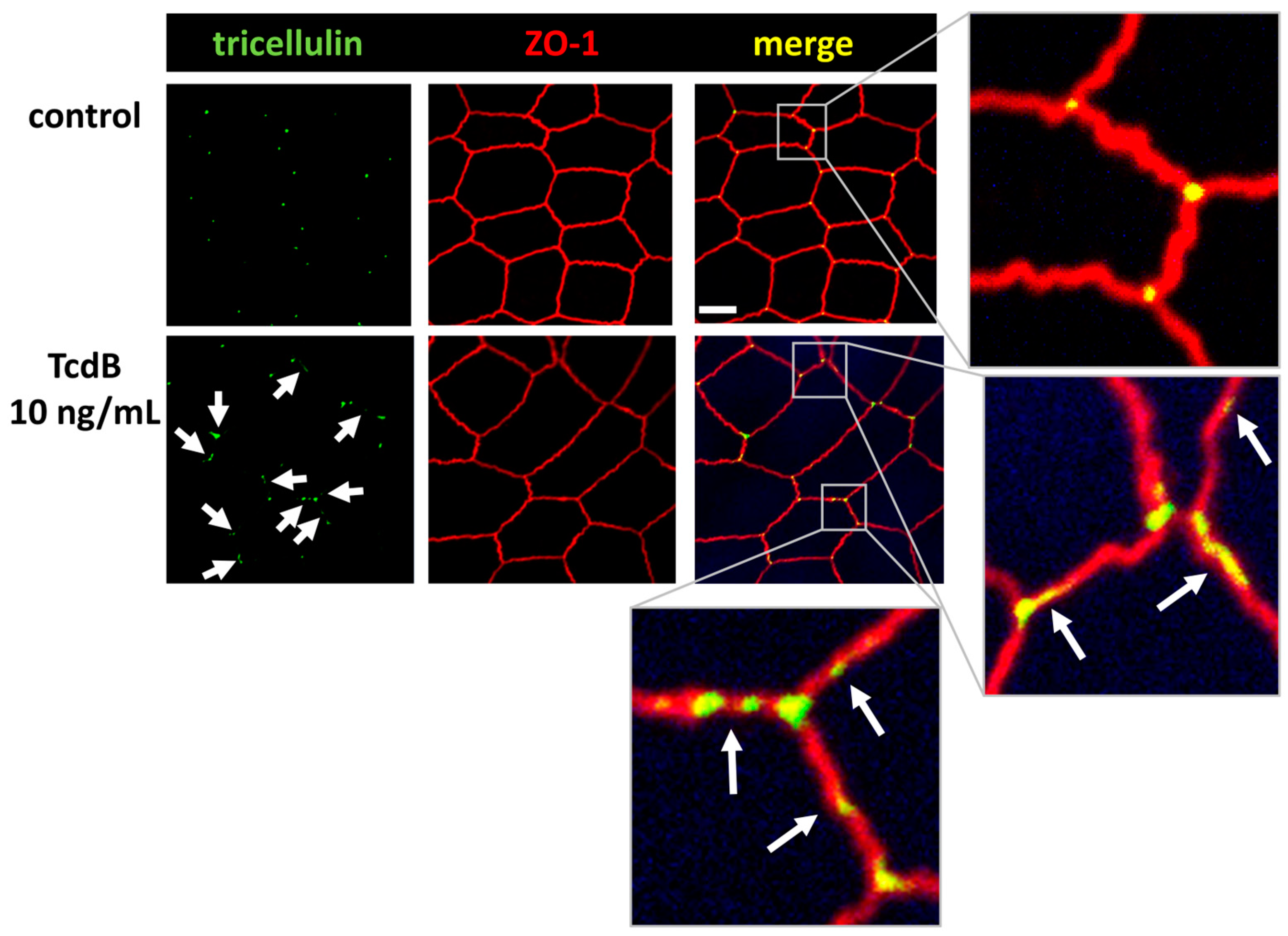
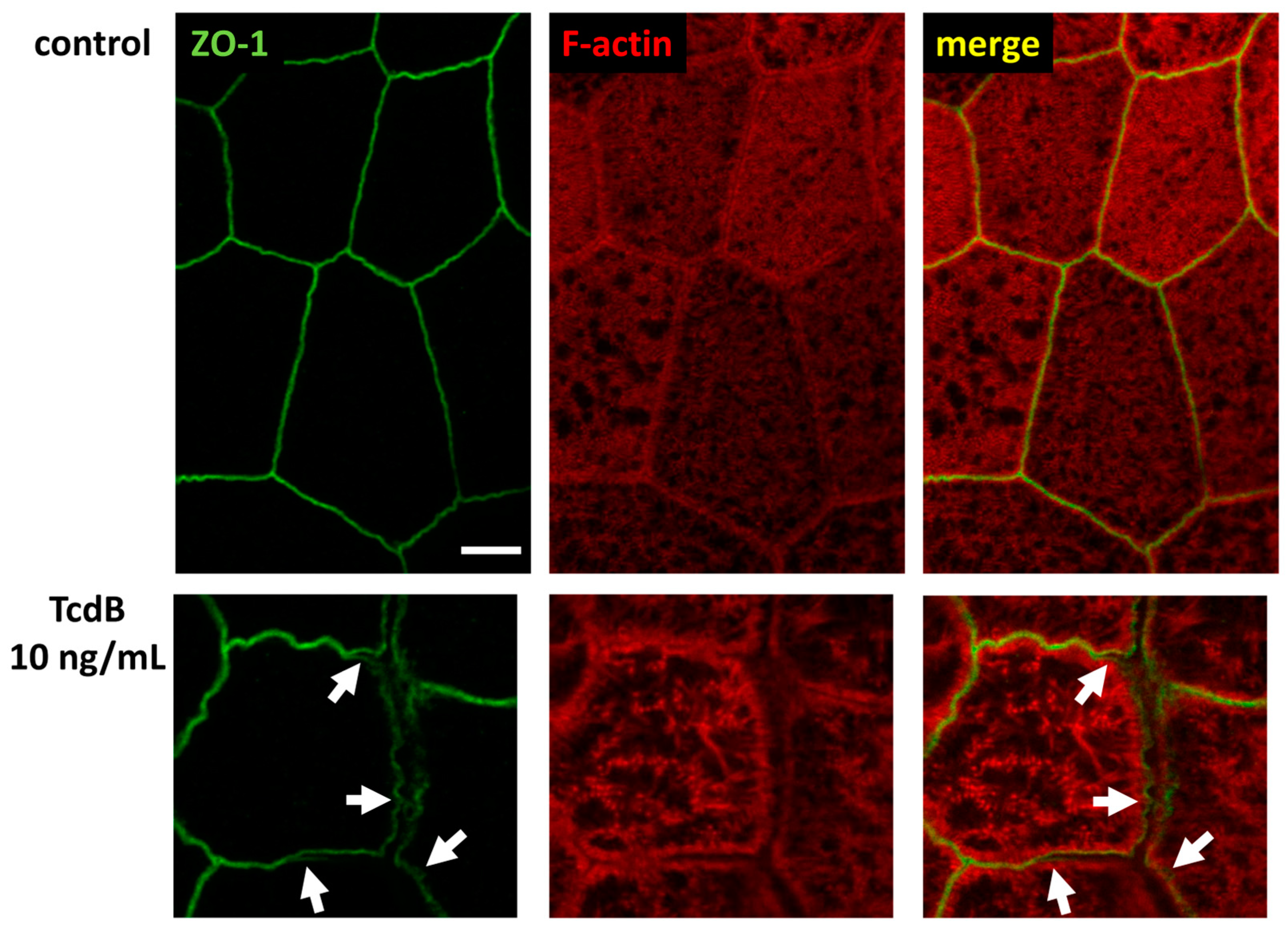
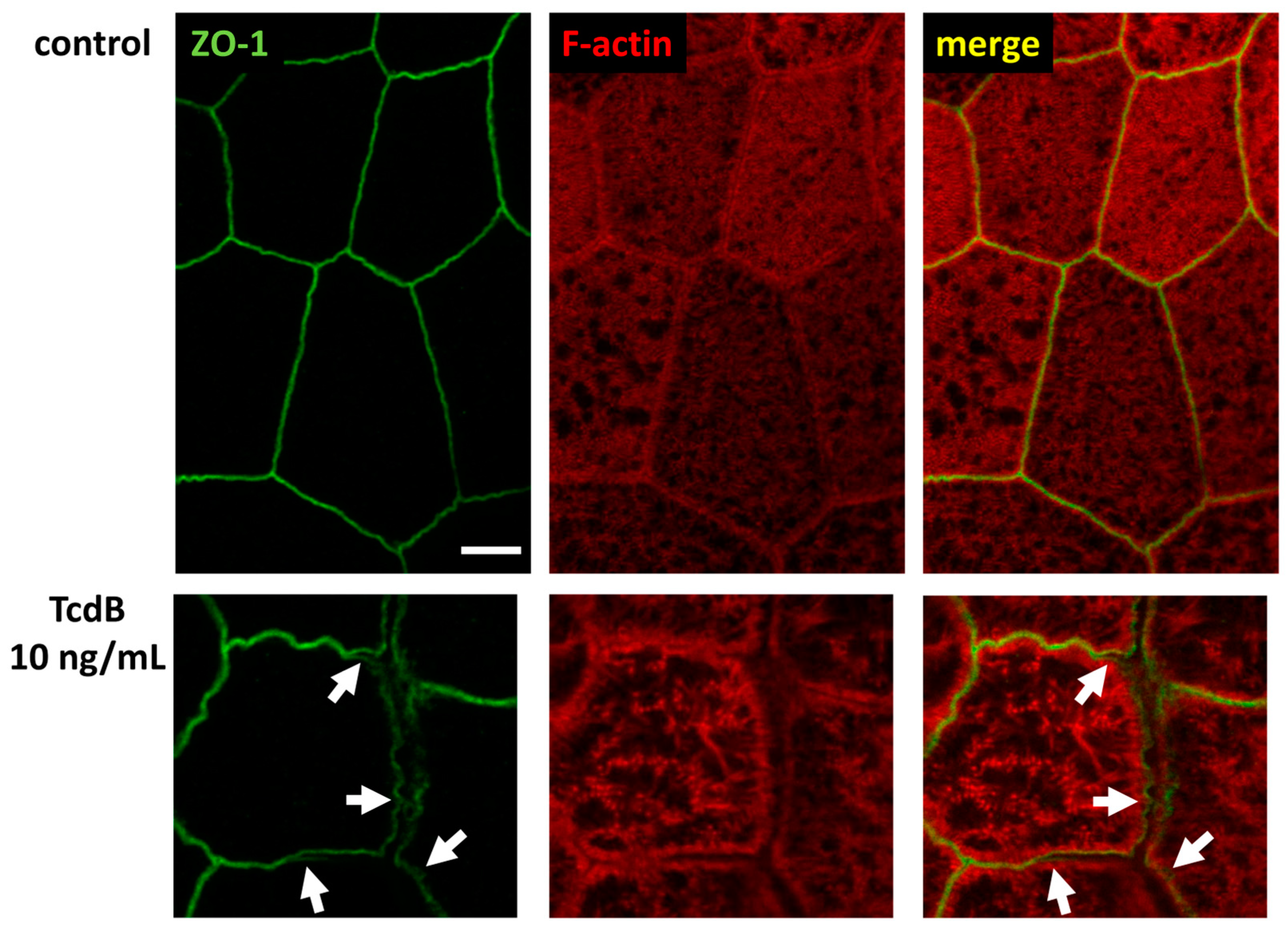
2.6. Tricellular Tight Junction Protein Delocalization in TcdB-Treated Epithelial Cells
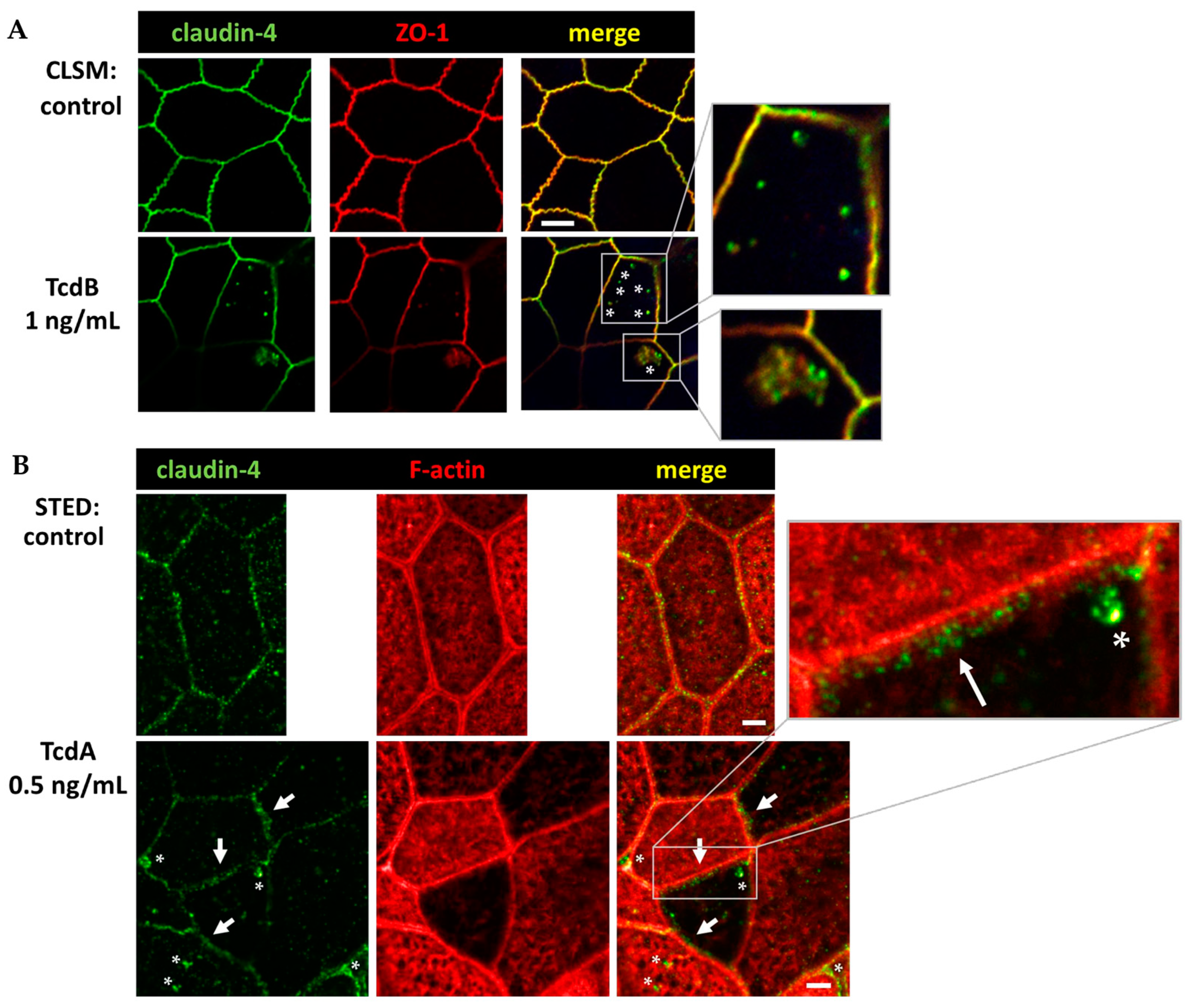
2.7. Conventional Laser-Scanning Microscopy and Super-Resolution Microscopy Revealed Subcellular Redistribution of Tight Junction Proteins after Exposure to Clostridial Toxins
3. Discussion
4. Methods
4.1. Generation of Intestinal Epithelial Organoids and Organoid Monolayers
4.2. Treatment of Cells and Studies on Epithelial Barrier Function
4.3. Analysis of Epithelial Cell Death Mechanisms
4.4. Tight Junction Protein Expression Evaluation
4.5. Analysis of Subcellular Tight Junction Protein Localization via Confocal Laser-Scanning Microscopy and Super-Resolution STED Microscopy
4.6. Flow Cytometry
4.7. RNA-Seq Expression Analysis
4.8. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kelly, C.P.; LaMont, J.T. Clostridium difficile—More difficult than ever. N. Engl. J. Med. 2008, 359, 1932–1940, Erratum in N. Engl. J. Med. 2010, 363, 1585. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, J.G.; Chang, T.W.; Gurwith, M.; Gorbach, S.L.; Onderdonk, A.B. Antibiotic-associated pseudomembranous colitis due to toxin-producing clostridia. N. Engl. J. Med. 1978, 298, 531–534. [Google Scholar] [CrossRef] [PubMed]
- Aas, J.; Gessert, C.E.; Bakken, J.S. Recurrent Clostridium difficile colitis: Case series involving 18 patients treated with donor stool administered via a nasogastric tube. Clin. Infect. Dis. 2003, 36, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Rao, K.; Young, V.B. Fecal microbiota transplantation for the management of Clostridium difficile infection. Infect. Dis. Clin. N. Am. 2015, 29, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Just, I.; Selzer, J.; Wilm, M.; von Eichel-Streiber, C.; Mann, M.; Aktories, K. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature. 1995, 375, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Just, I.; Selzer, J.; von Eichel-Streiber, C.; Aktories, K. The low molecular mass GTP-binding protein Rho is affected by toxin A from Clostridium difficile. J. Clin. Investig. 1995, 95, 1026–1031. [Google Scholar] [CrossRef]
- Popoff, M.R.; Rubin, E.J.; Gill, D.M.; Boquet, P. Actin-specific ADP-ribosyltransferase produced by a Clostridium difficile strain. Infect. Immun. 1988, 56, 2299–2306. [Google Scholar] [CrossRef]
- Gerding, D.N.; Johnson, S.; Rupnik, M.; Aktories, K. Clostridium difficile binary toxin CDT: Mechanism, epidemiology, and potential clinical importance. Gut Microbes. 2014, 5, 15–27. [Google Scholar] [CrossRef]
- Nusrat, A.; von Eichel-Streiber, C.; Turner, J.R.; Verkade, P.; Madara, J.L.; Parkos, C.A. Clostridium difficile toxins disrupt epithelial barrier function by altering membrane microdomain localization of tight junction proteins. Infect. Immun. 2001, 69, 1329–1336. [Google Scholar] [CrossRef]
- Heils, L.; Schneemann, M.; Gerhard, R.; Schulzke, J.D.; Bücker, R. CDT of Clostridioides difficile Induces MLC-Dependent Intestinal Barrier Dysfunction in HT-29/B6 Epithelial Cell Monolayers. Toxins 2023, 15, 54. [Google Scholar] [CrossRef]
- Hecht, G.; Pothoulakis, C.; LaMont, J.T.; Madara, J.L. Clostridium difficile toxin A perturbs cytoskeletal structure and tight junction permeability of cultured human intestinal epithelial monolayers. J. Clin. Investig. 1988, 82, 1516–1524. [Google Scholar] [CrossRef]
- Hecht, G.; Koutsouris, A.; Pothoulakis, C.; LaMont, J.T.; Madara, J.L. Clostridium difficile toxin B disrupts the barrier function of T84 monolayers. Gastroenterology 1992, 102, 416–423. [Google Scholar] [CrossRef]
- Aktories, K.; Schmidt, G.; Just, I. Rho GTPases as targets of bacterial protein toxins. Biol. Chem. 2000, 381, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Voth, D.E.; Ballard, J.D. Clostridium difficile toxins: Mechanism of action and role in disease. Clin. Microbiol. Rev. 2005, 18, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Shoshan, M.C.; Florin, I.; Thelestam, M. Activation of cellular phospholipase A2 by Clostridium difficile toxin B. J. Cell. Biochem. 1993, 52, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Bojarski, C.; Gitter, A.H.; Bendfeldt, K.; Mankertz, J.; Schmitz, H.; Wagner, S.; Fromm, M.; Schulzke, J.D. Permeability of human HT-29/B6 colonic epithelium as a function of apoptosis. J. Physiol. 2001, 535, 541–552. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, Q.; Wang, M.; Zhao, S.; Ma, J.; Luo, N.; Li, N.; Li, Y.; Xu, G.; Li, J. Interferon-gamma and tumor necrosis factor-alpha disrupt epithelial barrier function by altering lipid composition in membrane microdomains of tight junction. Clin. Immunol. 2008, 126, 67–80. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef]
- VanDussen, K.L.; Marinshaw, J.M.; Shaikh, N.; Miyoshi, H.; Moon, C.; Tarr, P.I.; Ciorba, M.A.; Stappenbeck, T.S. Development of an enhanced human gastrointestinal epithelial culture system to facilitate patient-based assays. Gut 2015, 64, 911–920. [Google Scholar] [CrossRef]
- Weiß, F.; Holthaus, D.; Kraft, M.; Klotz, C.; Schneemann, M.; Schulzke, J.D.; Krug, S.M. Human duodenal organ-oid-derived monolayers serve as a suitable barrier model for duodenal tissue. Ann. N. Y. Acad. Sci. 2022, 1515, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Lyerly, D.M.; Saum, K.E.; MacDonald, D.K.; Wilkins, T.D. Effects of Clostridium difficile toxins given intragastrically to animals. Infect. Immun. 1985, 47, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Feltis, B.A.; Wiesner, S.M.; Kim, A.S.; Erlandsen, S.L.; Lyerly, D.L.; Wilkins, T.D.; Wells, C.L. Clostridium difficile toxins A and B can alter epithelial permeability and promote bacterial paracellular migration through HT-29 enterocytes. Shock 2000, 14, 629–634. [Google Scholar] [CrossRef]
- Drudy, D.; Fanning, S.; Kyne, L. Toxin A-negative, toxin B-positive Clostridium difficile. Int. J. Infect. Dis. 2007, 11, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Kuehne, S.A.; Cartman, S.T.; Minton, N.P. Both, toxin A and toxin B, are important in Clostridium difficile infection. Gut Microbes 2011, 2, 252–255. [Google Scholar] [CrossRef] [PubMed]
- Alfa, M.J.; Kabani, A.; Lyerly, D.; Moncrief, S.; Neville, L.M.; Al-Barrak, A.; Harding, G.K.; Dyck, B.; Olekson, K.; Embil, J.M. Characterization of a toxin A-negative, toxin B-positive strain of Clostridium difficile responsible for a nosocomial outbreak of Clostridium difficile-associated diarrhea. J. Clin. Microbiol. 2000, 38, 2706–2714. [Google Scholar] [CrossRef]
- Holthaus, D.; Delgado-Betancourt, E.; Aebischer, T.; Seeber, F.; Klotz, C. Harmonization of Protocols for Multi-Species Organoid Platforms to Study the Intestinal Biology of Toxoplasma gondii and Other Protozoan Infections. Front. Cell Infect. Microbiol. 2021, 10, 610368. [Google Scholar] [CrossRef]
- Schmitz, H.; Barmeyer, C.; Fromm, M.; Runkel, N.; Foss, H.D.; Bentzel, C.J.; Riecken, E.O.; Schulzke, J.D. Altered tight junction structure contributes to the impaired epithelial barrier function in ulcerative colitis. Gastroenterology 1999, 116, 301–309. [Google Scholar] [CrossRef]
- Heller, F.; Florian, P.; Bojarski, C.; Richter, J.; Christ, M.; Hillenbrand, B.; Mankertz, J.; Gitter, A.H.; Bürgel, N.; Fromm, M.; et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 2005, 129, 550–564. [Google Scholar] [CrossRef]
- Zeissig, S.; Bürgel, N.; Günzel, D.; Richter, J.; Mankertz, J.; Wahnschaffe, U.; Kroesen, A.J.; Zeitz, M.; Fromm, M.; Schulzke, J.D. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007, 56, 61–72. [Google Scholar] [CrossRef]
- Bücker, R.; Schulz, E.; Günzel, D.; Bojarski, C.; Lee, I.F.M.; John, L.J.; Wiegand, S.; Janßen, T.; Wieler, L.H.; Dobrindt, U.; et al. α-Haemolysin of Escherichia coli in IBD: A Potentiator of Inflammatory Activity in the Colon. Gut 2014, 63, 1893–1901. [Google Scholar] [CrossRef]
- Beggs, M.R.; Young, K.; Pan, W.; O’Neill, D.D.; Saurette, M.; Plain, A.; Rievaj, J.; Doschak, M.R.; Cordat, E.; Dimke, H.; et al. Claudin-2 and claudin-12 form independent, complementary pores required to maintain calcium homeostasis. Proc. Natl. Acad. Sci. USA 2021, 118, e2111247118. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Sugimoto, K.; Inatomi, S.; Maeda, T.; Osanai, M.; Uchiyama, Y.; Yamamoto, Y.; Wada, T.; Kojima, T.; Yokozaki, H.; et al. Tight junction proteins claudin-2 and -12 are critical for vitamin D-dependent Ca2+ absorption between enterocytes. Mol. Biol. Cell. 2008, 19, 1912–1921. [Google Scholar] [CrossRef] [PubMed]
- Amasheh, S.; Meiri, N.; Gitter, A.H.; Schöneberg, T.; Mankertz, J.; Schulzke, J.D.; Fromm, M. Claudin-2 expression induces cation-selective channels in tight junctions of epithelial cells. J. Cell Sci. 2002, 115, 4969–4976. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, R.; Milatz, S.; Krug, S.M.; Oelrich, B.; Schulzke, J.D.; Amasheh, S.; Günzel, D.; Fromm, M. Claudin-2, a component of the tight junction, forms a paracellular water channel. J. Cell Sci. 2010, 123, 1913–1921. [Google Scholar] [CrossRef] [PubMed]
- Tsai, P.Y.; Zhang, B.; He, W.Q.; Zha, J.M.; Odenwald, M.A.; Singh, G.; Tamura, A.; Shen, L.; Sailer, A.; Yeruva, S.; et al. IL-22 Upregulates Epithelial Claudin-2 to Drive Diarrhea and Enteric Pathogen Clearance. Cell Host Microbe. 2017, 21, 671–681.e4. [Google Scholar] [CrossRef]
- Beutel, O.; Maraspini, R.; Pombo-García, K.; Martin-Lemaitre, C.; Honigmann, A. Phase Separation of Zonula Occludens Proteins Drives Formation of Tight Junctions. Cell 2019, 179, 923–936.e11. [Google Scholar] [CrossRef]
- Schulzke, J.D.; Ploeger, S.; Amasheh, M.; Fromm, A.; Zeissig, S.; Troeger, H.; Richter, J.; Bojarski, C.; Schumann, M.; Fromm, M. Epithelial tight junctions in intestinal inflammation. Ann. N. Y. Acad. Sci. 2009, 1165, 294–300. [Google Scholar] [CrossRef]
- Krug, S.M.; Bojarski, C.; Fromm, A.; Lee, I.M.; Dames, P.; Richter, J.F.; Turner, J.R.; Fromm, M.; Schulzke, J.D. Tricellulin Is Regulated via Interleukin-13-Receptor A2, Affects Macromolecule Uptake, and Is Decreased in Ulcerative Colitis. Mucosal Immunol. 2018, 11, 345–356. [Google Scholar] [CrossRef]
- May, M.; Wang, T.; Müller, M.; Genth, H. Difference in F-actin depolymerization induced by toxin B from the Clostridium difficile strain VPI 10463 and toxin B from the variant Clostridium difficile serotype F strain 1470. Toxins 2013, 5, 106–119. [Google Scholar] [CrossRef]
- Bücker, R.; Krug, S.M.; Moos, V.; Bojarski, C.; Schweiger, M.R.; Kerick, M.; Fromm, A.; Janßen, S.; Fromm, M.; Hering, N.A.; et al. Campylobacter jejuni impairs sodium transport and epithelial barrier function via cytokine release in human colon. Mucosal Immunol. 2018, 11, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Ok, M.T.; Liu, J.; Bliton, R.J.; Hinesley, C.M.; San Pedro, E.E.T.; Breau, K.A.; Gomez-Martinez, I.; Burclaff, J.; Magness, S.T. A leaky human colon model reveals uncoupled apical/basal cytotoxicity in early Clostridioides difficile toxin exposure. Am. J. Physiol. Gastrointest. Liver Physiol. 2023, 324, G262–G280. [Google Scholar] [CrossRef] [PubMed]
- Gülke, I.; Pfeifer, G.; Liese, J.; Fritz, M.; Hofmann, F.; Aktories, K.; Barth, H. Characterization of the Enzymatic Component of the ADP-Ribosyltransferase Toxin CDTa from Clostridium difficile. Infect. Immun. 2001, 69, 6004–6011. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Meléndez, A.; Cruz-López, F.; Morfin-Otero, R.; Maldonado-Garza, H.J.; Garza-González, E. An Update on Clostridioides difficile Binary Toxin. Toxins 2022, 14, 305. [Google Scholar] [CrossRef] [PubMed]
- López-Cárdenas, S.; Torres-Martos, E.; Mora-Delgado, J.; Sánchez-Calvo, J.M.; Santos-Peña, M.; Zapata López, Á.; Dolores López-Prieto, M.; Pérez-Cortés, S.; Carlos Alados, J. The Prognostic Value of Toxin B and Binary Toxin in Clostridioides difficile Infection. Gut Microbes. 2021, 13, 1884516. [Google Scholar] [CrossRef] [PubMed]
- Young, M.K.; Leslie, J.L.; Madden, G.R.; Lyerly, D.M.; Carman, R.J.; Lyerly, M.W.; Stewart, D.B.; Abhyankar, M.M.; Petri, W.A. Binary Toxin Expression by Clostridioides difficile Is Associated with Worse Disease. Open Forum Infect. Dis. 2022, 9, ofac001. [Google Scholar] [CrossRef]
- Stieglitz, F.; Gerhard, R.; Pich, A. The Binary Toxin of Clostridioides difficile Alters the Proteome and Phosphoproteome of HEp-2 Cells. Front. Microbiol. 2021, 12, 725612. [Google Scholar] [CrossRef]
- Tao, L.; Zhang, J.; Meraner, P.; Tovaglieri, A.; Wu, X.; Gerhard, R.; Zhang, X.; Stallcup, W.B.; Miao, J.; He, X.; et al. Frizzled Proteins Are Colonic Epithelial Receptors for C. difficile Toxin B. Nature 2016, 538, 350–355. [Google Scholar] [CrossRef]
- Yuan, P.; Zhang, H.; Cai, C.; Zhu, S.; Zhou, Y.; Yang, X.; He, R.; Li, C.; Guo, S.; Li, S.; et al. Chondroitin Sulfate Proteoglycan 4 Functions as the Cellular Receptor for Clostridium difficile Toxin B. Cell Res. 2015, 25, 157–168. [Google Scholar] [CrossRef]
- Hemmasi, S.; Czulkies, B.A.; Schorch, B.; Veit, A.; Aktories, K.; Papatheodorou, P. Interaction of the Clostridium difficile Binary Toxin CDT and Its Host Cell Receptor, Lipolysis-Stimulated Lipoprotein Receptor (LSR). J. Biol. Chem. 2015, 290, 14031–14044. [Google Scholar] [CrossRef]
- Masuda, S.; Oda, Y.; Sasaki, H.; Ikenouchi, J.; Higashi, T.; Akashi, M.; Nishi, E.; Furuse, M. LSR Defines Cell Corners for Tricellular Tight Junction Formation in Epithelial Cells. J. Cell Sci. 2011, 124, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Bergann, T.; Fromm, A.; Borden, S.A.; Fromm, M.; Schulzke, J.D. Glucocorticoid receptor is indispensable for phys-iological responses to aldosterone in epithelial Na+ channel induction via the mineralocorticoid receptor in a human colonic cell line. Eur. J. Cell Biol. 2011, 90, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Nattramilarasu, P.K.; Bücker, R.; Lobo de Sá, F.D.; Fromm, A.; Nagel, O.; Lee, I.M.; Butkevych, E.; Mousavi, S.; Genger, C.; Kløve, S.; et al. Campylobacter concisus Impairs Sodium Absorption in Colonic Epithelium via ENaC Dysfunction and Claudin-8 Disruption. Int. J. Mol. Sci. 2020, 21, 373. [Google Scholar] [CrossRef] [PubMed]
- Nieminen, M.; Stolpe, O.; Kuhring, M.; Weiner, J.; Pett, P.; Beule, D.; Holtgrewe, M. SODAR: Managing multiomics study data and metadata. Gigascience 2022, 12, giad052. [Google Scholar] [CrossRef]
- Heng, L. Fast but Inaccurate Adapter Trimmer for Illumina Reads; Broad Institute: Cambridge, MA, USA, 2014; Available online: https://github.com/lh3/trimadap (accessed on 1 May 2023).
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 May 2023).
- Sayols, S.; Scherzinger, D.; Klein, H. dupRadar: A Bioconductor package for the assessment of PCR artifacts in RNA-Seq data. BMC Bioinform. 2016, 17, 428. [Google Scholar] [CrossRef] [PubMed]
- DeLuca, D.S.; Levin, J.Z.; Sivachenko, A.; Fennell, T.; Nazaire, M.D.; Williams, C.; Reich, M.; Winckler, W.; Getz, G. RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics 2012, 28, 1530–1532. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Zyla, J.; Marczyk, M.; Domaszewska, T.; Kaufmann, S.H.E.; Polanska, J.; Weiner, J. Gene set enrichment for reproducible science: Comparison of CERNO and eight other algorithms. Bioinformatics 2019, 35, 5146–5154. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schneemann, M.; Heils, L.; Moos, V.; Weiß, F.; Krug, S.M.; Weiner, J.; Beule, D.; Gerhard, R.; Schulzke, J.-D.; Bücker, R. A Colonic Organoid Model Challenged with the Large Toxins of Clostridioides difficile TcdA and TcdB Exhibit Deregulated Tight Junction Proteins. Toxins 2023, 15, 643. https://doi.org/10.3390/toxins15110643
Schneemann M, Heils L, Moos V, Weiß F, Krug SM, Weiner J, Beule D, Gerhard R, Schulzke J-D, Bücker R. A Colonic Organoid Model Challenged with the Large Toxins of Clostridioides difficile TcdA and TcdB Exhibit Deregulated Tight Junction Proteins. Toxins. 2023; 15(11):643. https://doi.org/10.3390/toxins15110643
Chicago/Turabian StyleSchneemann, Martina, Lucas Heils, Verena Moos, Franziska Weiß, Susanne M. Krug, January Weiner, Dieter Beule, Ralf Gerhard, Jörg-Dieter Schulzke, and Roland Bücker. 2023. "A Colonic Organoid Model Challenged with the Large Toxins of Clostridioides difficile TcdA and TcdB Exhibit Deregulated Tight Junction Proteins" Toxins 15, no. 11: 643. https://doi.org/10.3390/toxins15110643
APA StyleSchneemann, M., Heils, L., Moos, V., Weiß, F., Krug, S. M., Weiner, J., Beule, D., Gerhard, R., Schulzke, J.-D., & Bücker, R. (2023). A Colonic Organoid Model Challenged with the Large Toxins of Clostridioides difficile TcdA and TcdB Exhibit Deregulated Tight Junction Proteins. Toxins, 15(11), 643. https://doi.org/10.3390/toxins15110643