A Potent Inhibitor of the Cystic Fibrosis Transmembrane Conductance Regulator Blocks Disease and Morbidity Due to Toxigenic Vibrio cholerae

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
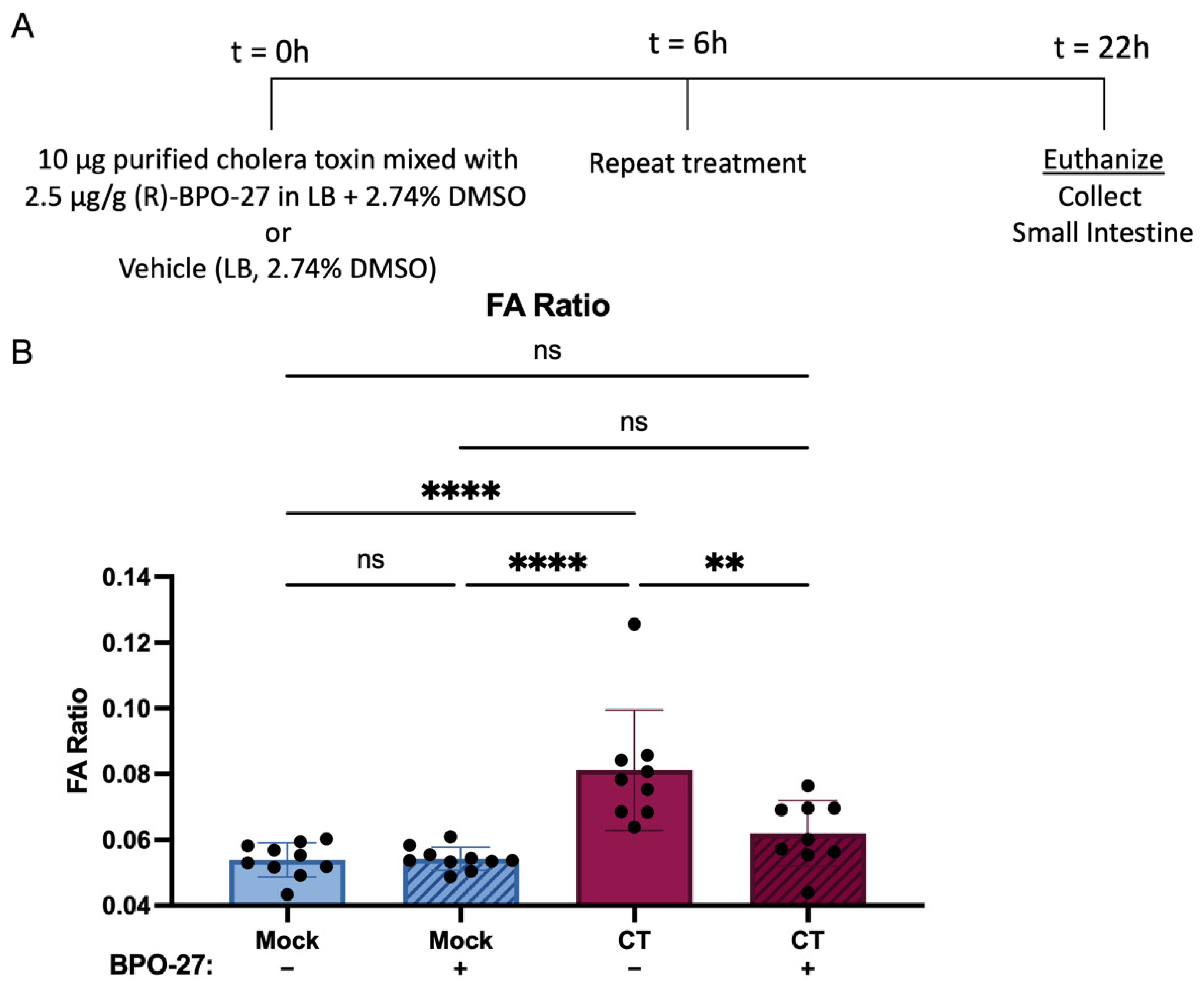
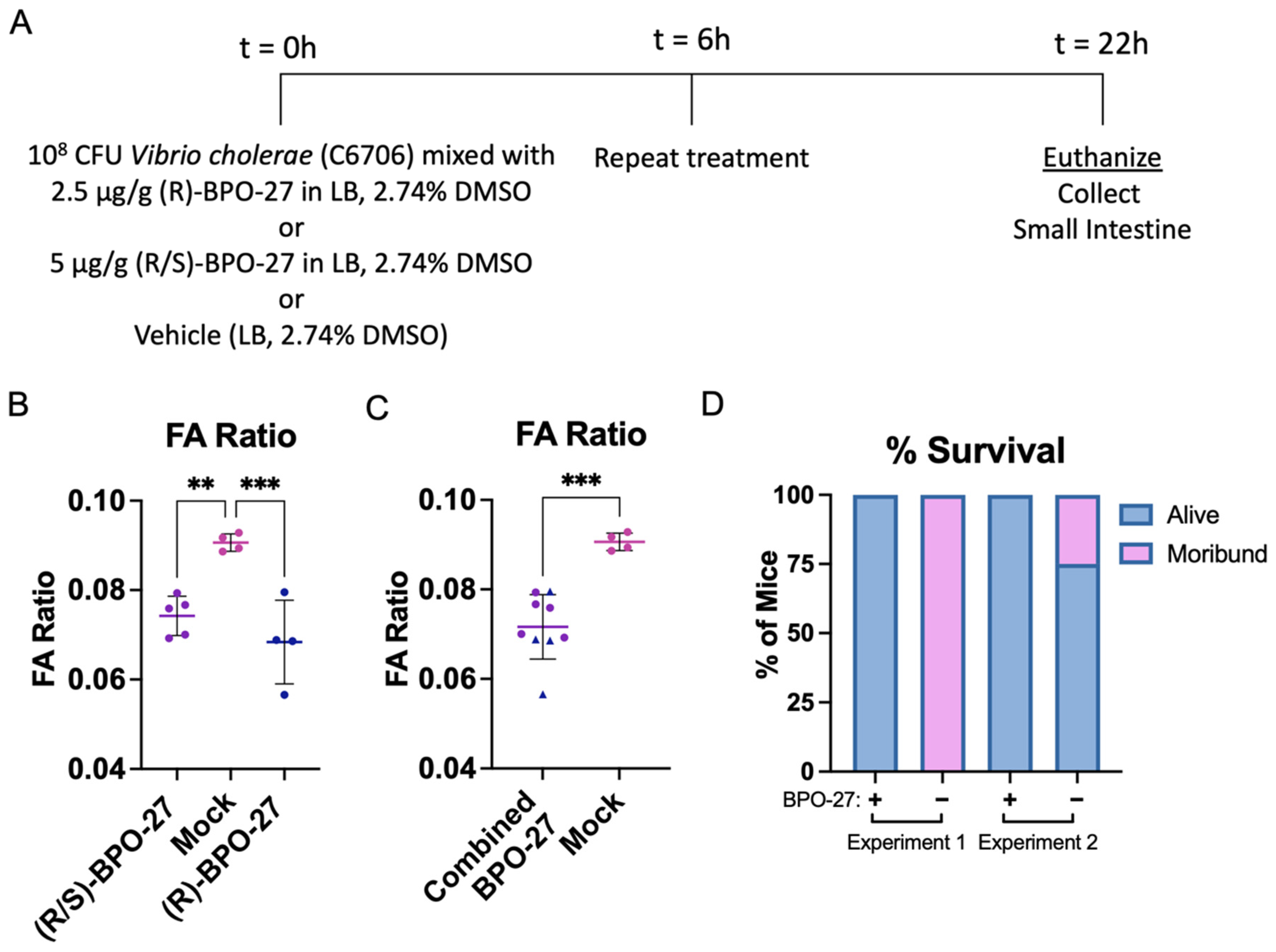
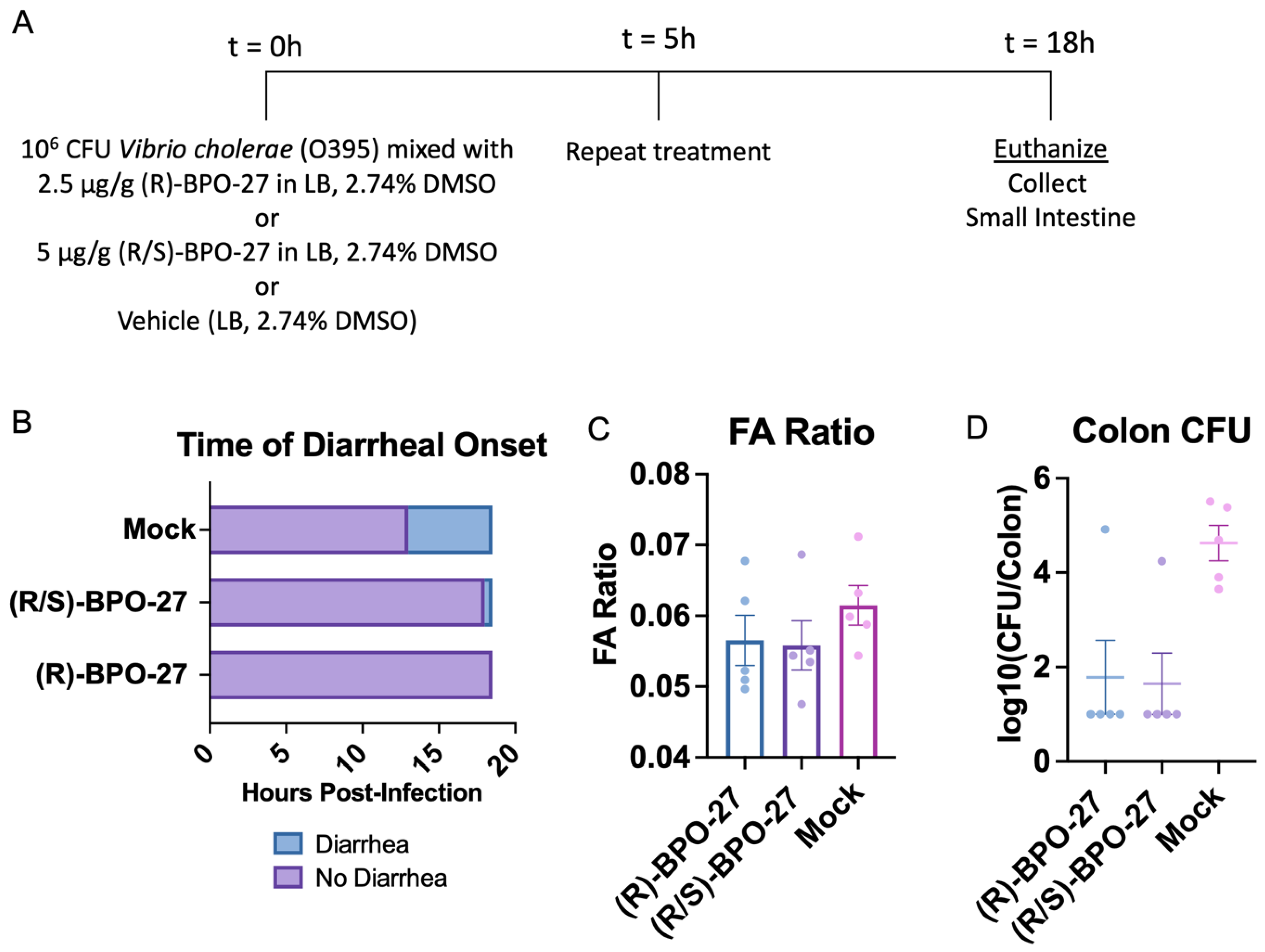
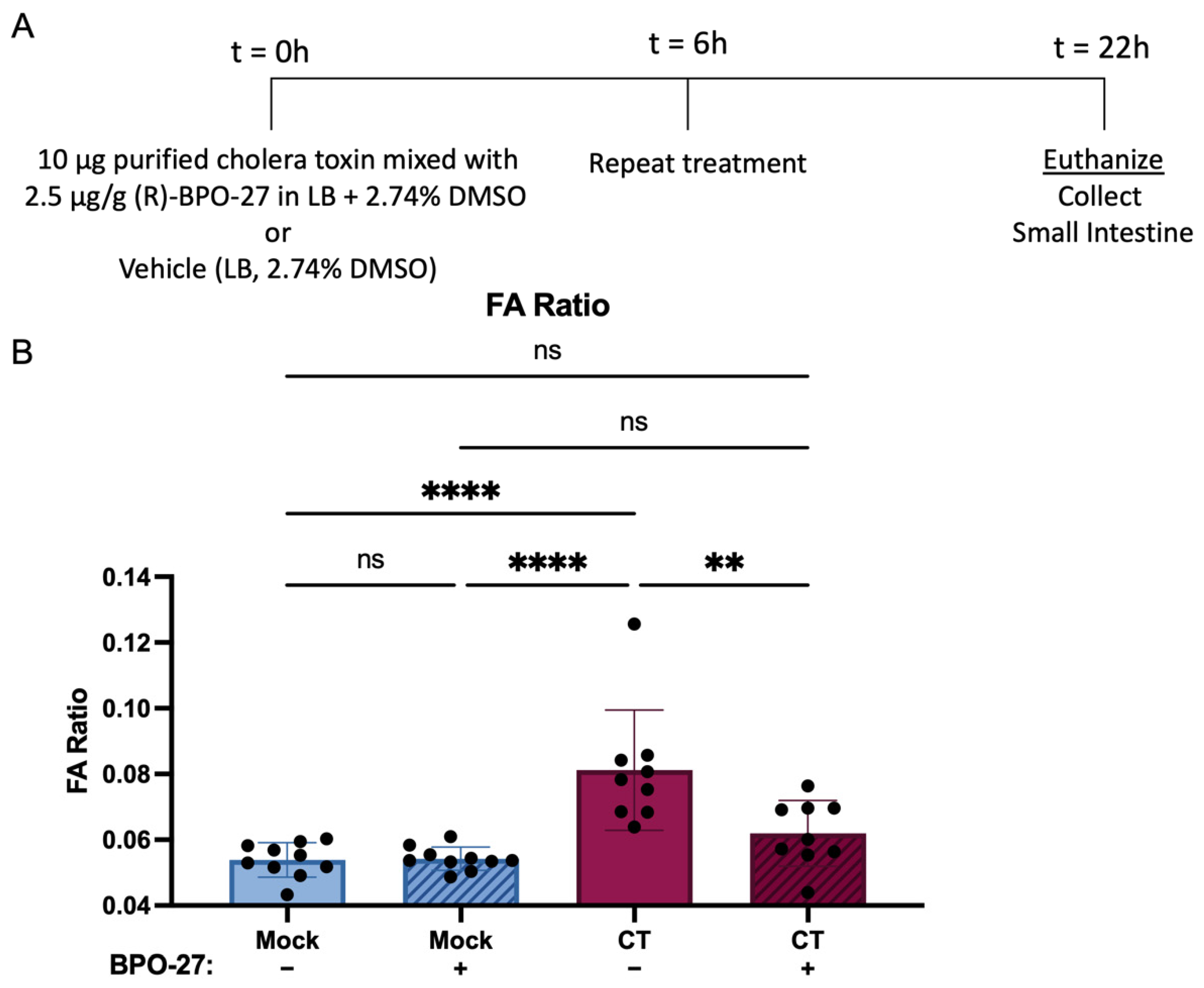
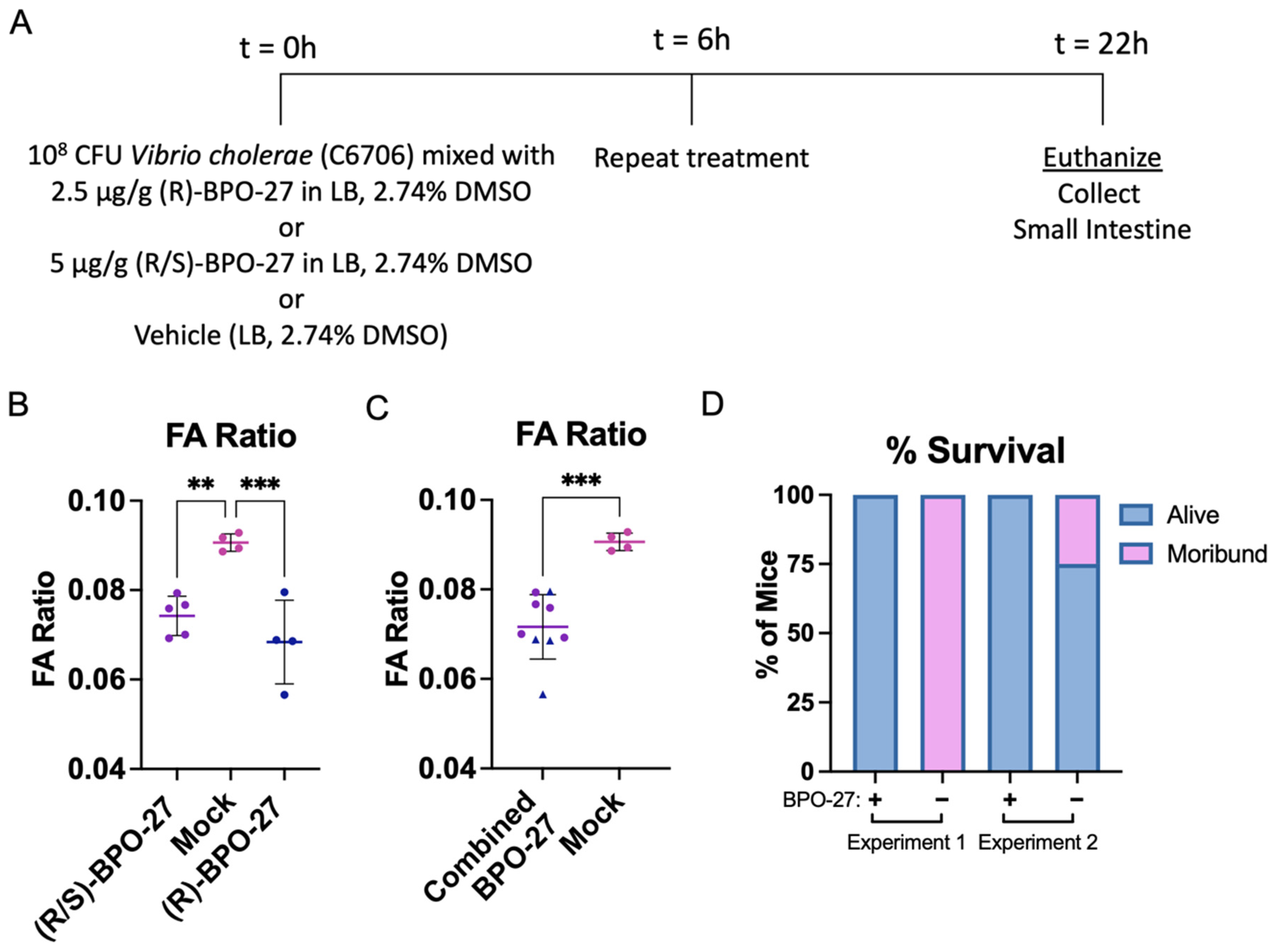
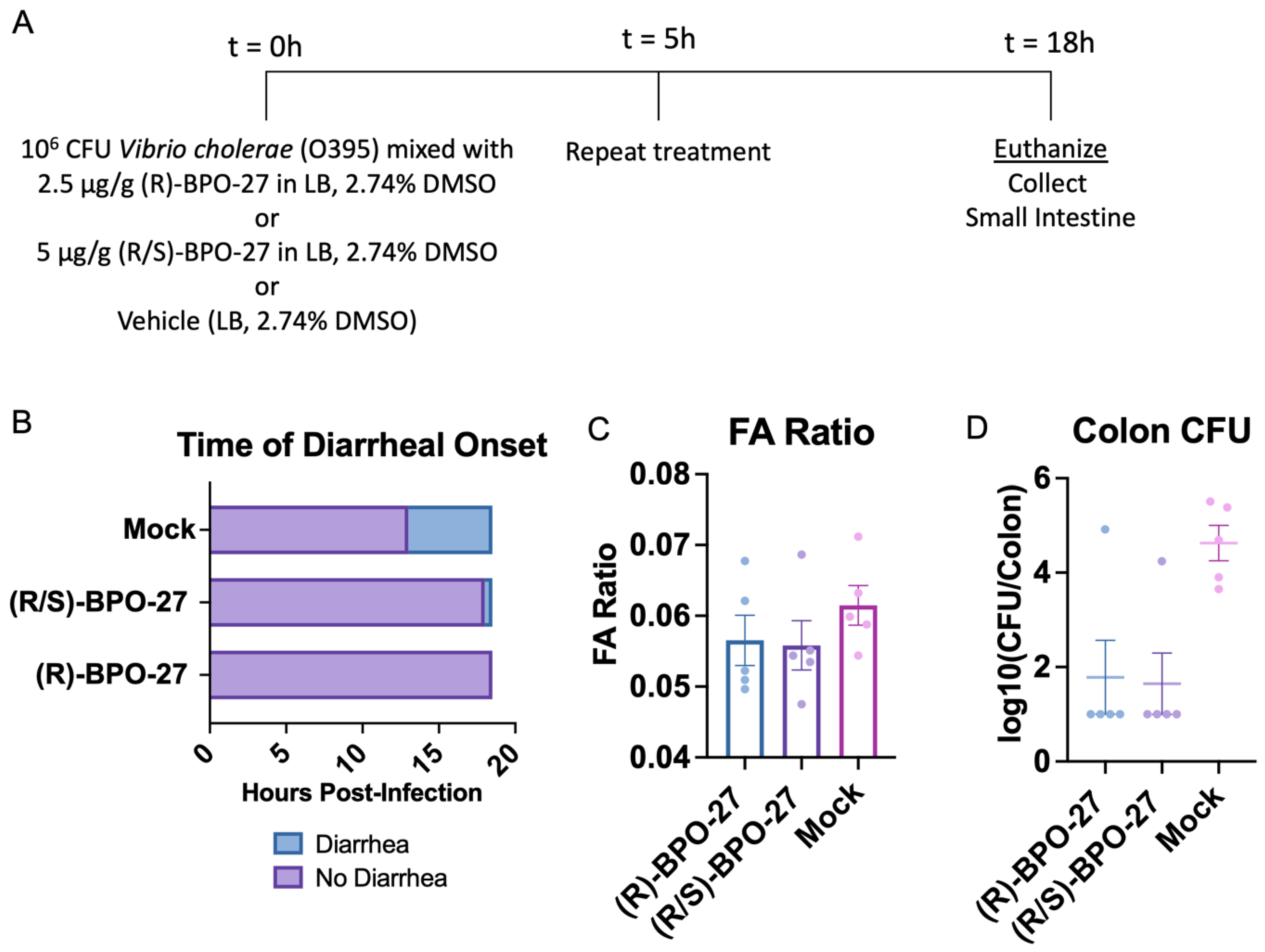
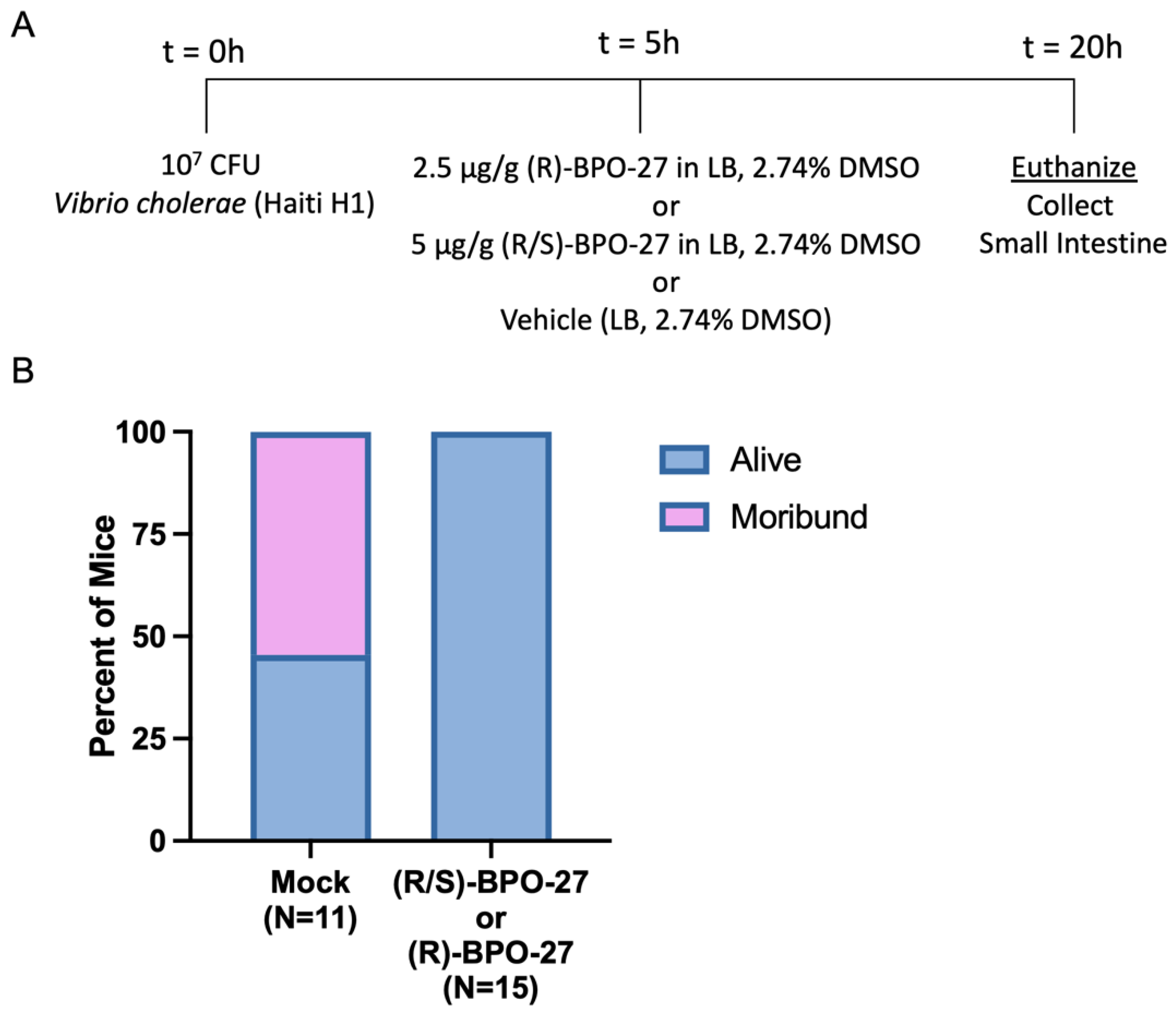
2.1. BPO-27 Treatment Prevents Morbidity and Mortality Associated with Oral Administration of Cholera Toxin (CT) or Toxigenic Vibrio cholerae
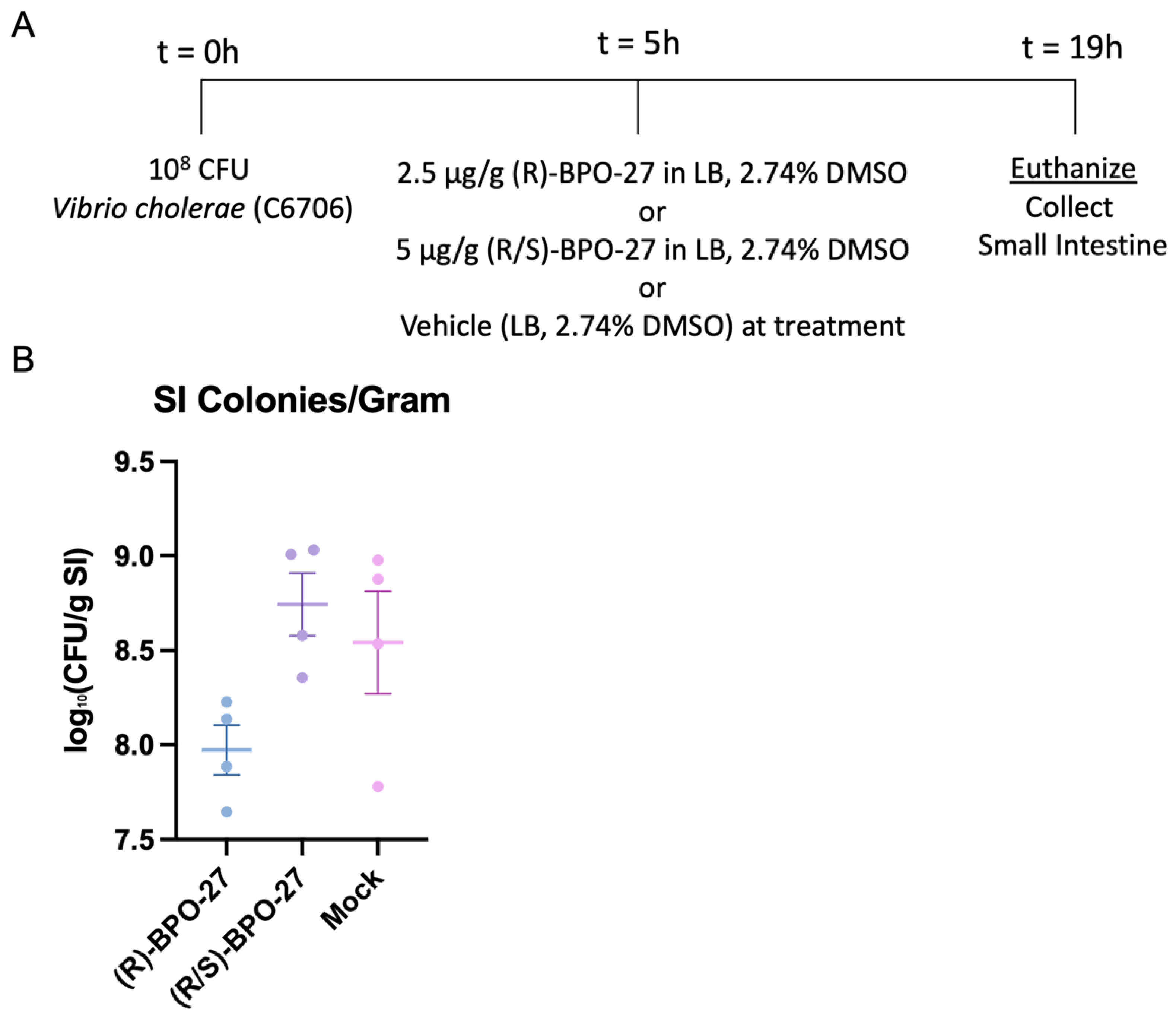
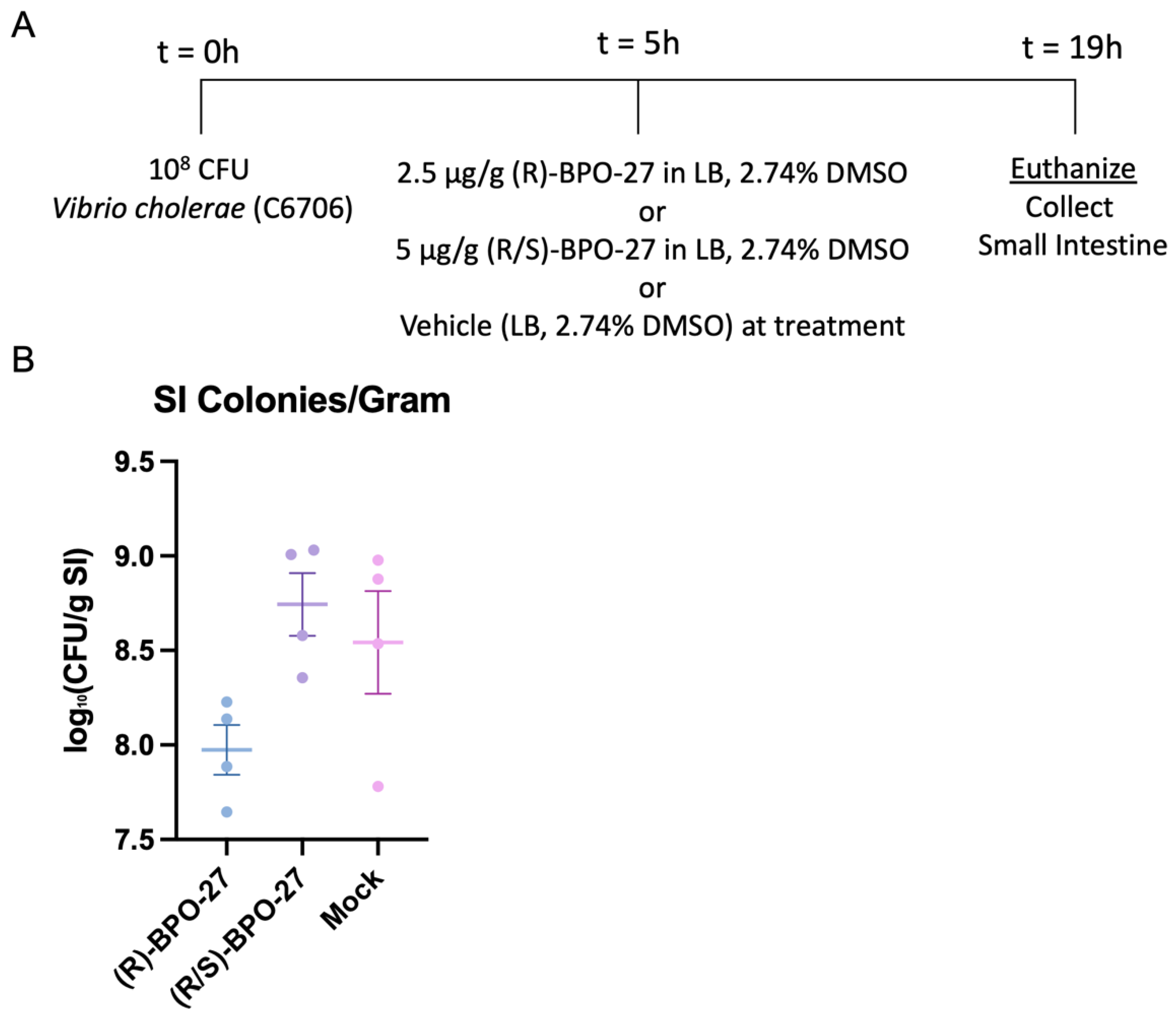
2.2. BPO-27 Treatment Can Suppress Intestinal Colonization of V. cholerae
2.3. BPO-27 Treatment Promotes Host Survival Even after Challenge by the Virulent “Variant’ Strain of V. cholerae Haiti-1
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Bacterial Strains
5.2. Drug Source and Properties
5.3. Suckling Mouse Experiments
5.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Harris, J.B.; LaRocque, R.C.; Qadri, F.; Ryan, E.T.; Calderwood, S.B. Cholera. Lancet 2012, 379, 2466–2476. [Google Scholar] [CrossRef] [Green Version]
- Sur, D.; Kanungo, S.; Sah, B.; Manna, B.; Ali, M.; Paisley, A.M.; Niyogi, S.K.; Park, J.K.; Sarkar, B.; Puri, M.K.; et al. Efficacy of a Low-Cost, Inactivated Whole-Cell Oral Cholera Vaccine: Results from 3 Years of Follow-Up of a Randomized, Controlled Trial. PLoS Negl. Trop. Dis. 2011, 5, e1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sack, D.A.; Tacket, C.O.; Cohen, M.; Sack, R.B.; Losonsky, G.A.; Shimko, J.; Nataro, J.P.; Edelman, R.; Levine, M.M.; Giannella, R.A.; et al. Validation of a Volunteer Model of Cholera with Frozen Bacteria as the Challenge. Infect. Immun. 1998, 66, 1968–1972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, J.W.; Whipp, S.C. Comparison of the mechanisms of action of cholera toxin and the heat-stable enterotoxins of Escherichia coli. Infect. Immun. 1995, 63, 1452–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrington, D.A.; Hall, R.H.; Losonsky, G.; Mekalanos, J.J.; Taylor, R.K.; Levine, M.M. Toxin, toxin-coregulated pili, and the toxR regulon are essential for Vibrio cholerae pathogenesis in humans. J. Exp. Med. 1988, 168, 1487–1492. [Google Scholar] [CrossRef]
- Chao, A.; De Sauvage, F.; Dong, Y.; Wagner, J.; Goeddel, D.; Gardner, P. Activation of intestinal CFTR Cl- channel by heat-stable enterotoxin and guanylin via cAMP-dependent protein kinase. EMBO J. 1994, 13, 1065–1072. [Google Scholar] [CrossRef]
- Waldor, M.K.; Mekalanos, J.J. Lysogenic Conversion by a Filamentous Phage Encoding Cholera Toxin. Science 1996, 272, 1910–1914. [Google Scholar] [CrossRef] [Green Version]
- Heidelberg, J.F.; Eisen, J.A.; Nelson, W.C.; Clayton, R.A.; Gwinn, M.L.; Dodson, R.J.; Haft, D.H.; Hickey, E.K.; Peterson, J.D.; Umayam, L.; et al. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 2000, 406, 477–483. [Google Scholar] [CrossRef] [Green Version]
- Faruque, S.M.; Albert, M.J.; Mekalanos, J.J. Epidemiology, Genetics, and Ecology of Toxigenic Vibrio cholerae. Microbiol. Mol. Biol. Rev. 1998, 62, 1301–1314. [Google Scholar] [CrossRef] [Green Version]
- Mutreja, A.; Kim, D.W.; Thomson, N.R.; Connor, T.R.; Lee, J.H.; Kariuki, S.; Croucher, N.J.; Choi, S.Y.; Harris, S.R.; Lebens, M.; et al. Evidence for several waves of global transmission in the seventh cholera pandemic. Nature 2011, 477, 462–465. [Google Scholar] [CrossRef] [Green Version]
- Chin, C.-S.; Sorenson, J.; Harris, J.B.; Robins, W.P.; Charles, R.C.; Jean-Charles, R.R.; Bullard, J.; Webster, D.R.; Kasarskis, A.; Peluso, P.; et al. The Origin of the Haitian Cholera Outbreak Strain. N. Engl. J. Med. 2011, 364, 33–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Mekhlafi, H. Yemen in a Time of Cholera: Current Situation and Challenges. Am. J. Trop. Med. Hyg. 2018, 98, 1558–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruxin, J.N. Magic bullet: The history of oral rehydration therapy. Med. Hist. 1994, 38, 363–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Field, M. Mechanisms of action of cholera and Escherichia coli enterotoxins. Am. J. Clin. Nutr. 1979, 32, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Thiagarajah, J.R.; Donowitz, M.; Verkman, A.S. Secretory diarrhoea: Mechanisms and emerging therapies. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 446–457. [Google Scholar] [CrossRef] [Green Version]
- Rhouma, M.; Fairbrother, J.M.; Beaudry, F.; Letellier, A. Post weaning diarrhea in pigs: Risk factors and non-colistin-based control strategies. Acta Vet. Scand. 2017, 59, 31. [Google Scholar] [CrossRef] [Green Version]
- Verkman, A.S.; Synder, D.; Tradtrantip, L.; Thiagarajah, J.R.; Anderson, M.O. CFTR Inhibitors. Curr. Pharm. Des. 2013, 19, 3529–3541. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Thiagarajah, J.R.; Yang, H.; Sonawane, N.D.; Folli, C.; Galietta, L.; Verkman, A. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin–induced intestinal fluid secretion. J. Clin. Investig. 2002, 110, 1651–1658. [Google Scholar] [CrossRef]
- Muanprasat, C.; Sonawane, N.; Salinas, D.; Taddei, A.; Galietta, L.J.; Verkman, A. Discovery of Glycine Hydrazide Pore-occluding CFTR Inhibitors: Mechanism, Structure-Activity Analysis, and In Vivo Efficacy. J. Gen. Physiol. 2004, 124, 125–137. [Google Scholar] [CrossRef] [Green Version]
- Sonawane, N.D.; Hu, J.; Muanprasat, C.; Verkman, A.S. Luminally active, nonabsorbable CFTR inhibitors as potential therapy to reduce intestinal fluid loss in cholera. FASEB J. 2005, 20, 130–132. [Google Scholar] [CrossRef]
- Snyder, D.S.; Tradtrantip, L.; Battula, S.; Yao, C.; Phuan, P.-W.; Fettinger, J.C.; Kurth, M.J.; Verkman, A.S. Absolute Configuration and Biological Properties of Enantiomers of CFTR Inhibitor BPO-27. ACS Med. Chem. Lett. 2013, 4, 456–459. [Google Scholar] [CrossRef]
- Kim, Y.; Anderson, M.O.; Park, J.; Lee, M.G.; Namkung, W.; Verkman, A.S. Benzopyrimido-pyrrolo-oxazine-dione (R)-BPO-27 Inhibits CFTR Chloride Channel Gating by Competition with ATP. Mol. Pharmacol. 2015, 88, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Cil, O.; Phuan, P.; Gillespie, A.M.; Lee, S.; Tradtrantip, L.; Yin, J.; Tse, M.; Zachos, N.C.; Lin, R.; Donowitz, M.; et al. Benzopyrimido-pyrrolo-oxazine-dione CFTR inhibitor (R)-BPO-27 for antisecretory therapy of diarrheas caused by bacterial enterotoxins. FASEB J. 2017, 31, 751–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klose, K.E. The suckling mouse model of cholera. Trends Microbiol. 2000, 8, 189–191. [Google Scholar] [CrossRef]
- Baselski, V.; Briggs, R.; Parker, C. Intestinal fluid accumulation induced by oral challenge with Vibrio cholerae or cholera toxin in infant mice. Infect. Immun. 1977, 15, 704–712. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.K.; Millert, V.L.; Furlong, D.B.; Mekalanost, J.J. Use of PhoA Gene Fusions to Identify a Pilus Colonization Factor Coor-dinately Regulated with Cholera Toxin. Proc. Natl. Acad. Sci. USA 1987, 84, 2833–2837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tauxe, R.V.; Blake, P.A. Epidemic cholera in Latin America. JAMA J. Am. Med. Assoc. 1992, 267, 1388–1390. [Google Scholar] [CrossRef]
- Matson, J.S. Infant Mouse Model of Vibrio cholerae Infection and Colonization. Methods Mol. Biol. 2018, 1839, 147–152. [Google Scholar] [CrossRef]
- Rivera-Chávez, F.; Mekalanos, J.J. Cholera toxin promotes pathogen acquisition of host-derived nutrients. Nature 2019, 572, 244–248. [Google Scholar] [CrossRef]
- Pierce, N.F.; Kaper, J.B.; Mekalanos, J.J.; Cray, W.C. Role of cholera toxin in enteric colonization by Vibrio cholerae O1 in rabbits. Infect. Immun. 1985, 50, 813–816. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Caro, F.; Robins, W.; Mekalanos, J.J. Antagonism toward the intestinal microbiota and its effect on Vibrio cholerae virulence. Science 2018, 359, 210–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, P.; Sinha, R.; Samanta, P.; Saha, D.R.; Koley, H.; Dutta, S.; Okamoto, K.; Ghosh, A.; Ramamurthy, T.; Mukhopadhyay, A.K. Haitian Variant Vibrio cholerae O1 Strains Manifest Higher Virulence in Animal Models. Front. Microbiol. 2019, 10, 111. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rivera-Chávez, F.; Meader, B.T.; Akosman, S.; Koprivica, V.; Mekalanos, J.J. A Potent Inhibitor of the Cystic Fibrosis Transmembrane Conductance Regulator Blocks Disease and Morbidity Due to Toxigenic Vibrio cholerae. Toxins 2022, 14, 225. https://doi.org/10.3390/toxins14030225
Rivera-Chávez F, Meader BT, Akosman S, Koprivica V, Mekalanos JJ. A Potent Inhibitor of the Cystic Fibrosis Transmembrane Conductance Regulator Blocks Disease and Morbidity Due to Toxigenic Vibrio cholerae. Toxins. 2022; 14(3):225. https://doi.org/10.3390/toxins14030225
Chicago/Turabian StyleRivera-Chávez, Fabian, Bradley T. Meader, Sinan Akosman, Vuk Koprivica, and John J. Mekalanos. 2022. "A Potent Inhibitor of the Cystic Fibrosis Transmembrane Conductance Regulator Blocks Disease and Morbidity Due to Toxigenic Vibrio cholerae" Toxins 14, no. 3: 225. https://doi.org/10.3390/toxins14030225
APA StyleRivera-Chávez, F., Meader, B. T., Akosman, S., Koprivica, V., & Mekalanos, J. J. (2022). A Potent Inhibitor of the Cystic Fibrosis Transmembrane Conductance Regulator Blocks Disease and Morbidity Due to Toxigenic Vibrio cholerae. Toxins, 14(3), 225. https://doi.org/10.3390/toxins14030225