4. Materials and Methods
4.1. Reagents and Instruments
All solvents were purified by distillation and, if necessary, they were dried using standard methods [
30]. Air- and moisture-sensitive reactions were carried out under a positive pressure of nitrogen using material previously oven-dried at 130 °C overnight. Reactions were monitored using thin-layer chromatography with 0.25 mm pre-coated silica gel plates. The plates were visualized under UV light at 366 and 254 nm, using an aqueous ceric ammonium molybdate solution or an ethanolic phosphomolybdic acid solution and heat as developing agents. Chromatography refers to flash column chromatography, which was performed on silica gel 60 (particle size 40−63 μm) with the given solvents.
1H/
13C NMR spectra were recorded at 298 °K, in the indicated solvent, at 300/75 MHz (Bruker Avance DPX300, Billerica, MA, USA) or 500/126 MHz (Bruker Avance DRX500).
1H and
13C chemical shifts (δ scale) are expressed in parts per million (ppm) downfield from tetramethylsilane and are referenced to residual proton or carbon in the NMR solvent (CHCl
3: δ 7.26/77.16; methanol-d
4: δ 3.31/40.00). A combination of COSY, edited HSQC, and HMBC experiments was used to assign the
1H and
13C chemical shifts of selected compounds. Accurate mass measurements (HRMS) were obtained using the electrospray ionization (ESI) mode on a Q-TOF premier mass spectrometer with an electrospray source from Waters (Manchester, UK).
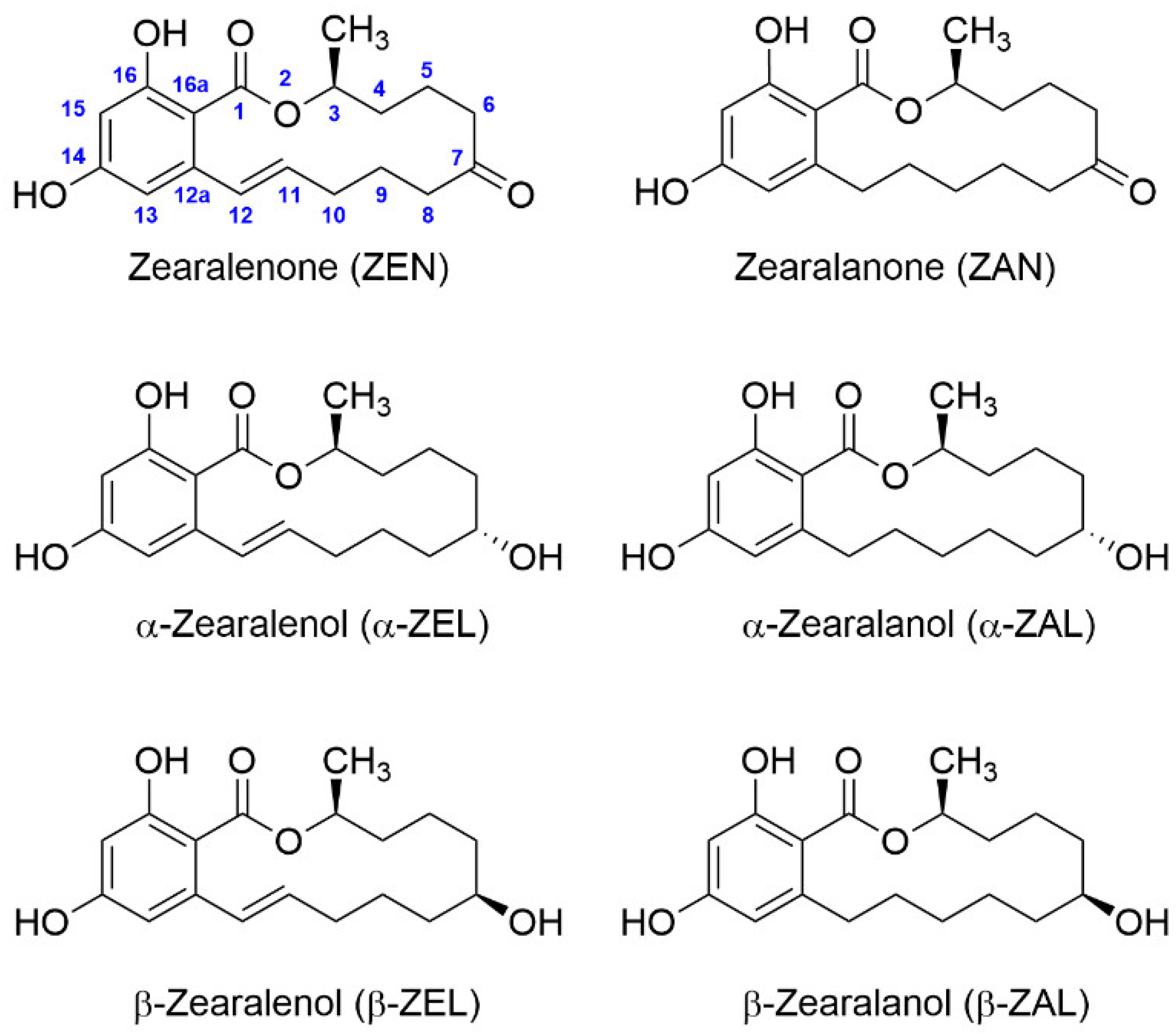
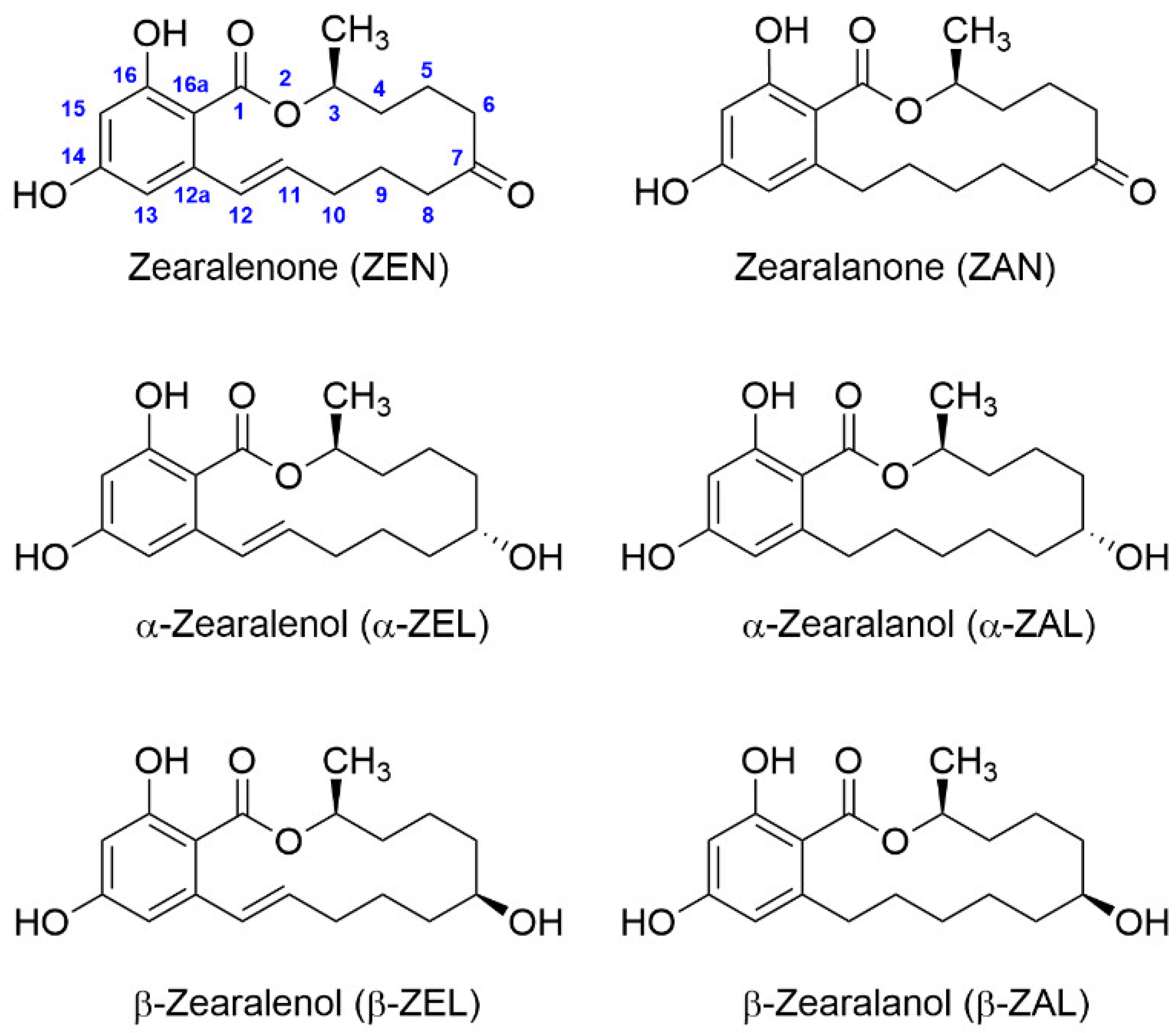
Standard ZEN ((4S,12E)-16,18-dihydroxy-4-methyl-3-oxabicyclo(12.4.0)octadeca-1(14),12,15,17-tetraene-2,8-dione, CAS registry number 17924-92-4, Mw 318.4) was purchased from Fermentek (Jerusalem, Israel). Metabolites (ZAN, α-ZEL, β-ZEL, α-ZAL, and β-ZAL) were obtained also from Fermentek. Standard solutions were prepared in anhydrous DMF, and the stock solutions were stored at −20 °C. PBS 10× solution (Fisher BioReagents BP399-20) was from Thermo Fisher Scientific (Waltham, MA, USA). Fraction V BSA, from Roche Applied Science (Mannheim, Germany) was employed to synthesize the immunizing bioconjugates. OVA, HRP, complete and incomplete Freund’s adjuvants, adult bovine serum (ABS), and o-phenylenediamine (OPD) were acquired from Merck (Darmstadt, Germany). Sephadex G-25 HiTrap® Desalting columns for protein–hapten conjugate purification were obtained from GE Healthcare (Uppsala, Sweden) and operated under an ÄKTA Purifier workstation also from GE Healthcare. A 5800 matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF/TOF) mass spectrometry apparatus from ABSciex (Framingham, MA, USA) was used for bioconjugate analysis. Immunoassays were carried out using Costar® 96-well flat-bottom high-binding polystyrene ELISA plates from Corning (Corning, NY, USA). Polyclonal goat anti-rabbit immunoglobulins antibody (GAR) for microplate coating and GAR conjugated to HRP (GAR-HRP) for competitive ELISA analysis were purchased from Rockland Immunochemicals Inc. (Pottstown, PA, USA) and BioRad (Madrid, Spain), respectively. Microplate wells were washed with an ELx405 washer and immunoassay absorbance values were read with a PowerWave HT microplate reader, both from BioTek Instruments (Winooski, VT, USA).
4.2. Solutions and Buffers
Coating buffer: 50 mM carbonate-bicarbonate buffer, pH 9.6; enzyme substrate solution: 2 mg/mL of OPD in 25 mM citrate and 62 mM phosphate buffer, pH 5.4, containing 0.012% (v/v) H2O2; HEPES buffer: 20 mM HEPES, pH 7.4; PB: 100 mM phosphate buffer, pH 7.4; PBS: 12 mM phosphate containing 137 mM NaCl and 2.7 mM KCl, pH 7.4; PBST: PBS containing 0.05% (v/v) Tween-20; secondary antibody solution: 104-fold diluted GAR-HRP in PBST containing 10% (v/v) ABS; washing solution: 150 mM NaCl containing 0.05% (v/v) Tween-20. All buffers and solutions were prepared in Milli-Q® water.
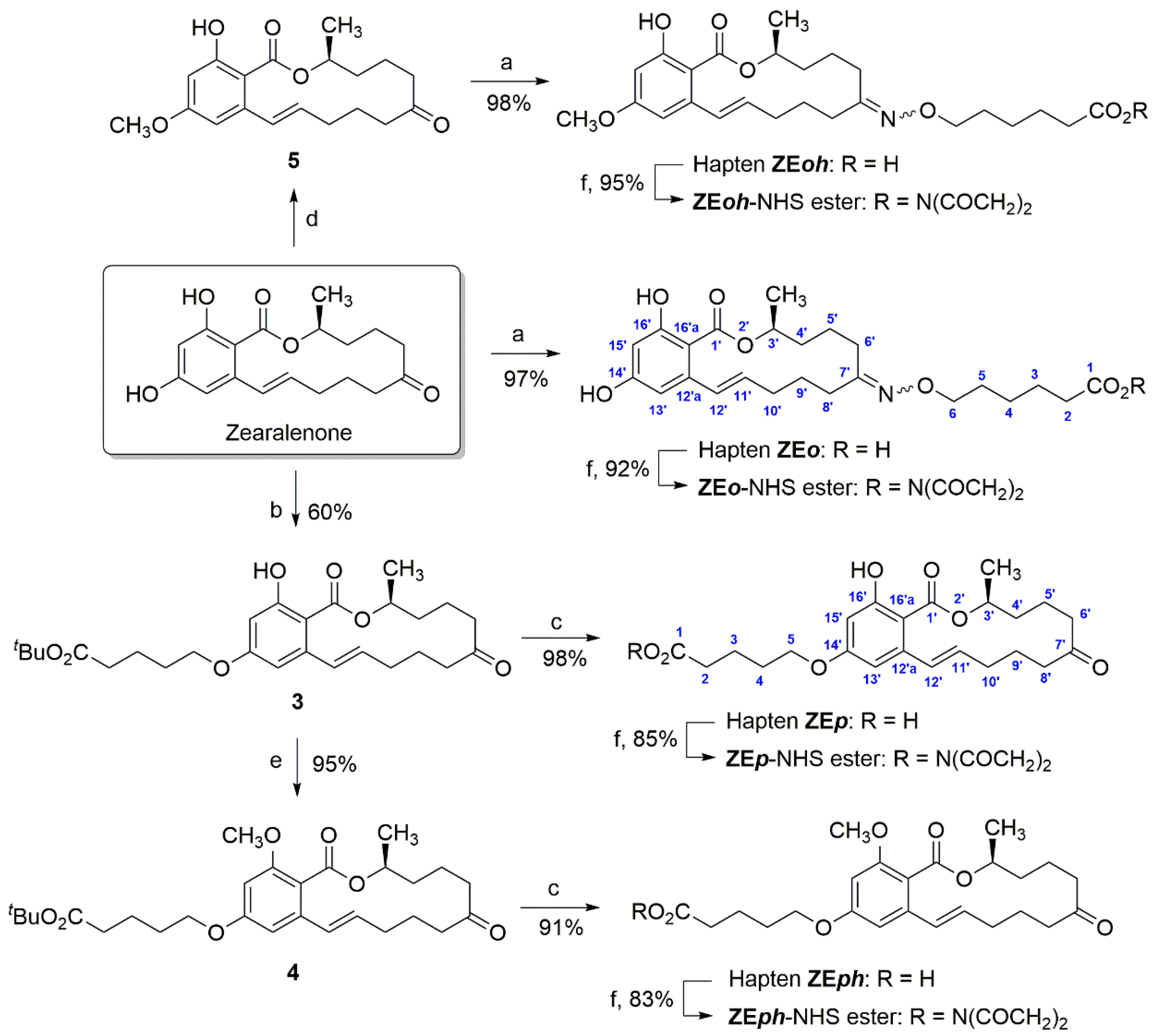
4.3. Synthesis of Immunizing Haptens
Preparation of 6-((((
S,
E)-14,16-dihydroxy-3-methyl-1-oxo-1,3,4,5,6,8,9,10-octahydro-7H-benzo[c][1]oxacyclotetradecin-7-ylidene)amino)oxy)hexanoic acid (hapten ZE
o). A solution of ZEN (12.8 mg, 40.2 µmol) and 6-(aminooxy)hexanoic acid hydrochloride [
23] (
1, 10 mg, 54.5 µmol) in anhydrous pyridine (370 µL) was stirred in the dark at room temperature for 24 h under nitrogen. The pyridine was removed with a stream of nitrogen and the resulting residue was dissolved in EtOAc, washed with water and brine, dried under anhydrous Na
2SO
4, and concentrated under a vacuum to give hapten ZE
o (17.5 mg, 97%)—a mixture of two geometrical isomers (
E and
Z) of the oximino moiety—as a viscous colorless oil.
1H NMR (300 MHz, methanol-d
4) δ 6.98 and 6.96 (each br d,
J = 15.5 Hz, 1H, H-12′), 6.39 and 6.36 (each d,
J = 2.5 Hz, 1H, H-13′), 6.22 and 6.21 (d,
J = 2.6 Hz, 1H, H-15′), 5.98 (dt,
J = 15.5, 6.4 Hz, 0.4H, H-11′), 5.75 (ddd,
J = 15.4, 9.4, 4.6 Hz, 0.6H, H-11′), 5.15 and 4.99 (each m, 1H, H-3′), 4.03 and 4.00 (each t,
J = 6.3 Hz, 2H, H
2-6), 2.84 (m, 0.6H, H-8′), 2.53 (dt,
J = 13.9, 7.2 Hz, 0.4H, H-6′), 2.40–2.00 (m, 7H), 1.98–1.40 (m, 12H), 1.38 and 1.36 (each d,
J = 6.2 Hz, 3H, Me-3′);
13C NMR (75 MHz, methanol-d
4), δ 177.6 (C-1), 172.4 and 172.4 (C-1′), 166.0 and 164.7 (C-14′), 163.7 and 163.3 (C-16′), 162.1 and 159.5 (C-7′), 145.0 and 143.7 (C-12′a), 133.9 and 132.9 (C-12′), 133.3 and 133.0 (C-11′), 109.4 and 108.5 (C-13′), 106.2 and 104.4 (C-16′a), 102.7 (C-15′), 74.3 and 73.9 (C-3′), 74.1 and 74.0 (C-6), 36.8 and 36.2 (C-4′), 35.6 and 31.2 (C-8′), 35.0 (C-2), 31.9 and 31.8 (C-10′), 30.1 and 29.9 (C-5), 28.4 and 27.5 (C-6′), 27.0 and 26.8 (C-4), 26.0 and 25.9 (C-3), 25.7 and 23.8 (C-9′), 23.3 and 23.2 (C-5′), 20.9 and 20.5 (Me-3′); HRMS (ESI)
m/
z calculated for C
24H
34NO
7 [M + H]
+ 448.2330, found 448.2324.
Preparation of
tert-butyl (
S,
E)-5-((16-hydroxy-3-methyl-1,7-dioxo-3,4,5,6,7,8,9,10-octahydro-1H-benzo[c][1]oxacyclotetradecin-14-yl)oxy)pentanoate (
3). A heterogeneous mixture of ZEN (32.7 mg, 0.103 mmol),
tert-butyl 5-bromovalerate [
25] (
2, 36.5 mg, 154 mmol), tetrabutylammonium iodide 3.6 mg, 9.5 µmol), and K
2CO
3 (21.9 mg, 0.158 mmol) in anhydrous acetone (2.2 mL) was stirred at 55 °C overnight under nitrogen. The reaction mixture was cooled to room temperature, diluted with Et
2O (50 mL) and washed with water and brine, dried over anhydrous MgSO
4, and concentrated at reduced pressure. The obtained residue (70 mg) was purified by column chromatography, using hexane/EtOAc 9:1 as eluent, to give the 14-alkylate derivative
3 (29.3 mg, 60%) as an oil, followed by the 14,16-dialkylated product (13.5 mg, 21%).
1H NMR (300 MHz, CDCl
3) δ 12.06 (s, 1H, 16′-OH), 7.01 (dd,
J = 15.3, 1.9 Hz, 1H, H-12′), 6.45 (dd,
J = 2.7, 0.5 Hz, 1H, H-13′), 6.36 (d,
J = 2.6 Hz, 1H, H-15′), 5.68 (ddd,
J = 15.2, 10.5, 3.7 Hz, 1H, H-11′), 5.00 (m, 1H, H-3′), 3.98 (d,
J = 5.8 Hz, 2H, H
2-5), 2.84 (ddd,
J = 18.8, 12.2, 2.7 Hz, 1H, H-8′), 2.60 (dt,
J = 12.6, 4.5 Hz, 1H, H-6′), 2.40–2.08 (m, 5H), 2.29 (t,
J = 6.9 Hz, 2H, H
2-2), 1.86–1.57 (m, 9H), 1.45 (s, 9H, CMe
3), 1.38 (d,
J = 6.1 Hz, 3H, Me-3′);
13C NMR (75 MHz, CDCl
3) δ 211.2 (C-7′), 172.9 (C-1), 171.5 (C-1′), 165.7 (C-16′), 163.6 (C-14′), 143.4 (C-12′a), 133.4 (C-11′), 132.5 (C-12′), 108.7 (C-15′), 103.6 (C-16′a), 100.5 (C-13′), 80.4 (CMe
3), 73.5 (C-3′), 67.7 (C-5), 44.1 (C-6′), 36.8 (C-8′), 35.2 (C-2), 34.9 (C-4′), 31.1 (C-10′), 28.5 (C-4), 28.3 (CMe
3), 22.4, 21.8, and 21.2 (C-3, C-5′, and C-9′), 21.0 (Me-3′); HRMS (ESI)
m/
z calculated for C
27H
39O
7 [M + H]
+ 475.2690, found 475.2670.
Preparation of (S,E)-5-((16-hydroxy-3-methyl-1,7-dioxo-3,4,5,6,7,8,9,10-octahydro-1H-benzo[c][1]oxacyclotetradecin-14-yl)oxy)pentanoic acid (hapten ZEp). Trifluoroacetic acid (0.8 mL) was dropwise added to a solution of tert-butyl ester 3 (13.5 mg, 28.4 μmol) in anhydrous CH2Cl2 (0.8 mL) cooled to 0 °C under nitrogen and the resultant solution was stirred for 1 h. The solvents were evaporated under vacuum and the remaining traces of trifluoroacetic acid were removed by repetitive evaporations with ethanol-free chloroform to afford hapten ZEp (11.6 mg, 98%) as an amorphous solid. 1H NMR (300 MHz, CDCl3), δ 12.09 (s, 1H, 16′-OH), 7.37 (br s, 1H, OH), 7.01 (dd, J = 15.3, 2.0 Hz, 1H, H-12′), 6.45 (d, J = 2.5 Hz, 1H, H-13′), 6.37 (d, J = 2.6 Hz, 1H, H-15′), 5.68 (ddd, J = 14.8, 10.4, 3.6 Hz, 1H, H-11′), 5.10–4.90 (m, 1H, H-3′), 3.99 (d, J = 5.4 Hz, 2H, H2-5), 2.86 (ddd, J = 18.5, 12.2, 2.6 Hz, 1H, H-8′), 2.68–2.55 (m, 1H, H-6′), 2.53–2.29 (m, 3H), 2.26–2.97 (m, 4H), 1.90–1.42 (m, 9H), 1.38 (d, J = 6.1 Hz, 3H, Me-3′); 13C NMR (75 MHz, CDCl3) δ 211.9 (C-7′), 171.5 (C-1′), 165.7 (C-16′), 163.6 (C-14′), 143.5 (C-12′a), 133.4 (C-11′), 132.5 (C-12′), 108.7 (C-15′), 103.7 (C-16′a), 100.5 (C-13′), 73.5 (C-3′), 67.6 (C-5), 43.1 (C-6′), 36.8 (C-8′), 34.9 (C-4′), 33.6 (C-2), 31.1 (C-10′), 28.4 (C-4), 22.4, 21.4, and 21.1 (C-3, C-5′, and C-9′), 21.0 (Me-3′); HRMS (ESI) m/z calculated for C23H31O7 [M + H]+ 419.2064, found 419.2073.
4.4. Synthesis of Heterologous Haptens
Preparation of 6-((((
S,
E)-16-hydroxy-14-methoxy-3-methyl-1-oxo-1,3,4,5,6,8,9,10-octahydro-7H-benzo[c][1]oxacyclotetradecin-7-ylidene)amino)oxy)hexanoic acid (hapten ZE
oh). A solution of 14-
O-methylzearalenone [
31] (
5, 7.5 mg, 22.6 μmol) and hydrochloride
1 (5.0 mg, 27.3 μmol) in anhydrous pyridine (200 μL) was stirred at room temperature for 24 h under nitrogen. After removing the pyridine with a stream of nitrogen, the residue was dissolved in EtOAc and successively washed with water, 0.1 M aqueous HCl, 5% aqueous NaHCO
3, and brine. Drying of the organic phase over anhydrous MgSO
4 and evaporation of the solvent under reduced pressure gave hapten ZE
oh (10.2 mg, 98%)—a mixture of
E and
Z oximes – as a colorless oil.
1H NMR (500 MHz, methanol-d
4) δ 6.96 and 6.93 (each br d,
J = 15.5 Hz, 1H, H-12′), 6.49 and 6.46 (each m, 1H, H-13′), 6.36 (m, 1H, H-15′), 6.04 and 5.79 (each m, 1H, H-11′), 5.16 and 5.01 (each m, 1H, H-3′), 4.03 and 4.00 (each t,
J = 6.4 Hz, 2H, H
2-6), 3.80 (s, 3H, OMe), 2.82 (m, 0.6H, H-8′), 2.49 (dt,
J = 14.3, 7.2 Hz, 0.4Hz, H-8′), 2.40–2.02 (m, 7H), 1.95–1.52 (m, 10H), 1.50–1.40 (m, 2H), 1.39 and 1.37 (each d,
J = 6.4 Hz, 3H, Me-3′);
13C NMR (126 MHz, methanol-d
4), δ 177.7 (C-1), 172.6 and 172.2 (C-1′), 165.9 and 165.3 (C-14′), 164.8 and 164.1 (C-16′), 162.2 and 159.5 (C-7′), 145.5 and 143.0 (C-12′a), 133.7 and 132.4 (C-12′), 133.6 and 133.5 (C-11′), 108.5 and 107.2 (C-13′), 107.9 and 105.5 (C-16′a), 100.9 and 100.8 (C-15′), 74.5 and 73.9 (C-3′), 74.1 and 74.0 (C-6), 55.9 (OMe), 36.8 and 36.2 (C-4′), 35.5 and 31.3 (C-8′), 35.1 (C-2), 31.9 and 31.8 (C-10′), 30.1 and 29.9 (C-5), 28.5 and 27.5 (C-6′), 27.0 and 26.8 (C-4), 26.0 (C-3), 25.6 and 23.8 (C-9′), 23.3 (C-5′), 20.9 and 20.5 (Me-3′); HRMS (ESI)
m/
z calculated for C
25H
36NO
7 [M + H]
+ 462.2486, found 462.2473.
Preparation of tert-butyl (S,E)-5-((16-methoxy-3-methyl-1,7-dioxo-3,4,5,6,7,8,9,10-octahydro-1H-benzo[c][1]oxacyclotetradecin-14-yl)oxy)pentanoate (4). A solution of Me2SO4 (6.6 mg, 4.9 μL, 52 μmol) in dry acetone (26 μL) was added to a mixture of tert-butyl ester 3 (13.8 mg, 29.1 μmol), and K2CO3 (6.0 mg, 43.4 μmol) in acetone (0.45 mL) at room temperature under nitrogen. The resulting mixture was stirred under the same conditions for 48 h, then diluted with water and extracted with EtOAc. The combined organic layers were washed with water and brine and dried under anhydrous Na2SO4. The residue that was left after evaporation of the solvent at reduced pressure was chromatographed on silica gel, using a 99:1 CHCl3/MeOH mixture as eluent, to give compound 4 (13.5 mg, 95%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 6.58 (d, J = 2.1 Hz, 1H, H-13′), 6.38 (dd, J = 15.5, 1.5 Hz, 1H, H-12′), 6.36 (d, J = 2.0 Hz, 1H, H-15′), 5.99 (ddd, J = 15.5, 10.0, 4.3 Hz, 1H, H-11′), 5.30 (ddd, J = 10.0, 6.4, 3.3 Hz, 1H, H-3′), 3.99 (t, J = 6.0 Hz, 2H, H2-5), 3.80 (s, 3H, OMe), 2.70 (ddd, J = 16.7, 11.2, 3.3 Hz, 1H, H-8′), 2.45-2.24 (m, 3H), 2.30 (t, J = 7.3 Hz, 2H, H2-2), 2.20–1.97 (m, 3H), 1.90–1.50 (m, 9H), 1.45 (s, 9H, CMe3), 1.34 (d, J = 6.3 Hz, 3H, Me-3′); 13C NMR (126 MHz, CDCl3) δ 211.6 (C-7′), 172.9 (C-1), 167.8 (C-1′), 160.9 (C-16′), 157.8 (C-14′), 136.9 (C-12′a), 133.3 (C-11′), 129.2 (C-12′), 116.4 (C-16′a), 102.1 (C-13′), 98.3 (C-15′), 80.4 (CMe3), 71.4 (C-3′), 67.8 (C-5), 56.1 (OMe), 44.3 (C-6′), 37.8 (C-8′), 35.3 (C-4′), 35.2 (C-2), 31.4 (C-10′), 28.7 (C-4), 28.3 (CMe3), 22.0, 21.8, and 21.5 (C-3, C-5′, and C-9′), 20.2 (Me-3′); HRMS (ESI) m/z calculated for C28H41O7 [M + H]+ 489.2847, found 489.2857.
Preparation of (S,E)-5-((16-methoxy-3-methyl-1,7-dioxo-3,4,5,6,7,8,9,10-octahydro-1H-benzo[c][1]oxacyclotetradecin-14-yl)oxy)pentanoic acid (hapten ZEph). This hapten was prepared as described above for hapten ZEp, using tert-butyl ester 4 (9.2 mg, 18.8 mmol), anhydrous CH2Cl2 (0.48 mL), and trifluoroacetic acid (0.50 mL). After workup of the reaction mixture, the obtained crude product was purified by chromatography on silica gel, using CHCl3/MeOH mixtures (from 99:1 to 95:5) as eluent, to afford hapten ZEph (7.4 mg, 91%) as an amorphous white solid. 1H NMR (300 MHz, CDCl3) δ 6.58 (d, J = 2.0 Hz, 1H, H-13′), 6.38 (dd, J = 15.5, 1.5 Hz, 1H, H-12′), 6.36 (d, J = 2.0 Hz, 1H, H-15′), 5.99 (ddd, J = 15.6, 9.9, 4.3 Hz, 1H, H-11′), 5.31 (m, 1H, H-3′), 4.01 (t, J = 7.0 Hz, 2H, H2-5), 3.80 (s, 3H, OMe), 2.70 (ddd, J = 17.6, 11.6, 3.4 Hz, 1H, H-8′), 2.49–2.25 (m, 5H), 2.20–1.97 (m, 3H), 1.89–1.50 (m, 9H), 1.34 (d, J = 6.2 Hz, 3H, Me-3′); 13C NMR (126 MHz, CDCl3) δ 211.8 (C-7′), 178.4 (C-1), 167.8 (C-1′), 160.8 (C-16′), 157.8 (C-14′), 137.0 (C-12′a), 133.3 (C-11′), 129.2 (C-12′), 116.4 (C-16′a), 102.1 (C-13′), 98.3 (C-15′), 71.4 (C-3′), 67.7 (C-4), 56.1 (OMe), 44.2 (C-6′), 37.8 (C-8′), 35.3 (C-4′), 33.5 (C-2), 31.4 (C-10′), 28.6 (C-4), 22.0 (C-5′), 21.5 (C-3 and C-9′), 20.2 (Me-3′); HRMS (ESI) m/z calculated for C24H33O7 [M + H]+ 433.2221, found 433.2217.
4.5. Hapten Activation
Preparation of the N-hydroxysuccinimidyl ester of hapten ZEo (ZEo-NHS ester). A solution of hapten ZEo (15.4 mg, 34.4 µmol), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (7.8 mg, 40.7 µmol, 1.2 equiv), and N-hydroxisuccinimide (6.0 mg, 52.1 µmol, 1.5 equiv) in anhydrous DMF (0.5 mL) was stirred at room temperature overnight under nitrogen. The reaction mixture was diluted with Et2O and successively washed with water, 5% aqueous NaHCO3, 1.5% aqueous LiCl, and brine. The organic layer was dried over anhydrous MgSO4 and concentrated under reduced pressure to give the N-hydroxysuccinimidyl ester of hapten ZEo, ZEo-NHS ester (15.5 mg, 92% of crude product)—a mixture of E and Z oximes—as a colorless oil, which was used immediately for the preparation of the corresponding protein and enzyme conjugates. 1H NMR (300 MHz, CDCl3) δ 12.00 and 11.91 (each s, 1H, 16′-OH), 7.07 and 7.00 (each br d, J = 15.6 Hz, 1H, H-12′), 6.40 and 6.39 (each d, J = 2.6 Hz, 1H, H-13′), 6.33 and 6.32 (d, J = 2.6 Hz, 1H, H-15′), 5.94 (dt, J = 15.7, 6.6 Hz, 0.4H, H-11′), 5.74 (ddd, J = 15.4, 8.7, 5.4 Hz, 0.6H, H-11′), 5.16 and 5.01 (each m, 1H, H-3′), 4.04 and 4.03 (each t, J = 6.4 Hz, 2H, H2-6), 2.83 (s, 4H, COCH2CH2CO), 2.63 and 2.62 (each t, J = 7.5 Hz, 2H, H2-2), 2.40–2.00 (m, 6H), 1.96–1.43 (m, 12H), 1.39 and 1.38 (each d, J = 6.2 Hz, 3H, Me-3′).
Preparation of the N-hydroxysuccinimidyl ester of hapten ZEp (ZEp-NHS ester). ZEp-NHS ester was prepared as described above for ZEo-NHS, using hapten ZEp (11.3 mg, 27 µmol), EDC·HCl (7.3 mg, 38.1 µmol, 1.4 equiv), NHS (4.8 mg, 41.7 µmol, 1.5 equiv), and DMF (0.5 mL). Crude ZEp-NHS ester (11.9 mg, 85%) was obtained as a foam. 1H NMR (300 MHz, CDCl3) δ 12.06 (s, 1H, 16′-OH), 7.01 (dd, J = 15.3, 2.0 Hz, 1H, H-12′), 6.45 (d, J = 2.5 Hz, 1H, H-13′), 6.37 (d, J = 2.5 Hz, 1H, H-15′), 5.69 (ddd, J = 15.3, 10.4, 3.7 Hz, 1H, H-11′), 5.05–4.94 (m, 1H, H-3′), 4.01 (d, J = 5.5 Hz, 2H, H2-5), 2.84 (s, 4H, COCH2CH2CO), 2.78–2.90 (m overlapped with signal at 2.84, 1H, H-8′), 2.70 (t, J = 7.0 Hz, 2H, H2-2), 2.64–2.55 (m, 1H, H-6′), 2.47–2.30 (m, 2H), 2.30–1.44 (m, 12H), 1.38 (d, J = 6.1 Hz, 3H, Me-3′).
Preparation of the N-hydroxysuccinimidyl ester of hapten ZEoh (ZEoh-NHS ester). ZEoh-NHS ester was prepared as described above for ZEo-NHS, using hapten ZEoh (9.8 mg, 21.2 µmol), EDC·HCl (5.0 mg, 26.1 µmol, 1.2 equiv), NHS (3.3 mg, 28.7 µmol, 1.4 equiv), and DMF (0.35 mL). Crude ZEoh-NHS ester (11.2 mg, 95%)—a mixture of E and Z oximes—was obtained as an oil. 1H NMR (500 MHz, CDCl3) δ 12.03 and 11.93 (each s, 1H, 16′-OH), 7.08 and 7.02 (dd, J = 15.7, 1.7 Hz, 1H, H-12′), 6.46 and 6.45 (each d, J = 2.6 Hz, 1H, H-13′), 6.39 and 6.38 (each d, J = 2.6 Hz, 1H, H-15′), 5.95 (ddd, J = 15.6, 7.4, 5.7 Hz, 0.4H, H-11′), 5.75 (ddd, J = 15.5, 9.5, 4.6 Hz, 0.6H, H-11′), 5.17 and 5.00 (each m, 1H, H-3′), 4.05 and 4.02 (each t, J = 6.4 Hz, 2H, H2-6), 3.82 and 3.81 (each s, 3H, OMe), 2.82 (br s, 4H, COCH2CH2CO), 2.63 and 2.62 (each t, J = 7.5 Hz, 2H, H2-2), 2.40–2.04 (m, 6H), 1.96–1.45 (m, 12H), 1.40 and 1.39 (each d, J = 6.3 Hz, 3H, Me-3′).
Preparation of the N-hydroxysuccinimidyl ester of hapten ZEph (ZEph-NHS ester). ZEph-NHS ester was prepared as described above for ZEo-NHS, using hapten ZEph (7.7 mg, 17.8 µmol), EDC·HCl (4.0 mg, 20.9 µmol, 1.2 equiv), NHS (3.0 mg, 26.1 µmol, 1.5 equiv), and DMF (0.25 mL). Crude ZEph-NHS ester (7.8 mg, 83%) was obtained as a foam. 1H NMR (500 MHz, CDCl3) δ 6.59 (d, J = 2.1 Hz, 1H, H-13′), 6.37 (dd, J = 15.6, 1.8 Hz, 1H, H-12′), 6.36 (d, J = 2.0 Hz, 1H, H-15′), 6.00 (ddd, J = 15.5, 10.0, 4.3 Hz, 1H, H-11′), 5.30 (ddt, J = 10.0, 6.3, 3.3 Hz, H-3′), 4.03 (t, J = 5.8 Hz, 2H, H2-5), 3.80 (s, 3H, OMe), 2.84 (s, 4H, COCH2CH2CO), 2.71 (t, J = 6.9 Hz, 2H, H2-2), 2.70 (ddd, J = 17.0, 11.1, 3.2 Hz, 1H, H-8′), 2.41 (ddd, J = 13.6, 9.4, 4.6 Hz, 1H, H-6′), 2.36–2.26 (m, 2H), 2.19–1.51 (m, 12H), 1.33 (d, J = 6.3 Hz, 3H, Me-3′).
4.6. Bioconjugate Preparation and Analysis
Protein conjugates were prepared by the active ester method. The purified hapten-NHS esters were dissolved in DMF to obtain 50 mM solutions. The four haptens were coupled to OVA and HRP, whereas only haptens ZE
o and ZE
p were conjugated to BSA. Protein solutions were prepared in PB at 15 mg/mL for BSA and OVA and at 3 mg/mL for HRP. Conjugation reactions were carried out overnight at room temperature in amber glass vials by drop wise adding the hapten-NHS solution over the protein solution under vigorous stirring. To prepare BSA conjugates, a 30-fold hapten-to-protein molar ratio was used whereas molar excess values around 12 were employed to prepare OVA and HRP conjugates. The conjugates were purified by size-exclusion chromatography using PB as eluent, with the exception of tracers HRP-ZE
o and HRP-ZE
oh that were purified with HEPES buffer. Fractions containing the bioconjugates were pooled and processed as described in previous studies [
32].
Analysis of hapten bioconjugates was performed by MALDI-TOF/TOF mass spectrometry as follows. Briefly, 100 μL of protein or enzyme conjugate solutions (
ca. 1 mg/mL and 0.5 mg/mL for BSA/OVA and HRP conjugates, respectively) were dialyzed against Milli-Q
® water. The final volume of each dialysis solution was ca. 200 μL. Then, 0.8 μL of every sample solution was spotted onto the MALDI plate. After, the droplets were air-dried at room temperature, and 0.8 μL of matrix (10 mg/mL of sinapinic acid (Bruker) in 70% MeCN, 0.1% TFA) was added and allowed to air-dry at room temperature. The resulting mixtures were analyzed by MALDI-TOF/TOF in positive linear mode (1500 shots every position) in a mass range of 12,000-100,000
m/
z, with laser intensity of 6000. Previously, the plate was calibrated with 1 μL of the TOF/TOF calibration mixture (ABSciex), in 13 positions. Every sample was calibrated by a ‘close external calibration’ method with a BSA, OVA, or HRP spectrum acquired in a close position. The analysis of the results was performed using the mMass program (v5.5.0, available at
http://www.mmass.org/, accessed on 3 February 2022).
4.7. Antibody Generation
Animal manipulation was carried out according to European Directive 2010/63EU and Spanish laws (RD118/2021 and law 32/2007) regarding the protection of experimental animals. A group of two female New Zealand white rabbits were immunized by subcutaneous injection of each BSA conjugate. Animals received, every three weeks, 300 μg of protein conjugate in a 1:1 water-in-oil emulsion (1 mL) between PBS and Freund’s adjuvant. Complete adjuvant was employed for the first injection, and it was substituted by incomplete adjuvant for subsequent boosts. Ten days after the fourth immunization, animals were exsanguinated, and the blood was left overnight at 4 °C for coagulation. The cells were separated from the serum by centrifugation during 20 min at 3000× g. The antisera were partially purified by salting out twice with one volume of a cold saturated (3.9 M) solution of ammonium sulfate. Antibodies were preserved as precipitates at 4 °C. For daily usage, a fraction of antiserum was diluted in PBS containing 1% BSA and 0.01% thimerosal.
4.8. Competitive ELISA
Plate microwells were washed four times with washing solution after each incubation step. For antibody-coated direct competitive assays, plates were coated by overnight incubation at 4 °C with 100 μL per well of GAR solution (1 μg/mL) in coating buffer. Then, 100 μL per well of antiserum dilution in PBST was added, and plates were incubated 1 h at room temperature. The competitive reaction was carried out at room temperature during 1 h by mixing 50 μL per well of standard solution in PBS and 50 μL per well of enzyme tracer solution in PBST. The retained enzyme activity was revealed during 10 min at room temperature by adding 100 μL per well of freshly prepared enzyme substrate solution. The chromogenic reaction was stopped with 100 μL per well of 1 M H2SO4.
For the conjugate-coated indirect competitive ELISA format, microplates were coated by overnight incubation at room temperature with 100 μL per well of bioconjugate solution in coating buffer. The competitive reaction was performed with 50 μL per well of standard solution in PBS and 50 μL per well of antibody dilution in PBST, and incubation at room temperature for 1 h. Next, 100 μL per well of secondary antibody solution was applied, and the plates were incubated again 1 h at room temperature. Finally, color was developed as for the previous format.
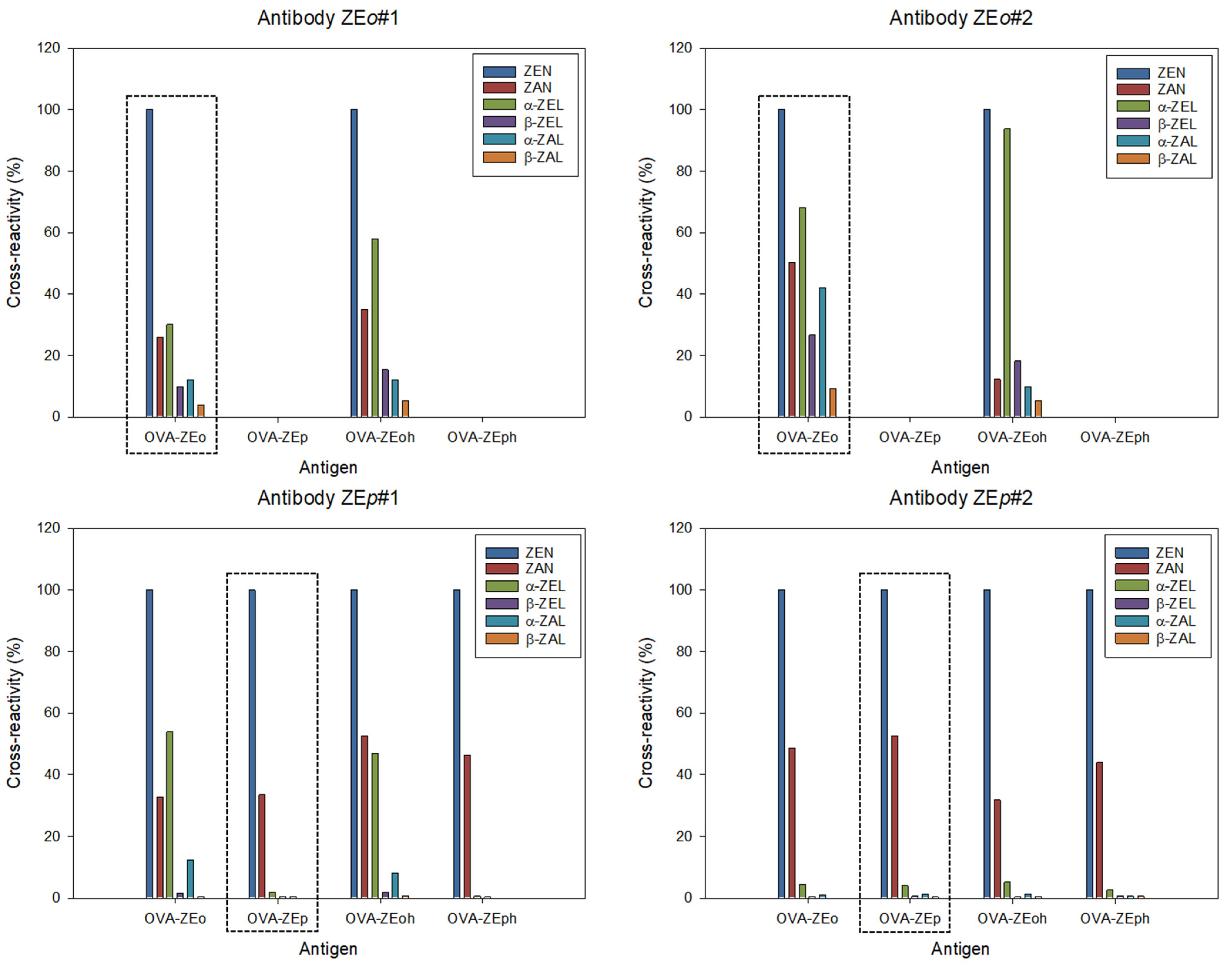
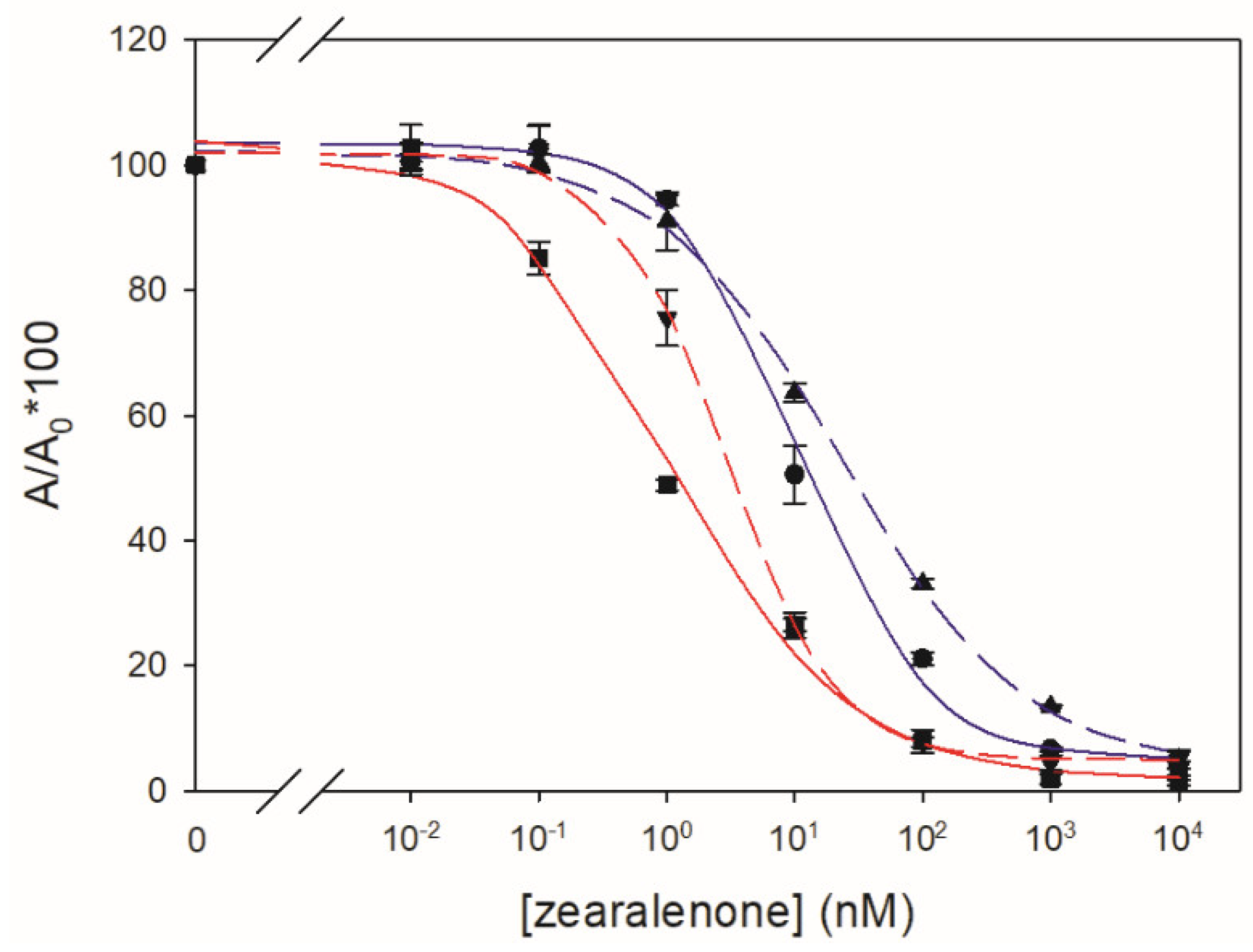
The absorbance of the chromogenic reaction products in the microwells was read at 492 nm with 650 nm as reference wavelength. Seven calibration solutions of each analyte were prepared in borosilicate glass vials by serial dilution in PBS from the most concentrated solution. A blank solution was assayed together with the standards. Inhibition curves were obtained by fitting the experimental values to a four-parameter logistic equation for standard curves using the SigmaPlot software from Systat Software Inc. (v14.5, San Jose, CA, USA, 2020). The Amax value corresponds to the maximum absorbance value obtained in the absence of analyte. The antibody apparent affinity was estimated as the half-maximum inhibition concentration (IC50) of the analyte. Cross-reactivity (%) was calculated from the quotient between the IC50 for ZEN and the IC50 for the assayed metabolite.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}