The Interplay between Uremic Toxins and Albumin, Membrane Transporters and Drug Interaction

 ,
,  , and
, and 
Abstract
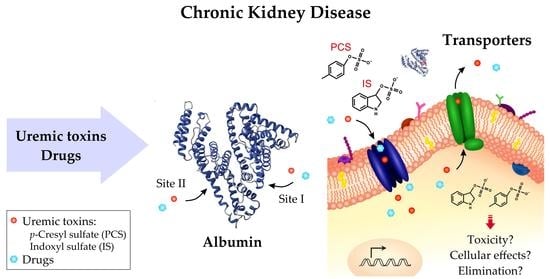
1. Introduction
2. Protein-Bound Uremic Toxins (PBUTs)
2.1. Indoxyl Sulfate (IS)
2.2. p-Cresyl Sulfate (PCS)
2.3. Indole-3-Acetic Acid (IAA)
2.4. 3-Carboxy-4-methyl-5-propyl-2-furanpropionate (CMPF)
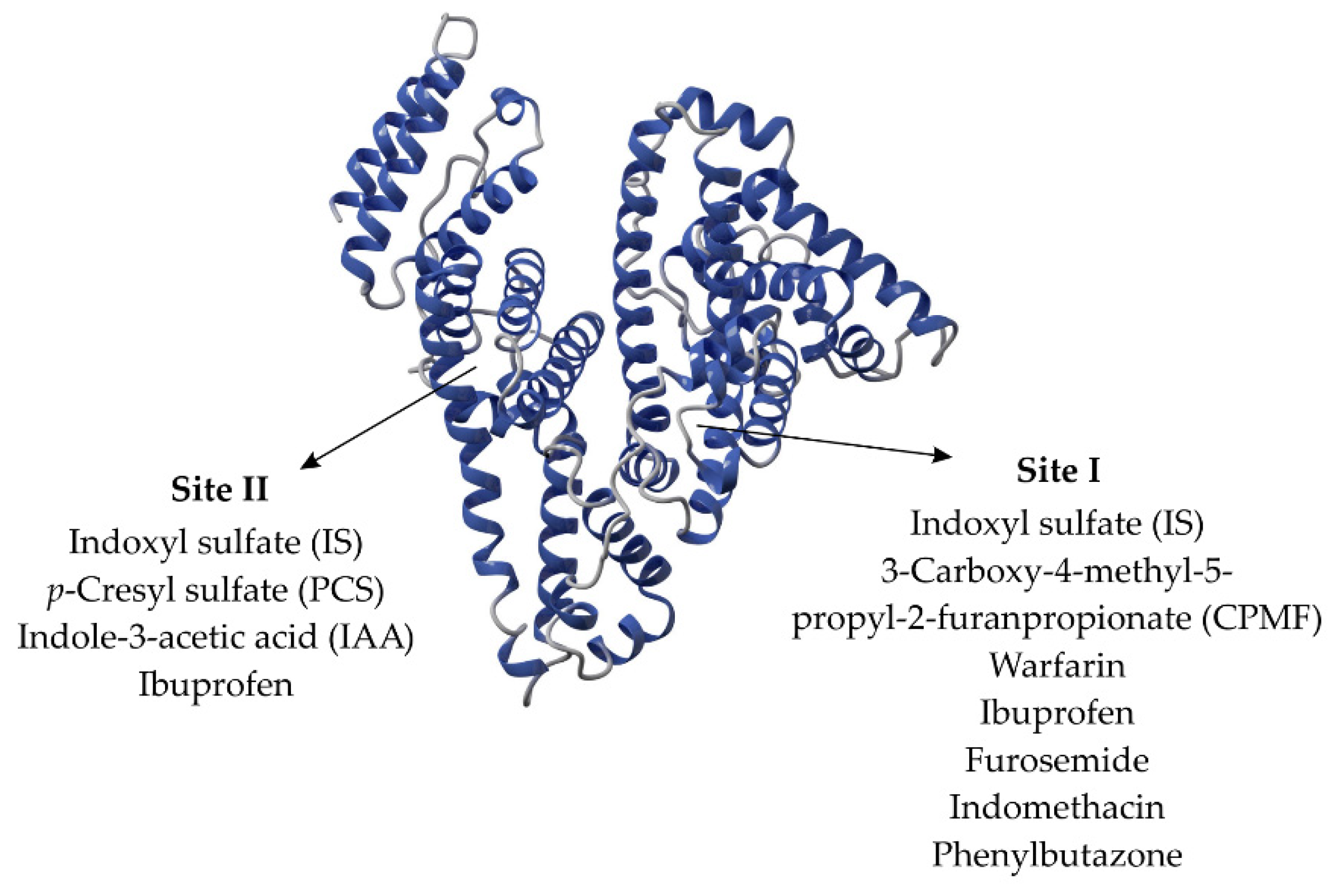
3. Interaction between Uremic Toxins and Albumin
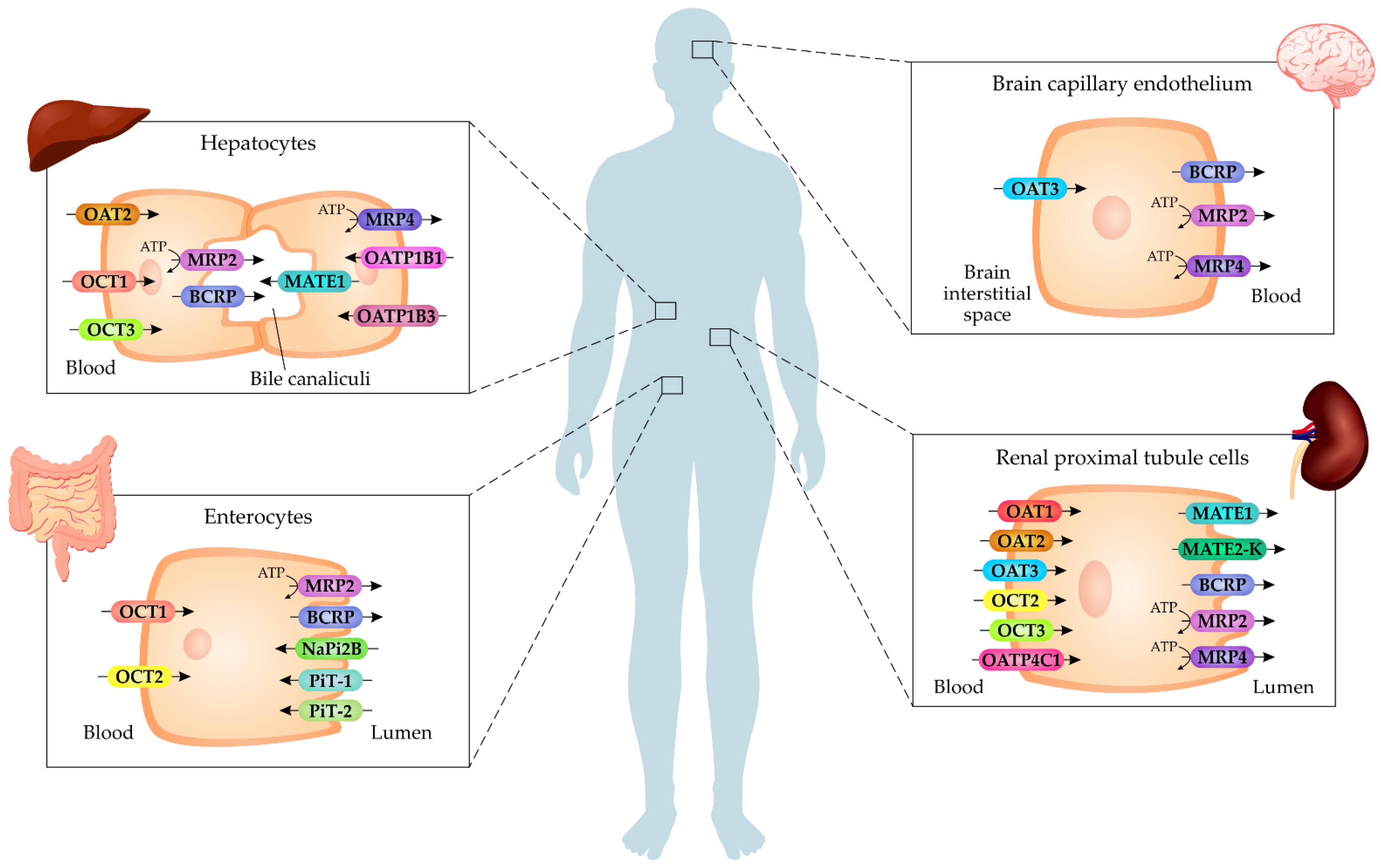
4. Cell Membrane Transporters of Uremic Toxins
4.1. Organic Anions Transporters (OATs)
4.2. Organic Cation Transporters (OCTs)
4.3. Organic Anion-Transporting Polypeptides (OATPs)
4.4. Inorganic Phosphate Transporters (PiTs)
4.5. Multidrug and Toxin Extrusion (MATE)
4.6. Breast Cancer Resistance Protein (BCRP)
4.7. Multidrug Resistance-Associated Proteins (MRPs)
5. Final Considerations
Author Contributions
Funding
Conflicts of Interest
References
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argilés, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [PubMed]
- Falconi, C.A.; da Cruz Junho, C.V.; Fogaça-Ruiz, F.; Vernier, I.C.S.; da Cunha, R.S.; Stinghen, A.E.M.; Carneiro-Ramos, M.S. Uremic Toxins: An Alarming Danger Concerning the Cardiovascular System. Front. Physiol. 2021, 12, 114460. [Google Scholar] [CrossRef]
- Li, X.S.; Obeid, S.; Klingenberg, R.; Gencer, B.; Mach, F.; Räber, L.; Windecker, S.; Rodondi, N.; Nanchen, D.; Muller, O.; et al. Gutmicrobiota-dependent trimethylamine N-oxide in acute coronary syndromes: A prognostic marker for incident cardiovascular events beyond traditional risk factors. Eur. Heart J. 2017, 38, 814–824. [Google Scholar] [CrossRef]
- Stubbs, J.R.; House, J.A.; Ocque, A.J.; Zhang, S.; Johnson, C.; Kimber, C.; Schmidt, K.; Gupta, A.; Wetmore, J.B.; Nolin, T.D.; et al. Serum Trimethylamine-N-Oxide is Elevated in CKD and Correlates with Coronary Atherosclerosis Burden. J. Am. Soc. Nephrol. 2016, 27, 305–313. [Google Scholar] [CrossRef]
- Barreto, F.C.; Stinghen, A.E.M.; De Oliveira, R.B.; Franco, A.T.B.; Moreno, A.N.; Barreto, D.V.; Pecoits-Filho, R.; Drüeke, T.B.; Massy, Z.A. The quest for a better understanding of chronic kidney disease complications: An update on uremic toxins. J. Bras. Nefrol. 2014, 36, 221–235. [Google Scholar] [CrossRef]
- Massy, Z.A.; Liabeuf, S. Middle-molecule uremic toxins and outcomes in chronic kidney disease. Contrib. Nephrol. 2017, 191, 8–17. [Google Scholar] [CrossRef]
- Van Gelder, M.K.; Middel, I.R.; Vernooij, R.W.M.; Bots, M.L.; Verhaar, M.C.; Masereeuw, R.; Grooteman, M.P.; Nubé, M.J.; van den Dorpel, M.A.; Blankestijn, P.J.; et al. Protein-Bound Uremic Toxins in Hemodialysis Patients Relate to Residual Kidney Function, Are Not Influenced by Convective Transport, and Do Not Relate to Outcome. Toxins 2020, 12, 234. [Google Scholar] [CrossRef]
- Han, H.; Zhu, J.; Zhu, Z.; Ni, J.; Du, R.; Dai, Y.; Chen, Y.; Wu, Z.; Lu, L.; Zhang, R. p-Cresyl Sulfate Aggravates Cardiac Dysfunction Associated with Chronic Kidney Disease by Enhancing Apoptosis of Cardiomyocytes. J. Am. Heart Assoc. 2015, 4, e001852. [Google Scholar] [CrossRef]
- Calaf, R.; Cerini, C.; Génovésio, C.; Verhaeghe, P.; Jourde-Chiche, N.; Bergé-Lefranc, D.; Gondouin, B.; Dou, L.; Morange, S.; Argilés, A.; et al. Determination of uremic solutes in biological fluids of chronic kidney disease patients by HPLC assay. J. Chromatogr. B 2011, 879, 2281–2286. [Google Scholar] [CrossRef]
- Niwa, T. Uremic toxicity of indoxyl sulfate. Nagoya J. Med. Sci. 2010, 72, 1–11. [Google Scholar]
- Ito, S.; Yoshida, M. Protein-bound uremic toxins: New culprits of cardiovascular events in chronic kidney disease patients. Toxins 2014, 6, 665–678. [Google Scholar] [CrossRef]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A. Serum Indoxyl Sulfate Is Associated with Vascular Disease and Mortality in Chronic Kidney Disease Patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef]
- Lekawanvijit, S. Cardiotoxicity of uremic toxins: A driver of cardiorenal syndrome. Toxins 2018, 10, 352. [Google Scholar] [CrossRef]
- Gao, H.; Liu, S. Role of uremic toxin indoxyl sulfate in the progression of cardiovascular disease. Life Sci. 2017, 185, 23–29. [Google Scholar] [CrossRef]
- Da Cunha, R.S.; Santos, A.F.; Barreto, F.C.; Stinghen, A.E.M. How do Uremic Toxins Affect the Endothelium? Toxins 2020, 12, 412. [Google Scholar] [CrossRef]
- Lekawanvijit, S.; Adrahtas, A.; Kelly, D.J.; Kompa, A.R.; Wang, B.H.; Krum, H. Does indoxyl sulfate, a uraemic toxin, have direct effects on cardiac fibroblasts and myocytes? Eur. Heart J. 2010, 31, 1771–1779. [Google Scholar] [CrossRef]
- Kim, H.Y.; Yoo, T.-H.; Cho, J.-Y.; Kim, H.C.; Lee, W.-W. Indoxyl sulfate–induced TNF-α is regulated by crosstalk between the aryl hydrocarbon receptor, NF-κB, and SOCS2 in human macrophages. FASEB J. 2019, 33, 10844–10858. [Google Scholar] [CrossRef]
- Yang, K.; Wang, C.; Nie, L.; Zhao, X.; Gu, J.; Guan, X.; Wang, S.; Xiao, T.; Xu, X.; He, T.; et al. Klotho Protects Against Indoxyl Sulphate-Induced Myocardial Hypertrophy. J. Am. Soc. Nephrol. 2015, 26, 2434–2446. [Google Scholar] [CrossRef]
- Nakano, T.; Katsuki, S.; Chen, M.; Decano, J.L.; Halu, A.; Lee, L.H.; Pestana, D.V.S.; Kum, A.S.T.; Kuromoto, R.K.; Golden, W.S.; et al. Uremic Toxin Indoxyl Sulfate Promotes Proinflammatory Macrophage Activation Via the Interplay of OATP2B1 and Dll4-Notch Signaling. Circulation 2019, 139, 78–96. [Google Scholar] [CrossRef]
- Gryp, T.; Vanholder, R.; Vaneechoutte, M.; Glorieux, G. p-Cresyl Sulfate. Toxins 2017, 9, 52. [Google Scholar] [CrossRef]
- Vanholder, R.; Schepers, E.; Pletinck, A.; Nagler, E.V.; Glorieux, G. The Uremic Toxicity of Indoxyl Sulfate and p-Cresyl Sulfate: A Systematic Review. J. Am. Soc. Nephrol. 2014, 25, 1897–1907. [Google Scholar] [CrossRef]
- Gondouin, B.; Cerini, C.; Dou, L.; Sallée, M.; Duval-Sabatier, A.; Pletinck, A.; Calaf, R.; Lacroix, R.; Jourde-Chiche, N.; Poitevin, S.; et al. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int. 2013, 84, 733–744. [Google Scholar] [CrossRef]
- Edamatsu, T.; Fujieda, A.; Itoh, Y. Phenyl sulfate, indoxyl sulfate and p-cresyl sulfate decrease glutathione level to render cells vulnerable to oxidative stress in renal tubular cells. PLoS ONE 2018, 13, e0193342. [Google Scholar] [CrossRef]
- Huang, T.H.; Yip, H.K.; Sun, C.K.; Chen, Y.L.; Yang, C.C.; Lee, F.Y. P-cresyl sulfate causes mitochondrial hyperfusion in H9C2 cardiomyoblasts. J. Cell. Mol. Med. 2020, 24, 8379–8390. [Google Scholar] [CrossRef]
- Glorieux, G.; Vanholder, R.; Van Biesen, W.; Pletinck, A.; Schepers, E.; Neirynck, N.; Speeckaert, M.; De Bacquer, D.; Verbeke, F. Free p-cresyl sulfate shows the highest association with cardiovascular outcome in chronic kidney disease. Nephrol. Dial. Transplant. 2021, 36, 998–1005. [Google Scholar] [CrossRef]
- Liabeuf, S.; Laville, S.M.; Glorieux, G.; Cheddani, L.; Brazier, F.; Beauport, D.T.; Valholder, R.; Choukroun, G.; Massy, Z.A. Difference in profiles of the gut-derived tryptophan metabolite indole acetic acid between transplanted and non-transplanted patients with chronic kidney disease. Int. J. Mol. Sci. 2020, 21, 2031. [Google Scholar] [CrossRef]
- Rysz, J.; Franczyk, B.; Ławiński, J.; Olszewski, R.; Ciałkowska-Rysz, A.; Gluba-Brzózka, A. The Impact of CKD on Uremic Toxins and Gut Microbiota. Toxins 2021, 13, 252. [Google Scholar] [CrossRef]
- Dou, L.; Poitevin, S.; Sallée, M.; Addi, T.; Gondouin, B.; McKay, N.; Denison, M.S.; Jourde-Chiche, N.; Duval-Sabatier, A.; Cerini, C.; et al. Aryl hydrocarbon receptor is activated in patients and mice with chronic kidney disease. Kidney Int. 2018, 93, 986–999. [Google Scholar] [CrossRef]
- Dou, L.; Sallée, M.; Cerini, C.; Poitevin, S.; Gondouin, B.; Jourde-Chiche, N.; Fallague, K.; Brunet, P.; Calaf, R.; Dussol, B.; et al. The cardiovascular effect of the uremic solute indole-3 acetic acid. J. Am. Soc. Nephrol. 2015, 26, 876–887. [Google Scholar] [CrossRef]
- Lamas, B.; Natividad, J.M.; Sokol, H. Aryl hydrocarbon receptor and intestinal immunity. Mucosal Immunol. 2018, 11, 1024–1038. [Google Scholar] [CrossRef]
- Satoh, M.; Hayashi, H.; Watanabe, M.; Ueda, K.; Yamato, H.; Yoshioka, T.; Motojima, M. Uremic Toxins Overload Accelerates Renal Damage in a Rat Model of Chronic Renal Failure. Nephron Exp. Nephrol. 2003, 95, e111–e118. [Google Scholar] [CrossRef] [PubMed]
- Meijers, B.K.I.; Bammens, B.; Verbeke, K.; Evenepoel, P. A review of albumin binding in CKD. Am. J. Kidney Dis. 2008, 51, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Takadate, A.; Otagiri, M. Characterization of Binding Site of Uremic Toxins on Human Serum Albumin. Biol. Pharm. Bull. 1995, 18, 1755–1761. [Google Scholar] [CrossRef] [PubMed]
- Mabuchi, H.; Nakahashi, H.; Hamajima, T.; Aikawa, I.; Oka, T. The Effect of Renal Transplantation on a Major Endogenous Ligand Retained in Uremic Serum. Am. J. Kidney Dis. 1989, 13, 49–54. [Google Scholar] [CrossRef]
- Koppe, L.; Poitout, V. CMPF: A Biomarker for Type 2 Diabetes Mellitus Progression? Trends Endocrinol. Metab. 2016, 27, 439–440. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Sinclair, A.J.; Faiza, M.; Li, D.; Han, X.; Yin, H.; Wang, Y. Furan fatty acids—Beneficial or harmful to health? Prog. Lipid Res. 2017, 68, 119–137. [Google Scholar] [CrossRef]
- Prentice, K.J.; Luu, L.; Allister, E.M.; Liu, Y.; Jun, L.S.; Sloop, K.W.; Hardy, A.B.; Wei, L.; Jia, W.; Fantus, I.G.; et al. The furan fatty acid metabolite CMPF is elevated in diabetes and induces β cell dysfunction. Cell Metab. 2014, 19, 653–666. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Iwao, Y.; Mera, K.; Watanabe, H.; Kadowaki, D.; Ishima, Y.; Chuang, V.T.G.; Sato, K.; Otagiri, M.; Maruyama, T. A uremic toxin, 3-carboxy-4-methyl-5-propyl-2-furanpropionate induces cell damage to proximal tubular cells via the generation of a radical intermediate. Biochem. Pharmacol. 2012, 84, 1207–1214. [Google Scholar] [CrossRef]
- Wratten, M.L.; Galaris, D.; Tetta, C.; Sevanian, A. Evolution of oxidative stress and inflammation during hemodialysis and their contribution to cardiovascular disease. Antioxid. Redox Signal. 2002, 4, 935–944. [Google Scholar] [CrossRef]
- Boelaert, J.; Lynen, F.; Glorieux, G.; Eloot, S.; Van Landschoot, M.; Waterloos, M.A.; Sandra, P.; Vanholder, R. A novel UPLC-MS-MS method for simultaneous determination of seven uremic retention toxins with cardiovascular relevance in chronic kidney disease patients. Anal. Bioanal. Chem. 2013, 405, 1937–1947. [Google Scholar] [CrossRef]
- Luce, M.; Bouchara, A.; Pastural, M.; Granjon, S.; Szelag, J.C.; Laville, M.; Arkouche, W.; Fouque, D.; Soulage, C.O.; Koppe, L. Is 3-carboxy-4-methyl-5-propyl-2-furanpropionate (CMPF) a clinically relevant uremic toxin in haemodialysis patients? Toxins 2018, 10, 205. [Google Scholar] [CrossRef] [PubMed]
- Fanali, G.; Di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human serum albumin: From bench to bedside. Mol. Asp. Med. 2012, 33, 209–290. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Tonelli, M.; Unsworth, L.D. Indoxyl and p-cresol sulfate binding with human serum albumin. Colloids Surf. A Physicochem. Eng. Asp. 2022, 635, 128042. [Google Scholar] [CrossRef]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; Bocedi, A.; Notari, S.; Fanali, G.; Fesce, R.; Fasano, M. Allosteric Modulation of Drug Binding to Human Serum Albumin. Mini-Rev. Med. Chem. 2006, 6, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Gundry, R.L.; Fu, Q.; Jelinek, C.A.; Van Eyk, J.E.; Cotter, R.J. Investigation of an albumin-enriched fraction of human serum and its albuminome. Proteom. Clin. Appl. 2007, 1, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Margarson, M.P.; Soni, N. Serum albumin: Touchstone or totem? Anaesthesia 1998, 53, 789–803. [Google Scholar] [CrossRef]
- Narazaki, R.; Otagiri, M. Covalent Binding of a Bucillamine Derivative with Albumin in Sera from Healthy Subjects and Patients with Various Diseases. Pharm. Res. 1997, 14, 351–353. [Google Scholar] [CrossRef]
- Kawakami, A.; Kubota, K.; Yamada, N.; Tagami, U.; Takehana, K.; Sonaka, I.; Suzuki, E.; Hirayama, K. Identification and characterization of oxidized human serum albumin: A slight structural change impairs its ligand-binding and antioxidant functions. FEBS J. 2006, 273, 3346–3357. [Google Scholar] [CrossRef]
- Oettl, K.; Stauber, R.E. Physiological and pathological changes in the redox state of human serum albumin critically influence its binding properties. Br. J. Pharmacol. 2007, 151, 580–590. [Google Scholar] [CrossRef]
- Duni, A.; Liakopoulos, V.; Roumeliotis, S.; Peschos, D.; Dounousi, E. Oxidative stress in the pathogenesis and evolution of chronic kidney disease: Untangling ariadne’s thread. Int. J. Mol. Sci. 2019, 20, 3711. [Google Scholar] [CrossRef] [PubMed]
- Anraku, M.; Yamasaki, K.; Maruyama, T.; Kragh-Hansen, U.; Otagiri, M. Effect of oxidative stress on the structure and function of human serum albumin. Pharm. Res. 2001, 18, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Himmelfarb, J.; McMonagle, E. Albumin is the major plasma protein target of oxidant stress in uremia. Kidney Int. 2001, 60, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Wratten, M.L.; Sereni, L.; Tetta, C. Oxidation of albumin is enhanced in the presence of uremic toxins. Ren. Fail. 2001, 23, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, Y.; Terawaki, H.; Terada, T.; Era, S. Albumin thiol oxidation and serum protein carbonyl formation are progressively enhanced with advancing stages of chronic kidney disease. Clin. Exp. Nephrol. 2009, 13, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Massy, Z.A.; Pietrement, C.; Touré, F. Reconsidering the Lack of Urea Toxicity in Dialysis Patients. Semin. Dial. 2016, 29, 333–337. [Google Scholar] [CrossRef]
- Jaisson, S.; Desmons, A.; Doué, M.; Gorisse, L.; Pietrement, C.; Gillery, P. Measurement of Homocitrulline, A Carbamylation-derived Product, in Serum and Tissues by LC-MS/MS. Curr. Protoc. Protein Sci. 2018, 92, e56. [Google Scholar] [CrossRef]
- Erill, S.; Calvo, R.; Carlos, R. Plasma protein carbamylation and decreased acidic drug protein binding in uremia. Clin. Pharmacol. Ther. 1980, 27, 612–618. [Google Scholar] [CrossRef]
- Viaene, L.; Annaert, P.; de Loor, H.; Poesen, R.; Evenepoel, P.; Meijers, B. Albumin is the main plasma binding protein for indoxyl sulfate and p-cresyl sulfate. Biopharm. Drug Dispos. 2013, 34, 165–175. [Google Scholar] [CrossRef]
- Watanabe, H.; Noguchi, T.; Miyamoto, Y.; Kadowaki, D.; Kotani, S.; Nakajima, M.; Miyamura, S.; Ishima, Y.; Otagiri, M.; Maruyama, T. Interaction between Two Sulfate-Conjugated Uremic Toxins, p-Cresyl Sulfate and Indoxyl Sulfate, during Binding with Human Serum Albumin. Drug Metab. Dispos. 2012, 40, 1423–1428. [Google Scholar] [CrossRef]
- Meijers, B.K.I.; De Loor, H.; Bammens, B.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. p-cresyl sulfate and indoxyl sulfate in hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1932–1938. [Google Scholar] [CrossRef] [PubMed]
- Hirata, K.; Ikeda, T.; Watanabe, H.; Maruyama, T.; Tanaka, M.; Chuang, V.T.G.; Uchida, Y.; Sakurama, K.; Nishi, K.; Yamasaki, K.; et al. The binding of aripiprazole to plasma proteins in chronic renal failure patients. Toxins 2021, 13, 811. [Google Scholar] [CrossRef] [PubMed]
- André, C.; Choukroun, G.; Bennis, Y.; Kamel, S.; Lemaire-Hurtel, A.S.; Masmoudi, K.; Bodeau, S.; Liabeuf, S. Potential interactions between uraemic toxins and drugs: An application in kidney transplant recipients treated with calcineurin inhibitors. Nephrol. Dial. Transplant. 2021, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.W. Increasing Dialysate Flow and Dialyzer Mass Transfer Area Coefficient to Increase the Clearance of Protein-bound Solutes. J. Am. Soc. Nephrol. 2004, 15, 1927–1935. [Google Scholar] [CrossRef]
- Böhringer, F.; Jankowski, V.; Gajjala, P.R.; Zidek, W.; Jankowski, J. Release of uremic retention solutes from protein binding by hypertonic predilution hemodiafiltration. ASAIO J. 2015, 61, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Ito, T.; Sato, M.; Goto, S.; Kazama, J.J.; Gejyo, F.; Narita, I. Adsorption of Protein-Bound Uremic Toxins Using Activated Carbon through Direct Hemoperfusion in vitro. Blood Purif. 2019, 48, 215–222. [Google Scholar] [CrossRef]
- Shi, Y.; Tian, H.; Wang, Y.; Shen, Y.; Zhu, Q.; Ding, F. Effect of ionic strength, pH and chemical displacers on the percentage protein binding of protein-bound uremic toxins. Blood Purif. 2019, 47, 351–360. [Google Scholar] [CrossRef]
- Tao, X.; Thijssen, S.; Kotanko, P.; Ho, C.H.; Henrie, M.; Stroup, E.; Handelman, G. Improved dialytic removal of protein-bound uraemic toxins with use of albumin binding competitors: An in vitro human whole blood study. Sci. Rep. 2016, 6, 23389. [Google Scholar] [CrossRef]
- Madero, M.; Cano, K.B.; Campos, I.; Tao, X.; Maheshwari, V.; Brown, J.; Cornejo, B.; Handelman, G.; Thijssen, S.; Kotanko, P. Removal of protein-bound uremic toxins during hemodialysis using a binding competitor. Clin. J. Am. Soc. Nephrol. 2019, 14, 394–402. [Google Scholar] [CrossRef]
- Van Biesen, W.; Eloot, S. Enhanced removal of protein-bound uremic toxins using displacers: Road to success? Clin. J. Am. Soc. Nephrol. 2019, 14, 324–326. [Google Scholar] [CrossRef]
- Shi, Y.; Tian, H.; Wang, Y.; Shen, Y.; Zhu, Q.; Ding, F. Removal of protein-bound uremic toxins by liposome-supported peritoneal dialysis. Perit. Dial. Int. 2019, 39, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Shen, Y.; Bi, X.; Li, J.; Chen, Y.; Zhu, Q.; Wang, Y.; Ding, F. Linoleic acid-modified liposomes for the removal of protein-bound toxins: An in vitro study. Int. J. Artif. Organs 2021, 44, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.Y.; Eraly, S.A.; Tsigelny, I.; Nigam, S.K. Interaction of organic cations with organic anion transporters. J. Biol. Chem. 2009, 284, 31422–31430. [Google Scholar] [CrossRef]
- Liu, H.C.; Goldenberg, A.; Chen, Y.; Lun, C.; Wu, W.; Bush, K.T.; Balac, N.; Rodriguez, P.; Abagyan, R.; Nigam, S.K. Molecular Properties of Drugs Interacting with SLC22 Transporters OAT1, OAT3, OCT1, and OCT2: A Machine-Learning Approach. J. Pharmacol. Exp. Ther. 2016, 359, 215–229. [Google Scholar] [CrossRef]
- Nigam, A.K.; Ojha, A.A.; Li, J.G.; Shi, D.; Bhatnagar, V.; Nigam, K.B.; Abagyan, R.; Nigam, S.K. Molecular Properties of Drugs Handled by Kidney OATs and Liver OATPs Revealed by Chemoinformatics and Machine Learning: Implications for Kidney and Liver Disease. Pharmaceutics 2021, 13, 1720. [Google Scholar] [CrossRef] [PubMed]
- Lowenstein, J.; Nigam, S.K. Uremic Toxins in Organ Crosstalk. Front. Med. 2021, 8, 592602. [Google Scholar] [CrossRef]
- Bleasby, K.; Castle, J.C.; Roberts, C.J.; Cheng, C.; Bailey, W.J.; Sina, J.F.; Kulkarni, A.V.; Hafey, M.J.; Evers, R.; Johnson, J.M.; et al. Expression profiles of 50 xenobiotic transporter genes in humans and pre-clinical species: A resource for investigations into drug disposition. Xenobiotica 2006, 36, 963–988. [Google Scholar] [CrossRef]
- Nigam, S.K.; Bush, K.T.; Martovetsky, G.; Ahn, S.-Y.; Liu, H.C.; Richard, E.; Bhatnagar, V.; Wu, W. The Organic Anion Transporter (OAT) Family: A Systems Biology Perspective. Physiol. Rev. 2015, 95, 83–123. [Google Scholar] [CrossRef]
- Roth, M.; Obaidat, A.; Hagenbuch, B. OATPs, OATs and OCTs: The organic anion and cation transporters of the SLCO and SLC22A gene superfamilies. Br. J. Pharmacol. 2012, 165, 1260–1287. [Google Scholar] [CrossRef]
- Breljak, D.; Ljubojević, M.; Hagos, Y.; Micek, V.; Balen Eror, D.; Vrhovac Madunić, I.; Brzica, H.; Karaica, D.; Radovic, N.; Kraus, O.; et al. Distribution of the organic anion transporters NaDC3 and OAT1-3 along the human nephron. Am. J. Physiol. Ren. Physiol. 2016, 311, F227–F238. [Google Scholar] [CrossRef]
- Bush, K.T.; Singh, P.; Nigam, S.K. Gut-derived uremic toxin handling in vivo requires OAT-mediated tubular secretion in chronic kidney disease. JCI Insight 2020, 5, e133817. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Nakao, Y.; Masuda, S.; Katsura, T.; Kamba, T.; Ogawa, O.; Inui, K.-I. Precise Comparison of Protein Localization Among OCT, OAT, and MATE in Human Kidney. J. Pharmacol. Sci. 2013, 102, 3302–3308. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.M.; Mac Laughlin, M.; Muller, A.; Brandoni, A.; Anzai, N.; Endou, H. Altered renal elimination of organic anions in rats with chronic renal failure. Biochim. Biophys. Acta 2005, 1740, 29–37. [Google Scholar] [CrossRef]
- Naud, J.; Roger, M.; Leblond, F.A.; Pichette, V. Effects of Chronic Renal Failure on Kidney Drug Transporters and Cytochrome P450 in Rats. Drug Metab. Dispos. 2011, 39, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Masuda, S.; Saito, H.; Inui, K.-I. Down-regulation of rat organic cation transporter rOCT2 by 5/6 nephrectomy. Kidney Int. 2002, 62, 514–524. [Google Scholar] [CrossRef]
- Deguchi, T.; Takemoto, M.; Uehara, N.; Lindup, W.E.; Suenaga, A.; Otagiri, M. Renal clearance of endogenous hippurate correlates with expression levels of renal organic anion transporters in uremic rats. J. Pharmacol. Exp. Ther. 2005, 314, 932–938. [Google Scholar] [CrossRef]
- Enomoto, A.; Takeda, M.; Tojo, A.; Sekine, T.; Cha, S.H.; Khamdang, S.; Takayama, F.; Aoyama, I.; Nakamura, S.; Endou, H.; et al. Role of Organic Anion Transporters in the Tubular Transport of Indoxyl Sulfate and the Induction of its Nephrotoxicity. J. Am. Soc. Nephrol. 2002, 13, 1711–1720. [Google Scholar] [CrossRef]
- Wikoff, W.R.; Nagle, M.A.; Kouznetsova, V.L.; Tsigelny, I.F.; Nigam, S.K. Untargeted Metabolomics Identifies Enterobiome Metabolites and Putative Uremic Toxins as Substrates of Organic Anion Transporter 1 (Oat1). J. Proteome Res. 2011, 10, 2842–2851. [Google Scholar] [CrossRef]
- Wu, W.; Bush, K.T.; Nigam, S.K. Key Role for the Organic Anion Transporters, OAT1 and OAT3, in the in vivo Handling of Uremic Toxins and Solutes. Sci. Rep. 2017, 7, 4939. [Google Scholar] [CrossRef]
- Nigam, S.K. The SLC22 Transporter Family: A Paradigm for the Impact of Drug Transporters on Metabolic Pathways, Signaling, and Disease. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 663–687. [Google Scholar] [CrossRef]
- Nagle, M.A.; Truong, D.M.; Dnyanmote, A.V.; Ahn, S.-Y.; Eraly, S.A.; Wu, W.; Nigam, S.K. Analysis of Three-dimensional Systems for Developing and Mature Kidneys Clarifies the Role of OAT1 and OAT3 in Antiviral Handling. J. Biol. Chem. 2011, 286, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, T.; Kouno, Y.; Terasaki, T.; Takadate, A.; Otagiri, M. Differential Contributions of rOat1 (Slc22a6) and rOat3 (Slc22a8) to the in Vivo Renal Uptake of Uremic Toxins in Rats. Pharm. Res. 2005, 22, 619–627. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Watanabe, H.; Noguchi, T.; Kotani, S.; Nakajima, M.; Kadowaki, D.; Otagiri, M.; Maruyama, T. Organic anion transporters play an important role in the uptake of p-cresyl sulfate, a uremic toxin, in the kidney. Nephrol. Dial. Transplant. 2011, 26, 2498–2502. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, T.; Ohtsuki, S.; Otagiri, M.; Takanaga, H.; Asaba, H.; Mori, S.; Terasaki, T. Major role of organic anion transporter 3 in the transport of indoxyl sulfate in the kidney. Kidney Int. 2002, 61, 1760–1768. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Tian, Y.; Fujita, T.; Ikeda, Y.; Kumagai, Y.; Kondo, T.; Tanabe, K.; Nakayama, H.; Horita, S.; Kusuhara, H.; et al. Inhibitory effects of p-aminohippurate and probenecid on the renal clearance of adefovir and benzylpenicillin as probe drugs for organic anion transporter (OAT) 1 and OAT3 in humans. Eur. J. Pharm. Sci. 2014, 59, 94–103. [Google Scholar] [CrossRef]
- Zou, L.; Matsson, P.; Stecula, A.; Ngo, H.X.; Zur, A.A.; Giacomini, K.M. Drug Metabolites Potently Inhibit Renal Organic Anion Transporters, OAT1 and OAT3. J. Pharm. Sci. 2021, 110, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Omura, K.; Motoki, K.; Sakai, M.; Chikamatsu, N.; Ashizawa, N.; Takada, T.; Iwanaga, T. Hypouricemic agents reduce indoxyl sulfate excretion by inhibiting the renal transporters OAT1/3 and ABCG2. Sci. Rep. 2021, 11, 7232. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, M.; Shimada, H.; Irino, Y.; Take, M.; Egashira, S. Inhibition of methotrexate uptake via organic anion transporters OAT1 and OAT3 by glucuronides of nonsteroidal anti-inflammatory drugs. Biol. Pharm. Bull. 2017, 40, 926–931. [Google Scholar] [CrossRef]
- Hotchkiss, A.G.; Gao, T.; Khan, U.; Berrigan, L.; Li, M.; Ingraham, L.; Pelis, R.M. Organic Anion Transporter 1 Is Inhibited by Multiple Mechanisms and Shows a Transport Mode Independent of Exchange. Drug Metab. Dispos. 2015, 43, 1847–1854. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Sakaguchi, Y.; Sugimoto, R.; Kaneko, K.; Iwata, H.; Kotani, S.; Nakajima, M.; Ishima, Y.; Otagiri, M.; Maruyama, T. Human organic anion transporters function as a high-capacity transporter for p-cresyl sulfate, a uremic toxin. Clin. Exp. Nephrol. 2013, 18, 814–820. [Google Scholar] [CrossRef]
- Favretto, G.; Souza, L.M.; Gregório, P.C.; Cunha, R.S.; Maciel, R.A.P.; Sassaki, G.L.; Toledo, M.G.; Pecoits-Filho, R.; Souza, W.M.; Stinghen, A.E.M. Role of Organic Anion Transporters in the Uptake of Protein-Bound Uremic Toxins by Human Endothelial Cells and Monocyte Chemoattractant Protein-1 Expression. J. Vasc. Res. 2017, 54, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Miyamoto, Y.; Enoki, Y.; Ishima, Y.; Kadowaki, D.; Kotani, S.; Nakajima, M.; Tanaka, M.; Matsushita, K.; Mori, Y.; et al. p-Cresyl sulfate, a uremic toxin, causes vascular endothelial and smooth muscle cell damages by inducing oxidative stress. Pharmacol. Res. Perspect. 2015, 3, e00092. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Osaka, M.; Higuchi, Y.; Nishijima, F.; Ishii, H.; Yoshida, M. Indoxyl sulfate induces leukocyte-endothelial interactions through up-regulation of E-selectin. J. Biol. Chem. 2010, 285, 38869–38875. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-H.; Kwak, K.-A.; Gil, H.-W.; Song, H.-Y.; Hong, S.-Y. Indoxyl sulfate promotes apoptosis in cultured osteoblast cells. BMC Pharmacol. Toxicol. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Mozar, A.; Louvet, L.; Godin, C.; Mentaverri, R.; Brazier, M.; Kamel, S.; Massy, Z.A. Indoxyl sulphate inhibits osteoclast differentiation and function. Nephrol. Dial. Transplant. 2012, 27, 2176–2181. [Google Scholar] [CrossRef]
- Enoki, Y.; Watanabe, H.; Arake, R.; Sugimoto, R.; Imafuku, T.; Tominaga, Y.; Ishima, Y.; Kotani, S.; Nakajima, M.; Tanaka, M.; et al. Indoxyl sulfate potentiates skeletal muscle atrophy by inducing the oxidative stress-mediated expression of myostatin and atrogin-1. Sci. Rep. 2016, 6, 32084. [Google Scholar] [CrossRef]
- Luo, S.-S.; Yu, C.-P.; Hsieh, Y.-W.; Chao, P.-D.L.; Sweet, D.H.; Hou, Y.-C.; Lin, S.-P. Effects of antibiotics on the pharmacokinetics of indoxyl sulfate, a nephro-cardiovascular toxin. Xenobiotica 2020, 50, 588–592. [Google Scholar] [CrossRef]
- VanWert, A.L.; Srimaroeng, C.; Sweet, D.H. Organic Anion Transporter 3 (Oat3/Slc22a8 ) Interacts with Carboxyfluoroquinolones, and Deletion Increases Systemic Exposure to Ciprofloxacin. Mol. Pharmacol. 2008, 74, 122–131. [Google Scholar] [CrossRef]
- Li, D.Y.; Wang, Z.; Jia, X.; Yan, D.; Shih, D.M.; Hazen, S.L.; Lusis, A.J.; Tang, W.H.W. Loop Diuretics Inhibit Renal Excretion of Trimethylamine N-Oxide. JACC Basic Transl. Sci. 2021, 6, 103–115. [Google Scholar] [CrossRef]
- Teft, W.A.; Morse, B.L.; Leake, B.F.; Wilson, A.; Mansell, S.E.; Hegele, R.A.; Ho, R.H.; Kim, R.B. Identification and Characterization of Trimethylamine-N-oxide Uptake and Efflux Transporters. Mol. Pharm. 2017, 14, 310–318. [Google Scholar] [CrossRef]
- Granados, J.C.; Richelle, A.; Gutierrez, J.M.; Zhang, P.; Zhang, X.; Bhatnagar, V.; Lewis, N.E.; Nigam, S.K. Coordinate regulation of systemic and kidney tryptophan metabolism by the drug transporters OAT1 and OAT3. J. Biol. Chem. 2021, 296, 100575. [Google Scholar] [CrossRef] [PubMed]
- André, C.; Mernissi, T.; Choukroun, G.; Bennis, Y.; Kamel, S.; Liabeuf, S.; Bodeau, S. The Prescription of Drugs That Inhibit Organic Anion Transporters 1 or 3 Is Associated with the Plasma Accumulation of Uremic Toxins in Kidney Transplant Recipients. Toxins 2021, 14, 15. [Google Scholar] [CrossRef] [PubMed]
- Lepist, E.-I.; Zhang, X.; Hao, J.; Huang, J.; Kosaka, A.; Birkus, G.; Murray, B.P.; Bannister, R.; Cihlar, T.; Huang, Y.; et al. Contribution of the organic anion transporter OAT2 to the renal active tubular secretion of creatinine and mechanism for serum creatinine elevations caused by cobicistat. Kidney Int. 2014, 86, 350–357. [Google Scholar] [CrossRef]
- Shen, H.; Liu, T.; Morse, B.L.; Zhao, Y.; Zhang, Y.; Qiu, X.; Chen, C.; Lewin, A.C.; Wang, X.T.; Liu, G.; et al. Characterization of Organic Anion Transporter 2 (SLC22A7): A Highly Efficient Transporter for Creatinine and Species-Dependent Renal Tubular Expression. Drug Metab. Dispos. 2015, 43, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Koepsell, H. Substrate recognition and translocation by polyspecific organic cation transporters. Biol. Chem. 2011, 392, 95–101. [Google Scholar] [CrossRef]
- Schophuizen, C.M.S.; Wilmer, M.J.; Jansen, J.; Gustavsson, L.; Hilgendorf, C.; Hoenderop, J.G.J.; Van Den Heuvel, L.P.; Masereeuw, R. Cationic uremic toxins affect human renal proximal tubule cell functioning through interaction with the organic cation transporter. Pflug. Arch. Eur. J. Physiol. 2013, 465, 1701–1714. [Google Scholar] [CrossRef]
- Scotcher, D.; Arya, V.; Yang, X.; Zhao, P.; Zhang, L.; Huang, S.M.; Rostami-Hodjegan, A.; Galetin, A. Mechanistic Models as Framework for Understanding Biomarker Disposition: Prediction of Creatinine-Drug Interactions. CPT Pharmacomet. Syst. Pharmacol. 2020, 9, 282–293. [Google Scholar] [CrossRef]
- Kimura, N.; Masuda, S.; Katsura, T.; Inui, K. ichi Transport of guanidine compounds by human organic cation transporters, hOCT1 and hOCT2. Biochem. Pharmacol. 2009, 77, 1429–1436. [Google Scholar] [CrossRef]
- Reznichenko, A.; Sinkeler, S.J.; Snieder, H.; van den Born, J.; de Borst, M.H.; Damman, J.; van Dijk, M.C.R.F.; van Goor, H.; Hepkema, B.G.; Hillebrands, J.L.; et al. SLC22A2 is associated with tubular creatinine secretion and bias of estimated GFR in renal transplantation. Physiol. Genom. 2013, 45, 201–209. [Google Scholar] [CrossRef]
- Urakami, Y.; Kimura, N.; Okuda, M.; Inui, K. Creatinine Transport by Basolateral Organic Cation Transporter hOCT2 in the Human Kidney. Pharm. Res. 2004, 21, 976–981. [Google Scholar] [CrossRef]
- Motohashi, H.; Sakurai, Y.; Saito, H.; Masuda, S.; Urakami, Y.; Goto, M.; Fukatsu, A.; Ogawa, O.; Inui, K. Gene expression levels and immunolocalization of organic ion transporters in the human kidney. J. Am. Soc. Nephrol. 2002, 13, 866–874. [Google Scholar] [CrossRef] [PubMed]
- Hacker, K.; Maas, R.; Kornhuber, J.; Fromm, M.F.; Zolk, O. Substrate-dependent inhibition of the human organic cation transporter OCT2: A comparison of metformin with experimental substrates. PLoS ONE 2015, 10, e0136451. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.W.K.; Hsueh, C.-H.; Zhao, P.; Meyer, T.W.; Zhang, L.; Huang, S.-M.; Giacomini, K.M. The Effect of Uremic Solutes on the Organic Cation Transporter 2. J. Pharm. Sci. 2017, 106, 2551–2557. [Google Scholar] [CrossRef] [PubMed]
- Winter, T.N.; Elmquist, W.F.; Fairbanks, C.A. OCT2 and MATE1 Provide Bidirectional Agmatine Transport. Mol. Pharm. 2011, 8, 133–142. [Google Scholar] [CrossRef]
- Gessner, A.; König, J.; Fromm, M.F. Contribution of multidrug and toxin extrusion protein 1 (MATE1) to renal secretion of trimethylamine-N-oxide (TMAO). Sci. Rep. 2018, 8, 6659. [Google Scholar] [CrossRef]
- Miyake, T.; Mizuno, T.; Mochizuki, T.; Kimura, M.; Matsuki, S.; Irie, S.; Ieiri, I.; Maeda, K.; Kusuhara, H. Involvement of Organic Cation Transporters in the Kinetics of Trimethylamine N-oxide. J. Pharm. Sci. 2017, 106, 2542–2550. [Google Scholar] [CrossRef]
- Nakada, T.; Kudo, T.; Kume, T.; Kusuhara, H.; Ito, K. Estimation of changes in serum creatinine and creatinine clearance caused by renal transporter inhibition in healthy subjects. Drug Metab. Pharmacokinet. 2019, 34, 233–238. [Google Scholar] [CrossRef]
- Ciarimboli, G.; Lancaster, C.S.; Schlatter, E.; Franke, R.M.; Sprowl, J.A.; Pavenstädt, H.; Massmann, V.; Guckel, D.; Mathijssen, R.H.J.; Yang, W.; et al. Proximal tubular secretion of creatinine by organic cation transporter OCT2 in cancer patients. Clin. Cancer Res. 2012, 18, 1101–1108. [Google Scholar] [CrossRef]
- Tanihara, Y.; Masuda, S.; Inui, K. ichi Inhibitory effects of vandetanib on creatinine transport via renal organic cation transporter OCT2. Eur. J. Pharm. Sci. 2021, 158, 105666. [Google Scholar] [CrossRef]
- Han, C.; Zheng, J.; Wang, F.; Lu, Q.; Chen, Q.; Hu, A.; Visentin, M.; Kullak-Ublick, G.A.; Gai, Z.; Chu, L. The Role of NF-kB in the Downregulation of Organic Cation Transporter 2 Expression and Renal Cation Secretion in Kidney Disease. Front. Med. 2022, 8, 800421. [Google Scholar] [CrossRef]
- Wang, Z.J.; Yin, O.Q.P.; Tomlinson, B.; Chow, M.S.S. OCT2 polymorphisms and in-vivo renal functional consequence: Studies with metformin and cimetidine. Pharmacogenet. Genom. 2008, 18, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Filipski, K.K.; Mathijssen, R.H.; Mikkelsen, T.S.; Schinkel, A.H.; Sparreboom, A. Contribution of organic cation transporter 2 (OCT2) to cisplatin-induced nephrotoxicity. Clin. Pharmacol. Ther. 2009, 86, 396–402. [Google Scholar] [CrossRef]
- Köttgen, A.; Pattaro, C.; Böger, C.A.; Fuchsberger, C.; Olden, M.; Glazer, N.L.; Parsa, A.; Gao, X.; Yang, Q.; Smith, A.V.; et al. New loci associated with kidney function and chronic kidney disease. Nat. Genet. 2010, 42, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Tachikawa, M.; Kasai, Y.; Takahashi, M.; Fujinawa, J.; Kitaichi, K.; Terasaki, T.; Hosoya, K.I. The blood-cerebrospinal fluid barrier is a major pathway of cerebral creatinine clearance: Involvement of transporter-mediated process. J. Neurochem. 2008, 107, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Hata, S.; Xiao, Y.; Murray, J.W.; Wolkoff, A.W. Topological assessment of Oatp1a1: A 12-transmembrane domain integral membrane protein with three N-linked carbohydrate chains. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G1052–G1059. [Google Scholar] [CrossRef] [PubMed]
- Hagenbuch, B.; Stieger, B. The SLCO (former SLC21) superfamily of transporters. Mol. Asp. Med. 2013, 34, 396–412. [Google Scholar] [CrossRef] [PubMed]
- El-Mesallamy, H.O.; Abdel Hamid, S.G.; Gad, M.Z. Oxidative stress and asymmetric dimethylarginine are associated with cardiovascular complications in hemodialysis patients: Improvements by L-arginine intake. Kidney Blood Press. Res. 2008, 31, 189–195. [Google Scholar] [CrossRef]
- Kajimoto, H.; Kai, H.; Aoki, H.; Yasuoka, S.; Anegawa, T.; Aoki, Y.; Ueda, S.; Okuda, S.; Imaizumi, T. Inhibition of eNOS phosphorylation mediates endothelial dysfunction in renal failure: New effect of asymmetric dimethylarginine. Kidney Int. 2012, 81, 762–768. [Google Scholar] [CrossRef]
- Shafi, T.; Hostetter, T.H.; Meyer, T.W.; Hwang, S.; Hai, X.; Melamed, M.L.; Banerjee, T.; Coresh, J.; Powe, N.R. Serum Asymmetric and Symmetric Dimethylarginine and Morbidity and Mortality in Hemodialysis Patients. Am. J. Kidney Dis. 2017, 70, 48–58. [Google Scholar] [CrossRef]
- Taghikhani, E.; Maas, R.; Fromm, M.F.; König, J. The renal transport protein OATP4C1 mediates uptake of the uremic toxin asymmetric dimethylarginine (ADMA) and efflux of cardioprotective L-homoarginine. PLoS ONE 2019, 14, e0213747. [Google Scholar] [CrossRef]
- Taghikhani, E.; Maas, R.; Taudte, R.V.; Gessner, A.; Fromm, M.F.; König, J. Vectorial transport of the arginine derivatives asymmetric dimethylarginine (ADMA) and l-homoarginine by OATP4C1 and P-glycoprotein studied in double-transfected MDCK cells. Amino Acids 2020, 52, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Toyohara, T.; Suzuki, T.; Morimoto, R.; Akiyama, Y.; Souma, T.; Shiwaku, H.O.; Takeuchi, Y.; Mishima, E.; Abe, M.; Tanemoto, M.; et al. SLCO4C1 Transporter Eliminates Uremic Toxins and Attenuates Hypertension and Renal Inflammation. J. Am. Soc. Nephrol. 2009, 20, 2546–2555. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, Y.; Kikuchi, K.; Saigusa, D.; Suzuki, T.; Takeuchi, Y.; Mishima, E.; Yamamoto, Y.; Ishida, A.; Sugawara, D.; Jinno, D.; et al. Indoxyl Sulfate Down-Regulates SLCO4C1 Transporter through Up-Regulation of GATA3. PLoS ONE 2013, 8, e66518. [Google Scholar] [CrossRef]
- Mikkaichi, T.; Suzuki, T.; Onogawa, T.; Tanemoto, M.; Mizutamari, H.; Okada, M.; Chaki, T.; Masuda, S.; Tokui, T.; Eto, N.; et al. Isolation and characterization of a digoxin transporter and its rat homologue expressed in the kidney. Proc. Natl. Acad. Sci. USA 2004, 101, 3569–3574. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.I.; Sugiura, T.; Okumura, H.; Umeda, S.; Nakamichi, N.; Watanabe, Y.; Suzuki, H.; Sunakawa, Y.; Shimada, K.; Kawara, K.; et al. Direct inhibition and down-regulation by uremic plasma components of hepatic uptake transporter for sn-38, an active metabolite of irinotecan, in humans. Pharm. Res. 2014, 31, 204–215. [Google Scholar] [CrossRef]
- Fujita, K.; Masuo, Y.; Okumura, H.; Watanabe, Y.; Suzuki, H.; Sunakawa, Y.; Shimada, K.; Kawara, K.; Akiyama, Y.; Kitamura, M.; et al. Increased Plasma Concentrations of Unbound SN-38, the Active Metabolite of Irinotecan, in Cancer Patients with Severe Renal Failure. Pharm. Res. 2016, 33, 269–282. [Google Scholar] [CrossRef]
- Bøttger, P.; Pedersen, L. Mapping of the minimal inorganic phosphate transporting unit of human PiT2 suggests a structure universal to PiT-related proteins from all kingdoms of life. BMC Biochem. 2011, 12, 21. [Google Scholar] [CrossRef]
- Forster, I.C.; Hernando, N.; Biber, J.; Murer, H. Phosphate transporters of the SLC20 and SLC34 families. Mol. Asp. Med. 2013, 34, 386–395. [Google Scholar] [CrossRef]
- Inden, M.; Iriyama, M.; Zennami, M.; Sekine, S.I.; Hara, A.; Yamada, M.; Hozumi, I. The type III transporters (PiT-1 and PiT-2) are the major sodium-dependent phosphate transporters in the mice and human brains. Brain Res. 2016, 1637, 128–136. [Google Scholar] [CrossRef]
- Ravera, S.; Virkki, L.V.; Murer, H.; Forster, I.C. Deciphering PiT transport kinetics and substrate specificity using electrophysiology and flux measurements. Am. J. Physiol. Cell Physiol. 2007, 293, 606–620. [Google Scholar] [CrossRef]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial calcification in chronic kidney disease: Key roles for calcium and phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef] [PubMed]
- Barreto, F.C.; Barreto, D.V.; Massy, Z.A.; Drüeke, T.B. Strategies for Phosphate Control in Patients With CKD. Kidney Int. Rep. 2019, 4, 1043–1056. [Google Scholar] [CrossRef] [PubMed]
- Six, I.; Maizel, J.; Barreto, F.C.; Rangrez, A.Y.; Dupont, S.; Slama, M.; Tribouilloy, C.; Choukroun, G.; Maziere, J.C.; Bode-Boeger, S.; et al. Effects of phosphate on vascular function under normal conditions and influence of the uraemic state. Cardiovasc. Res. 2012, 96, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Abbasian, N.; Burton, J.O.; Herbert, K.E.; Tregunna, B.-E.; Brown, J.R.; Ghaderi-Najafabadi, M.; Brunskill, N.J.; Goodall, A.H.; Bevington, A. Hyperphosphatemia, Phosphoprotein Phosphatases, and Microparticle Release in Vascular Endothelial Cells. J. Am. Soc. Nephrol. 2015, 26, 2152–2162. [Google Scholar] [CrossRef] [PubMed]
- Chavkin, N.W.; Chia, J.J.; Crouthamel, M.H.; Giachelli, C.M. Phosphate uptake-independent signaling functions of the type III sodium-dependent phosphate transporter, PiT-1, in vascular smooth muscle cells. Exp. Cell Res. 2015, 333, 39–48. [Google Scholar] [CrossRef]
- Li, X.; Yang, H.Y.; Giachelli, C.M. Role of the sodium-dependent phosphate cotransporter, Pit-1, in vascular smooth muscle cell calcification. Circ. Res. 2006, 98, 905–912. [Google Scholar] [CrossRef]
- Eto, N.; Miyata, Y.; Ohno, H.; Yamashita, T. Nicotinamide prevents the development of hyperphosphataemia by suppressing intestinal sodium-dependent phosphate transporter in rats with adenine-induced renal failure. Nephrol. Dial. Transplant. 2005, 20, 1378–1384. [Google Scholar] [CrossRef]
- Nies, A.T.; Damme, K.; Kruck, S.; Schaeffeler, E.; Schwab, M. Structure and function of multidrug and toxin extrusion proteins (MATEs) and their relevance to drug therapy and personalized medicine. Arch. Toxicol. 2016, 90, 1555–1584. [Google Scholar] [CrossRef]
- Tanihara, Y.; Masuda, S.; Sato, T.; Katsura, T.; Ogawa, O.; Inui, K.-i. Substrate specificity of MATE1 and MATE2-K, human multidrug and toxin extrusions/H+-organic cation antiporters. Biochem. Pharmacol. 2007, 74, 359–371. [Google Scholar] [CrossRef]
- Masuda, S.; Terada, T.; Yonezawa, A.; Tanihara, Y.; Kishimoto, K.; Katsura, T.; Ogawa, O.; Inui, K.I. Identification and functional characterization of a new human kidney-specific H+/organic cation antiporter, kidney-specific multidrug and toxin extrusion 2. J. Am. Soc. Nephrol. 2006, 17, 2127–2135. [Google Scholar] [CrossRef]
- Omote, S.; Matsuoka, N.; Arakawa, H.; Nakanishi, T.; Tamai, I. Effect of tyrosine kinase inhibitors on renal handling of creatinine by MATE1. Sci. Rep. 2018, 8, 9237. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, B.; Neumeister, C.; Schwantes, U.; Fromm, M.F.; König, J. Interplay of the Organic Cation Transporters OCT1 and OCT2 with the Apically Localized Export Protein MATE1 for the Polarized Transport of Trospium. Mol. Pharm. 2019, 16, 510–517. [Google Scholar] [CrossRef] [PubMed]
- George, B.; Wen, X.; Jaimes, E.A.; Joy, M.S.; Aleksunes, L.M. In vitro inhibition of renal oct2 and mate1 secretion by antiemetic drugs. Int. J. Mol. Sci. 2021, 22, 6439. [Google Scholar] [CrossRef]
- Nishihara, K.; Masuda, S.; Ji, L.; Katsura, T.; Inui, K.I. Pharmacokinetic significance of luminal multidrug and toxin extrusion 1 in chronic renal failure rats. Biochem. Pharmacol. 2007, 73, 1482–1490. [Google Scholar] [CrossRef]
- Shigeta, J.; Katayama, K.; Mitsuhashi, J.; Noguchi, K.; Sugimoto, Y. BCRP/ABCG2 confers anticancer drug resistance without covalent dimerization. Cancer Sci. 2010, 101, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lee, E.-W.; Cai, X.; Ni, Z.; Zhou, L.; Mao, Q. Membrane topology of the human breast cancer resistance protein (BCRP/ABCG2) determined by epitope insertion and immunofluorescence. Biochemistry 2008, 47, 13778–13787. [Google Scholar] [CrossRef][Green Version]
- Ferreira, R.J.; Bonito, C.A.; Cordeiro, M.N.D.S.; Ferreira, M.-J.U.; dos Santos, D.J.V.A. Structure-function relationships in ABCG2: Insights from molecular dynamics simulations and molecular docking studies. Sci. Rep. 2017, 7, 15534. [Google Scholar] [CrossRef]
- Wang, L.; Prasad, B.; Salphati, L.; Chu, X.; Gupta, A.; Hop, C.E.C.A.; Evers, R.; Unadkat, J.D. Interspecies Variability in Expression of Hepatobiliary Transporters across Human, Dog, Monkey, and Rat as Determined by Quantitative Proteomics. Drug Metab. Dispos. 2015, 43, 367–374. [Google Scholar] [CrossRef]
- Morimoto, K.; Tominaga, Y.; Agatsuma, Y.; Miyamoto, M.; Kashiwagura, S.; Takahashi, A.; Sano, Y.; Yano, K.; Kakinuma, C.; Ogihara, T.; et al. Intestinal secretion of indoxyl sulfate as a possible compensatory excretion pathway in chronic kidney disease. Biopharm. Drug Dispos. 2018, 39, 328–334. [Google Scholar] [CrossRef]
- Maliepaard, M.; Scheffer, G.L.; Faneyte, I.F.; Van Gastelen, M.A.; Pijnenborg, A.C.L.M.; Schinkel, A.H.; Van de Vijver, M.J.; Scheper, R.J.; Schellens, J.H.M. Subcellular localization and distribution of the Breast Resistance Protein Transporter in normal human tissues. Cancer Res. 2001, 61, 3458–3464. [Google Scholar]
- Cooray, H.C.; Blackmore, C.G.; Maskell, L.; Barrand, M.A. Localisation of breast cancer resistance protein in microvessel endothelium of human brain. Neuroreport 2002, 13, 2059–2063. [Google Scholar] [CrossRef]
- Lu, H.; Klaassen, C. Gender Differences in mRNA Expression of ATP-Binding Cassette Efflux and Bile Acid Transporters in Kidney, Liver, and Intestine of 5/6 Nephrectomized Rats. Drug Metab. Dispos. 2008, 36, 16–23. [Google Scholar] [CrossRef]
- Nagura, M.; Tamura, Y.; Kumagai, T.; Hosoyamada, M.; Uchida, S. Uric acid metabolism of kidney and intestine in a rat model of chronic kidney disease. Nucleosides Nucleotides Nucleic Acids 2016, 35, 550–558. [Google Scholar] [CrossRef]
- Mutsaers, H.A.M.; Caetano-Pinto, P.; Seegers, A.E.M.; Dankers, A.C.A.; van den Broek, P.H.H.; Wetzels, J.F.M.; van den Brand, J.A.J.G.; van den Heuvel, L.P.; Hoenderop, J.G.; Wilmer, M.J.G.; et al. Proximal tubular efflux transporters involved in renal excretion of p-cresyl sulfate and p-cresyl glucuronide: Implications for chronic kidney disease pathophysiology. Toxicol. In Vitro 2015, 29, 1868–1877. [Google Scholar] [CrossRef]
- Takada, T.; Yamamoto, T.; Matsuo, H.; Tan, J.K.; Ooyama, K.; Sakiyama, M.; Miyata, H.; Yamanashi, Y.; Toyoda, Y.; Higashino, T.; et al. Identification of ABCG2 as an Exporter of Uremic Toxin Indoxyl Sulfate in Mice and as a Crucial Factor Influencing CKD Progression. Sci. Rep. 2018, 8, 11147. [Google Scholar] [CrossRef]
- Jansen, J.; Fedecostante, M.; Wilmer, M.J.; Peters, J.G.; Kreuser, U.M.; van den Broek, P.H.; Mensink, R.A.; Boltje, T.J.; Stamatialis, D.; Wetzels, J.F.; et al. Bioengineered kidney tubules efficiently excrete uremic toxins. Sci. Rep. 2016, 6, 26715. [Google Scholar] [CrossRef]
- Dankers, A.C.A.; Mutsaers, H.A.M.; Dijkman, H.B.P.M.; van den Heuvel, L.P.; Hoenderop, J.G.; Sweep, F.C.G.J.; Russel, F.G.M.; Masereeuw, R. Hyperuricemia influences tryptophan metabolism via inhibition of multidrug resistance protein 4 (MRP4) and breast cancer resistance protein (BCRP). Biochim. Biophys. Acta 2013, 1832, 1715–1722. [Google Scholar] [CrossRef]
- Mutsaers, H.A.M.; van den Heuvel, L.P.; Ringens, L.H.J.; Dankers, A.C.A.; Russel, F.G.M.; Wetzels, J.F.M.; Hoenderop, J.G.; Masereeuw, R. Uremic toxins inhibit transport by breast cancer resistance protein and multidrug resistance protein 4 at clinically relevant concentrations. PLoS ONE 2011, 6, e18438. [Google Scholar] [CrossRef]
- Lu, Y.; Nakanishi, T.; Hosomi, A.; Komori, H.; Tamai, I. In-vitro evidence of enhanced breast cancer resistance protein-mediated intestinal urate secretion by uremic toxins in Caco-2 cells. J. Pharm. Pharmacol. 2015, 67, 170–177. [Google Scholar] [CrossRef]
- Nakayama, A.; Matsuo, H.; Takada, T.; Ichida, K.; Nakamura, T.; Ikebuchi, Y.; Ito, K.; Hosoya, T.; Kanai, Y.; Suzuki, H.; et al. ABCG2 is a High-Capacity Urate Transporter and its Genetic Impairment Increases Serum Uric Acid Levels in Humans. Nucleosides Nucleotides Nucleic Acids 2011, 30, 1091–1097. [Google Scholar] [CrossRef]
- Nakayama, A.; Matsuo, H.; Nakaoka, H.; Nakamura, T.; Nakashima, H.; Takada, Y.; Oikawa, Y.; Takada, T.; Sakiyama, M.; Shimizu, S.; et al. Common dysfunctional variants of ABCG2 have stronger impact on hyperuricemia progression than typical environmental risk factors. Sci. Rep. 2014, 4, 5227. [Google Scholar] [CrossRef]
- Chen, C.-J.; Tseng, C.-C.; Yen, J.-H.; Chang, J.-G.; Chou, W.-C.; Chu, H.-W.; Chang, S.-J.; Liao, W.-T. ABCG2 contributes to the development of gout and hyperuricemia in a genome-wide association study. Sci. Rep. 2018, 8, 3137. [Google Scholar] [CrossRef]
- Komori, H.; Yamada, K.; Tamai, I. Hyperuricemia enhances intracellular urate accumulation via down-regulation of cell-surface BCRP/ABCG2 expression in vascular endothelial cells. Biochim. Biophys. Acta Biomembr. 2018, 1860, 973–980. [Google Scholar] [CrossRef]
- Berggren, S.; Gall, C.; Wollnitz, N.; Ekelund, M.; Karlbom, U.; Hoogstraate, J.; Schrenk, D.; Lennernäs, H. Gene and Protein Expression of P-Glycoprotein, MRP1, MRP2, and CYP3A4 in the Small and Large Human Intestine. Mol. Pharm. 2007, 4, 252–257. [Google Scholar] [CrossRef]
- Gradhand, U.; Lang, T.; Schaeffeler, E.; Glaeser, H.; Tegude, H.; Klein, K.; Fritz, P.; Jedlitschky, G.; Kroemer, H.K.; Bachmakov, I.; et al. Variability in human hepatic MRP4 expression: Influence of cholestasis and genotype. Pharm. J. 2008, 8, 42–52. [Google Scholar] [CrossRef]
- Zhang, Y.; Han, H.; Elmquist, W.F.; Miller, D.W. Expression of various multidrug resistance-associated protein (MRP) homologues in brain microvessel endothelial cells. Brain Res. 2000, 876, 148–153. [Google Scholar] [CrossRef]
- Kubota, H.; Ishihara, H.; Langmann, T.; Schmitz, G.; Stieger, B.; Wieser, H.G.; Yonekawa, Y.; Frei, K. Distribution and functional activity of P-glycoprotein and multidrug resistance-associated proteins in human brain microvascular endothelial cells in hippocampal sclerosis. Epilepsy Res. 2006, 68, 213–228. [Google Scholar] [CrossRef]
- Naud, J.; Michaud, J.; Boisvert, C.; Desbiens, K.; Leblond, F.A.; Mitchell, A.; Jones, C.; Bonnardeaux, A.; Pichette, V. Down-Regulation of Intestinal Drug Transporters in Chronic Renal Failure in Rats. J. Pharmacol. Exp. Ther. 2007, 320, 978–985. [Google Scholar] [CrossRef]
- Gai, Z.; Chu, L.; Hiller, C.; Arsenijevic, D.; Penno, C.A.; Montani, J.-P.; Odermatt, A.; Kullak-Ublick, G.A. Effect of chronic renal failure on the hepatic, intestinal, and renal expression of bile acid transporters. Am. J. Physiol. Physiol. 2014, 306, F130–F137. [Google Scholar] [CrossRef]
- El-Sheikh, A.A.K.; van den Heuvel, J.J.M.W.; Koenderink, J.B.; Russel, F.G.M. Interaction of Nonsteroidal Anti-Inflammatory Drugs with Multidrug Resistance Protein (MRP)2/ABCC2- and MRP4/ABCC4-Mediated Methotrexate Transport. J. Pharmacol. Exp. Ther. 2007, 320, 229–235. [Google Scholar] [CrossRef]
- Reid, G.; Wielinga, P.; Zelcer, N.; van der Heijden, I.; Kuil, A.; de Haas, M.; Wijnholds, J.; Borst, P. The human multidrug resistance protein MRP4 functions as a prostaglandin efflux transporter and is inhibited by nonsteroidal antiinflammatory drugs. Proc. Natl. Acad. Sci. USA 2003, 100, 9244–9249. [Google Scholar] [CrossRef] [PubMed]
- Kawase, A.; Yamamoto, T.; Egashira, S.; Iwaki, M. Stereoselective inhibition of methotrexate excretion by glucuronides of nonsteroidal anti-inflammatory drugs via multidrug resistance proteins 2 and 4. J. Pharmacol. Exp. Ther. 2016, 356, 366–374. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Protein | Gene | Tissue Distribution | Uremic Toxin Interaction | Drug Interaction |
|---|---|---|---|---|
| OAT1 | SLC22A6 | Kidney | PCS, IS, kynurenic acid, hippuric acid | Probenecid, β-lactam antibiotics, nonsteroidal anti-inflammatory drugs |
| OAT2 | SLC22A7 | Kidney, liver | Creatinine | |
| OAT3 | SLC22A8 | Kidney | PCS, IS, kynurenic acid, hippuric acid | Probenecid, ciprofloxacin, β-lactam antibiotics, nonsteroidal anti-inflammatory drugs |
| OCT2 | SLC22A2 | Kidney | Creatinine, TMAO, methylguanidine, guanidine, putrescine | Metformin, cisplatin, cimetidine, vandetanib, trimethoprim |
| OCT3 | SLC22A3 | Choroid plexus, skeletal muscle, placenta, kidney | Creatinine | |
| OATP4C1 | SLCO4C1 | Kidney | ADMA | OATP4C1 expression is modulated by statins |
| PiT-1 | SLC20A1 | Endothelial cells, intestine, bones | Pi | |
| PiT-2 | SLC20A2 | Endothelial cells, intestine, bones, kidney | Pi | |
| NaPi2B | SLC34A2 | Intestine | Pi | Nicotinic acid and nicotinamide inhibit NaPi2B expression |
| MATE1 | SLC47A1 | Kidney, liver, heart | TMAO, creatinine, guanidine | Trimethoprim, trospium, ondansetron |
| MATE2-K | SLC47A2 | Kidney | Creatinine, guanidine | |
| BCRP | ABCG2 | Kidney, intestine, blood vessels, placenta | PCS, IS, kynurenic acid, TMAO, uric acid | Febuxostat |
| MRP2 | ABCC2 | Kidney, liver, intestine, brain capillary endothelium | TMAO | Methotrexate, nonsteroidal anti-inflammatory drugs |
| MRP4 | ABCC4 | Kidney, liver, intestine, brain capillary endothelium | TMAO | Methotrexate, nonsteroidal anti-inflammatory drugs |
| Protein | Experimental Model | Main Findings |
|---|---|---|
| OAT1 | OAT1-expressing HEK293 cells | Cell uptake of PCS [100] |
| Slc22a6-knockout mice | Increased plasma levels of PCS, IS, and kynurenine [89] | |
| Slc22a6-knockout mice | Increased plasma levels of PCS, IS, and IAA [88] | |
| Rats | Renal uptake of hippurate, IAA, and IS [92] | |
| Nephrectomized rats | Decreased protein levels in the kidneys [83] | |
| Nephrectomized rats | Decreased protein and mRNA levels in the kidneys [84] | |
| Nephrectomized rats | Decreased protein levels in the kidneys [86] | |
| Nephrectomized rats | No differences in protein levels in the kidney [85] | |
| Nephrectomized rats treated with IS | Increased protein levels in the renal tubules [87] | |
| OAT3 | OAT3-expressing HEK293 cells | Cell uptake of PCS [100] |
| Rats and Oat3-expressing oocytes | Renal uptake of IS [94] | |
| Slc22a8-knockout mice | Increased plasma levels of PCS, IS, CMPF, and TMAO [89] | |
| Rats | Renal uptake of IS and CMPF [92] | |
| Rats treated with IS | Decreased the renal clearance of IS through inhibition on the OAT3-mediated transport with ciprofloxacin [107] | |
| Nephrectomized rats | Decreased protein levels in the kidneys [86] | |
| Nephrectomized rats | No differences in protein levels in the kidney [85] | |
| OAT1/3 | HK-2 cells and rat renal cortical slices | Cell uptake of PCS, which was inhibited with probenecid, an inhibitor of OATs [93] |
| Endothelial cells | Cell uptake of PCS and IS, which was inhibited with probenecid [101] | |
| Endothelial cells | Probenecid attenuated the inductive effects of IS on the expression of E-selectin and monocytic cell adhesion [103] | |
| Endothelial cells and aortic smooth muscle cells | Probenecid reversed the inductive effect of PCS on MCP-1 expression in endothelial cells and on the expression of osteogenic differentiation genes in aortic smooth muscle cells [102] | |
| Osteoblasts | Probenecid restored IS-induced effects on cell viability and ROS levels [104] | |
| Myoblast cells | Probenecid reversed IS-induced effects on ROS levels and inflammatory cytokine expression [106] | |
| Human subjects | Subjects treated with probenecid had elevated IS and kynurenine levels [111] | |
| Kidney transplant patients | Increased plasma levels of IS, PCS and IAA in patients with a prescription of at least one drug which inhibits OAT1/OAT3 [112] | |
| OAT2 | MDCKII cells | Cell uptake of creatinine [113] |
| OAT2-transfected HEK cells | Cell uptake of creatinine [114] | |
| OCT2 | ciPTEC cells | Uptake of cationic uremic toxins, such as guanidine, methylguanidine, and putrescine [116] |
| HEK293 cells | Cell uptake of guanidine compounds [118] | |
| HEK293 cells | Cell uptake of creatinine [120] | |
| MDCKII and HEK cells | Cell uptake of TMAO and transcellular transport [125] | |
| HEK293 cells | Cell uptake of putrescine [124] | |
| OCT2-expressing HEK cells | Inhibited by creatinine, dimethylamine, malondialdehyde, trimethylamine, homocysteine, indoxyl-β-d-glucuronide, and glutathione disulfide [123] | |
| HEK293 cells | Vandetanib inhibited the uptake of creatinine [129] | |
| Slc22a2/1-double knockout mice and HeLa cells | Increased plasma levels of TMAO. In vitro, TMAO transport [110] | |
| Slc22a2/1-double knockout mice and Oct2-transfected HEK293 cells | Increased plasma levels of TMAO. In vitro and In vitro, TMAO uptake [126] | |
| Nephrectomized rats | Decreased protein levels in the kidney [85] | |
| Patients with CKD and nephrectomized rats | Decreased protein levels in the kidney [130] | |
| Patients with cancer undergoing treatment with cisplatin and HEK293 cells | Increased serum levels of creatinine. In vitro, creatinine uptake [128] | |
| Patients with end-stage renal disease | Relationship between SLC22A2 polymorphisms and phenotypes of net tubular creatinine secretion [119] | |
| OATP4C1 | MDCK cells | Transport of ADMA [141] |
| HEK293 cells | Cell uptake of ADMA [140] | |
| HK-2 cells and rats treated with IS | IS reduced the OATP4C1 expression [143] | |
| Transgenic mice overexpressing OATP4C1 in the kidneys | Decreased plasma levels of ADMA, guanidino succinate, and trans-aconitate [142] | |
| Nephrectomized rats | Decreased mRNA levels in the kidney [144] | |
| OATP1B1/3 | Human hepatocytes and HEK293 cells | Decreased mRNA levels in cells exposed to uremic plasma. Inhibited by uremic toxin mix (IS, indole acetate, hippuric acid, and CMPF) [145] |
| PiT-1/2 | Endothelial cells | Inhibition and knockout of PiT-1 reduced intracellular Pi concentrations [154] |
| PiT-1-expressing oocytes | Pi transport [150] | |
| VSMCs | Uptake of Pi, which at high levels induces osteochondrogenic differentiation of VSMCs [155] | |
| Human smooth muscle cells | Cell uptake of Pi [156] |
| Protein | Experimental Model | Main Findings |
|---|---|---|
| MATE1 | HEK293 cells | Creatinine and guanidine as substrates [159] |
| HEK293 cells | Creatinine as substrate [161] | |
| MDCKII and HEK cells | Transport of TMAO, which was suppressed by trimethoprim [125] | |
| Nephrectomized rats | Decreased protein levels in the kidneys [164] | |
| MATE2-K | HEK293 cells | Creatinine and guanidine as substrates [159] |
| BCRP | ciPTEC cells | BCRP inhibition increased intracellular PCS levels [174] |
| HeLa cells | TMAO transport [110] | |
| Membrane vesicles from MRP4-overexpressing HEK cells | Inhibited by hippuric acid, IS, and kynurenic acid [178] | |
| Caco-2 cells | Urate transport. IS reduced BCRP expression [179] | |
| Endothelial cells | Uric acid decreased the BCRP protein levels [183] | |
| Abcg2-knockout mice with adenine-induced CKD and membrane vesicles from HEK293A cells | Increased plasma levels and decreased renal elimination of IS. In vitro, IS transport [175] | |
| Abcg2-knockout mice and HEK293 cells | Kynurenic acid as substrate [177] | |
| Abcg2-knockout mice | Increased plasma levels and low urine levels of IS [175] | |
| Adenine-induced acute renal failure rats | Febuxostat, an BCRP inhibitor, decreased renal clearance of the IS [97] | |
| Nephrectomized rats | Decreased mRNA levels in the kidney [172] | |
| Nephrectomized rats | Decreased protein and mRNA levels in the kidney [173] | |
| MRP2 | HeLa cells | Performs cellular efflux of TMAO [110] |
| Nephrectomized rats | Increased protein and mRNA levels in the kidneys [84] | |
| Nephrectomized rats | Increased mRNA levels in the liver and the kidneys [172] | |
| Nephrectomized rats | Decreased protein levels in the intestine [188] | |
| MRP4 | Membrane vesicles from MRP4-overexpressing HEK cells | Inhibited by IS, hippuric acid, kynurenic acid, IAA, and phenylacetic acid [178] |
| ciPTEC cells | Inhibited by PCS and p-cresyl glucuronide [174] | |
| HeLa cells | Performs cellular efflux of TMAO [110] | |
| Nephrectomized rats | Increased protein and mRNA levels in the kidneys [84] | |
| Nephrectomized rats | No differences in mRNA levels in the kidney, liver, and intestine [172] | |
| Nephrectomized rats | No differences in mRNA levels in the kidney and the liver [189] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cunha, R.S.d.; Azevedo, C.A.B.; Falconi, C.A.; Ruiz, F.F.; Liabeuf, S.; Carneiro-Ramos, M.S.; Stinghen, A.E.M. The Interplay between Uremic Toxins and Albumin, Membrane Transporters and Drug Interaction. Toxins 2022, 14, 177. https://doi.org/10.3390/toxins14030177
Cunha RSd, Azevedo CAB, Falconi CA, Ruiz FF, Liabeuf S, Carneiro-Ramos MS, Stinghen AEM. The Interplay between Uremic Toxins and Albumin, Membrane Transporters and Drug Interaction. Toxins. 2022; 14(3):177. https://doi.org/10.3390/toxins14030177
Chicago/Turabian StyleCunha, Regiane Stafim da, Carolina Amaral Bueno Azevedo, Carlos Alexandre Falconi, Fernanda Fogaça Ruiz, Sophie Liabeuf, Marcela Sorelli Carneiro-Ramos, and Andréa Emilia Marques Stinghen. 2022. "The Interplay between Uremic Toxins and Albumin, Membrane Transporters and Drug Interaction" Toxins 14, no. 3: 177. https://doi.org/10.3390/toxins14030177
APA StyleCunha, R. S. d., Azevedo, C. A. B., Falconi, C. A., Ruiz, F. F., Liabeuf, S., Carneiro-Ramos, M. S., & Stinghen, A. E. M. (2022). The Interplay between Uremic Toxins and Albumin, Membrane Transporters and Drug Interaction. Toxins, 14(3), 177. https://doi.org/10.3390/toxins14030177