The Fast and the Furriest: Investigating the Rate of Selection on Mammalian Toxins

 , , and
, , and
Abstract
1. Introduction
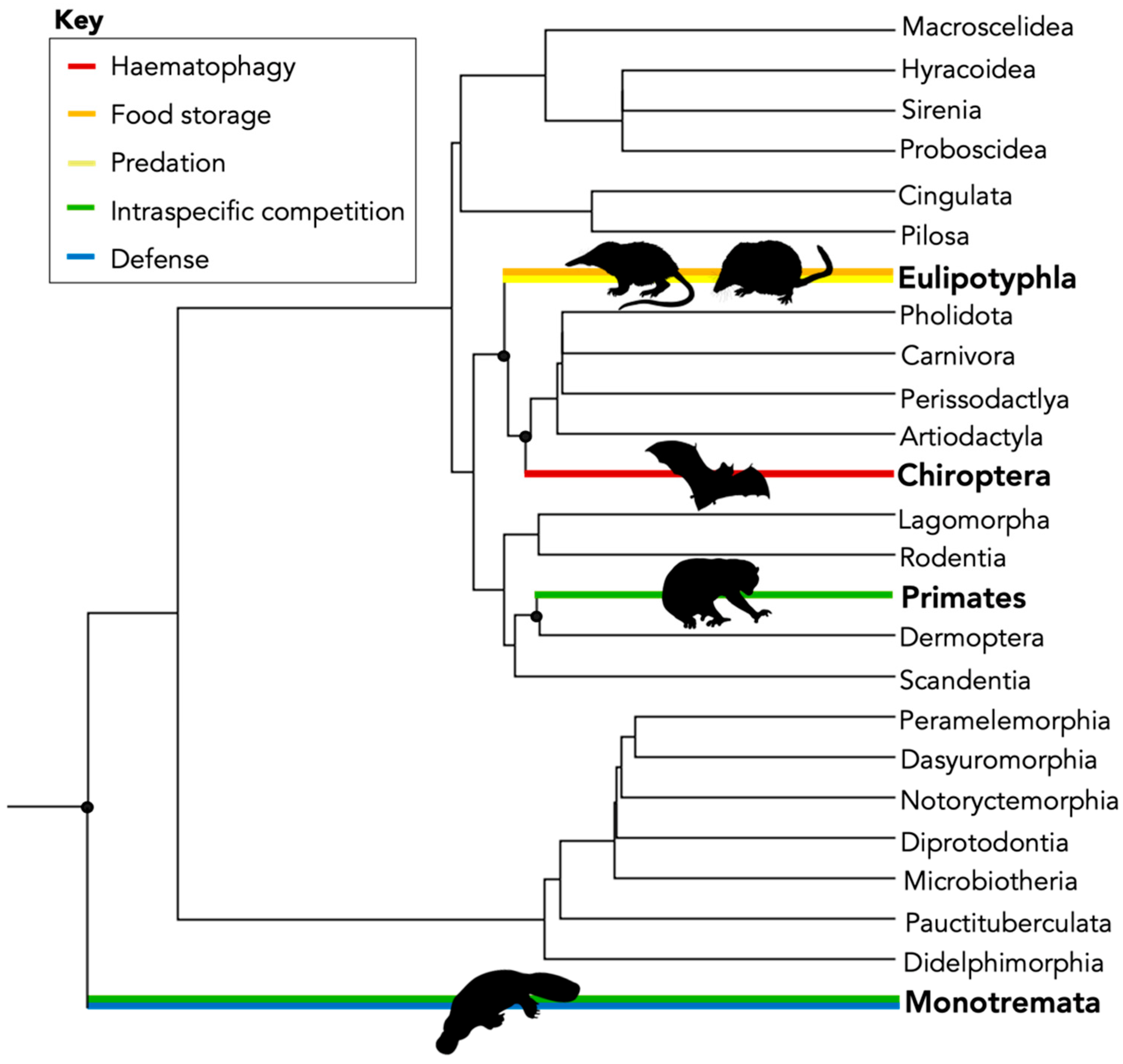
- What is the dN/dS ratio of mammal toxins and where does the Class fit within the ‘two-speed’ model of evolution proposed by Sunagar and Moran (2015) [9]?
- Are sites found under selection subject to pervasive selection (found across the whole phylogeny) or episodic selection (found within a subset of branches)? Is there evidence that different branches (toxin sequences) are under different rates of selection?
- Are any sites identified to be under diversifying selection within a toxin having a possible structural or chemical impact on the toxin?
2. Results
2.1. HyPhy DataMonkey
2.1.1. FUBAR and MEME Results
2.1.2. aBSREL Results
2.2. PAML Selection Results
2.2.1. Diversifying Selection Detected by Alternative Model
2.2.2. Purifying Selection Detected by Alternative Model
2.2.3. Alternative Models Rejected
2.3. Diversifying Sites Detected by Model 8 BEB
2.4. TreeSAAP Results
2.4.1. Properties Detected in Alignments by TreeSAAP
2.4.2. Alignments Which Had Significant Impact on Properties Identified by TreeSAAP
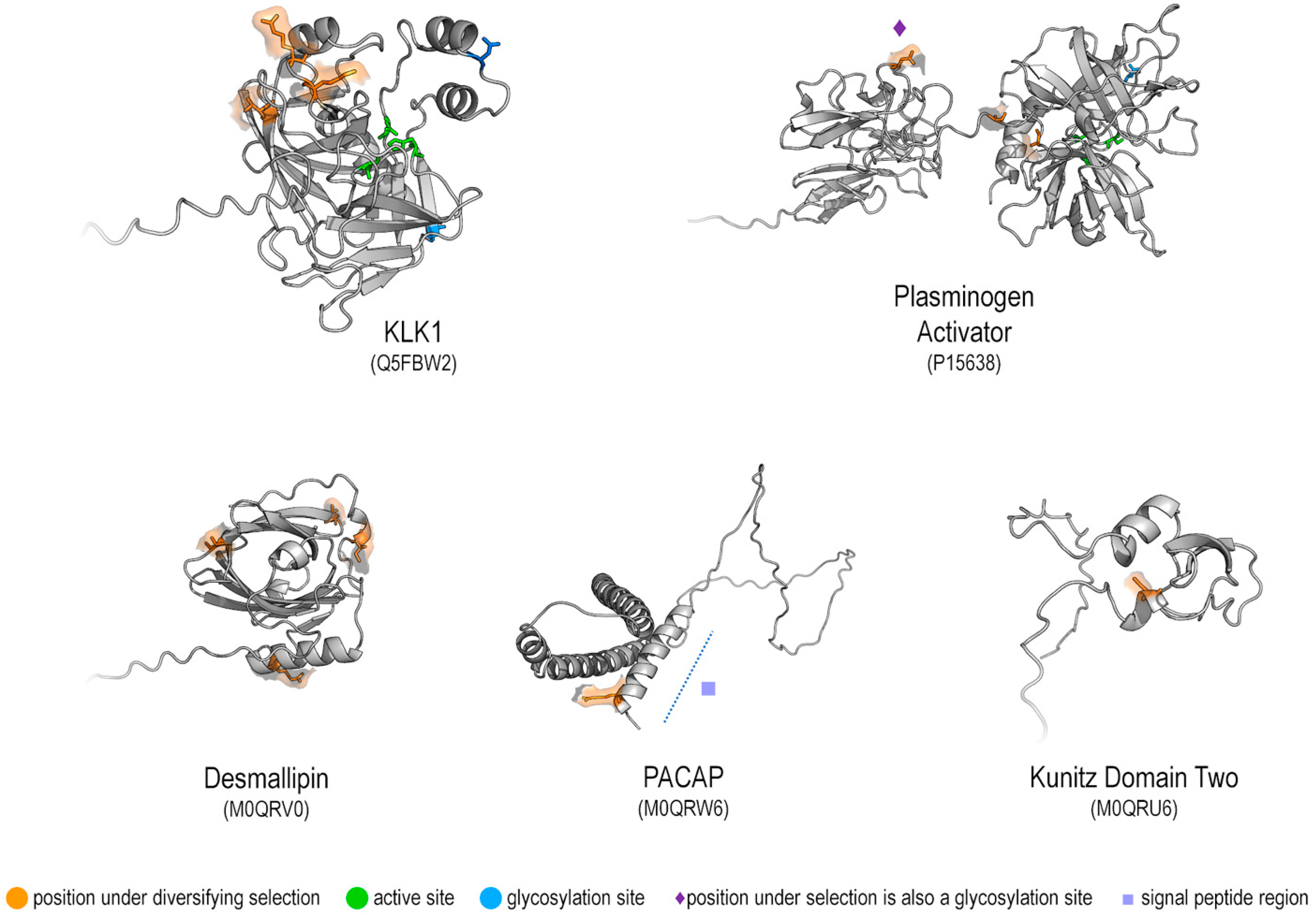
2.4.3. Location of Selection on the Protein Structure
3. Discussion
3.1. Mammal Toxins Are Acting under Diversifying Selection
3.2. Different Functions Have Different Rates of Selection on Mammal Toxins
3.3. Future Research
4. Conclusions
5. Materials and Methods
5.1. Data Collection
5.2. Alignments and Trimming
5.3. Datamonkey Selection Analysis
5.4. Site-Specific Selection
5.4.1. Fast Unconstrained Bayesian AppRoximation
5.4.2. Mixed Effects of Model Evolution
5.5. Branch Specific Selection
Adaptive Branch-Site Random Effective Likelihood
5.6. PAML Selection Tests
5.6.1. Tree Data
5.6.2. Selection Tests Using CodeML
5.7. Comparison of Toxin Sequences to Ancestral Group
5.7.1. Tree Selection on Amino Acid Properties
5.7.2. Visualisation of Results
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Surm, J.M.; Moran, Y. Insights into How Development and Life-History Dynamics Shape the Evolution of Venom. EvoDevo 2021, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Casewell, N.R.; Wüster, W.; Vonk, F.J.; Harrison, R.A.; Fry, B.G. Complex Cocktails: The Evolutionary Novelty of Venoms. Trends Ecol. Evol. 2013, 28, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Schendel, V.; Rash, L.D.; Jenner, R.A.; Undheim, E.A.B. The Diversity of Venom: The Importance of Behavior and Venom System Morphology in Understanding Its Ecology and Evolution. Toxins 2019, 11, 666. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.A. The Evolutionary Dynamics of Venom Toxins Made by Insects and Other Animals. Biochem. Soc. Trans. 2020, 48, 1353–1365. [Google Scholar] [CrossRef] [PubMed]
- Kryazhimskiy, S.; Plotkin, J.B. The Population Genetics of DN/DS. PLOS Genet. 2008, 4, e1000304. [Google Scholar] [CrossRef]
- Chang, D.; Duda, T.F., Jr. Extensive and Continuous Duplication Facilitates Rapid Evolution and Diversification of Gene Families. Mol. Biol. Evol. 2012, 29, 2019–2029. [Google Scholar] [CrossRef]
- Hargreaves, A.D.; Swain, M.T.; Hegarty, M.J.; Logan, D.W.; Mulley, J.F. Restriction and Recruitment—Gene Duplication and the Origin and Evolution of Snake Venom Toxins. Genome Biol. Evol. 2014, 6, 2088–2095. [Google Scholar] [CrossRef]
- Aird, S.D.; Aggarwal, S.; Villar-Briones, A.; Tin, M.M.-Y.; Terada, K.; Mikheyev, A.S. Snake Venoms Are Integrated Systems, but Abundant Venom Proteins Evolve More Rapidly. BMC Genom. 2015, 16, 647. [Google Scholar] [CrossRef]
- Sunagar, K.; Moran, Y. The Rise and Fall of an Evolutionary Innovation: Contrasting Strategies of Venom Evolution in Ancient and Young Animals. PLOS Genet. 2015, 11, e1005596. [Google Scholar] [CrossRef]
- Ligabue-Braun, R. Venom Use in Mammals: Evolutionary Aspects. In Evolution of Venomous Animals and Their Toxins; Gopalakrishnakone, P., Malhotra, A., Eds.; Springer: Dordrecht, The Netherlands, 2016; pp. 1–23. ISBN 978-94-007-6727-0. [Google Scholar]
- Rode-Margono, J.E.; Nekaris, K.A.-I. Cabinet of Curiosities: Venom Systems and Their Ecological Function in Mammals, with a Focus on Primates. Toxins 2015, 7, 2639. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, K.; Rychlik, L. Venom Use in Eulipotyphlans: An Evolutionary and Ecological Approach. Toxins 2021, 13, 231. [Google Scholar] [CrossRef]
- Warren, W.C.; Hillier, L.W.; Marshall Graves, J.A.; Birney, E.; Ponting, C.P.; Grützner, F.; Belov, K.; Miller, W.; Clarke, L.; Chinwalla, A.T.; et al. Genome Analysis of the Platypus Reveals Unique Signatures of Evolution. Nature 2008, 453, 175–183. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wong, E.S.W.; Nicol, S.; Warren, W.C.; Belov, K. Echidna Venom Gland Transcriptome Provides Insights into the Evolution of Monotreme Venom. PLOS ONE 2013, 8, e79092. [Google Scholar] [CrossRef] [PubMed]
- Casewell, N.R.; Petras, D.; Card, D.C.; Suranse, V.; Mychajliw, A.M.; Richards, D.; Koludarov, I.; Albulescu, L.-O.; Slagboom, J.; Hempel, B.-F.; et al. Solenodon Genome Reveals Convergent Evolution of Venom in Eulipotyphlan Mammals. Proc. Natl. Acad. Sci. 2019, 116, 25745–25755. [Google Scholar] [CrossRef]
- Hanf, Z.R.; Chavez, A.S. A Comprehensive Multi-Omic Approach Reveals a Relatively Simple Venom in a Diet Generalist, the Northern Short-Tailed Shrew, Blarina Brevicauda. Genome Biol. Evol. 2020, 12, 1148–1166. [Google Scholar] [CrossRef]
- Liao, Z.; Tang, X.; Chen, W.; Jiang, X.; Chen, Z.; He, K.; Li, Q.; Duan, Z.; He, X.; Kamau, P.M.; et al. Shrew’s Venom Quickly Causes Circulation Disorder, Analgesia and Hypokinesia. Cell. Mol. Life Sci. 2022, 79, 35. [Google Scholar] [CrossRef]
- Rychlik, L. CHANGES IN PREY SIZE PREFERENCES DURING SUCCESSIVE STAGES OF FORAGING IN THE MEDITERRANEAN WATER SHREW NEOMYS ANOMALUS. Behaviour 1999, 136, 345–365. [Google Scholar] [CrossRef]
- Kowalski, K.; Marciniak, P.; Rosiński, G.; Rychlik, L. Evaluation of the Physiological Activity of Venom from the Eurasian Water Shrew Neomys Fodiens. Front. Zool. 2017, 14, 46. [Google Scholar] [CrossRef] [PubMed]
- Camargo, I.; Alvarez-Castañeda, S.T. Analyses of Predation Behavior of the Desert Shrew Notiosorex Crawfordi. Mammalia 2018, 83, 276–280. [Google Scholar] [CrossRef]
- Lopez-Jurado, L.F.; Mateo, J.A. Evidence of Venom in the Canarian Shrew (Crocidura Canariensis): Immobilizing Effects on the Atlantic Lizard (Gallotia Atlantica). J. Zool. 1996, 239, 394–395. [Google Scholar] [CrossRef]
- Low, D.H.W.; Sunagar, K.; Undheim, E.A.B.; Ali, S.A.; Alagon, A.C.; Ruder, T.; Jackson, T.N.W.; Pineda Gonzalez, S.; King, G.F.; Jones, A.; et al. Dracula’s Children: Molecular Evolution of Vampire Bat Venom. J. Proteom. 2013, 89, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Tellgren-Roth, Å.; Dittmar, K.; Massey, S.E.; Kemi, C.; Tellgren-Roth, C.; Savolainen, P.; Lyons, L.A.; Liberles, D.A. Keeping the Blood Flowing—Plasminogen Activator Genes and Feeding Behavior in Vampire Bats. Naturwissenschaften 2009, 96, 39–47. [Google Scholar] [CrossRef]
- Hagey, L.R.; Fry, B.G.; Fitch-Snyder, H. Talking Defensively, a Dual Use for the Brachial Gland Exudate of Slow and Pygmy Lorises. In Primate Anti-Predator Strategies; Gursky, S.L., Nekaris, K.A.I., Eds.; Developments in Primatology: Progress and Prospects; Springer: Boston, MA, USA, 2007; pp. 253–272. ISBN 978-0-387-34807-0. [Google Scholar]
- Scheib, H.; Nekaris, K.A.-I.; Rode-Margono, J.; Ragnarsson, L.; Baumann, K.; Dobson, J.S.; Wirdateti, W.; Nouwens, A.; Nijman, V.; Martelli, P.; et al. The Toxicological Intersection between Allergen and Toxin: A Structural Comparison of the Cat Dander Allergenic Protein Fel D1 and the Slow Loris Brachial Gland Secretion Protein. Toxins 2020, 12, 86. [Google Scholar] [CrossRef]
- Madani, G.; Nekaris, K.A.-I. Anaphylactic Shock Following the Bite of a Wild Kayan Slow Loris (Nycticebus Kayan): Implications for Slow Loris Conservation. J. Venom. Anim. Toxins Trop. Dis. 2014, 20, 43. [Google Scholar] [CrossRef]
- Gardiner, M.; Weldon, A.; Poindexter, S.A.; Gibson, N.; Nekaris, K.A.I. Survey of Practitioners Handling Slow Lorises (Primates: Nycticebus): An Assessment of the Harmful Effects of Slow Loris Bites. J. Venom Res. 2018, 9, 1–7. [Google Scholar]
- Bal, A.K.; Giordano, A.J.; Gouda, S. Effects of a Bengal Slow Loris Nycticebus Bengalensis (Primates: Lorisidae) Bite: A Case Study from Murlen National Park, Mizoram, India. J. Threat. Taxa 2022, 14, 21449–21452. [Google Scholar] [CrossRef]
- Nekaris, K.A.-I.; Moore, R.S.; Rode, E.J.; Fry, B.G. Mad, Bad and Dangerous to Know: The Biochemistry, Ecology and Evolution of Slow Loris Venom. J. Venom. Anim. Toxins Trop. Dis. 2013, 19, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.L.; Parrott, M.L.; Handasyde, K.A.; Temple-Smith, P. Female Control of Reproductive Behaviour in the Platypus (Ornithorhynchusa anatinus), with Notes on Female Competition for Mating. Behaviour 2018, 155, 27–53. [Google Scholar] [CrossRef]
- Nekaris, K.A.I.; Campera, M.; Nijman, V.; Birot, H.; Rode-Margono, E.J.; Fry, B.G.; Weldon, A.; Wirdateti, W.; Imron, M.A. Slow Lorises Use Venom as a Weapon in Intraspecific Competition. Curr. Biol. 2020, 30, R1252–R1253. [Google Scholar] [CrossRef] [PubMed]
- Kakumanu, R.; Hodgson, W.C.; Ravi, R.; Alagon, A.; Harris, R.J.; Brust, A.; Alewood, P.F.; Kemp-Harper, B.K.; Fry, B.G. Vampire Venom: Vasodilatory Mechanisms of Vampire Bat (Desmodus rotundus) Blood Feeding. Toxins 2019, 11, 26. [Google Scholar] [CrossRef]
- Zhou, Y.; Shearwin-Whyatt, L.; Li, J.; Song, Z.; Hayakawa, T.; Stevens, D.; Fenelon, J.C.; Peel, E.; Cheng, Y.; Pajpach, F.; et al. Platypus and Echidna Genomes Reveal Mammalian Biology and Evolution. Nature 2021, 592, 756–762. [Google Scholar] [CrossRef]
- Hawkins, M.; Battaglia, A. Breeding Behaviour of the Platypus (Ornithorhynchus anatinus) in Captivity. Aust. J. Zool. 2009, 57, 283. [Google Scholar] [CrossRef]
- Whittington, C.M.; Papenfuss, A.T.; Locke, D.P.; Mardis, E.R.; Wilson, R.K.; Abubucker, S.; Mitreva, M.; Wong, E.S.; Hsu, A.L.; Kuchel, P.W.; et al. Novel Venom Gene Discovery in the Platypus. Genome Biol. 2010, 11, R95. [Google Scholar] [CrossRef]
- Kowalski, K.; Marciniak, P.; Rychlik, L. A New, Widespread Venomous Mammal Species: Hemolytic Activity of Sorex araneus Venom Is Similar to That of Neomys fodiens Venom. Zool. Lett. 2022, 8, 7. [Google Scholar] [CrossRef]
- Wong, E.S.W.; Morgenstern, D.; Mofiz, E.; Gombert, S.; Morris, K.M.; Temple-Smith, P.; Renfree, M.B.; Whittington, C.M.; King, G.F.; Warren, W.C.; et al. Proteomics and Deep Sequencing Comparison of Seasonally Active Venom Glands in the Platypus Reveals Novel Venom Peptides and Distinct Expression Profiles. Mol. Cell. Proteom. 2012, 11, 1354–1364. [Google Scholar] [CrossRef]
- Barua, A.; Koludarov, I.; Mikheyev, A.S. Co-Option of the Same Ancestral Gene Family Gave Rise to Mammalian and Reptilian Toxins. BMC Biol. 2021, 19, 268. [Google Scholar] [CrossRef]
- Tokuriki, N.; Stricher, F.; Schymkowitz, J.; Serrano, L.; Tawfik, D.S. The Stability Effects of Protein Mutations Appear to Be Universally Distributed. J. Mol. Biol. 2007, 369, 1318–1332. [Google Scholar] [CrossRef]
- Tokuriki, N.; Tawfik, D.S. Stability Effects of Mutations and Protein Evolvability. Curr. Opin. Struct. Biol. 2009, 19, 596–604. [Google Scholar] [CrossRef]
- Studer, R.A.; Dessailly, B.H.; Orengo, C.A. Residue Mutations and Their Impact on Protein Structure and Function: Detecting Beneficial and Pathogenic Changes. Biochem. J. 2013, 449, 581–594. [Google Scholar] [CrossRef]
- Roelants, K.; Champagne, D.; Scheib, H.; Tyndall, J.; King, G.; Nevalainen, T.; Norman, J.; Lewis, R.; Norton, R.; Renjifo, C.; et al. The Toxicogenomic Multiverse: Convergent Recruitment of Proteins Into Animal Venoms. Annu. Rev. Genom. Hum. Genet. 2009, 10, 483–511. [Google Scholar] [CrossRef]
- Aminetzach, Y.T.; Srouji, J.R.; Kong, C.Y.; Hoekstra, H.E. Convergent Evolution of Novel Protein Function in Shrew and Lizard Venom. Curr. Biol. 2009, 19, 1925–1931. [Google Scholar] [CrossRef]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in Health and Disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.Z.; Tablante, A.; Bartoli, F.; Beguin, S.; Hemker, H.C.; Apitz-Castro, R. Expression of Biological Activity of Draculin, the Anticoagulant Factor from Vampire Bat Saliva, Is Strictly Dependent on the Appropriate Glycosylation of the Native Molecule. Biochim. Biophys. Acta BBA—Gen. Subj. 1998, 1425, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Owji, H.; Nezafat, N.; Negahdaripour, M.; Hajiebrahimi, A.; Ghasemi, Y. A Comprehensive Review of Signal Peptides: Structure, Roles, and Applications. Eur. J. Cell Biol. 2018, 97, 422–441. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.J.; Arbuckle, K. Tempo and Mode of the Evolution of Venom and Poison in Tetrapods. Toxins 2016, 8, 193. [Google Scholar] [CrossRef]
- Botero-Castro, F.; Tilak, M.-K.; Justy, F.; Catzeflis, F.; Delsuc, F.; Douzery, E.J.P. In Cold Blood: Compositional Bias and Positive Selection Drive the High Evolutionary Rate of Vampire Bats Mitochondrial Genomes. Genome Biol. Evol. 2018, 10, 2218–2239. [Google Scholar] [CrossRef]
- Wong, E.S.W.; Belov, K. Venom Evolution through Gene Duplications. Gene 2012, 496, 1–7. [Google Scholar] [CrossRef]
- Casewell, N.R.; Jackson, T.N.W.; Laustsen, A.H.; Sunagar, K. Causes and Consequences of Snake Venom Variation. Trends Pharmacol. Sci. 2020, 41, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.D.; Baker, R.J. Secretory Gene Recruitments in Vampire Bat Salivary Adaptation and Potential Convergences With Sanguivorous Leeches. Front. Ecol. Evol. 2015, 3, 122. [Google Scholar] [CrossRef]
- Folinsbee, K.E. Evolution of Venom across Extant and Extinct Eulipotyphlans. Comptes Rendus Palevol. 2013, 12, 531–542. [Google Scholar] [CrossRef]
- Barua, A.; Mikheyev, A.S. An Ancient, Conserved Gene Regulatory Network Led to the Rise of Oral Venom Systems. Proc. Natl. Acad. Sci. USA 2021, 118, e2021311118. [Google Scholar] [CrossRef] [PubMed]
- Chaverri, G.; Ancillotto, L.; Russo, D. Social Communication in Bats. Biol. Rev. 2018, 93, 1938–1954. [Google Scholar] [CrossRef] [PubMed]
- Francischetti, I.M.B.; Assumpção, T.C.F.; Ma, D.; Li, Y.; Vicente, E.C.; Uieda, W.; Ribeiro, J.M.C. The “Vampirome”: Transcriptome and Proteome Analysis of the Principal and Accessory Submaxillary Glands of the Vampire Bat Desmodus Rotundus, a Vector of Human Rabies. J. Proteom. 2013, 82, 288–319. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Arévalo, L.; Brukman, N.G.; Cuasnicú, P.S.; Roldan, E.R.S. Evolutionary Analysis of Genes Coding for Cysteine-RIch Secretory Proteins (CRISPs) in Mammals. BMC Evol. Biol. 2020, 20, 67. [Google Scholar] [CrossRef]
- Sunagar, K.; Johnson, W.E.; O’Brien, S.J.; Vasconcelos, V.; Antunes, A. Evolution of CRISPs Associated with Toxicoferan-Reptilian Venom and Mammalian Reproduction. Mol. Biol. Evol. 2012, 29, 1807–1822. [Google Scholar] [CrossRef]
- Jackson, T.N.W.; Koludarov, I. How the Toxin Got Its Toxicity. Front. Pharmacol. 2020, 11, 574925. [Google Scholar] [CrossRef]
- Potter, J.H.T.; Davies, K.T.J.; Yohe, L.R.; Sanchez, M.K.R.; Rengifo, E.M.; Struebig, M.; Warren, K.; Tsagkogeorga, G.; Lim, B.K.; dos Reis, M.; et al. Dietary Diversification and Specialization in Neotropical Bats Facilitated by Early Molecular Evolution. Mol. Biol. Evol. 2021, 38, 3864–3883. [Google Scholar] [CrossRef]
- Zepeda Mendoza, M.L.; Xiong, Z.; Escalera-Zamudio, M.; Runge, A.K.; Thézé, J.; Streicker, D.; Frank, H.K.; Loza-Rubio, E.; Liu, S.; Ryder, O.A.; et al. Hologenomic Adaptations Underlying the Evolution of Sanguivory in the Common Vampire Bat. Nat. Ecol. Evol. 2018, 2, 659–668. [Google Scholar] [CrossRef]
- Moreno Santillán, D.D.; Lama, T.M.; Gutierrez Guerrero, Y.T.; Brown, A.M.; Donat, P.; Zhao, H.; Rossiter, S.J.; Yohe, L.R.; Potter, J.H.; Teeling, E.C.; et al. Large-Scale Genome Sampling Reveals Unique Immunity and Metabolic Adaptations in Bats. Mol. Ecol. 2021, 30, 6449–6467. [Google Scholar] [CrossRef] [PubMed]
- Vandewege, M.W.; Sotero-Caio, C.G.; Phillips, C.D. Positive Selection and Gene Expression Analyses from Salivary Glands Reveal Discrete Adaptations within the Ecologically Diverse Bat Family Phyllostomidae. Genome Biol. Evol. 2020, 12, 1419–1428. [Google Scholar] [CrossRef] [PubMed]
- Yarlagadda, K.; Razik, I.; Malhi, R.S.; Carter, G.G. Social Convergence of Gut Microbiomes in Vampire Bats. Biol. Lett. 2021, 17, 20210389. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Peigneur, S.; Gao, B.; Zhang, S.; Tytgat, J.; Zhu, S. Target-Driven Positive Selection at Hot Spots of Scorpion Toxins Uncovers Their Potential in Design of Insecticides. Mol. Biol. Evol. 2016, 33, 1907–1920. [Google Scholar] [CrossRef]
- Zhang, S.; Gao, B.; Zhu, S. Target-Driven Evolution of Scorpion Toxins. Sci. Rep. 2015, 5, 14973. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. TrimAl: A Tool for Automated Alignment Trimming in Large-Scale Phylogenetic Analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Delport, W.; Poon, A.F.Y.; Frost, S.D.W.; Kosakovsky Pond, S.L. Datamonkey 2010: A Suite of Phylogenetic Analysis Tools for Evolutionary Biology. Bioinformatics 2010, 26, 2455–2457. [Google Scholar] [CrossRef]
- Pond, S.L.K.; Frost, S.D.W. Datamonkey: Rapid Detection of Selective Pressure on Individual Sites of Codon Alignments. Bioinformatics 2005, 21, 2531–2533. [Google Scholar] [CrossRef]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A Modern Web Application for Characterizing Selective and Other Evolutionary Processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A Fast, Unconstrained Bayesian AppRoximation for Inferring Selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Pond, S.L.K. Detecting Individual Sites Subject to Episodic Diversifying Selection. PLOS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Murrell, B.; Fourment, M.; Frost, S.D.W.; Delport, W.; Scheffler, K. A Random Effects Branch-Site Model for Detecting Episodic Diversifying Selection. Mol. Biol. Evol. 2011, 28, 3033–3043. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Gao, F.; Chen, C.; Arab, D.A.; Du, Z.; He, Y.; Ho, S.Y.W. EasyCodeML: A Visual Tool for Analysis of Selection Using CodeML. Ecol. Evol. 2019, 9, 3891–3898. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Woolley, S.; Johnson, J.; Smith, M.J.; Crandall, K.A.; McClellan, D.A. TreeSAAP: Selection on Amino Acid Properties Using Phylogenetic Trees. Bioinformatics 2003, 19, 671–672. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly Accurate Protein Structure Prediction for the Human Proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Richardson, J.S. The Anatomy and Taxonomy of Protein Structure. In Advances in Protein Chemistry; Anfinsen, C.B., Edsall, J.T., Richards, F.M., Eds.; Academic Press: Cambridge, MA, USA, 1981; Volume 34, pp. 167–339. [Google Scholar]
- The UniProt Consortium UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Common Name | Order | Family | Genus | Species | Venom Status | Reference |
|---|---|---|---|---|---|---|
| Platypus | Monotremata | Ornithorhynchidae | Ornithorhynchus | anatinus | Confirmed 1 | [14] |
| Short-beaked echidna | Monotremata | Tachyglossidae | Tachyglossus | aculeatus | Vestigial 2 | [15] |
| Cuban solenodon | Eulipotyphla | Solenodontidae | Atopogale | cubana | Highly likely (evolutionary) | [16] |
| Hispaniolan solenodon | Eulipotyphla | Solenodontidae | Solenodon | paradoxus | Confirmed | |
| Northern short-tailed shrew | Eulipotyphla | Soricidae | Blarina | brevicauda | Confirmed | [17] |
| Southern short-tailed shrew | Eulipotyphla | Soricidae | Blarina | carolinensis | Highly likely (evolutionary) | |
| Elliot’s short-tailed shrew | Eulipotyphla | Soricidae | Blarina | hylophaga | Highly likely (evolutionary) | |
| Everglades short-tailed shrew | Eulipotyphla | Soricidae | Blarina | peninsulae | Highly likely (evolutionary) | |
| Asiatic short-tailed shrew | Eulipotyphla | Soricidae | Blarinella | quadraticauda | Confirmed | [18] |
| Indochinese short-tailed shrew | Eulipotyphla | Soricidae | Blarinella | griselda | Highly likely (evolutionary) | |
| Burmese short-tailed shrew | Eulipotyphla | Soricidae | Blarinella | wardi | Highly likely (evolutionary) | |
| Mediterranean water shrew | Eulipotyphla | Soricidae | Neomys | anomalus | Confirmed | [19] |
| Eurasian water shrew | Eulipotyphla | Soricidae | Neomys | fodiens | Confirmed | [20] |
| Transcaucasian water shrew | Eulipotyphla | Soricidae | Neomys | teres | Highly likely (evolutionary) | |
| Crawford’s grey shrew | Eulipotyphla | Soricidae | Notiosorex | crawfordi | Likely (behavioural observation) | [21] |
| Canarian shrew | Eulipotyphla | Soricidae | Crocidura | canariensis | Likely (behavioural observation) | [22] |
| Common vampire bat | Chiroptera | Phyllostomidae | Desmodus | rotundus | Confirmed | [23] |
| Hairy-legged vampire bat | Chiroptera | Phyllostomidae | Diphylla | ecaudata | Confirmed | [24] |
| White-winged vampire bat | Chiroptera | Phyllostomidae | Diaemus | youngi | Confirmed | |
| Pygmy slow loris | Primates | Lorisidae | Xanthonycticebus | pygamaeus | Confirmed | [25] |
| Greater slow loris | Primates | Lorisidae | Nycticebus | coucang | Confirmed | [25] |
| Javan slow loris | Primates | Lorisidae | Nycticebus | javanicus | Confirmed | [26] |
| Kayan slow loris | Primates | Lorisidae | Nycticebus | kayan | Confirmed | [27] |
| Bengal slow loris | Primates | Lorisidae | Nycticebus | bengalensis | Confirmed | [28,29] |
| Bornean slow loris | Primates | Lorisidae | Nycticebus | borneanus | Highly likely (evolutionary) | [26,27,30] |
| Philippine slow loris | Primates | Lorisidae | Nycticebus | menagensis | Highly likely (evolutionary) | |
| Sumatran slow loris | Primates | Lorisidae | Nycticebus | hilleri | Highly likely (evolutionary) | |
| Bangka slow loris | Primates | Lorisidae | Nycticebus | bancanus | Highly likely (evolutionary) | |
| Species | Toxin | FUBAR | MEME | CODEML | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ω > 1 | ω < 1 | ω > 1 | Model 0 Weighted Average | Model 2a Weighted Average | M2a Positive Sites >0.95 | M2a Positive Sites >0.99 | Model 8 Weighted Average | M8 Positive Sites >0.95 | M8 Positive Sites p > 0.99 | ||
| Blarina brevicauda | Kallikren-1 (KLK1) | 5 | 23 | 4 | 0.57984 | 0.8139 | N/A | N/A | 0.8075 | 13 | 3 |
| Solenodon paradoxus | Kallikren-1 (KLK1) | 8 | 16 | 6 | 0.58254 | 0.7298 | 1 | 2 | 0.7073 | 2 | 4 |
| Eupotiphyla (Blarina and Solenodon) | Kallikren-1 (KLK1) | 5 | 35 | 16 | 0.49379 | 0.6748 | 1 | 2 | 0.6260 | 3 | 2 |
| Desmodus rotundus, Blarina and Solenodon | Kallikren-1 (KLK1) | 3 | 47 | 21 | 0.43905 | 0.6339 | 1 | N/A | 0.5592 | 3 | 1 |
| Diphylla ecaudata, Desmodus and Diameus youngi | Plasminogen Activator | 15 | 8 | 4 | 1.0549 | 1.3046 | 10 | 4 | 1.3082 | 14 | 10 |
| Desmodus | Desmallipin | 22 | 8 | 15 | 1.09464 | 1.3216 | 3 | 7 | 1.3393 | 17 | 12 |
| Desmodus | PACAP | 3 | 5 | 2 | 0.90099 | 1.3239 | 4 | 1 | 1.3208 | 14 | N/A |
| Desmodus | CRiSP | 5 | 2 | 0 | 1.48836 | 2.0592 | 4 | N/A | 2.0806 | 4 | 2 |
| Desmodus and Ornithorhynchus anatinus | CRiSP | 7 | 2 | 0 | 0.34629 | 1.3843 | 3 | N/A | 1.2923 | 4 | 3 |
| Desmodus | Kunitz Domain One | 1 | 7 | 1 | 0.29323 | 1.4170 NS | 1 | N/A | 1.3985 NS | 2 | 2 |
| Desmodus | Kunitz Domain Two | 2 | 1 | 2 | 2.28040 NS | 2.6283 | 3 | 1 | 2.6283 | 4 | 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fitzpatrick, L.L.J.; Nijman, V.; Ligabue-Braun, R.; Nekaris, K.A.-I. The Fast and the Furriest: Investigating the Rate of Selection on Mammalian Toxins. Toxins 2022, 14, 842. https://doi.org/10.3390/toxins14120842
Fitzpatrick LLJ, Nijman V, Ligabue-Braun R, Nekaris KA-I. The Fast and the Furriest: Investigating the Rate of Selection on Mammalian Toxins. Toxins. 2022; 14(12):842. https://doi.org/10.3390/toxins14120842
Chicago/Turabian StyleFitzpatrick, Leah Lucy Joscelyne, Vincent Nijman, Rodrigo Ligabue-Braun, and K. Anne-Isola Nekaris. 2022. "The Fast and the Furriest: Investigating the Rate of Selection on Mammalian Toxins" Toxins 14, no. 12: 842. https://doi.org/10.3390/toxins14120842
APA StyleFitzpatrick, L. L. J., Nijman, V., Ligabue-Braun, R., & Nekaris, K. A.-I. (2022). The Fast and the Furriest: Investigating the Rate of Selection on Mammalian Toxins. Toxins, 14(12), 842. https://doi.org/10.3390/toxins14120842