Homeostasis in the Gut Microbiota in Chronic Kidney Disease


Abstract
1. Introduction
1.1. Mutualism between the Gut Microbiota in Healthy Individuals
1.2. Gut Microbiota Dysbiosis in Chronic Kidney Disease
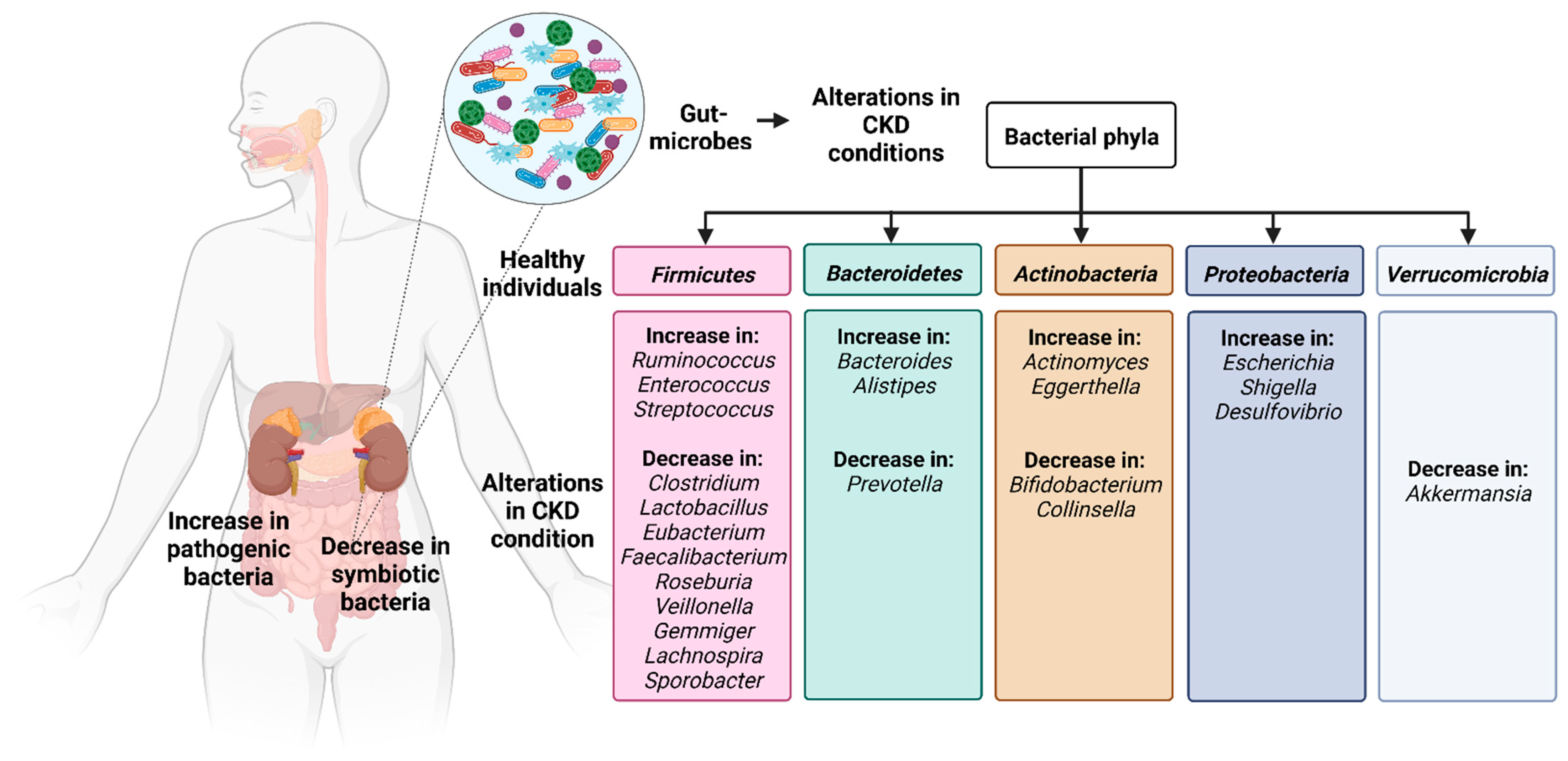
2. Alterations in the Intestinal Microflora in Chronic Kidney Disease
3. Effects of Alterations in the Intestinal Flora in Chronic Kidney Disease
3.1. Production of Gut-Derived Metabolites
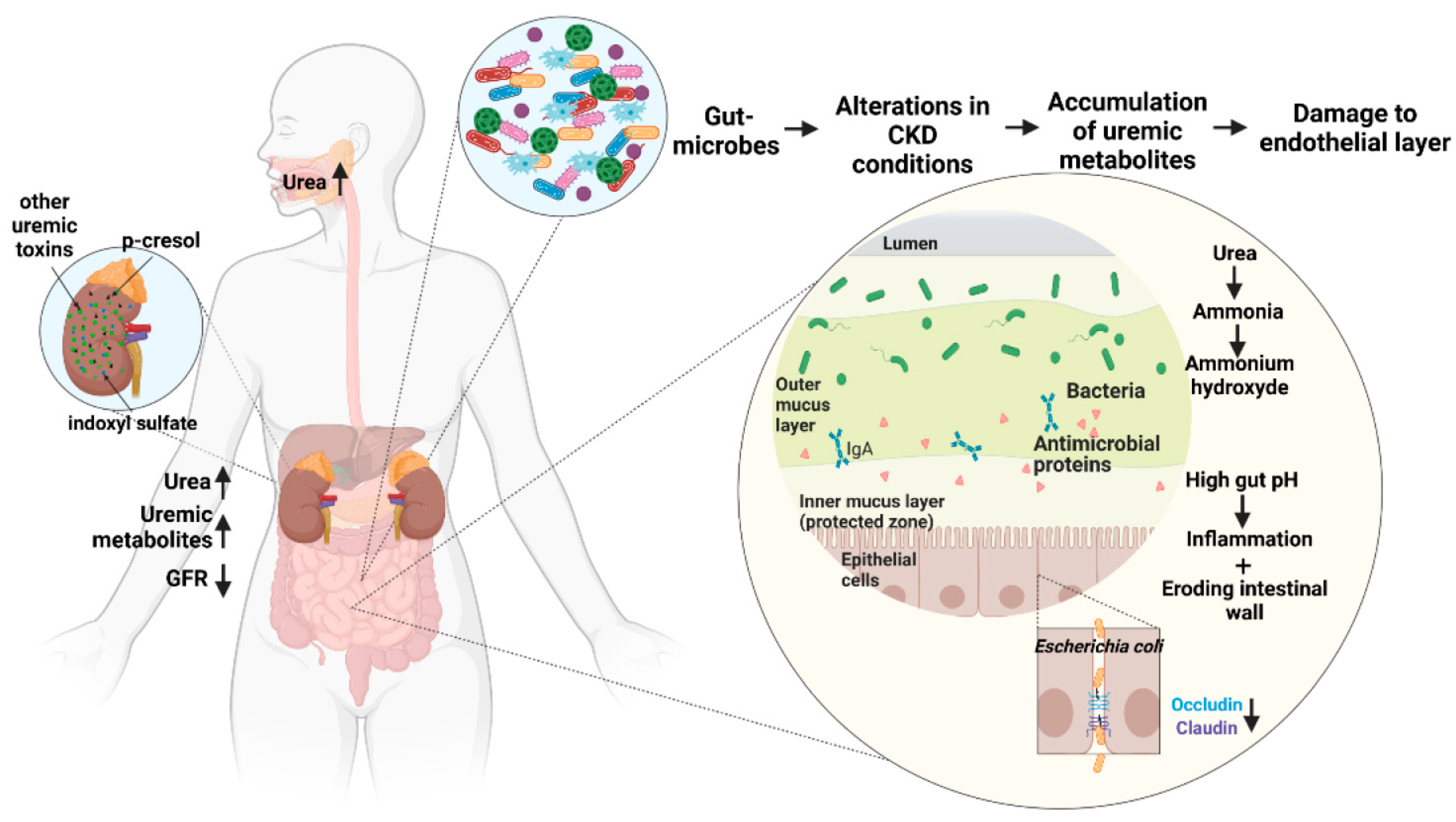
3.2. The Accumulation of Gut-Derived Metabolites
3.2.1. Small Water-Soluble Molecules
3.2.2. Gut-Derived, Protein-Bound Uremic Toxins (PBUTs)
3.2.3. Middle Molecules
3.3. Deimmunization: Inflammation and Immunosuppression
3.4. Damage to Intestinal Epithelial Barrier
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guldris, S.C.; Parra, E.G.; Amenos, A.C. Gut microbiota in chronic kidney disease. Nefrologia 2017, 37, 9–19. [Google Scholar] [CrossRef]
- Diez, J.; Ortiz, A. The need for a cardionephrology subspecialty. Clin. Kidney J. 2021, 14, 1491–1494. [Google Scholar] [CrossRef] [PubMed]
- Pickard, J.M.; Zeng, M.Y.; Caruso, R.; Nunez, G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 2017, 279, 70–89. [Google Scholar] [CrossRef] [PubMed]
- Ottman, N.; Geerlings, S.Y.; Aalvink, S.; de Vos, W.M.; Belzer, C. Action and function of Akkermansia muciniphila in microbiome ecology, health and disease. Best Pract. Res. Clin. Gastroenterol. 2017, 31, 637–642. [Google Scholar] [CrossRef]
- Backhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef]
- Mahmoodpoor, F.; Rahbar Saadat, Y.; Barzegari, A.; Ardalan, M.; Zununi Vahed, S. The impact of gut microbiota on kidney function and pathogenesis. Biomed Pharm. 2017, 93, 412–419. [Google Scholar] [CrossRef]
- Jenson, I. BACILLUS | Introduction. In Encyclopedia of Food Microbiology, 2nd ed.; Academic Press: Cambridge, MA, USA, 2014. [Google Scholar] [CrossRef]
- Castro-Gonzalez, J.M.; Castro, P.; Sandoval, H.; Castro-Sandoval, D. Probiotic lactobacilli precautions. Front. Microbiol. 2019, 10, 375. [Google Scholar] [CrossRef]
- Wexler, H.M. Bacteroides: The good, the bad, and the nitty-gritty. Clin. Microbiol. Rev. 2007, 20, 593–621. [Google Scholar] [CrossRef]
- Clemente, J.C.; Pehrsson, E.C.; Blaser, M.J.; Sandhu, K.; Gao, Z.; Wang, B.; Magris, M.; Hidalgo, G.; Contreras, M.; Noya-Alarcon, O.; et al. The microbiome of uncontacted Amerindians. Sci. Adv. 2015, 1, e1500183. [Google Scholar] [CrossRef]
- Gorvitovskaia, A.; Holmes, S.P.; Huse, S.M. Interpreting Prevotella and Bacteroides as biomarkers of diet and lifestyle. Microbiome 2016, 4, 15. [Google Scholar] [CrossRef]
- Iljazovic, A.; Roy, U.; Galvez, E.J.C.; Lesker, T.R.; Zhao, B.; Gronow, A.; Amend, L.; Will, S.E.; Hofmann, J.D.; Pils, M.C.; et al. Perturbation of the gut microbiome by Prevotella spp. enhances host susceptibility to mucosal inflammation. Mucosal Immunol. 2021, 14, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ma, Z.S. FBA ecological guild: Trio of firmicutes-bacteroidetes alliance against actinobacteria in human oral microbiome. Sci. Rep. 2020, 10, 287. [Google Scholar] [CrossRef]
- Stojanov, S.; Berlec, A.; Strukelj, B. The influence of probiotics on the firmicutes/bacteroidetes ratio in the treatment of obesity and inflammatory bowel disease. Microorganisms 2020, 8, 1715. [Google Scholar] [CrossRef] [PubMed]
- Vaiserman, A.; Romanenko, M.; Piven, L.; Moseiko, V.; Lushchak, O.; Kryzhanovska, N.; Guryanov, V.; Koliada, A. Differences in the gut Firmicutes to Bacteroidetes ratio across age groups in healthy Ukrainian population. BMC Microbiol. 2020, 20, 221. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Santisteban, M.M.; Rodriguez, V.; Li, E.; Ahmari, N.; Carvajal, J.M.; Zadeh, M.; Gong, M.; Qi, Y.; Zubcevic, J.; et al. Gut dysbiosis is linked to hypertension. Hypertension 2015, 65, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Kieffer, D.A.; Piccolo, B.D.; Vaziri, N.D.; Liu, S.; Lau, W.L.; Khazaeli, M.; Nazertehrani, S.; Moore, M.E.; Marco, M.L.; Martin, R.J.; et al. Resistant starch alters gut microbiome and metabolomic profiles concurrent with amelioration of chronic kidney disease in rats. Am. J. Physiol. Renal. Physiol. 2016, 310, F857–F871. [Google Scholar] [CrossRef]
- Marques, F.Z.; Nelson, E.; Chu, P.Y.; Horlock, D.; Fiedler, A.; Ziemann, M.; Tan, J.K.; Kuruppu, S.; Rajapakse, N.W.; El-Osta, A.; et al. High-fiber diet and acetate supplementation change the gut microbiota and prevent the development of hypertension and heart failure in hypertensive mice. Circulation 2017, 135, 964–977. [Google Scholar] [CrossRef]
- Binda, C.; Lopetuso, L.R.; Rizzatti, G.; Gibiino, G.; Cennamo, V.; Gasbarrini, A. Actinobacteria: A relevant minority for the maintenance of gut homeostasis. Dig. Liver Dis. 2018, 50, 421–428. [Google Scholar] [CrossRef]
- Shin, N.R.; Whon, T.W.; Bae, J.W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef]
- Fujio-Vejar, S.; Vasquez, Y.; Morales, P.; Magne, F.; Vera-Wolf, P.; Ugalde, J.A.; Navarrete, P.; Gotteland, M. The gut microbiota of healthy chilean subjects reveals a high abundance of the phylum verrucomicrobia. Front. Microbiol. 2017, 8, 1221. [Google Scholar] [CrossRef] [PubMed]
- Gomaa, E.Z. Human gut microbiota/microbiome in health and diseases: A review. Antonie Van Leeuwenhoek 2020, 113, 2019–2040. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; D’Souza, R.; Hong, S.T. The role of gut microbiota in the gut-brain axis: Current challenges and perspectives. Protein Cell 2013, 4, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Topping, D.L.; Clifton, P.M. Short-chain fatty acids and human colonic function: Roles of resistant starch and nonstarch polysaccharides. Physiol. Rev. 2001, 81, 1031–1064. [Google Scholar] [CrossRef]
- Valdes, A.M.; Walter, J.; Segal, E.; Spector, T.D. Role of the gut microbiota in nutrition and health. BMJ 2018, 361, k2179. [Google Scholar] [CrossRef]
- Tolhurst, G.; Heffron, H.; Lam, Y.S.; Parker, H.E.; Habib, A.M.; Diakogiannaki, E.; Cameron, J.; Grosse, J.; Reimann, F.; Gribble, F.M. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes 2012, 61, 364–371. [Google Scholar] [CrossRef]
- Carvalho, B.M.; Saad, M.J. Influence of gut microbiota on subclinical inflammation and insulin resistance. Mediat. Inflamm. 2013, 2013, 986734. [Google Scholar] [CrossRef]
- Sivaprakasam, S.; Prasad, P.D.; Singh, N. Benefits of short-chain fatty acids and their receptors in inflammation and carcinogenesis. Pharmacol. Ther. 2016, 164, 144–151. [Google Scholar] [CrossRef]
- Amabebe, E.; Robert, F.O.; Agbalalah, T.; Orubu, E.S.F. Microbial dysbiosis-induced obesity: Role of gut microbiota in homoeostasis of energy metabolism. Br. J. Nutr. 2020, 123, 1127–1137. [Google Scholar] [CrossRef]
- Hobby, G.P.; Karaduta, O.; Dusio, G.F.; Singh, M.; Zybailov, B.L.; Arthur, J.M. Chronic kidney disease and the gut microbiome. Am. J. Physiol. Renal. Physiol. 2019, 316, F1211–F1217. [Google Scholar] [CrossRef]
- Duttaroy, A.K. Role of gut microbiota and their metabolites on atherosclerosis, hypertension and human blood platelet function: A review. Nutrients 2021, 13, 144. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.J.; Andersen, K.; Stecher, B. The intestinal microbiota, a leaky gut, and abnormal immunity in kidney disease. Kidney Int. 2013, 83, 1010–1016. [Google Scholar] [CrossRef]
- Glorieux, G.; Gryp, T.; Perna, A. Gut-derived metabolites and their role in immune dysfunction in chronic kidney disease. Toxins 2020, 12, 245. [Google Scholar] [CrossRef]
- Mihajlovic, M.; Krebber, M.M.; Yang, Y.; Ahmed, S.; Lozovanu, V.; Andreeva, D.; Verhaar, M.C.; Masereeuw, R. Protein-bound uremic toxins induce reactive oxygen species-dependent and inflammasome-mediated il-1beta production in kidney proximal tubule cells. Biomedicines 2021, 9, 1326. [Google Scholar] [CrossRef] [PubMed]
- de Mello, V.D.; Paananen, J.; Lindstrom, J.; Lankinen, M.A.; Shi, L.; Kuusisto, J.; Pihlajamaki, J.; Auriola, S.; Lehtonen, M.; Rolandsson, O.; et al. Indolepropionic acid and novel lipid metabolites are associated with a lower risk of type 2 diabetes in the Finnish Diabetes Prevention Study. Sci. Rep. 2017, 7, 46337. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Pletinck, A.; Schepers, E.; Glorieux, G. Biochemical and clinical impact of organic uremic retention solutes: A comprehensive update. Toxins 2018, 10, 33. [Google Scholar] [CrossRef]
- Sabatino, A.; Regolisti, G.; Brusasco, I.; Cabassi, A.; Morabito, S.; Fiaccadori, E. Alterations of intestinal barrier and microbiota in chronic kidney disease. Nephrol. Dial. Transplant. 2015, 30, 924–933. [Google Scholar] [CrossRef]
- Li, F.; Wang, M.; Wang, J.; Li, R.; Zhang, Y. Alterations to the gut microbiota and their correlation with inflammatory factors in chronic kidney disease. Front. Cell Infect. Microbiol. 2019, 9, 206. [Google Scholar] [CrossRef]
- Hanninen, A.; Toivonen, R.; Poysti, S.; Belzer, C.; Plovier, H.; Ouwerkerk, J.P.; Emani, R.; Cani, P.D.; De Vos, W.M. Akkermansia muciniphila induces gut microbiota remodelling and controls islet autoimmunity in NOD mice. Gut 2018, 67, 1445–1453. [Google Scholar] [CrossRef]
- Kanbay, M.; Onal, E.M.; Afsar, B.; Dagel, T.; Yerlikaya, A.; Covic, A.; Vaziri, N.D. The crosstalk of gut microbiota and chronic kidney disease: Role of inflammation, proteinuria, hypertension, and diabetes mellitus. Int. Urol. Nephrol. 2018, 50, 1453–1466. [Google Scholar] [CrossRef]
- Wehedy, E.; Shatat, I.F.; Al Khodor, S. The human microbiome in chronic kidney disease: A double-edged sword. Front. Med. 2021, 8, 790783. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Ning, X.; Liu, B.; Dong, R.; Bai, M.; Sun, S. Specific alterations in gut microbiota in patients with chronic kidney disease: An updated systematic review. Ren. Fail. 2021, 43, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Felizardo, R.J.F.; de Almeida, D.C.; Pereira, R.L.; Watanabe, I.K.M.; Doimo, N.T.S.; Ribeiro, W.R.; Cenedeze, M.A.; Hiyane, M.I.; Amano, M.T.; Braga, T.T.; et al. Gut microbial metabolite butyrate protects against proteinuric kidney disease through epigenetic- and GPR109a-mediated mechanisms. FASEB J. 2019, 33, 11894–11908. [Google Scholar] [CrossRef]
- Brilli, L.L.; Swanhart, L.M.; de Caestecker, M.P.; Hukriede, N.A. HDAC inhibitors in kidney development and disease. Pediatr. Nephrol. 2013, 28, 1909–1921. [Google Scholar] [CrossRef]
- Hu, Q.; Wu, K.; Pan, W.; Zeng, Y.; Hu, K.; Chen, D.; Huang, X.; Zhang, Q. Intestinal flora alterations in patients with early chronic kidney disease: A case-control study among the Han population in southwestern China. J. Int. Med. Res. 2020, 48, 300060520926033. [Google Scholar] [CrossRef]
- Henke, M.T.; Kenny, D.J.; Cassilly, C.D.; Vlamakis, H.; Xavier, R.J.; Clardy, J. Ruminococcus gnavus, a member of the human gut microbiome associated with Crohn’s disease, produces an inflammatory polysaccharide. Proc. Natl. Acad. Sci. USA 2019, 116, 12672–12677. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.D.; Chen, C.Y.; Knox, N.C.; Marrie, R.A.; El-Gabalawy, H.; de Kievit, T.; Alfa, M.; Bernstein, C.N.; Van Domselaar, G. A comparative study of the gut microbiota in immune-mediated inflammatory diseases-does a common dysbiosis exist? Microbiome 2018, 6, 221. [Google Scholar] [CrossRef]
- Chung, S.; Barnes, J.L.; Astroth, K.S. Gastrointestinal microbiota in patients with chronic kidney disease: A systematic review. Adv. Nutr. 2019, 10, 888–901. [Google Scholar] [CrossRef]
- Chen, Y.J.; Wu, H.; Wu, S.D.; Lu, N.; Wang, Y.T.; Liu, H.N.; Dong, L.; Liu, T.T.; Shen, X.Z. Parasutterella, in association with irritable bowel syndrome and intestinal chronic inflammation. J. Gastroenterol. Hepatol. 2018, 33, 1844–1852. [Google Scholar] [CrossRef]
- Iwashita, Y.; Ohya, M.; Yashiro, M.; Sonou, T.; Kawakami, K.; Nakashima, Y.; Yano, T.; Iwashita, Y.; Mima, T.; Negi, S.; et al. Dietary changes involving bifidobacterium longum and other nutrients delays chronic kidney disease progression. Am. J. Nephrol. 2018, 47, 325–332. [Google Scholar] [CrossRef]
- Stearns-Kurosawa, D.J.; Osuchowski, M.F.; Valentine, C.; Kurosawa, S.; Remick, D.G. The pathogenesis of sepsis. Annu. Rev. Pathol. 2011, 6, 19–48. [Google Scholar] [CrossRef] [PubMed]
- Noce, A.; Marchetti, M.; Marrone, G.; Di Renzo, L.; Di Lauro, M.; Di Daniele, F.; Albanese, M.; Di Daniele, N.; De Lorenzo, A. Link between gut microbiota dysbiosis and chronic kidney disease. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 2057–2074. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.R.; Wang, Z.T.; Sun, J.J.; Yang, Y.Y.; Li, X.X.; Wang, X.R.; Shi, Y.; Zhu, Y.Y.; Wang, R.T.; Wang, M.N.; et al. Gut microbiota and diabetic kidney diseases: Pathogenesis and therapeutic perspectives. World J. Diabetes 2022, 13, 308–318. [Google Scholar] [CrossRef]
- Corb Aron, R.A.; Abid, A.; Vesa, C.M.; Nechifor, A.C.; Behl, T.; Ghitea, T.C.; Munteanu, M.A.; Fratila, O.; Andronie-Cioara, F.L.; Toma, M.M.; et al. Recognizing the benefits of pre-/probiotics in metabolic syndrome and type 2 diabetes mellitus considering the influence of Akkermansia muciniphila as a key gut bacterium. Microorganisms 2021, 9, 618. [Google Scholar] [CrossRef] [PubMed]
- Aronov, P.A.; Luo, F.J.; Plummer, N.S.; Quan, Z.; Holmes, S.; Hostetter, T.H.; Meyer, T.W. Colonic contribution to uremic solutes. J. Am. Soc. Nephrol. 2011, 22, 1769–1776. [Google Scholar] [CrossRef]
- Ramezani, A.; Raj, D.S. The gut microbiome, kidney disease, and targeted interventions. J. Am. Soc. Nephrol. 2014, 25, 657–670. [Google Scholar] [CrossRef]
- Rysz, J.; Franczyk, B.; Lawinski, J.; Olszewski, R.; Cialkowska-Rysz, A.; Gluba-Brzozka, A. The impact of CKD on uremic toxins and gut microbiota. Toxins 2021, 13, 252. [Google Scholar] [CrossRef]
- Dou, L.; Bertrand, E.; Cerini, C.; Faure, V.; Sampol, J.; Vanholder, R.; Berland, Y.; Brunet, P. The uremic solutes p-cresol and indoxyl sulfate inhibit endothelial proliferation and wound repair. Kidney Int. 2004, 65, 442–451. [Google Scholar] [CrossRef]
- Cao, X.S.; Chen, J.; Zou, J.Z.; Zhong, Y.H.; Teng, J.; Ji, J.; Chen, Z.W.; Liu, Z.H.; Shen, B.; Nie, Y.X.; et al. Association of indoxyl sulfate with heart failure among patients on hemodialysis. Clin. J. Am. Soc. Nephrol. 2015, 10, 111–119. [Google Scholar] [CrossRef]
- Vanholder, R.; Schepers, E.; Pletinck, A.; Nagler, E.V.; Glorieux, G. The uremic toxicity of indoxyl sulfate and p-cresyl sulfate: A systematic review. J. Am. Soc. Nephrol. 2014, 25, 1897–1907. [Google Scholar] [CrossRef]
- Wu, I.W.; Hsu, K.H.; Lee, C.C.; Sun, C.Y.; Hsu, H.J.; Tsai, C.J.; Tzen, C.Y.; Wang, Y.C.; Lin, C.Y.; Wu, M.S. p-Cresyl sulphate and indoxyl sulphate predict progression of chronic kidney disease. Nephrol. Dial. Transplant. 2011, 26, 938–947. [Google Scholar] [CrossRef] [PubMed]
- Lutz, W. A uremic peptide containing polyamine: Formation and possible role in uremic hypertriglyceridemia. Physiol. Chem. Phys. 1980, 12, 451–456. [Google Scholar] [PubMed]
- Farhadian, S.; Shareghi, B.; Saboury, A.A.; Babaheydari, A.K.; Raisi, F.; Heidari, E. Molecular aspects of the interaction of spermidine and alpha-chymotrypsin. Int. J. Biol. Macromol. 2016, 92, 523–532. [Google Scholar] [CrossRef]
- Dufour, C.; Dandrifosse, G.; Forget, P.; Vermesse, F.; Romain, N.; Lepoint, P. Spermine and spermidine induce intestinal maturation in the rat. Gastroenterology 1988, 95, 112–116. [Google Scholar] [CrossRef]
- Tang, W.H.; Wang, Z.; Kennedy, D.J.; Wu, Y.; Buffa, J.A.; Agatisa-Boyle, B.; Li, X.S.; Levison, B.S.; Hazen, S.L. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ. Res. 2015, 116, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, J.R.; House, J.A.; Ocque, A.J.; Zhang, S.; Johnson, C.; Kimber, C.; Schmidt, K.; Gupta, A.; Wetmore, J.B.; Nolin, T.D.; et al. Serum trimethylamine-N-oxide is elevated in CKD and correlates with coronary atherosclerosis burden. J. Am. Soc. Nephrol. 2016, 27, 305–313. [Google Scholar] [CrossRef]
- Santa Maria, J.; Vallance, P.; Charles, I.G.; Leiper, J.M. Identification of microbial dimethylarginine dimethylaminohydrolase enzymes. Mol. Microbiol. 1999, 33, 1278–1279. [Google Scholar] [CrossRef]
- Fiedler, L. The DDAH/ADMA pathway is a critical regulator of NO signalling in vascular homeostasis. Cell Adhes. Migr. 2008, 2, 149–150. [Google Scholar] [CrossRef][Green Version]
- Meinitzer, A.; Seelhorst, U.; Wellnitz, B.; Halwachs-Baumann, G.; Boehm, B.O.; Winkelmann, B.R.; Marz, W. Asymmetrical dimethylarginine independently predicts total and cardiovascular mortality in individuals with angiographic coronary artery disease (the Ludwigshafen Risk and Cardiovascular Health study). Clin. Chem. 2007, 53, 273–283. [Google Scholar] [CrossRef]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef]
- He, S.; Jiang, H.; Zhuo, C.; Jiang, W. Trimethylamine/trimethylamine-N-oxide as a key between diet and cardiovascular diseases. Cardiovasc. Toxicol. 2021, 21, 593–604. [Google Scholar] [CrossRef]
- Rath, S.; Rud, T.; Pieper, D.H.; Vital, M. Potential TMA-producing bacteria are ubiquitously found in mammalia. Front. Microbiol. 2019, 10, 2966. [Google Scholar] [CrossRef]
- Rath, S.; Heidrich, B.; Pieper, D.H.; Vital, M. Uncovering the trimethylamine-producing bacteria of the human gut microbiota. Microbiome 2017, 5, 54. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Y.; Ke, B.; Du, J. TMAO: How gut microbiota contributes to heart failure. Transl. Res. 2021, 228, 109–125. [Google Scholar] [CrossRef] [PubMed]
- Barr, W.G. Uric acid. In Clinical Methods: The History, Physical, and Laboratory Examinations, 3rd ed.; Walker, H.K., Hall, W.D., Hurst, J.W., Eds.; Butterworths: Boston, MA, USA, 1990. [Google Scholar]
- Ramezani, A.; Massy, Z.A.; Meijers, B.; Evenepoel, P.; Vanholder, R.; Raj, D.S. Role of the gut microbiome in uremia: A potential therapeutic target. Am. J. Kidney Dis. 2016, 67, 483–498. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhuang, J.; Tang, P.; Li, J.; Xiong, X.; Deng, H. The role of the gut microbiota in coronary heart disease. Curr. Atheroscler. Rep. 2020, 22, 77. [Google Scholar] [CrossRef]
- Xu, X.; Wang, H.; Guo, D.; Man, X.; Liu, J.; Li, J.; Luo, C.; Zhang, M.; Zhen, L.; Liu, X. Curcumin modulates gut microbiota and improves renal function in rats with uric acid nephropathy. Ren. Fail. 2021, 43, 1063–1075. [Google Scholar] [CrossRef]
- D’Apolito, M.; Du, X.; Zong, H.; Catucci, A.; Maiuri, L.; Trivisano, T.; Pettoello-Mantovani, M.; Campanozzi, A.; Raia, V.; Pessin, J.E.; et al. Urea-induced ROS generation causes insulin resistance in mice with chronic renal failure. J. Clin. Investig. 2010, 120, 203–213. [Google Scholar] [CrossRef]
- Koppe, L.; Nyam, E.; Vivot, K.; Manning Fox, J.E.; Dai, X.Q.; Nguyen, B.N.; Trudel, D.; Attane, C.; Moulle, V.S.; MacDonald, P.E.; et al. Urea impairs beta cell glycolysis and insulin secretion in chronic kidney disease. J. Clin. Investig. 2016, 126, 3598–3612. [Google Scholar] [CrossRef]
- D’Apolito, M.; Du, X.; Pisanelli, D.; Pettoello-Mantovani, M.; Campanozzi, A.; Giacco, F.; Maffione, A.B.; Colia, A.L.; Brownlee, M.; Giardino, I. Urea-induced ROS cause endothelial dysfunction in chronic renal failure. Atherosclerosis 2015, 239, 393–400. [Google Scholar] [CrossRef]
- Graboski, A.L.; Redinbo, M.R. Gut-derived protein-bound uremic toxins. Toxins 2020, 12, 590. [Google Scholar] [CrossRef] [PubMed]
- Rocchetti, M.T.; Cosola, C.; Ranieri, E.; Gesualdo, L. Protein-bound uremic toxins and immunity. Methods Mol. Biol. 2021, 2325, 215–227. [Google Scholar] [CrossRef] [PubMed]
- de Vos, W.M.; Tilg, H.; Van Hul, M.; Cani, P.D. Gut microbiome and health: Mechanistic insights. Gut 2022, 71, 1020–1032. [Google Scholar] [CrossRef]
- Yabuuchi, J.; Ueda, S.; Yamagishi, S.I.; Nohara, N.; Nagasawa, H.; Wakabayashi, K.; Matsui, T.; Yuichiro, H.; Kadoguchi, T.; Otsuka, T.; et al. Association of advanced glycation end products with sarcopenia and frailty in chronic kidney disease. Sci. Rep. 2020, 10, 17647. [Google Scholar] [CrossRef]
- Mallipattu, S.K.; Uribarri, J. Advanced glycation end product accumulation: A new enemy to target in chronic kidney disease? Curr. Opin. Nephrol. Hypertens. 2014, 23, 547–554. [Google Scholar] [CrossRef]
- Sasai, Y.; Iwakawa, K.; Yanagida, K.; Shen, Y.; Hosono, T.; Ariga, T.; Seki, T. Advanced glycation endproducts stimulate renal epithelial cells to release chemokines that recruit macrophages, leading to renal fibrosis. Biosci. Biotechnol. Biochem. 2012, 76, 1741–1745. [Google Scholar] [CrossRef][Green Version]
- Gryp, T.; Vanholder, R.; Vaneechoutte, M.; Glorieux, G. p-Cresyl Sulfate. Toxins 2017, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Qayed, M.; Michonneau, D.; Socie, G.; Waller, E.K. Indole derivatives, microbiome and graft versus host disease. Curr. Opin. Immunol. 2021, 70, 40–47. [Google Scholar] [CrossRef]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9, 3294. [Google Scholar] [CrossRef]
- Leong, S.C.; Sirich, T.L. Indoxyl sulfate—Review of toxicity and therapeutic strategies. Toxins 2016, 8, 358. [Google Scholar] [CrossRef]
- Lu, C.L.; Zheng, C.M.; Lu, K.C.; Liao, M.T.; Wu, K.L.; Ma, M.C. Indoxyl-sulfate-induced redox imbalance in chronic kidney disease. Antioxidants 2021, 10, 936. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Watanabe, H.; Imafuku, T.; Tokumaru, K.; Fujita, I.; Arimura, N.; Maeda, H.; Tanaka, M.; Matsushita, K.; Fukagawa, M.; et al. Indoxyl sulfate contributes to mTORC1-induced renal fibrosis via the OAT/NADPH Oxidase/ROS pathway. Toxins 2021, 13, 909. [Google Scholar] [CrossRef]
- Gryp, T.; Huys, G.R.B.; Joossens, M.; Van Biesen, W.; Glorieux, G.; Vaneechoutte, M. Isolation and quantification of uremic toxin precursor-generating gut bacteria in chronic kidney disease patients. Int. J. Mol. Sci. 2020, 21, 1986. [Google Scholar] [CrossRef]
- Zelante, T.; Puccetti, M.; Giovagnoli, S.; Romani, L. Regulation of host physiology and immunity by microbial indole-3-aldehyde. Curr. Opin. Immunol. 2021, 70, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Wesoly, R.; Weiler, U. Nutritional influences on skatole formation and skatole metabolism in the pig. Animals 2012, 2, 221–242. [Google Scholar] [CrossRef]
- Dou, L.; Sallee, M.; Cerini, C.; Poitevin, S.; Gondouin, B.; Jourde-Chiche, N.; Fallague, K.; Brunet, P.; Calaf, R.; Dussol, B.; et al. The cardiovascular effect of the uremic solute indole-3 acetic acid. J. Am. Soc. Nephrol. 2015, 26, 876–887. [Google Scholar] [CrossRef]
- Kaur, H.; Bose, C.; Mande, S.S. Tryptophan metabolism by gut microbiome and gut-brain-axis: An in silico analysis. Front. Neurosci. 2019, 13, 1365. [Google Scholar] [CrossRef]
- Cernaro, V.; Loddo, S.; Macaione, V.; Ferlazzo, V.T.; Cigala, R.M.; Crea, F.; De Stefano, C.; Genovese, A.R.R.; Gembillo, G.; Bolignano, D.; et al. RAS inhibition modulates kynurenine levels in a CKD population with and without type 2 diabetes mellitus. Int. Urol. Nephrol. 2020, 52, 1125–1133. [Google Scholar] [CrossRef]
- Robinson, C.M.; Hale, P.T.; Carlin, J.M. The role of IFN-gamma and TNF-alpha-responsive regulatory elements in the synergistic induction of indoleamine dioxygenase. J. Interferon Cytokine Res. 2005, 25, 20–30. [Google Scholar] [CrossRef]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [PubMed]
- Russell, W.R.; Duncan, S.H.; Scobbie, L.; Duncan, G.; Cantlay, L.; Calder, A.G.; Anderson, S.E.; Flint, H.J. Major phenylpropanoid-derived metabolites in the human gut can arise from microbial fermentation of protein. Mol. Nutr. Food Res. 2013, 57, 523–535. [Google Scholar] [CrossRef]
- Jankowski, J.; van der Giet, M.; Jankowski, V.; Schmidt, S.; Hemeier, M.; Mahn, B.; Giebing, G.; Tolle, M.; Luftmann, H.; Schluter, H.; et al. Increased plasma phenylacetic acid in patients with end-stage renal failure inhibits iNOS expression. J. Clin. Investig. 2003, 112, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Westhoff, T.H.; Krauser, P.; Ignatius, R.; Jankowski, J.; Jankowski, V.; Zidek, W.; van der Giet, M. The uraemic toxin phenylacetic acid impairs macrophage function. Nephrol. Dial. Transplant. 2008, 23, 3485–3493. [Google Scholar] [CrossRef] [PubMed]
- Wolley, M.; Jardine, M.; Hutchison, C.A. Exploring the clinical relevance of providing increased removal of large middle molecules. Clin. J. Am. Soc. Nephrol. 2018, 13, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Bach, M.; Asquith, M.; Lee, A.Y.; Akileswaran, L.; Stauffer, P.; Davin, S.; Pan, Y.; Cambronne, E.D.; Dorris, M.; et al. HLA-B27 and human beta2-microglobulin affect the gut microbiota of transgenic rats. PLoS ONE 2014, 9, e105684. [Google Scholar] [CrossRef]
- Chiou, S.J.; Wang, C.C.; Tseng, Y.S.; Lee, Y.J.; Chen, S.C.; Chou, C.H.; Chuang, L.Y.; Hong, Y.R.; Lu, C.Y.; Chiu, C.C.; et al. A novel role for beta2-microglobulin: A precursor of antibacterial chemokine in respiratory epithelial cells. Sci. Rep. 2016, 6, 31035. [Google Scholar] [CrossRef]
- Musial, K.; Zwolinska, D. New markers of cell migration and inflammation in children with chronic kidney disease. Biomarkers 2019, 24, 295–302. [Google Scholar] [CrossRef]
- Winchester, J.F.; Salsberg, J.A.; Levin, N.W. Beta-2 microglobulin in ESRD: An in-depth review. Adv. Ren. Replace. Ther. 2003, 10, 279–309. [Google Scholar] [CrossRef]
- Corlin, D.B.; Heegaard, N.H. Beta(2)-microglobulin amyloidosis. Subcell. Biochem. 2012, 65, 517–540. [Google Scholar] [CrossRef]
- Goltzman, D. Physiology of parathyroid hormone. Endocrinol. Metab. Clin. N. Am. 2018, 47, 743–758. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Yu, M.; Pal, S.; Tyagi, A.M.; Dar, H.; Adams, J.; Weitzmann, M.N.; Jones, R.M.; Pacifici, R. Parathyroid hormone-dependent bone formation requires butyrate production by intestinal microbiota. J. Clin. Investig. 2020, 130, 1767–1781. [Google Scholar] [CrossRef]
- Kermgard, E.; Chawla, N.K.; Wesseling-Perry, K. Gut microbiome, parathyroid hormone, and bone. Curr. Opin. Nephrol. Hypertens. 2021, 30, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Hruska, K.A.; Sugatani, T.; Agapova, O.; Fang, Y. The chronic kidney disease—Mineral bone disorder (CKD-MBD): Advances in pathophysiology. Bone 2017, 100, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Campbell, K.L.; Johnson, D.W.; Stanton, T.; Vesey, D.A.; Coombes, J.S.; Weston, K.S.; Hawley, C.M.; McWhinney, B.C.; Ungerer, J.P.; et al. Protein-bound uremic toxins, inflammation and oxidative stress: A cross-sectional study in stage 3–4 chronic kidney disease. Arch. Med. Res. 2014, 45, 309–317. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, P.; Jiang, H.H.; Cheng, S. Gut bacterial translocation contributes to microinflammation in experimental uremia. Dig. Dis. Sci. 2012, 57, 2856–2862. [Google Scholar] [CrossRef]
- Stenvinkel, P. Inflammation in end-stage renal disease—A fire that burns within. Contrib. Nephrol. 2005, 149, 185–199. [Google Scholar] [CrossRef]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef]
- Clarke, T.B.; Davis, K.M.; Lysenko, E.S.; Zhou, A.Y.; Yu, Y.; Weiser, J.N. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat. Med. 2010, 16, 228–231. [Google Scholar] [CrossRef]
- Vlacil, A.K.; Schuett, J.; Ruppert, V.; Soufi, M.; Oberoi, R.; Shahin, K.; Wachter, C.; Tschernig, T.; Lei, Y.; Liu, F.; et al. Deficiency of Nucleotide-binding oligomerization domain-containing proteins (NOD) 1 and 2 reduces atherosclerosis. Basic Res. Cardiol. 2020, 115, 47. [Google Scholar] [CrossRef]
- Kim, M.H.; Kang, S.G.; Park, J.H.; Yanagisawa, M.; Kim, C.H. Short-chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice. Gastroenterology 2013, 145, 396–406.e10. [Google Scholar] [CrossRef] [PubMed]
- Vinolo, M.A.; Rodrigues, H.G.; Festuccia, W.T.; Crisma, A.R.; Alves, V.S.; Martins, A.R.; Amaral, C.L.; Fiamoncini, J.; Hirabara, S.M.; Sato, F.T.; et al. Tributyrin attenuates obesity-associated inflammation and insulin resistance in high-fat-fed mice. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E272–E282. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Zhao, Y.Y.; Pahl, M.V. Altered intestinal microbial flora and impaired epithelial barrier structure and function in CKD: The nature, mechanisms, consequences and potential treatment. Nephrol. Dial. Transplant. 2016, 31, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D. CKD impairs barrier function and alters microbial flora of the intestine: A major link to inflammation and uremic toxicity. Curr. Opin. Nephrol. Hypertens. 2012, 21, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Piceno, Y.M.; DeSantis, T.Z.; Pahl, M.; Andersen, G.L.; Vaziri, N.D. Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am. J. Nephrol. 2014, 39, 230–237. [Google Scholar] [CrossRef]
- Konieczna, I.; Zarnowiec, P.; Kwinkowski, M.; Kolesinska, B.; Fraczyk, J.; Kaminski, Z.; Kaca, W. Bacterial urease and its role in long-lasting human diseases. Curr. Protein Pept. Sci. 2012, 13, 789–806. [Google Scholar] [CrossRef]
- Marcus, E.A.; Vagin, O.; Tokhtaeva, E.; Sachs, G.; Scott, D.R. Helicobacter pylori impedes acid-induced tightening of gastric epithelial junctions. Am. J. Physiol.-Gastrointest. Liver Physiol. 2013, 305, G731–G739. [Google Scholar] [CrossRef]
- Roxas, J.L.; Viswanathan, V.K. Modulation of intestinal paracellular transport by bacterial pathogens. Compr. Physiol. 2018, 8, 823–842. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Yuan, J.; Norris, K. Role of urea in intestinal barrier dysfunction and disruption of epithelial tight junction in chronic kidney disease. Am. J. Nephrol 2013, 37, 1–6. [Google Scholar] [CrossRef]
- Lau, W.L.; Chang, Y.; Vaziri, N.D. The consequences of altered microbiota in immune-related chronic kidney disease. Nephrol. Dial. Transplant. 2021, 36, 1791–1798. [Google Scholar] [CrossRef]
- Wang, H.B.; Wang, P.Y.; Wang, X.; Wan, Y.L.; Liu, Y.C. Butyrate enhances intestinal epithelial barrier function via up-regulation of tight junction protein Claudin-1 transcription. Dig. Dis. Sci. 2012, 57, 3126–3135. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Krieg, R.; Massey, H.D.; Carl, D.; Ghosh, S.; Gehr, T.W.B.; Ghosh, S.S. Sodium butyrate ameliorates insulin resistance and renal failure in CKD rats by modulating intestinal permeability and mucin expression. Nephrol. Dial. Transplant. 2019, 34, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Ikee, R.; Sasaki, N.; Yasuda, T.; Fukazawa, S. Chronic kidney disease, gut dysbiosis, and constipation: A burdensome triplet. Microorganisms 2020, 8, 1862. [Google Scholar] [CrossRef]
- O’Callaghan, A.A.; Dempsey, E.; Iyer, N.; Stiegeler, S.; Mercurio, K.; Corr, S.C. Intestinal metabolites influence macrophage phagocytosis and clearance of bacterial infection. Front. Cell Infect. Microbiol. 2021, 11, 622491. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Liu, H.; Jiang, H.; Wei, M.; Liang, S.; Wang, M.; Shi, K.; He, Q. Macrophages are involved in gut bacterial translocation and reversed by lactobacillus in experimental uremia. Dig. Dis. Sci. 2016, 61, 1534–1544. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, C.W.; Harrison, L.E.; Eldehni, M.T.; Jefferies, H.J.; Szeto, C.C.; John, S.G.; Sigrist, M.K.; Burton, J.O.; Hothi, D.; Korsheed, S.; et al. Circulating endotoxemia: A novel factor in systemic inflammation and cardiovascular disease in chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 133–141. [Google Scholar] [CrossRef]
- Hung, S.C.; Kuo, K.L.; Peng, C.H.; Wu, C.H.; Wang, Y.C.; Tarng, D.C. Association of fluid retention with anemia and clinical outcomes among patients with chronic kidney disease. J. Am. Heart Assoc. 2015, 4, e001480. [Google Scholar] [CrossRef]
- Meijers, B.; Evenepoel, P.; Anders, H.J. Intestinal microbiome and fitness in kidney disease. Nat. Rev. Nephrol. 2019, 15, 531–545. [Google Scholar] [CrossRef]
- Caplin, B.; Kumar, S.; Davenport, A. Patients’ perspective of haemodialysis-associated symptoms. Nephrol. Dial. Transplant. 2011, 26, 2656–2663. [Google Scholar] [CrossRef]
- Rossi, U.G.; Petrocelli, F.; Seitun, S.; Ferro, C. Nonocclusive mesenteric ischemia in a dialysis patient with extensive vascular calcification. Am. J. Kidney Dis. 2012, 60, 843–846. [Google Scholar] [CrossRef]
- Blikslager, A.T.; Moeser, A.J.; Gookin, J.L.; Jones, S.L.; Odle, J. Restoration of barrier function in injured intestinal mucosa. Physiol. Rev. 2007, 87, 545–564. [Google Scholar] [CrossRef] [PubMed]
- March, D.S.; Graham-Brown, M.P.; Stover, C.M.; Bishop, N.C.; Burton, J.O. Intestinal Barrier Disturbances in Haemodialysis Patients: Mechanisms, Consequences, and Therapeutic Options. Biomed. Res. Int. 2017, 2017, 5765417. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phyla | Prominent Members | Functions |
|---|---|---|
| Firmicutes | Ruminococcus, Clostridium, Lactobacillus, Anaerostipes, Eubacterium, Faecalibacterium, and Roseburia | - Break down complex carbohydrates in the gut that cannot be digested by endogenous enzymes [7] - SCFA production [8] - Production of antimicrobial, anti-carcinogenic and anti-inflammatory molecules and peptides [7] - Increase in firmicutes coincides with obesity [14] |
| Bacteroidetes | Bacteroides, Prevotella, Clostridiales and Xylanibacter | - Ferment complex carbohydrates and produce volatile fatty acids which are a source of energy for the host [9] - Promote the growth of mutualistic bacteria upon high-fiber consumption [11] - Metabolic changes in the microbiota, leading to reduced IL 18 production, mucosal inflammation, and potential systemic autoimmunity [12] - An increase in Bacteroidetes coincides with inflammatory bowel disease [14] |
| Actinobacteria | Bifidobacteria | - Essential for gut homeostasis - Probiotic [20] |
| Proteobacteria | Escherichia coli and Salmonella | - A dysbiotic increase leads to a compromised gut microbiota and inflammation [21] |
| Verrucomicrobia | Akkermansia muciniphila | - Improves gut barrier function and has anti-inflammatory properties [22] |
| Phyla | Changes in Microbes | Changes in Functions |
|---|---|---|
| Firmicutes | Lower Lactobacillus, Roseburia, Faecalibacterium, Prevotella, Gemmiger, Lachnospira, and Sporobacter Increase in Streptococcus, Clostridium III, Faecalicoccus | - Lower Lactobacillus is associated with hypertension and linked to adverse outcomes in patients with CKD [42]. - Less production of butyrate a compound known to protect kidneys [44]. - Ruminococcus promotes inflammatory bowel syndrome, produces inflammatory polysaccharides such as glucorhamnan [47]. - Higher protease production, lower saccharolysis [53]. - Leading to damaged local gastrointestinal tract function and aggravating inflammation [49]. |
| Bacteroidetes | Minor differences in Paraprevotella Alloprevotella | - Lower levels of Bacteroidetes are associated with lower SCFA production [16]. |
| Actinobacteria | Decrease in Bifidobacteria Increase in Eggerthella lenta and Actinomyces | - Supplementing Bifidobacterium reduced serum creatinine, urea nitrogen, and p-cresyl sulfate [51]. |
| Proteobacteria | Increase in Escherichia, Shigella, Desulfovibrio | - Inflammatory response, alteration of gut mucosal permeability and increasing the cell ratio of intestinal T helper 17 cells to T regulatory cells and promoting the LPS translocation [41]. |
| Verrucomicrobia | Decrease in Akkermansia muciniphila | - Proportionate reduction in functions such as gut-barrier function and thickness of the mucus [4] and the detoxification of hydrogen sulfide [39], the growth of bacteria-producing SCFAs and energy produced as a result of mucus degradation [40]. |
| Uremic Metabolite Class | Representative Molecules | Potential Microbes Involved |
|---|---|---|
| Small water-soluble molecules | Asymmetric dimethylarginine | Streptomyces coelicolor, Mycobacterium tuberculosis from the phylum Actinobacteria and Pseudomonas aeruginosa from the phylum Proteobacteria among others [65] |
| Trimethylamine-N-oxide | Clostridium XIVa strains, Eubacterium sp. strain AB3007 from the phylum Firmicutes and Gammaproteobacteria from the phylum Proteobacteria are likely candidates [67] | |
| Urea | Utilized by E. coli from the phylum Proteobacteria [71]. Escherichia-Shigella from the phylum Proteobacteria and Bacteroides from the phylum Bacteroidetes increase with the increase in urea [72]. | |
| Protein-bound uremic toxins | Advanced glycation end products | Utilized by the gut microbes E. coli from the phylum Proteobacteria, Intestinimonas spp from the phylum Firmicutes and Cloacibacillus from the phylum Synergistota and potentially Oscillibacter spp from the phylum Firmicutes [78] |
| P-cresyl sulfate | Bacteroidaceae, Clostridiaceae, Enterobacteriaceae, Ruminococcaceae, Veillonellaceae, Lactobacillaceae, Staphylococcaceae, Lachnospiraceae from the phylum Proteobacteria, Bifidobacteriaceae from the phylum Actinobacteria, Enterococcaceae, Eubacteriaceae from the phylum Firmicutes, Porphyromonadaceae from the phylum Bacteroidetes and Fusobacteriaceae from the phylum Fusobacteriota play a role in the production of p-cresyl sulfate [82] | |
| Indoxyl sulfate | Produced by E. coli from the phylum Proteobacteria [87], and Lactobacillus spp. from the phylum Firmicutes [88] | |
| indole acetic acid | Produced by numerous bacteria including E.coli from the phylum Proteobacteria, and is metabolized to Skatole by Clostridium from the phylum Firmicutes, Bacteroides from the phylum Bacteroidetes and to indole-3-aldehyde by Lactobacillus acidophilus, Lactobacillus murinus, Lactobacillus reuteri from the phylum Firmicutes, respectively [89,90,91] | |
| Kynurenines | In silico analysis predicts the phylum Actinobacteria, Firmicutes and Proteobacteria, and genus Bacteroides from the phylum Bacteroidetes, Fusobacteria from the phylum Fusobacteriota as kynurenine-producing gut microbes [97] | |
| Phenylacetic acid | Bacteroides thetaiotaomicron, Bacteroides eggerthii, Bacteroides ovatus, Bacteroides fragilis, Parabacteroides distasonis from the phylum Bacteroidetes and Eubacterium hallii, Clostridium bartlettii from the phylum Firmicutes lead to higher phenylacetic acid production [101] | |
| Middle molecules | β₂-microglobulin | An increase in Prevotella spp. And Bacteroides vulgatus from the phylum Bacteroidetes and a decrease in Rikenellaceae also from the phylum Bacteroidetes [101] |
| Parathyroid hormone | The exact microbes involved have not been established so far [112] |
| Microbes | Role in Inflammation and Immunosuppression |
|---|---|
| Clostridium spp. from the phylum Firmicutes | Induces FoxP3+ regulatory T cells [120] |
| Uncultivated segmented filamentous bacteria of the Clostridia family | Promotes T helper type 17 cell differentiation [120] |
| Akkermansia from the phylum Verrucomicrobiota | Higher levels of IL-10 [40] |
| Lower levels of Lactobacillus from the phylum Firmicutes | Higher levels of IL-4 and IL-10 [40] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhargava, S.; Merckelbach, E.; Noels, H.; Vohra, A.; Jankowski, J. Homeostasis in the Gut Microbiota in Chronic Kidney Disease. Toxins 2022, 14, 648. https://doi.org/10.3390/toxins14100648
Bhargava S, Merckelbach E, Noels H, Vohra A, Jankowski J. Homeostasis in the Gut Microbiota in Chronic Kidney Disease. Toxins. 2022; 14(10):648. https://doi.org/10.3390/toxins14100648
Chicago/Turabian StyleBhargava, Shruti, Erik Merckelbach, Heidi Noels, Ashima Vohra, and Joachim Jankowski. 2022. "Homeostasis in the Gut Microbiota in Chronic Kidney Disease" Toxins 14, no. 10: 648. https://doi.org/10.3390/toxins14100648
APA StyleBhargava, S., Merckelbach, E., Noels, H., Vohra, A., & Jankowski, J. (2022). Homeostasis in the Gut Microbiota in Chronic Kidney Disease. Toxins, 14(10), 648. https://doi.org/10.3390/toxins14100648