Extraction and LC-MS/MS Analysis of Ciguatoxins: A Semi-Targeted Approach Designed for Fish of Unknown Origin



Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CTX Congener | Formula | [M + H − 3H2O]+ | [M + H − 2H2O]+ | [M + H − H2O]+ | [M + H]+ | [M + NH4]+ | [M + Na]+ |
|---|---|---|---|---|---|---|---|
| CTX4A group 1 | |||||||
| CTX4A/B | C60H84O16 | 1007.55152 | 1025.56208 | 1043.57265 | 1061.58321 | 1078.60976 | 1083.56516 |
| M-seco-CTX4A/B | C60H86O17 | 1025.56208 | 1043.57265 | 1061.58321 | 1079.59378 | 1096.62033 | 1101.57572 |
| 52-epi-54-deoxyCTX1B 54-deoxyCTX1B | C60H86O18 | 1041.55700 | 1059.56756 | 1077.57813 | 1095.58869 | 1112.61524 | 1117.57064 |
| CTX1B 52-/54-epiCTX1B 52-epi-54-epiCTX1B 54-deoxy-50-hydroxyCTX1B | C60H86O19 | 1057.55191 | 1075.56248 | 1093.57304 | 1111.58361 | 1128.61016 | 1133.56555 |
| 7-oxoCTX1B | C60H86O20 | 1073.54683 | 1091.55739 | 1109.56796 | 1127.57852 | 1144.60507 | 1149.56047 |
| 7-hydroxyCTX1B | C60H88O20 | 1075.56248 | 1093.57304 | 1111.58361 | 1129.59417 | 1146.62072 | 1151.57612 |
| 4-hydroxy-7-oxoCTX1B | C60H88O21 | 1091.55739 | 1109.56796 | 1127.57852 | 1145.58909 | 1162.61564 | 1167.57103 |
| CTX3C group 1 | |||||||
| CTX3C/B | C57H82O16 | 969.53587 | 987.54643 | 1005.55700 | 1023.56756 | 1040.59411 | 1045.54951 |
| 51-hydroxyCTX3C | C57H82O17 | 985.53078 | 1003.54135 | 1021.55191 | 1039.56248 | 1056.58903 | 1061.54442 |
| M-seco-CTX3C 2-hydroxyCTX3C | C57H84O17 | 987.54643 | 1005.55700 | 1023.56756 | 1041.57813 | 1058.60468 | 1063.56007 |
| M-seco-CTX3C methyl acetal | C58H86O17 | 1001.56208 | 1019.57265 | 1037.58321 | 1055.59378 | 1072.62033 | 1077.57572 |
| 51-hydroxy-2-oxoCTX3C | C57H82O18 | 1001.52570 | 1019.53626 | 1037.54683 | 1055.55739 | 1072.58394 | 1077.53934 |
| 2,3-dihydroxyCTX3C | C57H84O18 | 1003.54135 | 1021.55191 | 1039.56248 | 1057.57304 | 1074.59959 | 1079.55499 |
| A-seco-51-hydroxyCTX3C | C57H86O18 | 1005.55700 | 1023.56756 | 1041.57813 | 1059.58869 | 1076.61524 | 1081.57064 |
| 2,3,51-trihydroxyCTX3C | C57H84O19 | 1019.53626 | 1037.54683 | 1055.55739 | 1073.56796 | 1090.59451 | 1095.54990 |
| C-CTX group 2 | |||||||
| C-CTX-1/2 | C62H92O19 | 1087.59886 | 1105.60943 | 1123.61999 | 1141.63056 | 1158.65711 | 1163.61250 |
| C-CTX-3/4 | C62H94O19 | 1089.61451 | 1107.62508 | 1125.63564 | 1143.64621 | 1160.67276 | 1165.62815 |
| C-CTX reaction product 8 | C61H88O18 | 1055.57265 | 1073.58321 | 1091.59378 | 1109.60434 | 1126.63089 | 1131.58629 |
| C-CTX reaction product 9 | C61H90O18 | 1057.58830 | 1075.59886 | 1093.60943 | 1111.61999 | 1128.64654 | 1133.60194 |
| C-CTX-1127 | C61H90O19 * | 1073.6 | 1091.6 | 1109.6 | 1127.6 | 1144.6 | 1149.6 |
| C-CTX-1157 | C62H92O20 * | 1103.6 | 1121.6 | 1139.6 | 1157.6 | 1174.6 | 1179.6 |
| I-CTX group 3 | |||||||
| I-CTX-1/2 | C62H92O19 | 1087.59886 | 1105.60943 | 1123.61999 | 1141.63056 | 1158.65711 | 1163.61250 |
| I-CTX-3/4 | C62H92O20 | 1103.59378 | 1121.60434 | 1139.61491 | 1157.62547 | 1174.65202 | 1179.60742 |
| I-CTX-5 | C62H90O19 | 1085.58321 | 1103.59378 | 1121.60434 | 1139.61491 | 1156.64146 | 1161.59685 |
| I-CTX-6 | C62H90O20 | 1101.57813 | 1119.58869 | 1137.59926 | 1155.60982 | 1172.63637 | 1177.59177 |
2. Results and Discussion
2.1. Extraction Protocol Development
2.2. Method Validation
2.2.1. Recovery Rates, Matrix Effects, Extraction Efficiency, and Sample Stability
2.2.2. Limit of Detection, Limit of Quantitation, and Linearity
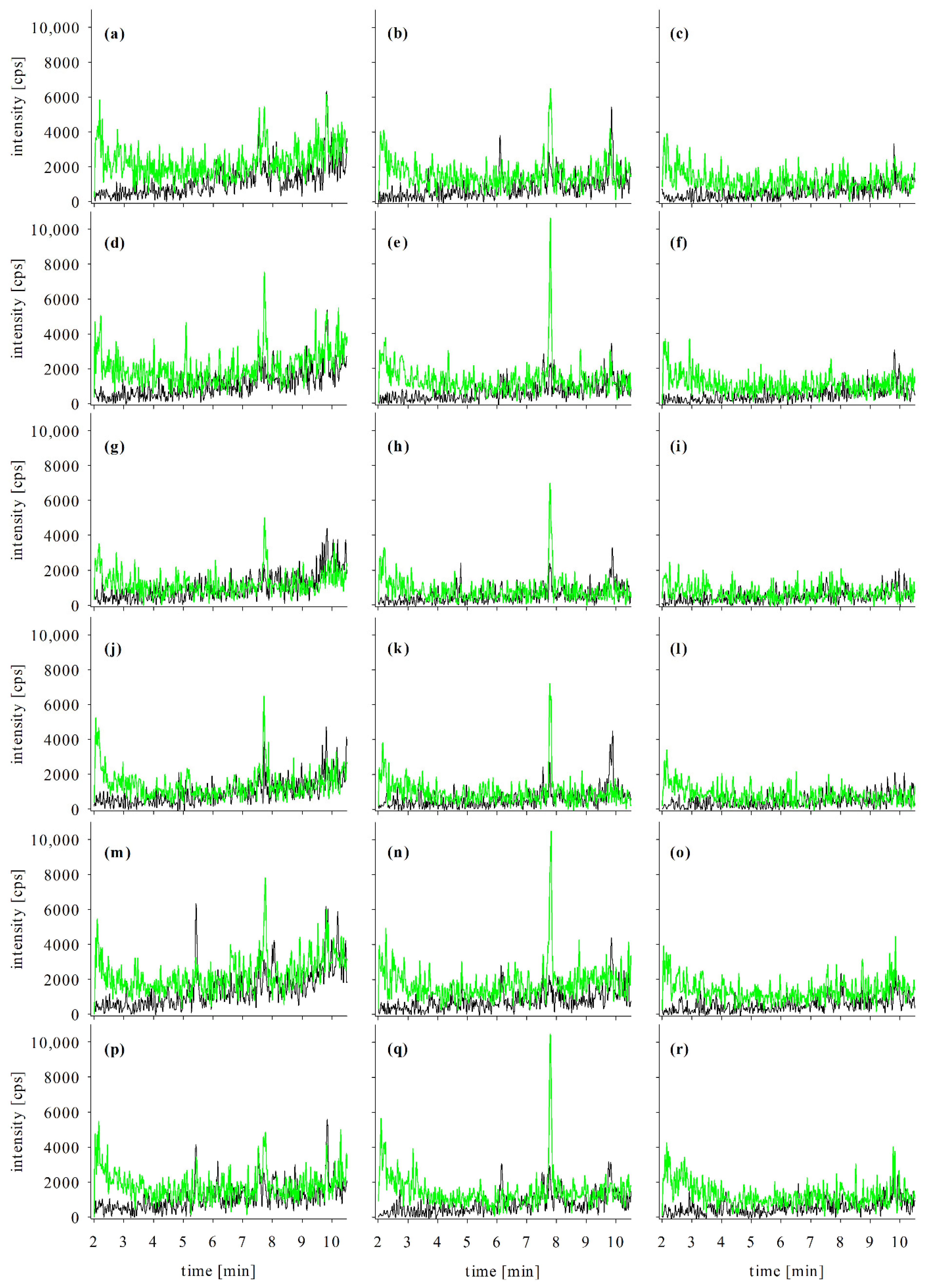
2.2.3. Blank Matrix Samples
2.2.4. Repeatability
2.3. Confirmation Analyses in Naturally Contaminated Samples
2.3.1. Generation and Fragmentation of Ammonium Adducts
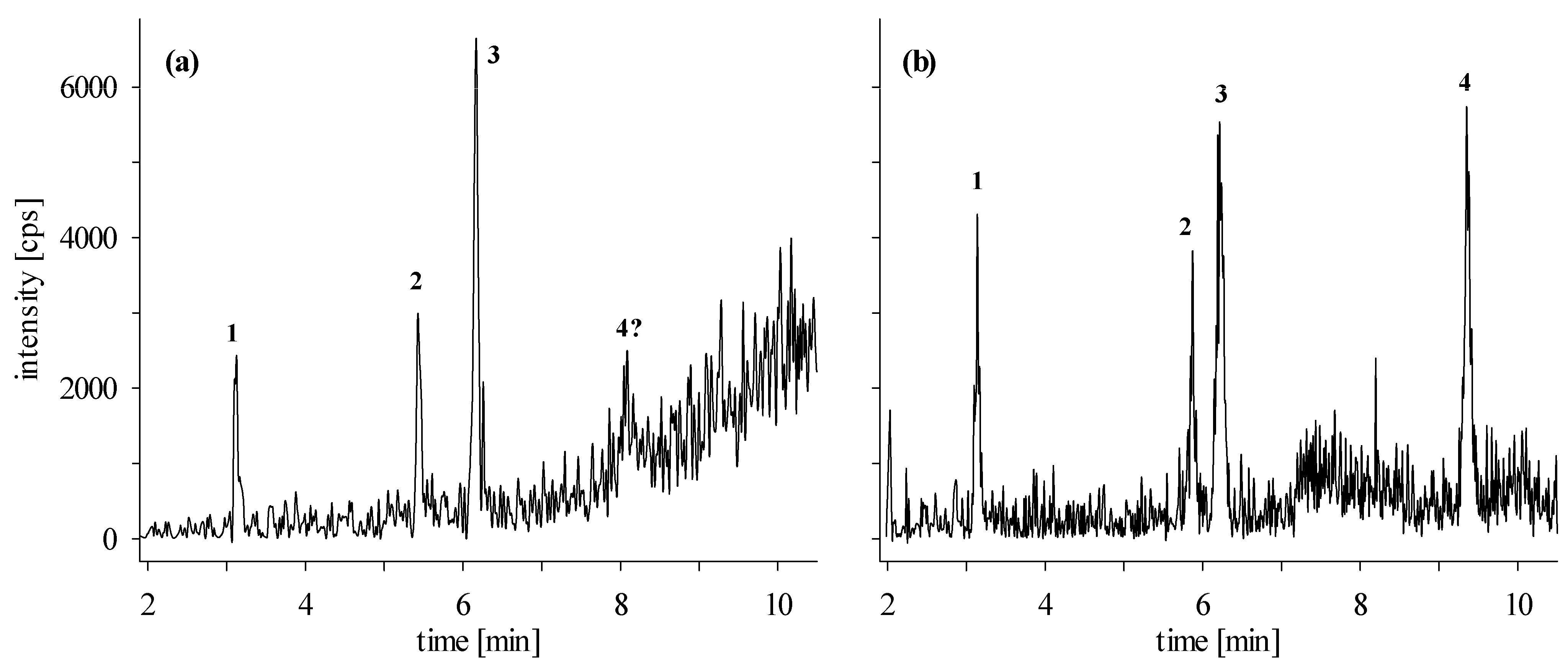
2.3.2. Confirmation Analyses in Naturally Contaminated Samples
2.4. Extract Suitability for In Vitro Assay (N2a-Assay) Analysis
3. Conclusions
4. Materials and Methods
4.1. Reagents and Materials
4.2. Sample Preparation—Enzyme Protocol
4.2.1. Sample Pre-Treatment and Extraction
4.2.2. Defatting
4.2.3. Reversed-Phase (RP) SPE
4.2.4. Normal-Phase (NP) SPE
4.3. LC-MS/MS Analysis
4.3.1. Analysis of Sodium Adducts [M + Na]+
4.3.2. Analysis of Ammonium Adducts [M + NH4]+—Low-Resolution
4.3.3. Analysis of Ammonium Adducts [M + NH4]+—High-Resolution
4.4. Method Validation
4.4.1. Recovery Rates, Matrix Effects, Extraction Efficiency, and Sample Stability
4.4.2. Limit of Detection, Limit of Quantitation, and Linearity
4.4.3. Method Precision—Repeatability
4.5. Extract Performance in the N2a-Assay
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A









| Day | 0 | 6 | 13 | 19 | 26 | 0 | 6 | 13 | 19 | 26 |
|---|---|---|---|---|---|---|---|---|---|---|
| ME | RR | |||||||||
| CTX1B | ||||||||||
| snapper, fillet | 75 ± 4 | 75 ± 2 | 69 ± 3 | 86 ± 9 | 85 ± 4 | 47 ± 2 | 43 ± 1 | 40 ± 6 | 47 ± 5 | 46 ± 2 |
| snapper, freeze-dried | 76 ± 3 | 67 ± 2 | 71 ± 4 | 81 ± 7 | 83 ± 1 | 47 ± 1 | 40 ± 0 | 44 ± 5 | 48 ± 3 | 45 ± 4 |
| parrotfish, fillet | 92 ± 4 | 85 ± 3 | 89 ± 7 | 100 ± 3 | 97 ± 3 | 62 ± 6 | 62 ± 2 | 55 ± 5 | 62 ± 3 | 55 ± 3 |
| parrotfish, freeze-dried | 90 ± 1 | 86 ± 4 | 84 ± 5 | 95 ± 6 | 97 ± 10 | 57 ± 5 | 56 ± 4 | 54 ± 3 | 62 ± 3 | 53 ± 6 |
| grouper, fillet | 66 ± 5 | 54 ± 3 | 61 ± 1 | 77 ± 2 | 62 ± 1 | 39 ± 2 | 36 ± 2 | 36 ± 2 | 41 ± 0 | 34 ± 2 |
| grouper, freeze-dried | 74 ± 4 | 67 ± 4 | 66 ± 5 | 82 ± 5 | 82 ± 8 | 45 ± 2 | 41 ± 3 | 40 ± 2 | 50 ± 3 | 48 ± 1 |
| 52-epi-54-deoxyCTX1B | ||||||||||
| snapper, fillet | 100 ± 5 | 96 ± 2 | 81 ± 11 | 99 ± 6 | 88 ± 10 | 63 ± 1 | 53 ± 4 | 47 ± 6 | 61 ± 5 | 46 ± 3 |
| snapper, freeze-dried | 84 ± 1 | 78 ± 4 | 78 ± 2 | 95 ± 1 | 83 ± 6 | 57 ± 3 | 53 ± 4 | 54 ± 5 | 60 ± 1 | 53 ± 4 |
| parrotfish, fillet | 93 ± 6 | 94 ± 5 | 93 ± 7 | 98 ± 4 | 96 ± 2 | 66 ± 3 | 59 ± 0 | 62 ± 9 | 69 ± 2 | 59 ± 3 |
| parrotfish, freeze-dried | 103 ± 4 | 103 ± 2 | 95 ± 2 | 97 ± 10 | 104 ± 3 | 71 ± 2 | 65 ± 1 | 64 ± 5 | 77 ± 2 | 65 ± 8 |
| grouper, fillet | 70 ± 2 | 60 ± 7 | 57 ± 0 | 70 ± 2 | 62 ± 5 | 35 ± 2 | 33 ± 1 | 33 ± 2 | 43 ± 2 | 35 ± 2 |
| grouper, freeze-dried | 66 ± 7 | 69 ± 1 | 66 ± 10 | 85 ± 6 | 75 ± 4 | 52 ± 2 | 43 ± 3 | 38 ± 1 | 55 ± 3 | 44 ± 2 |
| 54-deoxyCTX1B | ||||||||||
| snapper, fillet | 73 ± 2 | 73 ± 1 | 69 ± 2 | 85 ± 6 | 82 ± 1 | 42 ± 2 | 43 ± 1 | 40 ± 2 | 47 ± 0 | 46 ± 3 |
| snapper, freeze-dried | 79 ± 6 | 68 ± 2 | 65 ± 1 | 84 ± 5 | 88 ± 3 | 48 ± 3 | 45 ± 2 | 47 ± 5 | 55 ± 2 | 49 ± 1 |
| parrotfish, fillet | 90 ± 1 | 83 ± 2 | 84 ± 2 | 97 ± 7 | 104 ± 3 | 64 ± 6 | 64 ± 1 | 58 ± 1 | 67 ± 1 | 58 ± 5 |
| parrotfish, freeze-dried | 93 ± 3 | 87 ± 1 | 89 ± 2 | 96 ± 7 | 101 ± 1 | 69 ± 2 | 66 ± 1 | 61 ± 7 | 72 ± 2 | 64 ± 4 |
| grouper, fillet | 66 ± 3 | 63 ± 2 | 62 ± 4 | 76 ± 2 | 62 ± 13 | 35 ± 1 | 36 ± 2 | 37 ± 3 | 43 ± 1 | 39 ± 3 |
| grouper, freeze-dried | 72 ± 5 | 73 ± 2 | 72 ± 5 | 86 ± 1 | 81 ± 7 | 48 ± 2 | 47 ± 1 | 45 ± 3 | 55 ± 3 | 52 ± 3 |
| CTX3C—filtrate | ||||||||||
| snapper, fillet | 95 ± 6 | 94 ± 3 | 87 ± 12 | 110 ± 6 | 102 ± 3 | 36 ± 6 | 38 ± 4 | 34 ± 2 | 38 ± 1 | 41 ± 2 |
| snapper, freeze-dried | 98 ± 1 | 102 ± 2 | 100 ± 11 | 121 ± 17 | 114 ± 3 | 40 ± 4 | 41 ± 1 | 41 ± 0 | 46 ± 1 | 49 ± 3 |
| parrotfish, fillet | 116 ± 5 | 112 ± 7 | 102 ± 6 | 101 ± 4 | 130 ± 11 | 48 ± 5 | 47 ± 1 | 41 ± 2 | 52 ± 1 | 52 ± 6 |
| parrotfish, freeze-dried | 109 ± 7 | 118 ± 10 | 107 ± 6 | 112 ± 13 | 128 ± 11 | 53 ± 6 | 56 ± 1 | 50 ± 3 | 61 ± 4 | 61 ± 2 |
| grouper, fillet | 85 ± 6 | 90 ± 4 | 84 ± 3 | 100 ± 10 | 98 ± 4 | 40 ± 3 | 38 ± 3 | 35 ± 2 | 40 ± 2 | 42 ± 2 |
| grouper, freeze-dried | 98 ± 4 | 113 ± 4 | 100 ± 4 | 107 ± 10 | 125 ± 10 | 45 ± 5 | 42 ± 7 | 41 ± 3 | 49 ± 3 | 49 ± 2 |
| CTX3C—eluate | ||||||||||
| snapper, fillet | 94 ± 3 | 91 ± 4 | 83 ± 9 | 100 ± 11 | 82 ± 7 | 25 ± 0 | 25 ± 3 | 24 ± 3 | 27 ± 2 | 21 ± 6 |
| snapper, freeze-dried | 95 ± 9 | 88 ± 5 | 88 ± 4 | 100 ± 4 | 93 ± 4 | 28 ± 2 | 28 ± 1 | 26 ± 3 | 27 ± 1 | 24 ± 3 |
| parrotfish, fillet | 104 ± 4 | 102 ± 5 | 101 ± 3 | 107 ± 5 | 97 ± 17 | 35 ± 1 | 32 ± 2 | 29 ± 3 | 31 ± 1 | 27 ± 3 |
| parrotfish, freeze-dried | 99 ± 3 | 101 ± 5 | 96 ± 8 | 105 ± 7 | 93 ± 7 | 35 ± 2 | 34 ± 1 | 31 ± 6 | 32 ± 0 | 26 ± 6 |
| grouper, fillet | 72 ± 5 | 61 ± 4 | 65 ± 5 | 79 ± 6 | 63 ± 1 | 25 ± 1 | 21 ± 1 | 23 ± 1 | 22 ± 3 | 20 ± 1 |
| grouper, freeze-dried | 89 ± 3 | 79 ± 2 | 81 ± 5 | 97 ± 2 | 75 ± 10 | 30 ± 1 | 27 ± 1 | 28 ± 4 | 31 ± 1 | 29 ± 7 |
References
- Bagnis, R.; Chanteau, S.; Chungue, E.; Hurtel, J.M.; Yasumoto, T.; Inoue, A. Origins of ciguatera fish poisoning: A new dinoflagellate, Gambierdiscus toxicus Adachi and Fukuyo, definitively involved as a causal agent. Toxicon 1980, 18, 199–208. [Google Scholar] [CrossRef]
- Chinain, M.; Gatti, C.M.I.; Roué, M.; Darius, H.T. Ciguatera-causing dinoflagellates in the genera Gambierdiscus and Fukuyoa: Distribution, ecophysiology and toxicology. In Dinoflagellates; Subba Rao, D.V., Ed.; Nova Science Publishers, Inc.: New York, NY, USA, 2020; pp. 405–457. [Google Scholar]
- Satake, M.; Ishibashi, Y.; Legrand, A.-M.; Yasumoto, T. Isolation and structure of ciguatoxin-4A, a new ciguatoxin precursor, from cultures of dinoflagellate Gambierdiscus toxicus and parrotfish Scarus gibbus. Biosci. Biotechnol. Biochem. 1996, 60, 2103–2105. [Google Scholar] [CrossRef] [Green Version]
- Satake, M.; Murata, M.; Yasumoto, T. The structure of CTX3C; a ciguatoxin congener isolated from cultured Gambierdiscus toxicus. Tetrahedron. Lett. 1993, 34, 1975–1978. [Google Scholar] [CrossRef]
- Available online: https://pubchem.ncbi.nlm.nih.gov/compound/5311333 (accessed on 26 August 2021).
- Available online: https://pubchem.ncbi.nlm.nih.gov/compound/23427919 (accessed on 26 August 2021).
- FAO; WHO. Report of the Expert Meeting on Ciguatera Poisoning; Food Safety and Quality No. 9; FAO: Rome, Italy, 2020. [Google Scholar] [CrossRef]
- Diogène, J.; Reverté, L.; Rambla-Alegre, M.; del Río, V.; de la Iglesia, P.; Campàs, M.; Palacios, O.; Flores, C.; Caixach, J.; Ralijaona, C.; et al. Identification of ciguatoxins in a shark involved in a fatal food poisoning in the Indian Ocean. Sci. Rep. 2017, 7, 8240. [Google Scholar] [CrossRef]
- Hamilton, B.; Hurbungs, M.; Jones, A.; Lewis, R.J. Multiple ciguatoxins present in Indian Ocean reef fish. Toxicon 2002, 40, 1347–1353. [Google Scholar] [CrossRef]
- Hamilton, B.; Hurbungs, M.; Vernoux, J.-P.; Jones, A.; Lewis, R.J. Isolation and characterisation of Indian Ocean ciguatoxin. Toxicon 2002, 40, 685–693. [Google Scholar] [CrossRef]
- Chinain, M.; Gatti, C.M.i.; Darius, H.T.; Quod, J.P.; Tester, P.A. Ciguatera poisonings: A global review of occurrences and trends. Harmful Algae 2021, 102, 101873. [Google Scholar] [CrossRef]
- Fish and Fishery Products—Hazards and Controls Guidance, 4th ed.; U.S. Food and Drug Administration, Department of Health and Human Services: Collega Park, MD, USA, 2021.
- Dickey, R.W.; Plakas, S.M. Ciguatera: A public health perspective. Toxicon 2010, 56, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Yasumoto, T.; Igarashi, T.; Legrand, A.M.; Cruchet, P.; Chinain, M.; Fujita, T.; Naoki, H. Structural elucidation of ciguatoxin congeners by fast-atom bombardment tandem mass spectroscopy. J. Am. Chem. Soc. 2000, 122, 4988–4989. [Google Scholar] [CrossRef]
- Abraham, A.; Jester, E.L.E.; Granade, H.R.; Plakas, S.M.; Dickey, R.W. Caribbean ciguatoxin profile in raw and cooked fish implicated in ciguatera. Food Chem. 2012, 131, 192–198. [Google Scholar] [CrossRef]
- Kryuchkov, F.; Robertson, A.; Miles, C.O.; Mudge, E.M.; Uhlig, S. LC–HRMS and chemical derivatization strategies for the structure elucidation of Caribbean ciguatoxins: Identification of C-CTX-3 and -4. Mar. Drugs 2020, 18, 182. [Google Scholar] [CrossRef] [Green Version]
- Pottier, I.; Vernoux, J.P.; Jones, A.; Lewis, R.J. Analysis of toxin profiles in three different fish species causing ciguatera fish poisoning in Guadeloupe; French West Indies. Food Addit. Contam. 2002, 19, 1034–1042. [Google Scholar] [CrossRef]
- Regulation (EC) No 853/2004 of the European Parliament and of the Council of 29 April 2004 Laying Down Specific Hygiene Rules for Food of Animal Origin; Last Amendment by Commission Delegated Regulation (EU) 2020/2192 of 7 December 2020; European Union: Brussels, Belgium, 2020.
- Commission Implementing Regulation (EU) 2019/627 of 15 March 2019 Laying Down Uniform Practical Arrangements for the Performance of Official Controls on Products of Animal Origin Intended for Human Consumption in Accordance with Regulation (EU) 2017/625 of the European Parliament and of the Council and Amending Commission Regulation (EC) No 2074/2005 as Regards Official Controls; Last Amendment by Commission Delegated Regulation (EU) 2020/2108 of 16 December 2020; European Union: Brussels, Belgium, 2020.
- Caillaud, A.; de la Iglesia, P.; Darius, H.T.; Pauillac, S.; Aligizaki, K.; Fraga, S.; Chinain, M.; Diogene, J. Update on methodologies available for ciguatoxin determination: Perspectives to confront the onset of ciguatera fish poisoning in Europe. Mar. Drugs 2010, 8, 1838–1907. [Google Scholar] [CrossRef] [PubMed]
- Harwood, D.T.; Murray, S.; Boundy, M.J. Sample Preparation Prior to Marine Toxin Analysis. Comp. Anal. C 2017, 78, 89–136. [Google Scholar] [CrossRef]
- Dickey, R.W. Ciguatera Toxins: Chemistry, Toxicology, and Detection. In Seafood and Freshwater Toxins, 2nd ed.; Botana, L.M., Ed.; CRC Press: Boca Raton, FL, USA, 2008; pp. 479–500. [Google Scholar]
- Nagae, M.; Igarashi, T.; Mizukoshi, K.; Yasumoto, T.; Kuniyoshi, K.; Oshiro, N. Development and validation of an LC-MS/MS method for the ultra-trace analysis of Pacific ciguatoxins in fish. J. AOAC Int. 2021, 1–10. [Google Scholar] [CrossRef]
- Yogi, K.; Sakugawa, S.; Oshiro, N.; Ikehara, T.; Sugiyama, K.; Yasumoto, T. Determination of toxins involved in ciguatera fish poisoning in the Pacific by LC/MS. J. AOAC Int. 2014, 97, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Estevez, P.; Castro, D.; Manuel Leao, J.; Yasumoto, T.; Dickey, R.; Gago-Martinez, A. Implementation of liquid chromatography tandem mass spectrometry for the analysis of ciguatera fish poisoning in contaminated fish samples from Atlantic coasts. Food Chem. 2019, 280, 8–14. [Google Scholar] [CrossRef]
- Dechraoui Bottein, M.-Y.; Wang, Z.; Ramsdell, J.S. Toxicokinetics of the ciguatoxin P-CTX-1 in rats after intraperitoneal or oral administration. Toxicology 2011, 284, 1–6. [Google Scholar] [CrossRef]
- Hahn, S.T.; Capra, M.F.; Walsh, T.P. Ciguatoxin-protein association in skeletal muscle of Spanish mackerel (Scomberomorus commersoni). Toxicon 1992, 30, 843–852. [Google Scholar] [CrossRef]
- Vernoux, J.P.; Lahlou, N.; Abbad el Andaloussi, S.; Riyeche, N.; Magras, L.P. A study of the distribution of ciguatoxin in individual Caribbean fish. Acta Trop. 1985, 42, 225–233. [Google Scholar] [PubMed]
- Kristinsson, H.G.; Rasco, B.A. Fish protein hydrolysates: Production, biochemical, and functional properties. Crit. Rev. Food Sci. 2000, 40, 43–81. [Google Scholar] [CrossRef]
- Oshiro, N.; Tomikawa, T.; Kuniyoshi, K.; Ishikawa, A.; Toyofuku, H.; Kojima, T.; Asakura, H. LC–MS/MS analysis of ciguatoxins revealing the regional and species distinction of fish in the tropical Western Pacific. J. Mar. Sci. Eng. 2021, 9, 299. [Google Scholar] [CrossRef]
- Lewis, R.J.; Yang, A.; Jones, A. Rapid extraction combined with LC-tandem mass spectrometry (CREM-LC/MS/MS) for the determination of ciguatoxins in ciguateric fish flesh. Toxicon 2009, 54, 62–66. [Google Scholar] [CrossRef]
- Stewart, I.; Eaglesham, G.K.; Poole, S.; Graham, G.; Paulo, C.; Wickramasinghe, W.; Sadler, R.; Shaw, G.R. Establishing a public health analytical service based on chemical methods for detecting and quantifying Pacific ciguatoxin in fish samples. Toxicon 2010, 56, 804–812. [Google Scholar] [CrossRef]
- Loeffler, C.R.; Tartaglione, L.; Friedemann, M.; Spielmeyer, A.; Kappenstein, O.; Bodi, D. Ciguatera Mini Review: 21st Century Environmental Challenges and the Interdisciplinary Research Efforts Rising to Meet Them. Int. J. Environ. Res. Public Health 2021, 18, 3027. [Google Scholar] [CrossRef]
- Kaufmann, A.; Butcher, P.; Maden, K.; Walker, S.; Widmer, M. Semi-targeted residue screening in complex matrices with liquid chromatography coupled to high resolution mass spectrometry: Current possibilities and limitations. Analyst 2011, 136, 1898–1909. [Google Scholar] [CrossRef]
- Estevez, P.; Leao, J.M.; Yasumoto, T.; Dickey, R.W.; Gago-Martinez, A. Caribbean Ciguatoxin-1 stability under strongly acidic conditions: Characterisation of a new C-CTX1 methoxy congener. Food Addit. Contam. Part. A 2020, 37, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://pubchem.ncbi.nlm.nih.gov/compound/176 (accessed on 26 August 2021).
- Available online: https://pubchem.ncbi.nlm.nih.gov/compound/284 (accessed on 26 August 2021).
- Trummal, A.; Lipping, L.; Kaljurand, I.; Koppel, I.A.; Leito, I. Acidity of Strong Acids in Water and Dimethyl Sulfoxide. J. Phys. Chem. A 2016, 120, 3663–3669. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.J.; Mak, Y.L.; Murphy, M.B.; Lam, J.C.W.; Chan, W.H.; Wang, M.; Chan, L.L.; Lam, P.K.S. Validation of an accelerated solvent extraction liquid chromatography–tandem mass spectrometry method for Pacific ciguatoxin-1 in fish flesh and comparison with the mouse neuroblastoma assay. Anal. Bioanal. Chem. 2011, 400, 3165–3175. [Google Scholar] [CrossRef] [PubMed]
- Kohli, G.S.; Haslauer, K.; Sarowar, C.; Kretzschmar, A.L.; Boulter, M.; Harwood, D.T.; Laczka, O.; Murray, S.A. Qualitative and quantitative assessment of the presence of ciguatoxin, P-CTX-1B, in Spanish Mackerel (Scomberomorus commerson) from waters in New South Wales (Australia). Toxicol. Rep. 2017, 4, 328–334. [Google Scholar] [CrossRef]
- Castro, D.; Manger, R.; Vilariño, O.; Gago-Martínez, A. Evaluation of matrix issues in the applicability of the Neuro-2a cell based assay on the detection of CTX in fish samples. Toxins 2020, 12, 308. [Google Scholar] [CrossRef]
- AOAC International. Appendix F: Guidelines for Standard Method Performance Requirements. 2016. Available online: https://www.aoac.org/wp-content/uploads/2019/08/app_f.pdf (accessed on 7 September 2021).
- Document Nº SANTE/12682/2019: Analytical Quality Control and Method Validation Procedures for Pesticide Residues Analysis in Food and Feed; Implemented by 1 January 2020; European Union: Brussels, Belgium, 2020.
- Ha, D.V.; Uesugi, A.; Uchida, H.; Ky, P.X.; Minh, D.Q.; Watanabe, R.; Matsushima, R.; Oikawa, H.; Nagai, S.; Iwataki, M.; et al. Identification of causative ciguatoxins in red snappers Lutjanus bohar implicated in ciguatera fish poisonings in Vietnam. Toxins 2018, 10, 420. [Google Scholar] [CrossRef] [Green Version]
- Lehane, L.; Lewis, R.J. Ciguatera: Recent advances but the risk remains. Int. J. Food Microbiol. 2000, 61, 91–125. [Google Scholar] [CrossRef]
- Lewis, R.J.; Vernoux, J.P.; Brereton, I.M. Structure of Caribbean ciguatoxin isolated from Caranx latus. J. Am. Chem. Soc. 1998, 120, 5914–5920. [Google Scholar] [CrossRef]
- Wenzl, T.; Haedrich, J.; Schaechtele, A.; Robouch, P.; Stroka, J. Guidance Document on the Estimation of LOD and LOQ for Measurements in the Field of Contaminants in Feed and Food; EUR 28099; European Union: Luxembourg, 2016; ISBN 978-992-979-61768-61763. [Google Scholar] [CrossRef]
- Sibat, M.; Herrenknecht, C.; Darius, H.T.; Roué, M.; Chinain, M.; Hess, P. Detection of pacific ciguatoxins using liquid chromatography coupled to either low or high resolution mass spectrometry (LC-MS/MS). J. Chromatogr. A 2018, 1571, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Yogi, K.; Oshiro, N.; Inafuku, Y.; Hirama, M.; Yasumoto, T. Detailed LC-MS/MS analysis of ciguatoxins revealing distinct regional and species characteristics in fish and causative alga from the pacific. Anal. Chem. 2011, 83, 8886–8891. [Google Scholar] [CrossRef] [PubMed]
- Estevez, P.; Castro, D.; Pequeño-Valtierra, A.; Leao, J.M.; Vilariño, O.; Diogène, J.; Gago-Martinez, A. An attempt to characterize the ciguatoxin profile in Seriola fasciata causing ciguatera fish poisoning in Macaronesia. Toxins 2019, 11, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshiro, N.; Nagasawa, H.; Kuniyoshi, K.; Kobayashi, N.; Sugita-Konishi, Y.; Asakura, H.; Yasumoto, T. Characteristic distribution of ciguatoxins in the edible parts of a grouper, Variola louti. Toxins 2021, 13, 218. [Google Scholar] [CrossRef] [PubMed]
- Comission Implementing Regulation (EU) 2021/808 of 22 March 2021 on the Performance of Analytical Methods for Residues of Pharmacologically Active Substances Used in Food-Producing Animals and on the Interpretation of Results as well as on the Methods to be Used for Sampling and Repealing Decisions 2002/657/EC and 98/179/EC; Last Amendment by Commission Delegated Regulation (EU) 2021/810 of 20 May 2021; European Union: Brussels, Belgium, 2021.
- Szpot, P.; Buszewicz, G.; Jurek, T.; Teresiński, G. Fragmentation patterns involving ammonium adduct fragment ions: A comparison of the determination of metaldehyde in human blood by HPLC-QqQ-MS/MS and UHPLC-Q-TOF-MS. J. Chromatogr. B 2018, 1085, 104–109. [Google Scholar] [CrossRef]
- Estevez, P.; Sibat, M.; Leão-Martins, J.M.; Reis Costa, P.; Gago-Martínez, A.; Hess, P. Liquid chromatography coupled to high-resolution mass spectrometry for the confirmation of Caribbean ciguatoxin-1 as the main toxin responsible for ciguatera poisoning caused by fish from European Atlantic coasts. Toxins 2020, 12, 267. [Google Scholar] [CrossRef] [Green Version]
- Ikehara, T.; Kuniyoshi, K.; Oshiro, N.; Yasumoto, T. Biooxidation of ciguatoxins leads to species-specific toxin profiles. Toxins 2017, 9, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Henao, J.A.; García-Álvarez, N.; Fernández, A.; Saavedra, P.; Silva Sergent, F.; Padilla, D.; Acosta-Hernández, B.; Martel Suárez, M.; Diogène, J.; Real, F. Predictive score and probability of CTX-like toxicity in fish samples from the official control of ciguatera in the Canary Islands. Sci. Total Environ. 2019, 673, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Caillaud, A.; Eixarch, H.; de la Iglesia, P.; Rodriguez, M.; Dominguez, L.; Andree, K.B.; Diogene, J. Towards the standardisation of the neuroblastoma (neuro-2a) cell-based assay for ciguatoxin-like toxicity detection in fish: Application to fish caught in the Canary Islands. Food Addit Contam Part. A Chem. Anal. Control. Expo. Risk Assess. 2012, 29, 1000–1010. [Google Scholar] [CrossRef] [PubMed]
- Friedemann, M. Ciguatera fish poisoning outbreaks from 2012 to 2017 in Germany caused by snappers from India, Indonesia, and Vietnam. J. Consum. Prot. Food Saf. 2019, 14, 71–80. [Google Scholar] [CrossRef]
- Loeffler, C.R.; Bodi, D.; Tartaglione, L.; Dell’Aversano, C.; Preiss-Weigert, A. Improving in vitro ciguatoxin and brevetoxin detection: Selecting neuroblastoma (Neuro-2a) cells with lower sensitivity to ouabain and veratridine (OV-LS). Harmful Algae 2021, 103, 101994. [Google Scholar] [CrossRef]
- Caban, M.; Migowska, N.; Stepnowski, P.; Kwiatkowski, M.; Kumirska, J. Matrix effects and recovery calculations in analyses of pharmaceuticals based on the determination of β-blockers and β-agonists in environmental samples. J. Chromatogr. A 2012, 1258, 117–127. [Google Scholar] [CrossRef]
- DIN Deutsches Institut für Normung e.V. DIN 38402-51: German Standard Methods for the Examination of Water, Waste Water and Sludge; General Information (Group A); Calibration of Analytical Methods, Evaluation of Analytical Results and Linear Calibration Functions Used to Determine the Performance Characteristics of Analytical Methods (A 51); DIN Deutsches Institut für Normung e.V: Berlin, Germany, 1986. [Google Scholar]
- ISO 3534-2:2006: Statistics—Vocabulary and Symbols—Part 2: Applied Statistics; ISO: Geneva, Switzerland, 2006.
- Manger, R.L.; Leja, L.S.; Lee, S.Y.; Hungerford, J.M.; Hokama, Y.; Dickey, R.W.; Granade, H.R.; Lewis, R.; Yasumoto, T.; Wekell, M.M. Detection of sodium channel toxins: Directed cytotoxicity assays of purified ciguatoxins, brevetoxins, saxitoxins, and seafood extracts. J. AOAC Int. 1995, 78, 521–527. [Google Scholar] [CrossRef]
- Manger, R.L.; Leja, L.S.; Lee, S.Y.; Hungerford, J.M.; Wekell, M.M. Tetrazolium-based cell bioassay for neurotoxins active on voltage-sensitive sodium channels: Semiautomated assay for saxitoxins, brevetoxins, and ciguatoxins. Anal. Biochem. 1993, 214, 190–194. [Google Scholar] [CrossRef]





| Day | 0 | 6 | 13 | 19 | 26 | 0 | 6 | 13 | 19 | 26 | 0 | 6 | 13 | 19 | 26 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ME | RR | EE | |||||||||||||
| CTX1B | |||||||||||||||
| snapper, fillet | 75 | 75 | 69 | 86 | 85 | 47 | 43 | 40 | 47 | 46 | 64 | 57 | 58 | 54 | 54 |
| snapper, freeze-dried | 76 | 67 | 71 | 81 | 83 | 47 | 40 | 44 | 48 | 45 | 62 | 60 | 61 | 60 | 54 |
| parrotfish, fillet | 92 | 85 | 89 | 100 | 97 | 62 | 62 | 55 | 62 | 55 | 68 | 72 | 62 | 61 | 57 |
| parrotfish, freeze-dried | 90 | 86 | 84 | 95 | 97 | 57 | 56 | 54 | 62 | 53 | 63 | 65 | 65 | 65 | 54 |
| grouper, fillet | 66 | 54 | 61 | 77 | 62 | 39 | 36 | 36 | 41 | 34 | 59 | 66 | 59 | 53 | 56 |
| grouper, freeze-dried | 74 | 67 | 66 | 82 | 82 | 45 | 41 | 40 | 50 | 48 | 61 | 61 | 61 | 60 | 59 |
| 52-epi-54-deoxyCTX1B | |||||||||||||||
| snapper, fillet | 100 | 96 | 81 | 99 | 88 | 63 | 53 | 47 | 61 | 46 | 63 | 55 | 59 | 61 | 52 |
| snapper, freeze-dried | 84 | 78 | 78 | 95 | 83 | 57 | 53 | 54 | 60 | 53 | 67 | 67 | 69 | 64 | 63 |
| parrotfish, fillet | 93 | 94 | 93 | 98 | 96 | 66 | 59 | 62 | 69 | 59 | 72 | 63 | 67 | 71 | 62 |
| parrotfish, freeze-dried | 103 | 103 | 95 | 97 | 104 | 71 | 65 | 64 | 77 | 65 | 69 | 63 | 67 | 80 | 62 |
| grouper, fillet | 70 | 60 | 57 | 70 | 62 | 35 | 33 | 33 | 43 | 35 | 50 | 55 | 57 | 61 | 56 |
| grouper, freeze-dried | 66 | 69 | 66 | 85 | 75 | 52 | 43 | 38 | 55 | 44 | 79 | 62 | 58 | 65 | 59 |
| 54-deoxyCTX1B | |||||||||||||||
| snapper, fillet | 73 | 73 | 69 | 85 | 82 | 42 | 43 | 40 | 47 | 46 | 58 | 59 | 59 | 55 | 57 |
| snapper, freeze-dried | 79 | 68 | 65 | 84 | 88 | 48 | 45 | 47 | 55 | 49 | 61 | 66 | 71 | 66 | 56 |
| parrotfish, fillet | 90 | 83 | 84 | 97 | 104 | 64 | 64 | 58 | 67 | 58 | 71 | 77 | 68 | 69 | 56 |
| parrotfish, freeze-dried | 93 | 87 | 89 | 96 | 101 | 69 | 66 | 61 | 72 | 64 | 84 | 76 | 68 | 75 | 63 |
| grouper, fillet | 66 | 63 | 62 | 76 | 62 | 35 | 36 | 37 | 43 | 39 | 53 | 58 | 60 | 56 | 63 |
| grouper, freeze-dried | 72 | 73 | 72 | 86 | 81 | 48 | 47 | 45 | 55 | 52 | 66 | 65 | 62 | 64 | 64 |
| CTX3C—filtrate | |||||||||||||||
| snapper, fillet | 95 | 94 | 87 | 110 | 102 | 36 | 38 | 34 | 38 | 41 | 38 | 41 | 39 | 35 | 40 |
| snapper, freeze-dried | 98 | 102 | 100 | 121 | 114 | 40 | 41 | 41 | 46 | 49 | 41 | 40 | 41 | 38 | 43 |
| parrotfish, fillet | 116 | 112 | 102 | 101 | 130 | 48 | 47 | 41 | 52 | 52 | 42 | 42 | 41 | 52 | 40 |
| parrotfish, freeze-dried | 109 | 118 | 107 | 112 | 128 | 53 | 56 | 50 | 61 | 61 | 49 | 47 | 46 | 55 | 47 |
| grouper, fillet | 85 | 90 | 84 | 100 | 98 | 40 | 38 | 35 | 40 | 42 | 47 | 42 | 42 | 40 | 43 |
| grouper, freeze-dried | 98 | 113 | 100 | 107 | 125 | 45 | 42 | 41 | 49 | 49 | 46 | 37 | 41 | 46 | 39 |
| CTX3C—eluate | |||||||||||||||
| snapper, fillet | 94 | 91 | 83 | 100 | 82 | 25 | 25 | 24 | 27 | 21 | 26 | 27 | 29 | 27 | 26 |
| snapper, freeze-dried | 95 | 88 | 88 | 100 | 93 | 28 | 28 | 26 | 27 | 24 | 30 | 32 | 30 | 27 | 26 |
| parrotfish, fillet | 104 | 102 | 101 | 107 | 97 | 35 | 32 | 29 | 31 | 27 | 33 | 31 | 28 | 29 | 28 |
| parrotfish, freeze-dried | 99 | 101 | 96 | 105 | 93 | 35 | 34 | 31 | 32 | 26 | 35 | 33 | 32 | 31 | 28 |
| grouper, fillet | 72 | 61 | 65 | 79 | 63 | 25 | 21 | 23 | 22 | 20 | 34 | 35 | 36 | 28 | 32 |
| grouper, freeze-dried | 89 | 79 | 81 | 97 | 75 | 30 | 27 | 28 | 31 | 29 | 34 | 34 | 34 | 32 | 39 |
| CTX3C—sum | |||||||||||||||
| snapper, fillet | 60 | 63 | 58 | 65 | 62 | 64 | 68 | 68 | 62 | 66 | |||||
| snapper, freeze-dried | 68 | 69 | 68 | 73 | 73 | 71 | 72 | 71 | 65 | 69 | |||||
| parrotfish, fillet | 83 | 79 | 70 | 83 | 80 | 75 | 73 | 69 | 81 | 68 | |||||
| parrotfish, freeze-dried | 88 | 89 | 81 | 93 | 86 | 84 | 81 | 79 | 85 | 75 | |||||
| grouper, fillet | 65 | 59 | 58 | 62 | 62 | 81 | 76 | 78 | 67 | 74 | |||||
| grouper, freeze-dried | 76 | 69 | 69 | 80 | 79 | 80 | 71 | 75 | 78 | 78 | |||||
| min compared to t = 0 * | 82 | 81 | 87 | 84 | 83 | 74 | 89 | 73 | 79 | 74 | 81 | 74 | |||
| max compared to t = 0 * | 115 | 102 | 130 | 127 | 107 | 106 | 123 | 123 | 113 | 117 | 124 | 120 |
| Parameter | CTX1B | 52-epi-54-deoxyCTX1B | 54-deoxyCTX1B | CTX3C | |
|---|---|---|---|---|---|
| linearity ranges, methanol [µg L−1] | 0.075–10 | 0.2–10 | 0.1–7 | 0.2–20 | |
| linearity ranges, matrix 1 [µg L−1] | 0.1–5 | 0.2–5 | 0.2–5 | 0.2–10 | |
| LOD methanol [µg L−1] | 0.02 | 0.05 | 0.03 | 0.06 | |
| LOQ methanol [µg L−1] | 0.075 | 0.2 | 0.1 | 0.2 | |
| CTX3C eluate | CTX3C filtrate | ||||
| LOD fillet [µg kg−1], full 2 | 0.02 | 0.02 | 0.02 | 0.08 | 0.04 |
| LOD fillet [µg kg−1], reduced 3 | 0.01 | 0.01 | 0.01 | (0.04) | 0.02 |
| LOQ fillet [µg kg−1], full 2 | 0.04 | 0.1 | 0.04 | 0.4 | 0.2 |
| LOQ fillet [µg kg−1], reduced 3 | (0.02) | (0.05) | (0.02) | (0.2) | (0.1) |
| LOD freeze-dried [µg kg−1], full 2 | 0.2 | 0.2 | 0.1 | 0.4 | 0.2 |
| LOD freeze-dried [µg kg−1], reduced 3 | 0.1 | 0.1 | (0.05) | (0.2) | 0.2 |
| LOQ freeze-dried [µg kg−1], full 2 | 0.4 | 1 | 0.4 | 0.8 | 0.8 |
| LOQ freeze-dried [µg kg−1], reduced 3 | (0.2) | (0.5) | 0.1 | (0.4) | (0.4) |
| Congener | [14] | [49] | This Study 1 | This Study 2 |
|---|---|---|---|---|
| 2,3,51-trihydroxyCTX3C | 4.52 | |||
| 4-hydroxy-7-oxoCTX1B | 4.58 | |||
| 7-oxoCTX1B | 4.71 | |||
| 7-hydroxyCTX1B | 4.71 | |||
| A-seco-51-hydroxyCTX3C | 4.75 | |||
| CTX1B | 4.95 | 2.6 | 3.1 | 3.0 |
| M-seco-CTX3C | 6.21 | 4.7 | ||
| 2,3-dihydroxyCTX3C | 7.80 | 5.9 | ||
| 51-hydroxyCTX3C | 7.24 | 6.1 | ||
| M-seco-CTX4A/B | 7.19 | 6.6 | ||
| 52-epi-54-deoxyCTX1B | 7.87 | 6.7 | 5.9 | 6.2 |
| 51-hydroxy-2-oxoCTX3C | 8.15 | |||
| 54-deoxyCTX1B | 8.30 | 7.2 | 6.2 | 6.6 |
| 2-hydroxyCTX3C | 8.94 | 7.3 | ||
| M-seco-CTX3C methyl acetal | 10.5 | |||
| CTX3B | 14.50 | 11.5 | ||
| CTX3C | 15.44 | 11.8 | 9.4 | 10.0 |
| CTX4A | 15.30 | 12.9 | ||
| CTX4B | 13.3 |
| Congener | [16] | [8] | [9] | [50] 1 | [50] 2 | This Study 3 | This Study 4 |
|---|---|---|---|---|---|---|---|
| C-CTX-3/4 | 3.70/4.11 | ||||||
| C-CTX-1/2 | 6.4 | 62 | 4.6 | 9.4 | |||
| C-CTX reaction product 9 | 8.0 | ||||||
| C-CTX reaction product 8 | 9.82 | ||||||
| C-CTX-1127 | 10.6 | ||||||
| C-CTX-1157 | 7.6 | ||||||
| I-CTX-3/4 | 5.49/5.60 | ||||||
| I-CTX-6 | 5.82 | ||||||
| I-CTX-1/2 | 6.49/6.60 | 62.5 | |||||
| I-CTX-5 | 6.95 | ||||||
| CTX1B | 59 | 3.7 | 3.1 | 3.0 | |||
| 52-epi-54-deoxyCTX1B | 7.4 | 5.9 | 6.2 | ||||
| 54-deoxyCTX1B | 7.8 | 6.2 | 6.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spielmeyer, A.; Loeffler, C.R.; Bodi, D. Extraction and LC-MS/MS Analysis of Ciguatoxins: A Semi-Targeted Approach Designed for Fish of Unknown Origin. Toxins 2021, 13, 630. https://doi.org/10.3390/toxins13090630
Spielmeyer A, Loeffler CR, Bodi D. Extraction and LC-MS/MS Analysis of Ciguatoxins: A Semi-Targeted Approach Designed for Fish of Unknown Origin. Toxins. 2021; 13(9):630. https://doi.org/10.3390/toxins13090630
Chicago/Turabian StyleSpielmeyer, Astrid, Christopher R. Loeffler, and Dorina Bodi. 2021. "Extraction and LC-MS/MS Analysis of Ciguatoxins: A Semi-Targeted Approach Designed for Fish of Unknown Origin" Toxins 13, no. 9: 630. https://doi.org/10.3390/toxins13090630
APA StyleSpielmeyer, A., Loeffler, C. R., & Bodi, D. (2021). Extraction and LC-MS/MS Analysis of Ciguatoxins: A Semi-Targeted Approach Designed for Fish of Unknown Origin. Toxins, 13(9), 630. https://doi.org/10.3390/toxins13090630