Real-World Dosing of OnabotulinumtoxinA and IncobotulinumtoxinA for Cervical Dystonia and Blepharospasm: Results from TRUDOSE and TRUDOSE II

Abstract
:1. Introduction
2. Results
2.1. Patient Disposition and Demographics
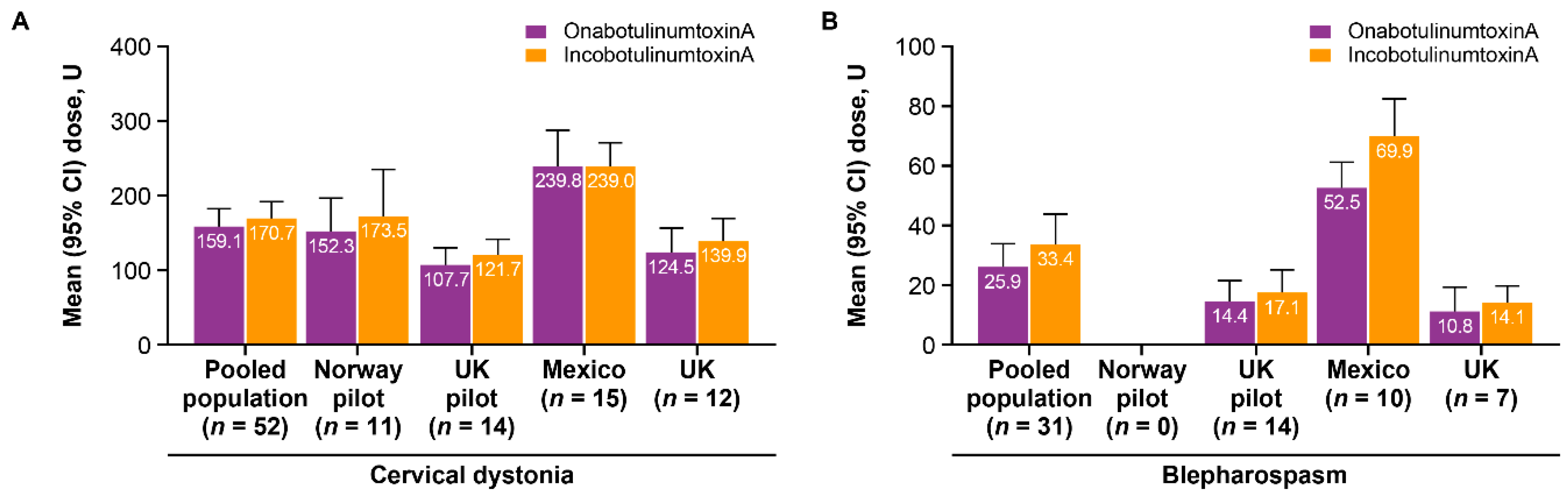
2.2. Treatment Characteristics
2.3. Safety and Tolerability
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Study Design
5.2. Patients
5.3. Study Measures
5.4. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dressler, D. Botulinum toxin drugs: Brief history and outlook. J. Neural Transm. 2016, 123, 277–279. [Google Scholar] [CrossRef]
- Kumar, R.; Dhaliwal, H.P.; Kukreja, R.V.; Singh, B.R. The botulinum toxin as a therapeutic agent: Molecular structure and mechanism of action in motor and sensory systems. Semin. Neurol. 2016, 36, 10–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabbari, B. History of botulinum toxin treatment in movement disorders. Tremor Other Hyperkinet. Mov. 2016, 6, 394. [Google Scholar] [CrossRef]
- Marchetti, A.; Magar, R.; Findley, L.; Larsen, J.P.; Pirtosek, Z.; Ruzicka, E.; Jech, R.; Slawek, J.; Ahmed, F. Retrospective evaluation of the dose of Dysport and BOTOX in the management of cervical dystonia and blepharospasm: The REAL DOSE study. Mov. Disord. 2005, 20, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K.R.; Ranoux, D.; Wissel, J. Using translational medicine to understand clinical differences between botulinum toxin formulations. Eur. J. Neurol. 2006, 13 (Suppl. S4), 10–19. [Google Scholar] [CrossRef] [PubMed]
- Albanese, A.; Abbruzzese, G.; Dressler, D.; Duzynski, W.; Khatkova, S.; Marti, M.J.; Mir, P.; Montecucco, C.; Moro, E.; Pinter, M.; et al. Practical guidance for CD management involving treatment of botulinum toxin: A consensus statement. J. Neurol. 2015, 262, 2201–2213. [Google Scholar] [CrossRef] [PubMed]
- Karp, B.I.; Alter, K. Botulinum toxin treatment of blepharospasm, orofacial/oromandibular dystonia, and hemifacial spasm. Semin. Neurol. 2016, 36, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Brin, M.F.; James, C.; Maltman, J. Botulinum toxin type A products are not interchangeable: A review of the evidence. Biologics 2014, 8, 227–241. [Google Scholar] [CrossRef] [Green Version]
- Kutschenko, A.; Manig, A.; Reinert, M.C.; Mönnich, A.; Liebetanz, D. In-vivo comparison of the neurotoxic potencies of incobotulinumtoxinA, onabotulinumtoxinA, and abobotulinumtoxinA. Neurosci. Lett. 2016, 627, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Dressler, D.; Tacik, P.; Adib Saberi, F. Botulinum toxin therapy of cervical dystonia: Comparing onabotulinumtoxinA (Botox®) and incobotulinumtoxinA (Xeomin®). J. Neural Transm. 2014, 121, 29–31. [Google Scholar] [CrossRef] [Green Version]
- Benecke, R.; Jost, W.H.; Kanovsky, P.; Ruzicka, E.; Comes, G.; Grafe, S. A new botulinum toxin type A free of complexing proteins for treatment of cervical dystonia. Neurology 2005, 64, 1949–1951. [Google Scholar] [CrossRef]
- Saad, J.; Gourdeau, A. A direct comparison of onabotulinumtoxinA (Botox) and incobotulinumtoxinA (Xeomin) in the treatment of benign essential blepharospasm: A split-face technique. J. Neuroophthalmol. 2014, 34, 233–236. [Google Scholar] [CrossRef]
- Roggenkämper, P.; Jost, W.H.; Bihari, K.; Comes, G.; Grafe, S.; for the N. T. Blepharospasm Study Team. Efficacy and safety of a new Botulinum Toxin Type A free of complexing proteins in the treatment of blepharospasm. J. Neural Transm. 2006, 113, 303–312. [Google Scholar] [CrossRef]
- Banegas, R.A.; Farache, F.; Rancati, A.; Chain, M.; Gallagher, C.J.; Chapman, M.A.; Caulkins, C.A. The South American Glabellar Experience Study (SAGE): A multicenter retrospective analysis of real-world treatment patterns following the introduction of incobotulinumtoxinA in Argentina. Aesthet. Surg. J. 2013, 33, 1039–1045. [Google Scholar] [CrossRef] [Green Version]
- Chapman, M.A.; Barron, R.; Tanis, D.C.; Gill, C.E.; Charles, P.D. Comparison of botulinum neurotoxin preparations for the treatment of cervical dystonia. Clin. Ther. 2007, 29, 1325–1337. [Google Scholar] [CrossRef]
- Monti, S.; Grosso, V.; Todoerti, M.; Caporali, R. Randomized controlled trials and real-world data: Differences and similarities to untangle literature data. Rheumatology 2018, 57, vii54–vii58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, R.E.; Anderson, S.A.; Dal Pan, G.J.; Gray, G.W.; Gross, T.; Hunter, N.L.; LaVange, L.; Marinac-Dabic, D.; Marks, P.W.; Robb, M.A.; et al. Real-world evidence—What is it and what can it tell us. N. Engl. J. Med. 2016, 375, 2293–2297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kollewe, K.; Mohammadi, B.; Köhler, S.; Pickenbrock, H.; Dengler, R.; Dressler, D. Blepharospasm: Long-term treatment with either Botox®, Xeomin® or Dysport®. J. Neural Transm. 2015, 122, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.Y.; Kim, J.W.; Kim, H.T.; Chung, S.J.; Kim, J.M.; Cho, J.W.; Lee, J.Y.; Lee, H.N.; You, S.; Oh, E.; et al. Dysport and Botox at a ratio of 2.5:1 units in cervical dystonia: A double-blind, randomized study. Mov. Disord. 2015, 30, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Rystedt, A.; Zetterberg, L.; Burman, J.; Nyholm, D.; Johansson, A. A comparison of Botox 100 U/mL and Dysport 100 U/mL using dose conversion ratio 1:3 and 1:1.7 in the treatment of cervical dystonia: A double-blind, randomized, crossover trial. Clin. Neuropharmacol. 2015, 38, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Nuhoğlu, F.; Özdemir, F.E.; Karademir, Z.; Eltutar, K. OnabotulinumtoxinA (Botox®) and abobotulinumtoxinA (Dysport®) in treating essential blepharospasm: Long term results. J. Acad. Res. Med. 2016, 6, 110–114. [Google Scholar] [CrossRef]
- Dashtipour, K.; Chen, J.J.; Espay, A.J.; Mari, Z.; Ondo, W. OnabotulinumtoxinA and abobotulinumtoxinA dose conversion: A systematic literature review. Mov. Disord. Clin. Pract. 2016, 3, 109–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dressler, D.; Altenmueller, E.; Bhidayasiri, R.; Bohlega, S.; Chana, P.; Chung, T.M.; Frucht, S.; Garcia-Ruiz, P.J.; Kaelin, A.; Kaji, R.; et al. Strategies for treatment of dystonia. J. Neural Transm. 2016, 123, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Camargo, C.H.; Cattai, L.; Teive, H.A. Pain relief in cervical dystonia with botulinum toxin treatment. Toxins 2015, 7, 2321–2335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brin, M.F.; Blitzer, A. Botulinum toxin: Dangerous terminology errors. J. R. Soc. Med. 1993, 86, 493–494. [Google Scholar]
- Wabbels, B.; Reichel, G.; Fulford-Smith, A.; Wright, N.; Roggenkämper, P. Double-blind, randomised, parallel group pilot study comparing two botulinum toxin type A products for the treatment of blepharospasm. J. Neural Transm. 2011, 118, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Dressler, D. Routine use of Xeomin in patients previously treated with Botox: Long term results. Eur. J. Neurol. 2009, 16 (Suppl. S2), 2–5. [Google Scholar] [CrossRef]
- Wilson, A.J.; Chang, B.; Taglienti, A.J.; Chin, B.C.; Chang, C.S.; Folsom, N.; Percec, I. A quantitative analysis of onabotulinumtoxinA, abobotulinumtoxinA, and incobotulinumtoxinA: A randomized, double-blind, prospective clinical trial of comparative dynamic strain reduction. Plast. Reconstr. Surg. 2016, 137, 1424–1433. [Google Scholar] [CrossRef] [Green Version]
- Thomas, A.J.; Larson, M.O.; Braden, S.; Cannon, R.B.; Ward, P.D. Effect of 3 commercially available botulinum toxin neuromodulators on facial synkinesis: A randomized clinical trial. JAMA Facial Plast. Surg. 2018, 20, 141–147. [Google Scholar] [CrossRef]
- Lamanna, C.; Spero, L.; Schantz, E.J. Dependence of time to death on molecular size of botulinum toxin. Infect. Immun. 1970, 1, 423–424. [Google Scholar] [CrossRef] [Green Version]
- Hunt, T.; Clarke, K. Potency evaluation of a formulated drug product containing 150-kd botulinum neurotoxin type A. Clin. Neuropharmacol. 2009, 32, 28–31. [Google Scholar] [CrossRef]
- Dressler, D.; Mander, G.; Fink, K. Measuring the potency labelling of onabotulinumtoxinA (Botox®) and incobotulinumtoxinA (Xeomin®) in an LD50 assay. J. Neural Transm. 2012, 119, 13–15. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.; Nicholson, G.; Ardila, M.C.; Satorius, A.; Broide, R.S.; Clarke, K.; Hunt, T.; Francis, J. Comparative evaluation of the potency and antigenicity of two distinct BoNT/A-derived formulations. J. Neural Transm. 2013, 120, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Rupp, D.; Nicholson, G.; Canty, D.; Wang, J.; Rheaume, C.; Le, L.; Steward, L.E.; Washburn, M.; Jacky, B.P.; Broide, R.S.; et al. OnabotulinumtoxinA displays greater biological activity compared to incobotulinumtoxinA, demonstrating non-interchangeability in both in vitro and in vivo assays. Toxins 2020, 12, 393. [Google Scholar] [CrossRef] [PubMed]
- Poulain, B.; Trevidic, P.; Clavé, M.; Aharoni, C.; Baspeyras, M.; Bui, P.; Cartier, H.; Charavel, M.H.; Coulon, P.; Dahan, S.; et al. Clinical equivalence of conventional OnabotulinumtoxinA (900 KDa) and IncobotulinumtoxinA (neurotoxin free from complexing proteins—150 KDa): 2013 multidisciplinary French consensus in aesthetics. J. Drugs Dermatol. 2013, 12, 1434–1446. [Google Scholar] [PubMed]
- Jandhyala, R. Relative potency of incobotulinumtoxinA vs onabotulinumtoxinA a meta-analysis of key evidence. J. Drugs Dermatol. 2012, 11, 731–736. [Google Scholar]
- BOTOX® (OnabotulinumtoxinA) for Injection, for Intramuscular, Intradetrusor, or Intradermal Use; Allergan plc: Irvine, CA, USA, 2016.
- Chundury, R.V.; Couch, S.M.; Holds, J.B. Comparison of preferences between onabotulinumtoxinA (Botox) and incobotulinumtoxinA (Xeomin) in the treatment of benign essential blepharospasm. Ophthal. Plast. Reconstr. Surg. 2013, 29, 205–207. [Google Scholar] [CrossRef]
- Dressler, D.; Tacik, P.; Adib Saberi, F. Botulinum toxin therapy of cervical dystonia: Duration of therapeutic effects. J. Neural Transm. 2015, 122, 297–300. [Google Scholar] [CrossRef]
- Charles, P.D.; Adler, C.H.; Stacy, M.; Comella, C.; Jankovic, J.; Manack Adams, A.; Schwartz, M.; Brin, M.F. Cervical dystonia and pain: Characteristics and treatment patterns from CD PROBE (Cervical Dystonia Patient Registry for Observation of OnabotulinumtoxinA Efficacy). J. Neurol. 2014, 261, 1309–1319. [Google Scholar] [CrossRef] [Green Version]
- Jankovic, J.; Adler, C.H.; Charles, D.; Comella, C.; Stacy, M.; Schwartz, M.; Manack Adams, A.; Brin, M.F. Primary results from the cervical dystonia patient registry for observation of onabotulinumtoxina efficacy (CD PROBE). J. Neurol. Sci. 2015, 349, 84–93. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TRUDOSE Pilot | TRUDOSE II | |
|---|---|---|
| Patients screened, n | 116 | 89 |
| Ineligible patients that did not meet inclusion/exclusion criteria, n | 77 | 45 |
| Insufficient treatment time | 62 | 20 |
| Lack of confirmed diagnosis ≥ 2 years before the switch | 11 | 0 |
| Surgical procedure involving bone or muscle for disease management | 4 | 7 |
| Enrollment in another clinical trial | 0 | 7 |
| Changes in adjuvant therapy for dystonia that could affect disease severity or result in increased adverse events | 0 | 7 |
| Evaluable patients, n | 39 | 44 |
| Characteristic | TRUDOSE Pilot (n = 39) | TRUDOSE II (n = 44) | Pooled Data (n = 83) |
|---|---|---|---|
| Mean (SD) age at enrollment, y | 63.5 (12.8) | 65.4 (11.4) | 64.5 (12.1) |
| Median (range) age, y | 65.0 (33.0–88.0) | 64.0 (32.0–≥90.0) * | 65.0 (32.0–≥90.0) * |
| Female, n (%) | 28 (71.8) | 32 (72.7) | 60 (72.3) |
| Condition, n (%) | |||
| Cervical dystonia | 25 (64.1) | 27 (61.4) | 52 (62.7) |
| Blepharospasm | 14 (35.9) | 17 (38.6) | 31 (37.3) |
| Time since onset of condition, n (%) | |||
| >5 years | 36 (92.3) | 44 (100.0) | 80 (96.4) |
| 2–5 years | 3 (7.7) | 0 (0.0) | 3 (3.6) |
| Patient Group | Mean Dose Ratio Using Last Dose | Mean Dose Ratio Using Total Dose | ||||
|---|---|---|---|---|---|---|
| TRUDOSE Pilot (n = 39) | TRUDOSE II (n = 44) | Pooled Data (n = 83) | TRUDOSE Pilot (n = 39) | TRUDOSE II (n = 44) | Pooled Data (n = 83) | |
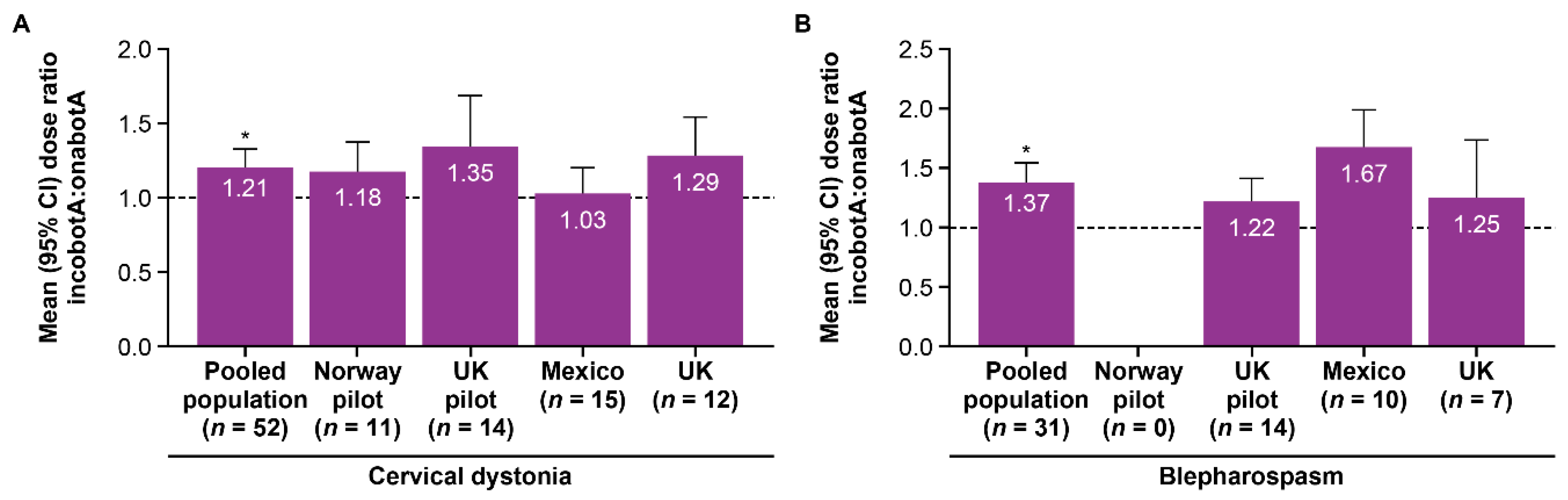
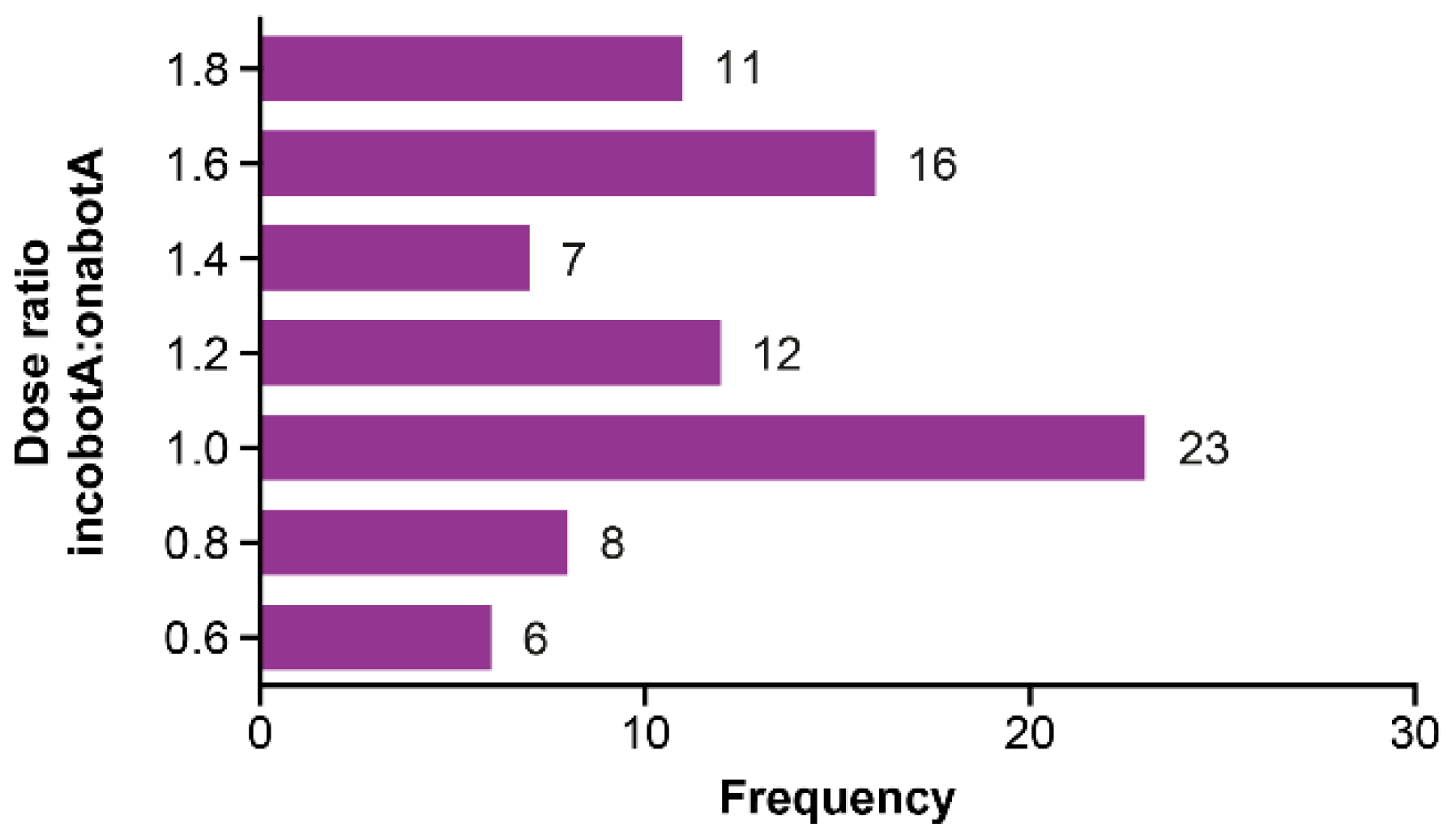
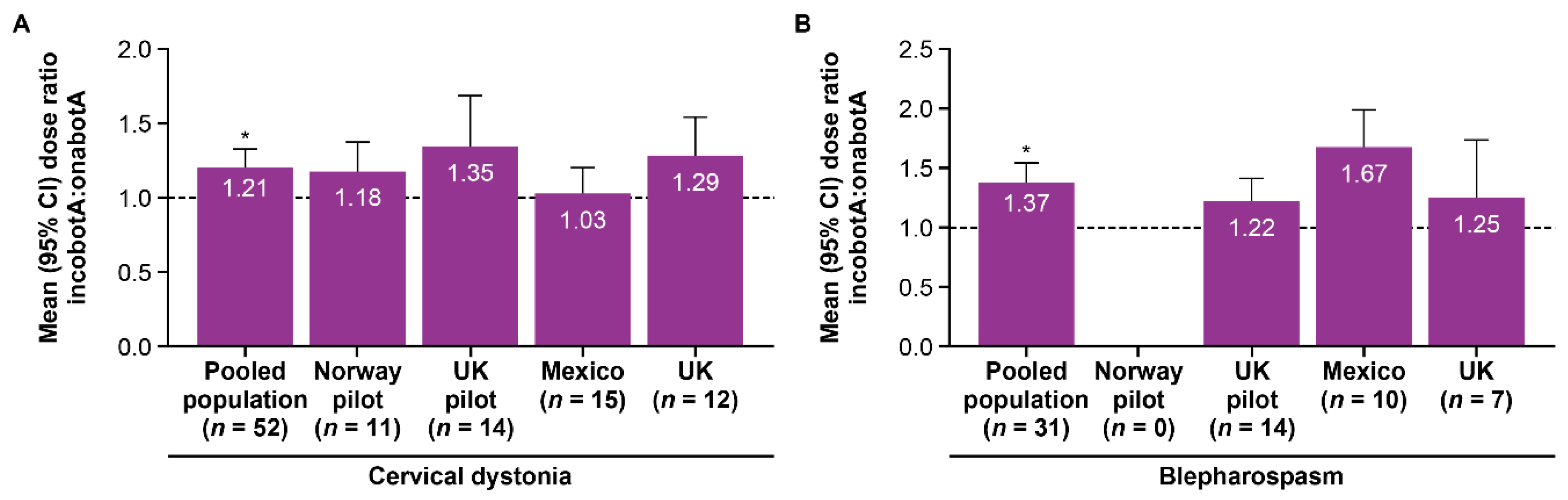
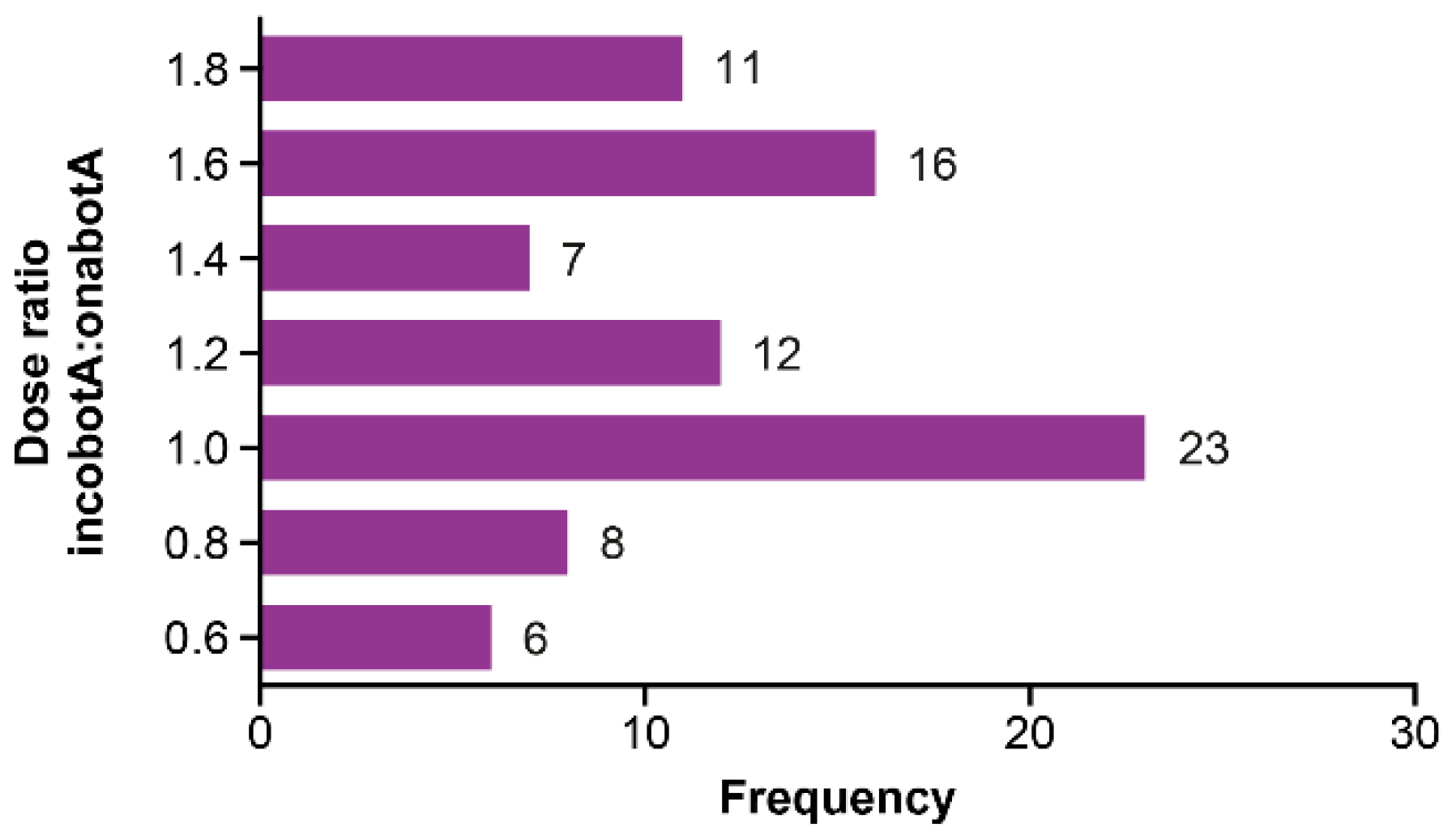
| Cervical dystonia | 1.27 | 1.14 | 1.21 † | 1.15 | 1.10 | 1.12 † |
| Blepharospasm | 1.22 | 1.50 | 1.37 † | 1.20 | 1.34 | 1.28 † |
| All patients | 1.25 | 1.28 | 1.27 † | 1.17 | 1.19 | 1.18 † |
| Characteristic | TRUDOSE Pilot (n = 39) | TRUDOSE II (n = 44) | ||
|---|---|---|---|---|
| OnabotulinumtoxinA | IncobotulinumtoxinA | OnabotulinumtoxinA | IncobotulinumtoxinA | |
| Cervical dystonia | ||||
| Mean (SD) dosage per visit, U | 127.3 (55.9) | 144.5 (69.5) | 188.6 (92.0) | 194.9 (72.2) |
| Median (range) dosage, U | 120.0 (41.4–257.1) | 132.5 (42.5–366.0) | 200 (70.9–487.1) | 203.8 (81.8–376.7) |
| Mean (SD) total dose per patient per year, U/year | 444.7 (347.2–542.2) * | 536.3 (409.6–663.0) *,† | 706.2 (374.9) | 837.3 (336.7) † |
| Inter-injection intervals, week | 15.6 | 14.5 | 14.6 | 14.4 |
| Blepharospasm | ||||
| Mean (SD) dosage per treatment, U | 14.4 (12.2) | 17.1 (13.8) | 35.3 (23.1) | 46.8 (31.4) |
| Median (range) dosage, U | 11.2 (5.4–52.9) | 12.0 (6.3–56.3) | 40.0 (6.0–65.0) | 46.0 (7.5–89.1) |
| Mean (SD) total dose per patient per year, U/year | 50.4 (21.2–79.6) * | 64.0 (33.3–94.7) *,† | 133.2 (93.5) | 207.2 (145.1) † |
| Inter-injection intervals, week | 16.3 | 14.2 | 16.2 | 13.9 |
| Pooled Population (n = 92) | ||
|---|---|---|
| Events, n | Patients, n (%) | |
| Any adverse event | 30 | 22 (23.9) |
| Serious adverse event | 0 | 0 |
| Severe adverse event | 0 | 0 |
| Adverse events by treatment received | ||
| OnabotulinumtoxinA | 15 | 12 (13.0) |
| IncobotulinumtoxinA | 15 | 13 (14.1) |
| Adverse events by underlying condition | ||
| Cervical dystonia (n = 58) | 17 | 12 (20.7) |
| Blepharospasm (n = 34) | 13 | 10 (29.4) |
| Adverse Event, n | Pooled Population (n = 92) | |
|---|---|---|
| OnabotulinumtoxinA | IncobotulinumtoxinA | |
| Cervical dystonia | ||
| Dysphagia | 4 | 2 |
| Head drop | 2 | 2 |
| Cervical extension weakness | 1 | 1 |
| Injection site pain | 1 | 2 |
| Blepharospasm | ||
| Ptosis | 3 | 4 |
| Bruising | 0 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kent, R.; Robertson, A.; Quiñones Aguilar, S.; Tzoulis, C.; Maltman, J. Real-World Dosing of OnabotulinumtoxinA and IncobotulinumtoxinA for Cervical Dystonia and Blepharospasm: Results from TRUDOSE and TRUDOSE II. Toxins 2021, 13, 488. https://doi.org/10.3390/toxins13070488
Kent R, Robertson A, Quiñones Aguilar S, Tzoulis C, Maltman J. Real-World Dosing of OnabotulinumtoxinA and IncobotulinumtoxinA for Cervical Dystonia and Blepharospasm: Results from TRUDOSE and TRUDOSE II. Toxins. 2021; 13(7):488. https://doi.org/10.3390/toxins13070488
Chicago/Turabian StyleKent, Ruth, Adrian Robertson, Sandra Quiñones Aguilar, Charalampos Tzoulis, and John Maltman. 2021. "Real-World Dosing of OnabotulinumtoxinA and IncobotulinumtoxinA for Cervical Dystonia and Blepharospasm: Results from TRUDOSE and TRUDOSE II" Toxins 13, no. 7: 488. https://doi.org/10.3390/toxins13070488
APA StyleKent, R., Robertson, A., Quiñones Aguilar, S., Tzoulis, C., & Maltman, J. (2021). Real-World Dosing of OnabotulinumtoxinA and IncobotulinumtoxinA for Cervical Dystonia and Blepharospasm: Results from TRUDOSE and TRUDOSE II. Toxins, 13(7), 488. https://doi.org/10.3390/toxins13070488