Huntingtin and Its Role in Mechanisms of RNA-Mediated Toxicity


Abstract
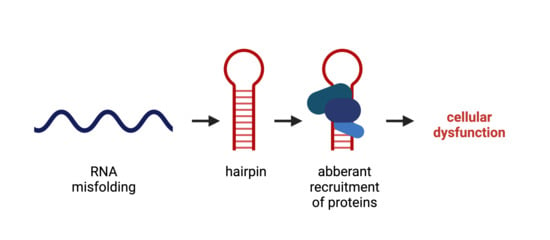
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
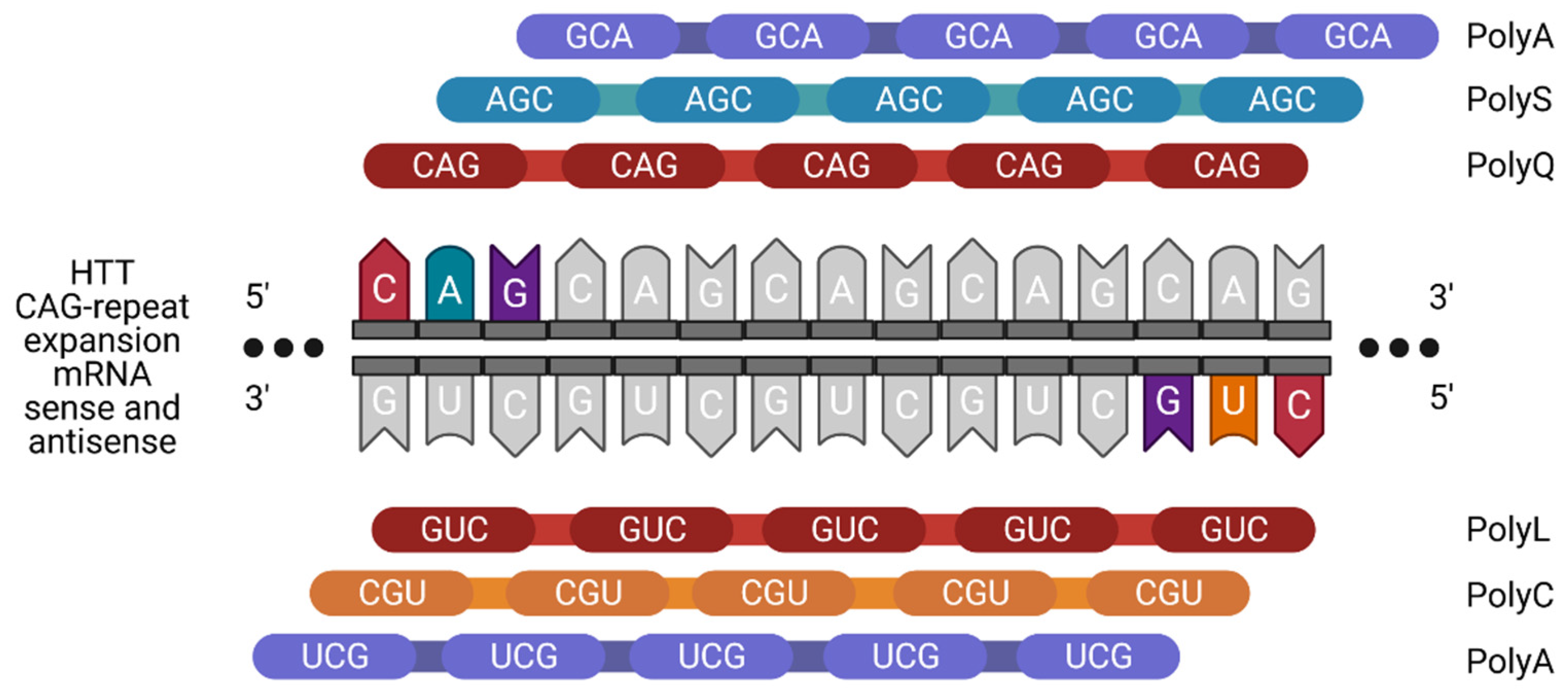
2. RNA Structure and Misfolding
3. RNA Localization and RNA Granules
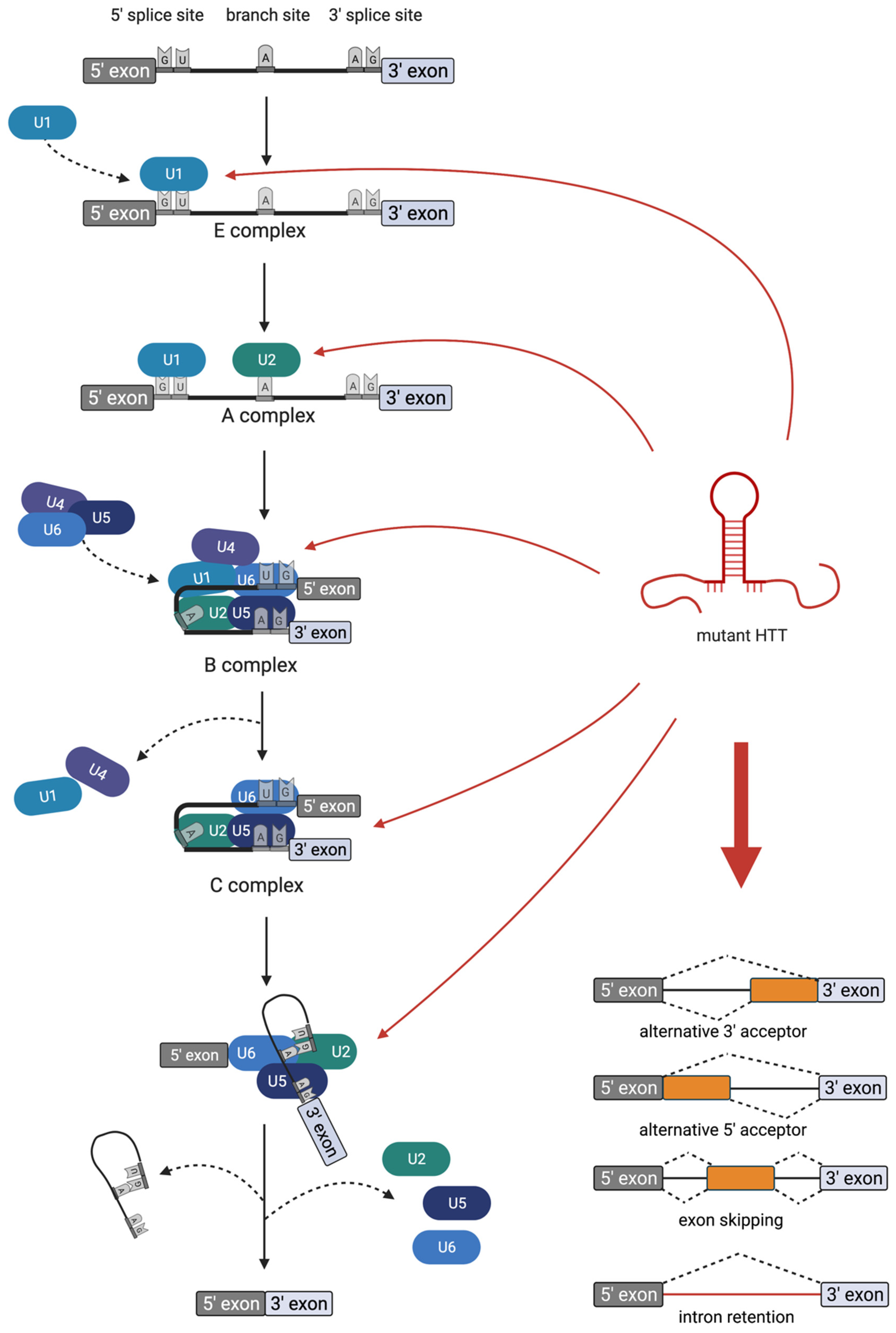
4. Deregulated Splicing
5. Aberrant Translation
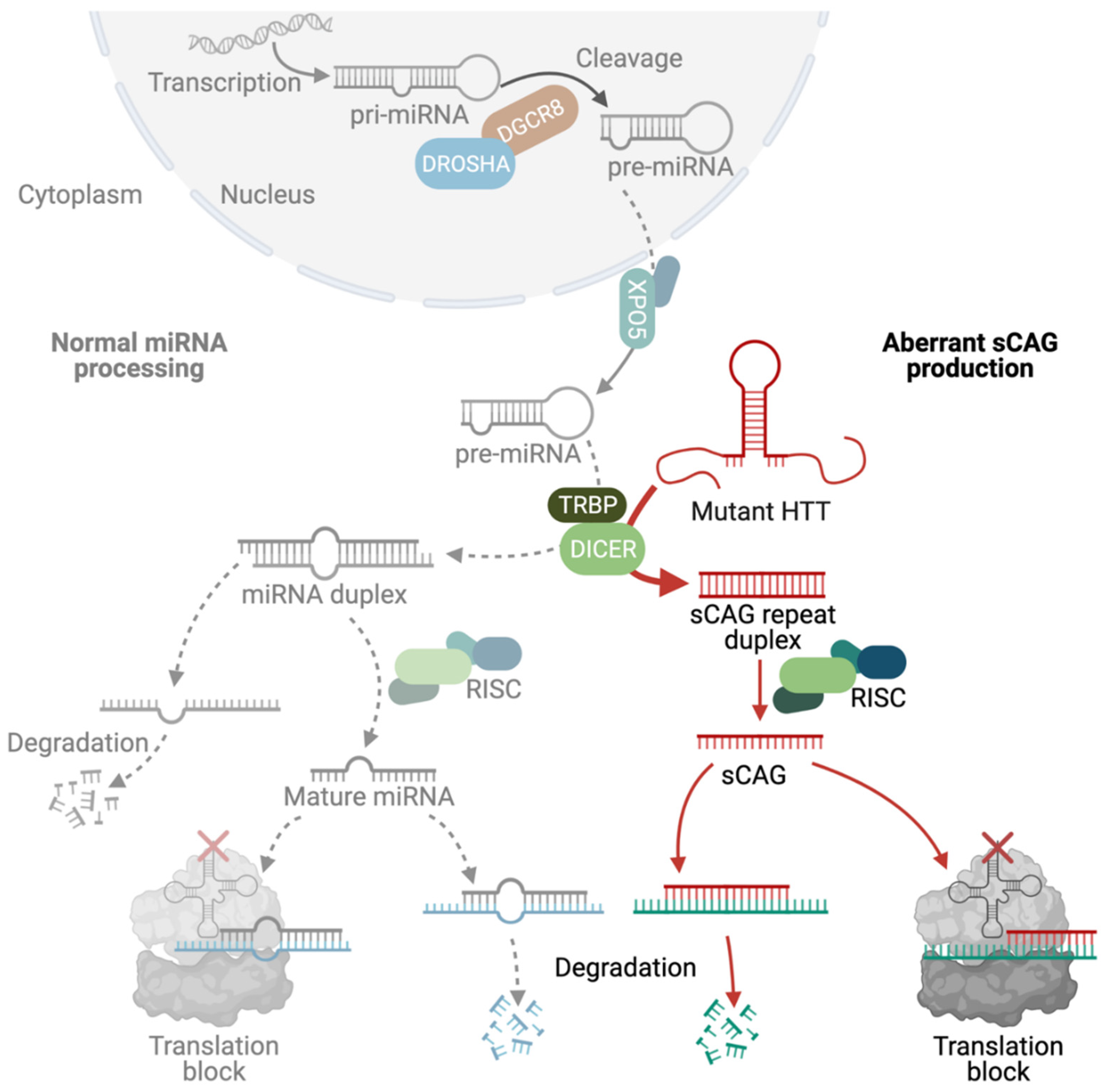
6. Deregulation of the microRNA Machinery
7. Mitochondrial RNA
8. Compounds
9. Antisense Oligonucleotides
10. Clinical Trials and Novel Therapies
11. Concluding Remarks and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kozlowski, P.; de Mezer, M.; Krzyzosiak, W.J. Trinucleotide repeats in human genome and exome. Nucleic Acids Res. 2010, 38, 4027–4039. [Google Scholar] [CrossRef]
- Fan, H.; Chu, J.Y. A brief review of short tandem repeat mutation. Genom. Proteom. Bioinform. 2007, 5, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Krzyzosiak, W.J.; Sobczak, K.; Wojciechowska, M.; Fiszer, A.; Mykowska, A.; Kozlowski, P. Triplet repeat RNA structure and its role as pathogenic agent and therapeutic target. Nucleic Acids Res. 2012, 40, 11–26. [Google Scholar] [CrossRef] [Green Version]
- Lopez Castel, A.; Cleary, J.D.; Pearson, C.E. Repeat instability as the basis for human diseases and as a potential target for therapy. Nat. Rev. Mol. Cell Biol. 2010, 11, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Galka-Marciniak, P.; Urbanek, M.O.; Krzyzosiak, W.J. Triplet repeats in transcripts: Structural insights into RNA toxicity. Biol. Chem. 2012, 393, 1299–1315. [Google Scholar] [CrossRef]
- Jasinska, A.; Krzyzosiak, W.J. Repetitive sequences that shape the human transcriptome. FEBS Lett. 2004, 567, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Craufurd, D.; Thompson, J.C.; Snowden, J.S. Behavioral Changes in Huntington Disease. Cogn. Behav. Neurol. 2001, 14, 219–226. [Google Scholar]
- Lipe, H.; Schultz, A.; Bird, T.D. Risk factors for suicide in huntingtons disease: A retrospective case controlled study. Am. J. Med. Genet. 1993, 48, 231–233. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, J.S.; Ready, R.E.; Hamilton, J.M.; Mega, M.S.; Cummings, J.L. Neuropsychiatric aspects of Huntington’s disease. J. Neurol. Neurosurg. Psychiatry 2001, 71, 310–314. [Google Scholar] [CrossRef] [Green Version]
- Wyant, K.J.; Ridder, A.J.; Dayalu, P. Huntington’s Disease—Update on Treatments. Curr. Neurol. Neurosci. Rep. 2017, 17, 33. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Alegre, P.; Afifi, A.K. Clinical characteristics of childhood-onset (juvenile) Huntington disease: Report of 12 patients and review of the literature. J. Child. Neurol. 2006, 21, 223–229. [Google Scholar] [CrossRef]
- Cattaneo, E.; Zuccato, C.; Tartari, M. Normal huntingtin function: An alternative approach to Huntington’s disease. Nat. Rev. Neurosci. 2005, 6, 919–930. [Google Scholar] [CrossRef]
- Labbadia, J.; Morimoto, R.I. Huntington’s disease: Underlying molecular mechanisms and emerging concepts. Trends Biochem. Sci. 2013, 38, 378–385. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Katada, S.; Onodera, O. Polyglutamine diseases: Where does toxicity come from? what is toxicity? where are we going? J. Mol. Cell Biol. 2010, 2, 180–191. [Google Scholar] [CrossRef]
- Pan, Y.; Zhu, Y.; Yang, W.; Tycksen, E.; Liu, S.; Palucki, J.; Zhu, L.; Sasaki, Y.; Sharma, M.K.; Kim, A.H.; et al. The role of Twist1 in mutant huntingtin-induced transcriptional alterations and neurotoxicity. J. Biol. Chem. 2018, 293, 11850–11866. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Zhong, S.; Zhang, R.; Kang, K.; Zhang, X.; Xu, Y.; Zhao, C.; Zhao, M. Functional analysis of brain derived neurotrophic factor (BDNF) in Huntington’s disease. Aging (Albany N.Y.) 2021, 13, 6103–6114. [Google Scholar] [CrossRef]
- Park, H.; Poo, M.-m. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 2013, 14, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Canals, J.M.; Pineda, J.R.; Torres-Peraza, J.F.; Bosch, M.; Martín-Ibañez, R.; Munoz, M.T.; Mengod, G.; Ernfors, P.; Alberch, J. Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal de-generation in Huntington’s disease. J. Neurosci. 2004, 24, 7727–7739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.; Zhao, T.; Li, X.J.; Li, S. Mutant Huntingtin Impairs BDNF Release from Astrocytes by Disrupting Conversion of Rab3a-GTP into Rab3a-GDP. J. Neurosci. 2016, 36, 8790–8801. [Google Scholar] [CrossRef] [PubMed]
- El-Daher, M.T.; Hangen, E.; Bruyère, J.; Poizat, G.; Al-Ramahi, I.; Pardo, R.; Bourg, N.; Souquere, S.; Mayet, C.; Pierron, G.; et al. Huntingtin proteolysis releases non-polyQ fragments that cause toxicity through dynamin 1 dysregulation. EMBO J. 2015, 34, 2255–2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinsztein, D.C. Lessons from animal models of Huntington’s disease. Trends Genet. 2002, 18, 202–209. [Google Scholar] [CrossRef]
- Perutz, M.F. Glutamine repeats and neurodegenerative diseases: Molecular aspects. Trends Biochem. Sci. 1999, 24, 58–63. [Google Scholar] [CrossRef]
- Cummings, C.J.; Zoghbi, H.Y. Trinucleotide repeats: Mechanisms and pathophysiology. Annu Rev. Genom. Hum. Genet. 2000, 1, 281–328. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A. Polyglutamine pathogenesis: Emergence of unifying mechanisms for Huntington’s disease and related disorders. Neuron 2002, 35, 819–822. [Google Scholar] [CrossRef] [Green Version]
- Michalik, A.; Van Broeckhoven, C. Pathogenesis of polyglutamine disorders: Aggregation revisited. Hum. Mol. Genet. 2003, 12 (Suppl. 2), R173–R186. [Google Scholar] [CrossRef] [Green Version]
- Hsu, R.J.; Hsiao, K.M.; Lin, M.J.; Li, C.Y.; Wang, L.C.; Chen, L.K.; Pan, H. Long tract of untranslated CAG repeats is deleterious in transgenic mice. PLoS ONE 2011, 6, e16417. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, K.T.; O’Keefe, L.V.; Samaraweera, S.E.; van Eyk, C.L.; McLeod, C.J.; Maloney, C.A.; Dang, T.H.; Suter, C.M.; Richards, R.I. Double-stranded RNA is pathogenic in Drosophila models of expanded repeat neurodegenerative diseases. Hum. Mol. Genet. 2011, 20, 3757–3768. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.C.; Chen, K.Y.; Pan, H.; Wu, C.C.; Chen, P.H.; Liao, Y.T.; Li, C.; Huang, M.L.; Hsiao, K.M. Muscleblind participates in RNA toxicity of expanded CAG and CUG repeats in Caenorhabditis elegans. Cell. Mol. Life Sci. 2011, 68, 1255–1267. [Google Scholar] [CrossRef]
- Bano, D.; Zanetti, F.; Mende, Y.; Nicotera, P. Neurodegenerative processes in Huntington’s disease. Cell Death Dis. 2011, 2, e228. [Google Scholar] [CrossRef]
- Jimenez-Sanchez, M.; Thomson, F.; Zavodszky, E.; Rubinsztein, D.C. Autophagy and polyglutamine diseases. Prog. Neurobiol. 2012, 97, 67–82. [Google Scholar] [CrossRef] [Green Version]
- Bernat, V.; Disney, M.D. RNA Structures as Mediators of Neurological Diseases and as Drug Targets. Neuron 2015, 87, 28–46. [Google Scholar] [CrossRef] [Green Version]
- Nalavade, R.; Griesche, N.; Ryan, D.P.; Hildebrand, S.; Krauss, S. Mechanisms of RNA-induced toxicity in CAG repeat disorders. Cell Death Dis. 2013, 4, e752. [Google Scholar] [CrossRef] [Green Version]
- Schilling, J.; Griesche, N.; Krauß, S. Mechanisms of RNA-Induced Toxicity in Diseases Characterised by CAG Repeat Expansions; John Wiley & Sons, Ltd.: Chichester, UK, 2016. [Google Scholar]
- Mankodi, A.; Teng-Umnuay, P.; Krym, M.; Henderson, D.; Swanson, M.; Thornton, C.A. Ribonuclear inclusions in skeletal muscle in myotonic dystrophy types 1 and 2. Ann. Neurol. 2003, 54, 760–768. [Google Scholar] [CrossRef]
- de Mezer, M.; Wojciechowska, M.; Napierala, M.; Sobczak, K.; Krzyzosiak, W.J. Mutant CAG repeats of Huntingtin transcript fold into hairpins, form nuclear foci and are targets for RNA interference. Nucleic Acids Res. 2011, 39, 3852–3863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbanek, M.O.; Fiszer, A.; Krzyzosiak, W.J. Reduction of Huntington’s Disease RNA Foci by CAG Repeat-Targeting Reagents. Front. Cell Neurosci. 2017, 11, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojciechowska, M.; Krzyzosiak, W.J. Cellular toxicity of expanded RNA repeats: Focus on RNA foci. Hum. Mol. Genet. 2011, 20, 3811–3821. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Onisko, B.; Napoli, J.L. The nuclear transcription factor RARalpha associates with neuronal RNA granules and suppresses translation. J. Biol. Chem. 2008, 283, 20841–20847. [Google Scholar] [CrossRef] [Green Version]
- De Graeve, F.; Besse, F. Neuronal RNP granules: From physiological to pathological assemblies. Biol. Chem. 2018, 399, 623–635. [Google Scholar] [CrossRef]
- Moujaber, O.; Stochaj, U. Cytoplasmic RNA Granules in Somatic Maintenance. Gerontology 2018, 64, 485–494. [Google Scholar] [CrossRef]
- Savas, J.N.; Ma, B.; Deinhardt, K.; Culver, B.P.; Restituito, S.; Wu, L.; Belasco, J.G.; Chao, M.V.; Tanese, N. A role for huntington disease protein in dendritic RNA granules. J. Biol. Chem. 2010, 285, 13142–13153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinz, A.; Nabariya, D.K.; Krauß, S. Huntington’s Disease and Neurodegeneration. In Handbook of Neurotoxicity; Springer: Cham, Switzerland, 2021. [Google Scholar]
- Barbee, S.A.; Estes, P.S.; Cziko, A.M.; Hillebrand, J.; Luedeman, R.A.; Coller, J.M.; Johnson, N.; Howlett, I.C.; Geng, C.; Ueda, R.; et al. Staufen- and FMRP-containing neuronal RNPs are structurally and functionally related to somatic P bodies. Neuron 2006, 52, 997–1009. [Google Scholar] [CrossRef] [Green Version]
- Cougot, N.; Bhattacharyya, S.N.; Tapia-Arancibia, L.; Bordonne, R.; Filipowicz, W.; Bertrand, E.; Rage, F. Dendrites of mammalian neurons contain specialized P-body-like structures that respond to neuronal activation. J. Neurosci. 2008, 28, 13793–13804. [Google Scholar] [CrossRef] [Green Version]
- Zeitelhofer, M.; Karra, D.; Macchi, P.; Tolino, M.; Thomas, S.; Schwarz, M.; Kiebler, M.; Dahm, R. Dynamic interaction between P-bodies and transport ribonucleoprotein particles in dendrites of mature hippocampal neurons. J. Neurosci. 2008, 28, 7555–7562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolozin, B. Regulated protein aggregation: Stress granules and neurodegeneration. Mol. Neurodegener. 2012, 7, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kedersha, N.; Anderson, P. Mammalian stress granules and processing bodies. Methods Enzym. 2007, 431, 61–81. [Google Scholar] [CrossRef]
- Buchan, J.R.; Parker, R. Eukaryotic stress granules: The ins and outs of translation. Mol. Cell 2009, 36, 932–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanderweyde, T.; Youmans, K.; Liu-Yesucevitz, L.; Wolozin, B. Role of stress granules and RNA-binding proteins in neurodegeneration: A mini-review. Gerontology 2013, 59, 524–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukong, K.E.; Chang, K.W.; Khandjian, E.W.; Richard, S. RNA-binding proteins in human genetic disease. Trends Genet. 2008, 24, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, N.C.; Wang, Y.D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; Molliex, A.; et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013, 495, 467–473. [Google Scholar] [CrossRef]
- Schilling, J.; Broemer, M.; Atanassov, I.; Duernberger, Y.; Vorberg, I.; Dieterich, C.; Dagane, A.; Dittmar, G.; Wanker, E.; van Roon-Mom, W.; et al. Deregulated Splicing Is a Major Mechanism of RNA-Induced Toxicity in Huntington’s Disease. J. Mol. Biol. 2019. [Google Scholar] [CrossRef]
- Gipson, T.A.; Neueder, A.; Wexler, N.S.; Bates, G.P.; Housman, D. Aberrantly spliced HTT, a new player in Huntington’s disease pathogenesis. RNA Biol. 2013, 10, 1647–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sathasivam, K.; Neueder, A.; Gipson, T.A.; Landles, C.; Benjamin, A.C.; Bondulich, M.K.; Smith, D.L.; Faull, R.L.; Roos, R.A.; Howland, D.; et al. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. USA 2013, 110, 2366–2370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franich, N.R.; Hickey, M.A.; Zhu, C.; Osborne, G.F.; Ali, N.; Chu, T.; Bove, N.H.; Lemesre, V.; Lerner, R.P.; Zeitlin, S.O.; et al. Phenotype onset in Huntington’s disease knock-in mice is correlated with the incomplete splicing of the mutant huntingtin gene. J. Neurosci. Res. 2019, 97, 1590–1605. [Google Scholar] [CrossRef]
- Neueder, A.; Landles, C.; Ghosh, R.; Howland, D.; Myers, R.H.; Faull, R.L.M.; Tabrizi, S.J.; Bates, G.P. The pathogenic exon 1 HTT protein is produced by incomplete splicing in Huntington’s disease patients. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Neueder, A.; Dumas, A.A.; Benjamin, A.C.; Bates, G.P. Regulatory mechanisms of incomplete huntingtin mRNA splicing. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Mason, M.A.; Gomez-Paredes, C.; Sathasivam, K.; Neueder, A.; Papadopoulou, A.S.; Bates, G.P. Silencing Srsf6 does not modulate incomplete splicing of the huntingtin gene in Huntington’s disease models. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Mykowska, A.; Sobczak, K.; Wojciechowska, M.; Kozlowski, P.; Krzyzosiak, W.J. CAG repeats mimic CUG repeats in the misregulation of alternative splicing. Nucleic Acids Res. 2011, 39, 8938–8951. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Park, J.W.; Ramachandran, S.; Zhang, Y.; Tseng, Y.T.; Shen, S.; Waldvogel, H.J.; Curtis, M.A.; Faull, R.L.; Troncoso, J.C.; et al. Transcriptome sequencing reveals aberrant alternative splicing in Huntington’s disease. Hum. Mol. Genet. 2016, 25, 3454–3466. [Google Scholar] [CrossRef] [Green Version]
- Elorza, A.; Márquez, Y.; Cabrera, J.R.; Sánchez-Trincado, J.L.; Santos-Galindo, M.; Hernández, I.H.; Díaz-Hernández, J.I.; García-Escudero, R.; Irimia, M.; Lucas, J.J. Huntington’s disease-specific mis-splicing captured by human-mouse intersect-RNA-seq unveils pathogenic effectors and reduced splicing factors. bioRxiv 2020. [Google Scholar] [CrossRef]
- Krauss, S.; Griesche, N.; Jastrzebska, E.; Chen, C.; Rutschow, D.; Achmuller, C.; Dorn, S.; Boesch, S.M.; Lalowski, M.; Wanker, E.; et al. Translation of HTT mRNA with expanded CAG repeats is regulated by the MID1-PP2A protein complex. Nat. Commun. 2013, 4, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meroni, G.; Diez-Roux, G. TRIM/RBCC, a novel class of ‘single protein RING finger’ E3 ubiquitin ligases. Bioessays 2005, 27, 1147–1157. [Google Scholar] [CrossRef]
- Trockenbacher, A.; Suckow, V.; Foerster, J.; Winter, J.; Krauss, S.; Ropers, H.H.; Schneider, R.; Schweiger, S. MID1, mutated in Opitz syndrome, encodes an ubiquitin ligase that targets phosphatase 2A for degradation. Nat. Genet. 2001, 29, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Knutzen, C.A.; Krauss, S.; Schweiger, S.; Chiang, G.G. Control of mTORC1 signaling by the Opitz syndrome protein MID1. Proc. Natl. Acad. Sci. USA 2011, 108, 8680–8685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nojima, H.; Tokunaga, C.; Eguchi, S.; Oshiro, N.; Hidayat, S.; Yoshino, K.-i.; Hara, K.; Tanaka, N.; Avruch, J.; Yonezawa, K. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J. Biol. Chem. 2003, 278, 15461–15464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griesche, N.; Schilling, J.; Weber, S.; Rohm, M.; Pesch, V.; Matthes, F.; Auburger, G.; Krauss, S. Regulation of mRNA translation by MID1: A common mechanism of expanded CAG repeat RNAs. Front. Cell. Neurosci. 2016. [Google Scholar] [CrossRef] [Green Version]
- Zu, T.; Gibbens, B.; Doty, N.S.; Gomes-Pereira, M.; Huguet, A.; Stone, M.D.; Margolis, J.; Peterson, M.; Markowski, T.W.; Ingram, M.A.; et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. USA 2011, 108, 260–265. [Google Scholar] [CrossRef] [Green Version]
- Banez-Coronel, M.; Ayhan, F.; Tarabochia, A.D.; Zu, T.; Perez, B.A.; Tusi, S.K.; Pletnikova, O.; Borchelt, D.R.; Ross, C.A.; Margolis, R.L.; et al. RAN Translation in Huntington Disease. Neuron 2015, 88, 667–677. [Google Scholar] [CrossRef] [Green Version]
- Rudich, P.D.; Watkins, S.; Lamitina, T. PolyQ-independent toxicity associated with novel translational products from CAG repeat expansions. PLoS ONE 2020, 15, e0227464. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Yang, H.; Huang, L.; Chen, L.; Qin, Z.; Li, S.; Li, X.-J. Lack of RAN-mediated toxicity in Huntington’s disease knock-in mice. Proc. Natl. Acad. Sci. USA 2020, 117, 4411–4417. [Google Scholar] [CrossRef]
- Eshraghi, M.; Karunadharma, P.P.; Blin, J.; Shahani, N.; Ricci, E.P.; Michel, A.; Urban, N.T.; Galli, N.; Sharma, M.; Ramírez-Jarquín, U.N.; et al. Mutant Huntingtin stalls ribosomes and represses protein synthesis in a cellular model of Huntington disease. Nat. Commun. 2021, 12, 1–20. [Google Scholar] [CrossRef]
- Weydt, P.; Pineda, V.V.; Torrence, A.E.; Libby, R.T.; Satterfield, T.F.; Lazarowski, E.R.; Gilbert, M.L.; Morton, G.J.; Bammler, T.K.; Strand, A.D.; et al. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1α in Huntington’s disease neurodegeneration. Cell Metab. 2006, 4, 349–362. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.; Jeong, H.; Borovecki, F.; Parkhurst, C.N.; Tanese, N.; Krainc, D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 2006, 127, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Jesse, S.; Bayer, H.; Alupei, M.C.; Zügel, M.; Mulaw, M.; Tuorto, F.; Malmsheimer, S.; Singh, K.; Steinacker, J.; Schumann, U.; et al. Ribosomal transcription is regulated by PGC-1alpha and disturbed in Huntington’s disease. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, E.P.; Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigo, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cech, T.R.; Steitz, J.A. The noncoding RNA revolution-trashing old rules to forge new ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of novel genes coding for small expressed RNAs. Science 2001, 294, 853–858. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.C.; Ambros, V. An extensive class of small RNAs in Caenorhabditis elegans. Science 2001, 294, 862–864. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Radmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef]
- Han, J.; Lee, Y.; Yeom, K.H.; Kim, Y.K.; Jin, H.; Kim, V.N. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004, 18, 3016–3027. [Google Scholar] [CrossRef] [Green Version]
- Yi, R.; Qin, Y.; Macara, I.G.; Cullen, B.R. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003, 17, 3011–3016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohnsack, M.T.; Czaplinski, K.; Gorlich, D. Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA 2004, 10, 185–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, F.; Ye, X.; Liu, X.; Fincher, L.; McKearin, D.; Liu, Q. Dicer-1 and R3D1-L catalyze microRNA maturation in Drosophila. Genes Dev. 2005, 19, 1674–1679. [Google Scholar] [CrossRef] [Green Version]
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Norman, J.; Cooch, N.; Nishikura, K.; Shiekhattar, R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005, 436, 740–744. [Google Scholar] [CrossRef]
- Haase, A.D.; Jaskiewicz, L.; Zhang, H.; Laine, S.; Sack, R.; Gatignol, A.; Filipowicz, W. TRBP, a regulator of cellular PKR and HIV-1 virus expression, interacts with Dicer and functions in RNA silencing. EMBO Rep. 2005, 6, 961–967. [Google Scholar] [CrossRef]
- Ketting, R.F.; Fischer, S.E.; Bernstein, E.; Sijen, T.; Hannon, G.J.; Plasterk, R.H. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev. 2001, 15, 2654–2659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filipowicz, W. RNAi: The nuts and bolts of the RISC machine. Cell 2005, 122, 17–20. [Google Scholar] [CrossRef] [Green Version]
- Robb, G.B.; Rana, T.M. RNA helicase A interacts with RISC in human cells and functions in RISC loading. Mol. Cell 2007, 26, 523–537. [Google Scholar] [CrossRef]
- Valencia-Sanchez, M.A.; Liu, J.; Hannon, G.J.; Parker, R. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 2006, 20, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Parker, R.; Song, H. The enzymes and control of eukaryotic mRNA turnover. Nat. Struct. Mol. Biol. 2004, 11, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Petersen, C.P.; Bordeleau, M.E.; Pelletier, J.; Sharp, P.A. Short RNAs repress translation after initiation in mammalian cells. Mol. Cell 2006, 21, 533–542. [Google Scholar] [CrossRef]
- Krol, J.; Fiszer, A.; Mykowska, A.; Sobczak, K.; de Mezer, M.; Krzyzosiak, W.J. Ribonuclease dicer cleaves triplet repeat hairpins into shorter repeats that silence specific targets. Mol. Cell 2007, 25, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.W.; Rudnicki, D.D.; Yu, L.; Margolis, R.L. A natural antisense transcript at the Huntington’s disease repeat locus regulates HTT expression. Hum. Mol. Genet. 2011, 20, 3467–3477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banez-Coronel, M.; Porta, S.; Kagerbauer, B.; Mateu-Huertas, E.; Pantano, L.; Ferrer, I.; Guzman, M.; Estivill, X.; Marti, E. A pathogenic mechanism in Huntington’s disease involves small CAG-repeated RNAs with neurotoxic activity. PLoS Genet. 2012, 8, e1002481. [Google Scholar] [CrossRef] [Green Version]
- Marti, E. RNA toxicity induced by expanded CAG repeats in Huntington’s disease. Brain Pathol. 2016, 26, 779–786. [Google Scholar] [CrossRef]
- Peng, S.; Guo, P.; Lin, X.; An, Y.; Sze, K.H.; Lau, M.H.Y.; Chen, Z.S.; Wang, Q.; Li, W.; Sun, J.K.; et al. CAG RNAs induce DNA damage and apoptosis by silencing NUDT16 expression in polyglutamine degeneration. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Marti, E.; Pantano, L.; Banez-Coronel, M.; Llorens, F.; Minones-Moyano, E.; Porta, S.; Sumoy, L.; Ferrer, I.; Estivill, X. A myriad of miRNA variants in control and Huntington’s disease brain regions detected by massively parallel sequencing. Nucleic Acids Res. 2010, 38, 7219–7235. [Google Scholar] [CrossRef]
- Krauss, S.; Evert, B.O. The Role of MicroRNAs in Spinocerebellar Ataxia Type 3. J. Mol. Biol. 2019, 431, 1729–1742. [Google Scholar] [CrossRef]
- Krauss, S.; Nalavade, R.; Weber, S.; Carter, K.; Evert, B.O. Upregulation of miR-25 and miR-181 Family Members Correlates with Reduced Expression of ATXN3 in Lymphocytes from SCA3 Patients. Microrna 2019, 8, 76–85. [Google Scholar] [CrossRef]
- Shi, Y.; Huang, F.; Tang, B.; Li, J.; Wang, J.; Shen, L.; Xia, K.; Jiang, H. MicroRNA profiling in the serums of SCA3/MJD patients. Int. J. Neurosci. 2014, 124, 97–101. [Google Scholar] [CrossRef]
- Savas, J.N.; Makusky, A.; Ottosen, S.; Baillat, D.; Then, F.; Krainc, D.; Shiekhattar, R.; Markey, S.P.; Tanese, N. Huntington’s disease protein contributes to RNA-mediated gene silencing through association with Argonaute and P bodies. Proc. Natl. Acad. Sci. USA 2008, 105, 10820–10825. [Google Scholar] [CrossRef] [Green Version]
- Unterbruner, K.; Matthes, F.; Schilling, J.; Nalavade, R.; Weber, S.; Winter, J.; Krauss, S. MicroRNAs miR-19, miR-340, miR-374 and miR-542 regulate MID1 protein expression. PLoS ONE 2018, 13, e0190437. [Google Scholar] [CrossRef] [Green Version]
- Khani-Habibabadi, F.; Askari, S.; Zahiri, J.; Javan, M.; Behmanesh, M. Novel BDNF-regulatory microRNAs in neurodegenerative disorders pathogenesis: An in silico study. Comput. Biol. Chem. 2019, 83, 107153. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Cong, S. Bioinformatic analysis of microRNA expression in Huntington’s disease. Mol. Med. Rep. 2018, 18, 2857–2865. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.L.; Weissman, L.; Bohr, V.A.; Mattson, M.P. Mitochondrial DNA damage and repair in neurodegenerative disorders. DNA Repair 2008, 7, 1110–1120. [Google Scholar] [CrossRef] [Green Version]
- Polidori, M.C.; Mecocci, P.; Browne, S.E.; Senin, U.; Beal, M.F. Oxidative damage to mitochondrial DNA in Huntington’s disease parietal cortex. Neurosci. Lett. 1999, 272, 53–56. [Google Scholar] [CrossRef]
- Siddiqui, A.; Rivera-Sanchez, S.; Castro Mdel, R.; Acevedo-Torres, K.; Rane, A.; Torres-Ramos, C.A.; Nicholls, D.G.; Andersen, J.K.; Ayala-Torres, S. Mitochondrial DNA damage is associated with reduced mitochondrial bioenergetics in Huntington’s disease. Free Radic. Biol. Med. 2012, 53, 1478–1488. [Google Scholar] [CrossRef] [Green Version]
- Brustovetsky, N. Mutant Huntingtin and Elusive Defects in Oxidative Metabolism and Mitochondrial Calcium Handling. Mol. Neurobiol. 2016, 53, 2944–2953. [Google Scholar] [CrossRef] [Green Version]
- Bae, B.I.; Xu, H.; Igarashi, S.; Fujimuro, M.; Agrawal, N.; Taya, Y.; Hayward, S.D.; Moran, T.H.; Montell, C.; Ross, C.A.; et al. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron 2005, 47, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Fenster, R.J.; Pineda, S.S.; Gibbs, W.S.; Mohammadi, S.; Davila-Velderrain, J.; Garcia, F.J.; Therrien, M.; Novis, H.S.; Gao, F.; et al. Cell Type-Specific Transcriptomics Reveals that Mutant Huntingtin Leads to Mitochondrial RNA Release and Neuronal Innate Immune Activation. Neuron 2020, 107, 891–908. [Google Scholar] [CrossRef]
- Ghosh, R.; Wood-Kaczmar, A.; Dobson, L.; Smith, E.J.; Sirinathsinghji, E.C.; Kriston-Vizi, J.; Hargreaves, I.P.; Heaton, R.; Herrmann, F.; Abramov, A.Y.; et al. Expression of mutant exon 1 huntingtin fragments in human neural stem cells and neurons causes inclusion formation and mitochondrial dysfunction. FASEB J. 2020, 34, 8139–8154. [Google Scholar] [CrossRef] [Green Version]
- Arnoux, I.; Willam, M.; Griesche, N.; Krummeich, J.; Watari, H.; Offermann, N.; Weber, S.; Narayan Dey, P.; Chen, C.; Monteiro, O.; et al. Metformin reverses early cortical network dysfunction and behavior changes in Huntington’s disease. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Gantois, I.; Khoutorsky, A.; Popic, J.; Aguilar-Valles, A.; Freemantle, E.; Cao, R.; Sharma, V.; Pooters, T.; Nagpal, A.; Skalecka, A.; et al. Metformin ameliorates core deficits in a mouse model of fragile X syndrome. Nat. Med. 2017, 23, 674–677. [Google Scholar] [CrossRef] [PubMed]
- Matthes, F.; Massari, S.; Bochicchio, A.; Schorpp, K.; Schilling, J.; Weber, S.; Offermann, N.; Desantis, J.; Wanker, E.E.; Carloni, P.; et al. Reducing mutant Huntingtin protein expression in living cells by a newly identified RNA CAG binder. ACS Chem. Neurosci. 2018, 9, 1399–1408. [Google Scholar] [CrossRef] [Green Version]
- Khan, E.; Tawani, A.; Mishra, S.K.; Verma, A.K.; Upadhyay, A.; Kumar, M.; Sandhir, R.; Mishra, A.; Kumar, A. Myricetin Reduces Toxic Level of CAG Repeats RNA in Huntington’s Disease (HD) and Spino Cerebellar Ataxia (SCAs). ACS Chem. Biol. 2018, 13, 180–188. [Google Scholar] [CrossRef]
- Wurster, C.D.; Ludolph, A.C. Antisense oligonucleotides in neurological disorders. Adv. Neurol. Disord. 2018, 11, 1756286418776932. [Google Scholar] [CrossRef] [Green Version]
- Marwick, C. First “antisense” drug will treat CMV retinitis. JAMA 1998, 280, 871. [Google Scholar] [CrossRef]
- Kordasiewicz, H.B.; Stanek, L.M.; Wancewicz, E.V.; Mazur, C.; McAlonis, M.M.; Pytel, K.A.; Artates, J.W.; Weiss, A.; Cheng, S.H.; Shihabuddin, L.S.; et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron 2012, 74, 1031–1044. [Google Scholar] [CrossRef] [Green Version]
- Ostergaard, M.E.; Southwell, A.L.; Kordasiewicz, H.; Watt, A.T.; Skotte, N.H.; Doty, C.N.; Vaid, K.; Villanueva, E.B.; Swayze, E.E.; Bennett, C.F.; et al. Rational design of antisense oligonucleotides targeting single nucleotide polymorphisms for potent and allele selective suppression of mutant Huntingtin in the CNS. Nucleic Acids Res. 2013, 41, 9634–9650. [Google Scholar] [CrossRef] [Green Version]
- Kourkouta, E.; Weij, R.; Gonzalez-Barriga, A.; Mulder, M.; Verheul, R.; Bosgra, S.; Groenendaal, B.; Puolivali, J.; Toivanen, J.; van Deutekom, J.C.T.; et al. Suppression of Mutant Protein Expression in SCA3 and SCA1 Mice Using a CAG Repeat-Targeting Antisense Oligonucleotide. Mol. Nucleic Acids 2019, 17, 601–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evers, M.M.; Pepers, B.A.; van Deutekom, J.C.; Mulders, S.A.; den Dunnen, J.T.; Aartsma-Rus, A.; van Ommen, G.J.; van Roon-Mom, W.M. Targeting several CAG expansion diseases by a single antisense oligonucleotide. PLoS ONE 2011, 6, e24308. [Google Scholar] [CrossRef]
- Datson, N.A.; Gonzalez-Barriga, A.; Kourkouta, E.; Weij, R.; van de Giessen, J.; Mulders, S.; Kontkanen, O.; Heikkinen, T.; Lehtimaki, K.; van Deutekom, J.C. The expanded CAG repeat in the huntingtin gene as target for therapeutic RNA modulation throughout the HD mouse brain. PLoS ONE 2017, 12, e0171127. [Google Scholar] [CrossRef]
- Deeprose, C.; Rosser, A. Looking Back, Looking Foward. EHDN News. 2021. Available online: http://www.ehdn.org/wp-content/uploads/2021/07/EHDN-NL-Jul2021-final.pdf (accessed on 8 July 2021).
- Sciences, W.L. Wave Life Sciences Provides Update on Phase 1b/2a PRECISION-HD Trials. Available online: https://ir.wavelifesciences.com/news-releases/news-release-details/wave-life-sciences-provides-update-phase-1b2a-precision-hd (accessed on 8 July 2021).
- Miniarikova, J.; Zanella, I.; Huseinovic, A.; van der Zon, T.; Hanemaaijer, E.; Martier, R.; Koornneef, A.; Southwell, A.L.; Hayden, M.R.; van Deventer, S.J.; et al. Design, Characterization, and Lead Selection of Therapeutic miRNAs Targeting Huntingtin for Development of Gene Therapy for Huntington’s Disease. Mol. Ther. Nucleic Acids 2016, 5, e297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franich, N.R.; Fitzsimons, H.L.; Fong, D.M.; Klugmann, M.; During, M.J.; Young, D. AAV vector-mediated RNAi of mutant huntingtin expression is neuroprotective in a novel genetic rat model of Huntington’s disease. Mol. Ther. 2008, 16, 947–956. [Google Scholar] [CrossRef] [PubMed]
- McBride, J.L.; Pitzer, M.R.; Boudreau, R.L.; Dufour, B.; Hobbs, T.; Ojeda, S.R.; Davidson, B.L. Preclinical safety of RNAi-mediated HTT suppression in the rhesus macaque as a potential therapy for Huntington’s disease. Mol. Ther. 2011, 19, 2152–2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahara, T.; Yanagi, H.; Yura, T.; Mori, K. Endoplasmic reticulum stress-induced mRNA splicing permits synthesis of transcription factor Hac1p/Ern4p that activates the unfolded protein response. Mol. Biol. Cell 1997, 8, 1845–1862. [Google Scholar] [CrossRef] [Green Version]
- Mesitov, M.V.; Soldatov, R.A.; Zaichenko, D.M.; Malakho, S.G.; Klementyeva, T.S.; Sokolovskaya, A.A.; Kubatiev, A.A.; Mironov, A.A.; Moskovtsev, A.A. Differential processing of small RNAs during endoplasmic reticulum stress. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Barnat, M.; Capizzi, M.; Aparicio, E.; Boluda, S.; Wennagel, D.; Kacher, R.; Kassem, R.; Lenoir, S.; Agasse, F.; Braz, B.Y.; et al. Huntington’s disease alters human neurodevelopment. Science 2020, 369, 787–793. [Google Scholar] [CrossRef]
- Li, L.B.; Yu, Z.; Teng, X.; Bonini, N.M. RNA toxicity is a component of ataxin-3 degeneration in Drosophila. Nature 2008, 453, 1107–1111. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heinz, A.; Nabariya, D.K.; Krauss, S. Huntingtin and Its Role in Mechanisms of RNA-Mediated Toxicity. Toxins 2021, 13, 487. https://doi.org/10.3390/toxins13070487
Heinz A, Nabariya DK, Krauss S. Huntingtin and Its Role in Mechanisms of RNA-Mediated Toxicity. Toxins. 2021; 13(7):487. https://doi.org/10.3390/toxins13070487
Chicago/Turabian StyleHeinz, Annika, Deepti Kailash Nabariya, and Sybille Krauss. 2021. "Huntingtin and Its Role in Mechanisms of RNA-Mediated Toxicity" Toxins 13, no. 7: 487. https://doi.org/10.3390/toxins13070487
APA StyleHeinz, A., Nabariya, D. K., & Krauss, S. (2021). Huntingtin and Its Role in Mechanisms of RNA-Mediated Toxicity. Toxins, 13(7), 487. https://doi.org/10.3390/toxins13070487