The Impact of CKD on Uremic Toxins and Gut Microbiota



Abstract
1. Introduction
2. Gut Microbiome in Health
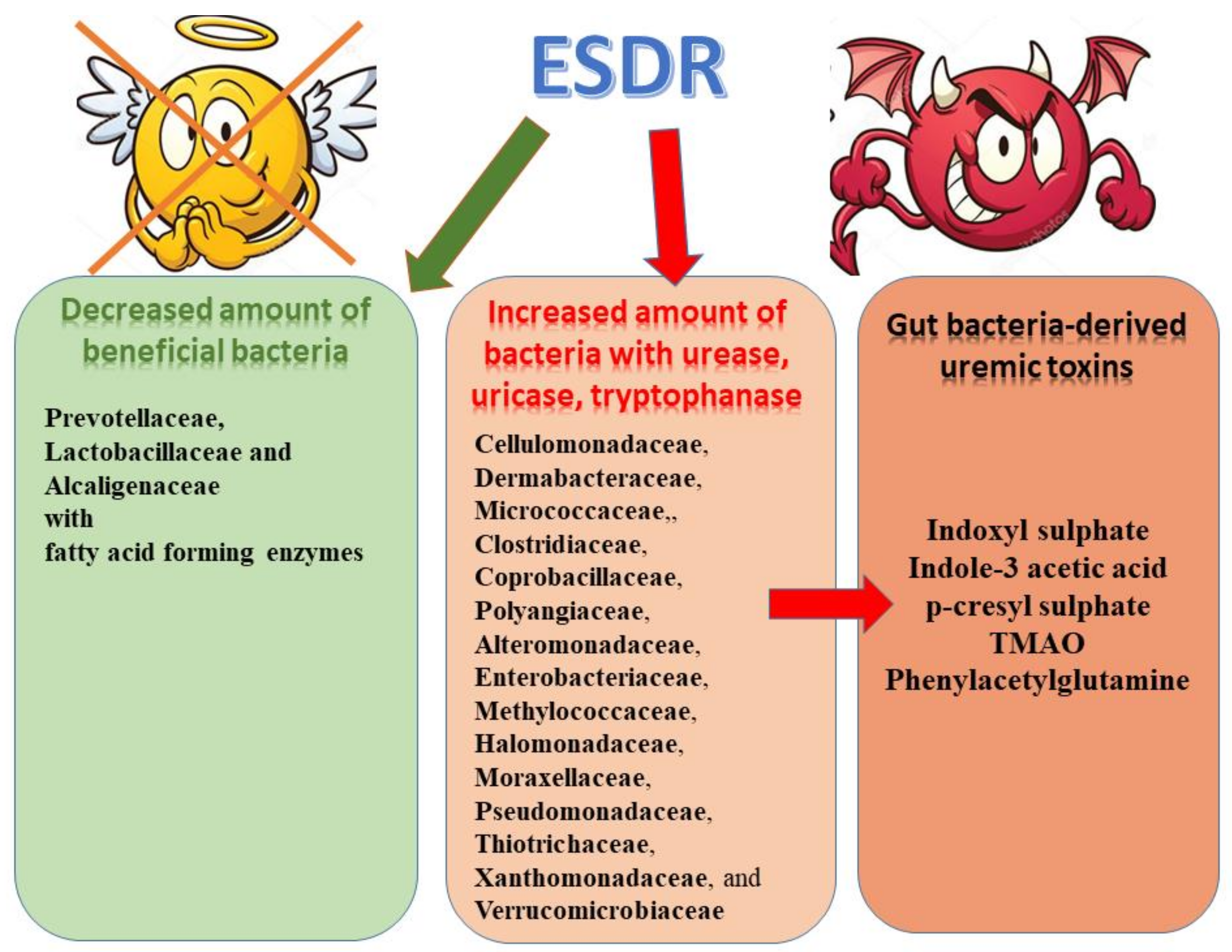
3. Gut Microbiome in CKD
4. Adverse Effects of Gut-Derived Uremic Toxins and Microbial Metabolites
5. Therapeutic Potential
5.1. Probiotics
5.2. Prebiotics
5.3. Synbiotics
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABI | ankle brachial index |
| BAR-GTN | brachial artery response to glyceryl trinitrate |
| CAD | cardiovascular disease |
| CAPD | continuous ambulatory peritoneal dialysis |
| cIMT | carotid intima-media thickness |
| CKD | chronic kidney disease |
| Cl | confidence interval |
| COX-2 | cycloxygenase-2 |
| CRF | chronic renal failure |
| CRP | C-reactive protein |
| EMT | tubular epithelial-to-mesenchymal transition |
| ESKD | end-stage kidney disease |
| ESRD | end-stage renal disease |
| FGF 23 | fibroblast growth factor 23 |
| GBM | glomerular basement membrane |
| HD | hemodialysis |
| HR | hazard ratio |
| HUVEC | human umbilical vein endothelial cells |
| IAA | indole acetic acid |
| IL-6 | interleukin-6 |
| iNOS | inducible nitric oxide synthase |
| IS | indoxyl sulphate |
| IRI | ischemia reperfusion injury |
| LPS | lipopolysaccharide |
| MAPK | mitogen-activated protein kinase |
| NO | nitric oxide |
| OAT1, OAT3 | organic anion transporters |
| OCL | osteoclast |
| OR | odds ratio |
| OTUs | operational taxonomic units |
| PAG | Phenylacetylglutamine |
| PBUT | protein-bound uremic toxins |
| pCG | p-cresyl glucuronide |
| PCS | p-cresyl sulphate |
| PDGF | platelet-derived growth factor |
| PTA | percutaneous transluminal angioplasty |
| PTH | parathormon |
| PWV | pulse wave velocity |
| SDGT | Sodium-dependent glucose cotransporters |
| RAAS | renal renin-angiotensin-aldosterone system |
| ROS | reactive oxygen species |
| RTR | renal transplant recipients |
| SCFA | Short chain fatty acids |
| TGF-β1 | transforming growth factor β1 |
| TIMP | tissue inhibitor of matrix proteinase |
| TLRs | toll-like receptors |
| TMAO | trimethylamine N-oxide |
| TNF-α | tumor necrosis factor-α |
| VMSC | vascular smooth muscle cell |
References
- Ramezani, A.; Massy, Z.A.; Meijers, B.; Evenepoel, P.; Vanholder, R.; Raj, D.S. Role of the Gut Microbiome in Uremia: A Potential Therapeutic Target. Am. J. Kidney Dis. 2016, 67, 483–498. [Google Scholar] [CrossRef]
- Ramezani, A.; Raj, D.S. The Gut Microbiome, Kidney Disease, and Targeted Interventions. J. Am. Soc. Nephrol. 2014, 25, 657–670. [Google Scholar] [CrossRef]
- Li, D.Y.; Tang, W.H.W. Contributory Role of Gut Microbiota and Their Metabolites toward Cardiovascular Complications in Chronic Kidney Disease. Semin. Nephrol. 2018, 38, 193–205. [Google Scholar] [CrossRef]
- Chen, Y.-Y.; Chen, D.-Q.; Chen, L.; Liu, J.-R.; Vaziri, N.D.; Guo, Y.; Zhao, Y.-Y. Microbiome–metabolome reveals the contribution of gut–kidney axis on kidney disease. J. Transl. Med. 2019, 17, 5. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Wong, J.; Pahl, M.; Piceno, Y.M.; Yuan, J.; DeSantis, T.Z.; Ni, Z.; Nguyen, T.H.; Andersen, G.L. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013, 83, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Piceno, Y.M.; DeSantis, T.Z.; Pahl, M.; Andersen, G.L.; Vaziri, N.D. Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am. J. Nephrol. 2014, 39, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Hida, M.; Aiba, Y.; Sawamura, S.; Suzuki, N.; Satoh, T.; Koga, Y. Inhibition of the accumulation of uremic toxins in the blood and their precursors in the feces after oral administration of Lebenin, a lactic acid bacteria preparation, to uremic patients undergoing hemodialysis. Nephron 1996, 74, 349–355. [Google Scholar] [CrossRef]
- Meyer, T.W.; Hostetter, T.H. Uremia. N. Engl. J. Med. 2007, 357, 1316–1325. [Google Scholar] [CrossRef] [PubMed]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef]
- Wang, X.; Yang, S.; Li, S.; Zhao, L.; Hao, Y.; Qin, J.; Zhang, L.; Zhang, C.; Bian, W.; Zuo, L.; et al. Aberrant gut microbiota alters host metabolome and impacts renal failure in humans and rodents. Gut 2020, 69, 2131–2142. [Google Scholar] [CrossRef]
- Mishima, E.; Fukuda, S.; Mukawa, C.; Yuri, A.; Kanemitsu, Y.; Matsumoto, Y.; Akiyama, Y.; Fukuda, N.N.; Tsukamoto, H.; Asaji, K.; et al. Evaluation of the impact of gut microbiota on uremic solute accumulation by a CE-TOFMS-based metabolomics approach. Kidney Int. 2017, 92, 634–645. [Google Scholar] [CrossRef]
- Kang, J.Y. The gastrointestinal tract in uremia. Dig. Dis. Sci. 1993, 38, 257–268. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Dure-Smith, B.; Miller, R.; Mirahmadi, M.K. Pathology of gastrointestinal tract in chronic hemodialysis patients: An autopsy study of 78 cases. Am. J. Gastroenterol. 1985, 80, 608–611. [Google Scholar]
- Walser, M.; Bodenlos, L.J. Urea metabolism in man. J. Clin. Investig. 1959, 38, 1617–1626. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.S.; Smith, C.P. Urea nitrogen salvage mechanisms and their relevance to ruminants, non-ruminants and man. Nutr. Res. Rev. 2005, 18, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Hatch, M.; Vaziri, N.D. Enhanced enteric excretion of urate in rats with chronic renal failure. Clin. Sci. 1994, 86, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, M.; Magnusson, K.E.; Sundqvist, T.; Denneberg, T. Impaired intestinal barrier function measured by differently sized polyethylene glycols in patients with chronic renal failure. Gut 1991, 32, 754–759. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Goshtasbi, N.; Yuan, J.; Jellbauer, S.; Moradi, H.; Raffatellu, M.; Kalantar-Zadeh, K. Uremic plasma impairs barrier function and depletes the tight junction protein constituents of intestinal epithelium. Am. J. Nephrol. 2012, 36, 438–443. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Yuan, J.; Norris, K. Role of urea in intestinal barrier dysfunction and disruption of epithelial tight junction in chronic kidney disease. Am. J. Nephrol. 2013, 37, 1–6. [Google Scholar] [CrossRef]
- Guldris, S.C.; Parra, E.G.; Amenόs, A.C. Gut microbiota in chronic kidney disease. Nefrología 2017, 37, 9–19. [Google Scholar] [CrossRef]
- Ritz, E. Intestinal-renal syndrome: Mirage or reality? Blood Purif. 2011, 31, 70–76. [Google Scholar] [CrossRef]
- Sabatino, A.; Regolisti, G.; Brusasco, I.; Cabassi, A.; Morabito, S.; Fiaccadori, E. Alterations of intestinal barrier and microbiota in chronic kidney disease. Nephrol. Dial. Transplant. 2014, 30, 924–933. [Google Scholar] [CrossRef] [PubMed]
- De Almeida Duarte, J.B.; de Aguilar-Nascimento, J.E.; Nascimento, M.; Nochi, R.J., Jr. Bacterial translocation in experimental uremia. Urol. Res. 2004, 32, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, D.C.; Dignass, A.U. Intestinal barrier function. Curr. Opin. Clin. Nutr. Metab. Care 2002, 5, 685–694. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Yuan, J.; Nazertehrani, S.; Ni, Z.; Liu, S. Chronic kidney disease causes disruption of gastric and small intestinal epithelial tight junction. Am. J. Nephrol. 2013, 38, 99–103. [Google Scholar] [CrossRef]
- Jernberg, C.; Löfmark, S.; Edlund, C.; Jansson, J.K. Long-term impacts of antibiotic exposure on the human intestinal microbiota. Microbiology 2010, 156, 3216–3223. [Google Scholar] [CrossRef]
- Wu, M.J.; Chang, C.S.; Cheng, C.H.; Chen, C.H.; Lee, W.C.; Hsu, Y.H.; Shu, K.H.; Tang, M.J. Colonic transit time in long-term dialysis patients. Am. J. Kidney Dis. 2004, 44, 322–327. [Google Scholar] [CrossRef]
- Devlin, A.S.; Marcobal, A.; Dodd, D.; Nayfach, S.; Plummer, N.; Meyer, T.; Pollard, K.S.; Sonnenburg, J.L.; Fischbach, M.A. Modulation of a Circulating Uremic Solute via Rational Genetic Manipulation of the Gut Microbiota. Cell Host Microbe 2016, 20, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Gevers, D.; Knight, R.; Petrosino, J.F.; Huang, K.; McGuire, A.L.; Birren, B.W.; Nelson, K.E.; White, O.; Methé, B.A.; Huttenhower, C. The Human Microbiome Project: A community resource for the healthy human microbiome. PLoS Biol. 2012, 10, e1001377. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Gordon, J.I. Honor thy symbionts. Proc. Natl. Acad. Sci. USA 2003, 100, 10452–10459. [Google Scholar] [CrossRef]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinal microbial flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef]
- Tremaroli, V.; Bäckhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Odenwald, M.A.; Turner, J.R. The intestinal epithelial barrier: A therapeutic target? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 9–21. [Google Scholar] [CrossRef]
- Ulluwishewa, D.; Anderson, R.C.; McNabb, W.C.; Moughan, P.J.; Wells, J.M.; Roy, N.C. Regulation of tight junction permeability by intestinal bacteria and dietary components. J. Nutr. 2011, 141, 769–776. [Google Scholar] [CrossRef]
- Cario, E.; Gerken, G.; Podolsky, D.K. Toll-like receptor 2 controls mucosal inflammation by regulating epithelial barrier function. Gastroenterology 2007, 132, 1359–1374. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.J. Intestinal flora and endogenous vitamin synthesis. Eur. J. Cancer Prev. 1997, 6 (Suppl. S1), S43–S45. [Google Scholar] [CrossRef]
- Hylemon, P.B.; Harder, J. Biotransformation of monoterpenes, bile acids, and other isoprenoids in anaerobic ecosystems. FEMS Microbiol. Rev. 1998, 22, 475–488. [Google Scholar] [CrossRef]
- Duncan, S.H.; Richardson, A.J.; Kaul, P.; Holmes, R.P.; Allison, M.J.; Stewart, C.S. Oxalobacter formigenes and Its Potential Role in Human Health. Appl. Environ. Microbiol. 2002, 68, 3841–3847. [Google Scholar] [CrossRef]
- Cummings, J.H.; Macfarlane, G.T. Role of intestinal bacteria in nutrient metabolism. JPEN J. Parenter. Enter. Nutr. 1997, 21, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Blaut, M.; Clavel, T. Metabolic diversity of the intestinal microbiota: Implications for health and disease. J. Nutr. 2007, 137, 751s–755s. [Google Scholar] [CrossRef]
- Tao, Y.; Drabik, K.A.; Waypa, T.S.; Musch, M.W.; Alverdy, J.C.; Schneewind, O.; Chang, E.B.; Petrof, E.O. Soluble factors from Lactobacillus GG activate MAPKs and induce cytoprotective heat shock proteins in intestinal epithelial cells. Am. J. Physiol. Cell Physiol. 2006, 290, C1018–C1030. [Google Scholar] [CrossRef]
- Ewaschuk, J.B.; Diaz, H.; Meddings, L.; Diederichs, B.; Dmytrash, A.; Backer, J.; Looijer-van Langen, M.; Madsen, K.L. Secreted bioactive factors from Bifidobacterium infantis enhance epithelial cell barrier function. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G1025–G1034. [Google Scholar] [CrossRef]
- Mattar, A.F.; Teitelbaum, D.H.; Drongowski, R.A.; Yongyi, F.; Harmon, C.M.; Coran, A.G. Probiotics up-regulate MUC-2 mucin gene expression in a Caco-2 cell-culture model. Pediatr. Surg. Int. 2002, 18, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Schlee, M.; Harder, J.; Köten, B.; Stange, E.F.; Wehkamp, J.; Fellermann, K. Probiotic lactobacilli and VSL#3 induce enterocyte beta-defensin 2. Clin. Exp. Immunol. 2008, 151, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Rakoff-Nahoum, S.; Paglino, J.; Eslami-Varzaneh, F.; Edberg, S.; Medzhitov, R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 2004, 118, 229–241. [Google Scholar] [CrossRef]
- Layden, B.T.; Angueira, A.R.; Brodsky, M.; Durai, V.; Lowe, W.L., Jr. Short chain fatty acids and their receptors: New metabolic targets. Transl. Res. 2013, 161, 131–140. [Google Scholar] [CrossRef]
- Andrade-Oliveira, V.; Amano, M.T.; Correa-Costa, M.; Castoldi, A.; Felizardo, R.J.; de Almeida, D.C.; Bassi, E.J.; Moraes-Vieira, P.M.; Hiyane, M.I.; Rodas, A.C.; et al. Gut Bacteria Products Prevent AKI Induced by Ischemia-Reperfusion. J. Am. Soc. Nephrol. 2015, 26, 1877–1888. [Google Scholar] [CrossRef]
- Barrows, I.R.; Ramezani, A.; Raj, D.S. Gut Feeling in AKI: The Long Arm of Short-Chain Fatty Acids. J. Am. Soc. Nephrol. 2015, 26, 1755–1757. [Google Scholar] [CrossRef] [PubMed][Green Version]
- De Andrade, L.S.; Ramos, C.I.; Cuppari, L. The cross-talk between the kidney and the gut: Implications for chronic kidney disease. Nutrire 2017, 42, 27. [Google Scholar] [CrossRef]
- Simenhoff, M.L.; Dunn, S.R.; Zollner, G.P.; Fitzpatrick, M.E.; Emery, S.M.; Sandine, W.E.; Ayres, J.W. Biomodulation of the toxic and nutritional effects of small bowel bacterial overgrowth in end-stage kidney disease using freeze-dried Lactobacillus acidophilus. Min. Electrolyte Metab. 1996, 22, 92–96. [Google Scholar]
- Moco, S.; Martin, F.P.; Rezzi, S. Metabolomics view on gut microbiome modulation by polyphenol-rich foods. J. Proteome Res. 2012, 11, 4781–4790. [Google Scholar] [CrossRef]
- Hugon, P.; Ramasamy, D.; Lagier, J.-C.; Rivet, R.; Couderc, C.; Raoult, D.; Fournier, P.-E. Non contiguous-finished genome sequence and description of Alistipes obesi sp. nov. Stand. Genom. Sci. 2013, 7, 427–439. [Google Scholar] [CrossRef]
- Dawson, L.F.; Stabler, R.A.; Wren, B.W. Assessing the role of p-cresol tolerance in Clostridium difficile. J. Med. Microbiol. 2008, 57, 745–749. [Google Scholar] [CrossRef] [PubMed]
- Nakabayashi, I.; Nakamura, M.; Kawakami, K.; Ohta, T.; Kato, I.; Uchida, K.; Yoshida, M. Effects of synbiotic treatment on serum level of p-cresol in haemodialysis patients: A preliminary study. Nephrol. Dial. Transplant. 2011, 26, 1094–1098. [Google Scholar] [CrossRef]
- Joossens, M.; Faust, K.; Gryp, T.; Nguyen, A.T.L.; Wang, J.; Eloot, S.; Schepers, E.; Dhondt, A.; Pletinck, A.; Vieira-Silva, S.; et al. Gut microbiota dynamics and uraemic toxins: One size does not fit all. Gut 2019, 68, 2257–2260. [Google Scholar] [CrossRef]
- Poesen, R.; Windey, K.; Neven, E.; Kuypers, D.; De Preter, V.; Augustijns, P.; D’Haese, P.; Evenepoel, P.; Verbeke, K.; Meijers, B. The Influence of CKD on Colonic Microbial Metabolism. J. Am. Soc. Nephrol. 2016, 27, 1389–1399. [Google Scholar] [CrossRef] [PubMed]
- Gryp, T.; De Paepe, K.; Vanholder, R.; Kerckhof, F.-M.; Van Biesen, W.; Van de Wiele, T.; Verbeke, F.; Speeckaert, M.; Joossens, M.; Couttenye, M.M.; et al. Gut microbiota generation of protein-bound uremic toxins and related metabolites is not altered at different stages of chronic kidney disease. Kidney Int. 2020, 97, 1230–1242. [Google Scholar] [CrossRef] [PubMed]
- Bammens, B.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. Evidence for impaired assimilation of protein in chronic renal failure. Kidney Int. 2003, 64, 2196–2203. [Google Scholar] [CrossRef]
- Krishnamurthy, V.M.; Wei, G.; Baird, B.C.; Murtaugh, M.; Chonchol, M.B.; Raphael, K.L.; Greene, T.; Beddhu, S. High dietary fiber intake is associated with decreased inflammation and all-cause mortality in patients with chronic kidney disease. Kidney Int. 2012, 81, 300–306. [Google Scholar] [CrossRef]
- Wandersman, C.; Delepelaire, P. Bacterial iron sources: From siderophores to hemophores. Annu. Rev. Microbiol. 2004, 58, 611–647. [Google Scholar] [CrossRef]
- Qureshi, A.R.; Alvestrand, A.; Danielsson, A.; Divino-Filho, J.C.; Gutierrez, A.; Lindholm, B.; Bergström, J. Factors predicting malnutrition in hemodialysis patients: A cross-sectional study. Kidney Int. 1998, 53, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, L.B. Role of the intestinal tract in the elimination of uric acid. Arthritis Rheumatol. 1965, 8, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Zhao, Y.Y.; Pahl, M.V. Altered intestinal microbial flora and impaired epithelial barrier structure and function in CKD: The nature, mechanisms, consequences and potential treatment. Nephrol. Dial. Transplant. 2016, 31, 737–746. [Google Scholar] [CrossRef]
- Kanbay, M.; Onal, E.M.; Afsar, B.; Dagel, T.; Yerlikaya, A.; Covic, A.; Vaziri, N.D. The crosstalk of gut microbiota and chronic kidney disease: Role of inflammation, proteinuria, hypertension, and diabetes mellitus. Int. Urol. Nephrol. 2018, 50, 1453–1466. [Google Scholar] [CrossRef]
- Satoh, M.; Hayashi, H.; Watanabe, M.; Ueda, K.; Yamato, H.; Yoshioka, T.; Motojima, M. Uremic toxins overload accelerates renal damage in a rat model of chronic renal failure. Nephron Exp. Nephrol. 2003, 95, e111–e118. [Google Scholar] [CrossRef]
- Motojima, M.; Hosokawa, A.; Yamato, H.; Muraki, T.; Yoshioka, T. Uraemic toxins induce proximal tubular injury via organic anion transporter 1-mediated uptake. Br. J. Pharm. 2002, 135, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Bertrand, E.; Cerini, C.; Faure, V.; Sampol, J.; Vanholder, R.; Berland, Y.; Brunet, P. The uremic solutes p-cresol and indoxyl sulfate inhibit endothelial proliferation and wound repair. Kidney Int. 2004, 65, 442–451. [Google Scholar] [CrossRef]
- Schepers, E.; Meert, N.; Glorieux, G.; Goeman, J.; Van der Eycken, J.; Vanholder, R. P-cresylsulphate, the main in vivo metabolite of p-cresol, activates leucocyte free radical production. Nephrol. Dial. Transplant. 2007, 22, 592–596. [Google Scholar] [CrossRef]
- Adesso, S.; Popolo, A.; Bianco, G.; Sorrentino, R.; Pinto, A.; Autore, G.; Marzocco, S. The uremic toxin indoxyl sulphate enhances macrophage response to LPS. PLoS ONE 2013, 8, e76778. [Google Scholar] [CrossRef] [PubMed]
- Gibson, S.A.; McFarlan, C.; Hay, S.; MacFarlane, G.T. Significance of microflora in proteolysis in the colon. Appl. Environ. Microbiol. 1989, 55, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Campbell, K.; Johnson, D.; Stanton, T.; Pascoe, E.; Hawley, C.; Dimeski, G.; McWhinney, B.; Ungerer, J.; Isbel, N. Uraemic toxins and cardiovascular disease across the chronic kidney disease spectrum: An observational study. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Wu, I.W.; Hsu, K.H.; Lee, C.C.; Sun, C.Y.; Hsu, H.J.; Tsai, C.J.; Tzen, C.Y.; Wang, Y.C.; Lin, C.Y.; Wu, M.S. p-Cresyl sulphate and indoxyl sulphate predict progression of chronic kidney disease. Nephrol. Dial. Transplant. 2011, 26, 938–947. [Google Scholar] [CrossRef] [PubMed]
- Liabeuf, S.; Barreto, D.V.; Barreto, F.C.; Meert, N.; Glorieux, G.; Schepers, E.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A. Free p-cresylsulphate is a predictor of mortality in patients at different stages of chronic kidney disease. Nephrol. Dial. Transplant. 2010, 25, 1183–1191. [Google Scholar] [CrossRef]
- Liabeuf, S.; Glorieux, G.; Lenglet, A.; Diouf, M.; Schepers, E.; Desjardins, L.; Choukroun, G.; Vanholder, R.; Massy, Z.A. Does p-cresylglucuronide have the same impact on mortality as other protein-bound uremic toxins? PLoS ONE 2013, 8, e67168. [Google Scholar] [CrossRef]
- Bammens, B.; Evenepoel, P.; Keuleers, H.; Verbeke, K.; Vanrenterghem, Y. Free serum concentrations of the protein-bound retention solute p-cresol predict mortality in hemodialysis patients. Kidney Int. 2006, 69, 1081–1087. [Google Scholar] [CrossRef]
- Dou, L.; Sallée, M.; Cerini, C.; Poitevin, S.; Gondouin, B.; Jourde-Chiche, N.; Fallague, K.; Brunet, P.; Calaf, R.; Dussol, B.; et al. The cardiovascular effect of the uremic solute indole-3 acetic acid. J. Am. Soc. Nephrol. 2015, 26, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Chuang, C.K.; Jayakumar, T.; Liu, H.L.; Pan, C.F.; Wang, T.J.; Chen, H.H.; Wu, C.J. Serum p-cresyl sulfate predicts cardiovascular disease and mortality in elderly hemodialysis patients. Arch. Med. Sci. 2013, 9, 662–668. [Google Scholar] [CrossRef]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Glorieux, G.; De Smet, R.; Lameire, N. New insights in uremic toxins. Kidney Int. Suppl. 2003, S6–S10. [Google Scholar] [CrossRef]
- Gryp, T.; Vanholder, R.; Vaneechoutte, M.; Glorieux, G. p-Cresyl Sulfate. Toxins 2017, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Mair, R.D.; Sirich, T.L.; Plummer, N.S.; Meyer, T.W. Characteristics of Colon-Derived Uremic Solutes. Clin. J. Am. Soc. Nephrol. 2018, 13, 1398–1404. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argilés, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef]
- De Loor, H.; Bammens, B.; Evenepoel, P.; De Preter, V.; Verbeke, K. Gas chromatographic-mass spectrometric analysis for measurement of p-cresol and its conjugated metabolites in uremic and normal serum. Clin. Chem. 2005, 51, 1535–1538. [Google Scholar] [CrossRef]
- Lin, C.J.; Chen, H.H.; Pan, C.F.; Chuang, C.K.; Wang, T.J.; Sun, F.J.; Wu, C.J. p-Cresylsulfate and indoxyl sulfate level at different stages of chronic kidney disease. J. Clin. Lab. Anal. 2011, 25, 191–197. [Google Scholar] [CrossRef]
- Nigam, S.K.; Wu, W.; Bush, K.T.; Hoenig, M.P.; Blantz, R.C.; Bhatnagar, V. Handling of Drugs, Metabolites, and Uremic Toxins by Kidney Proximal Tubule Drug Transporters. Clin. J. Am. Soc. Nephrol. 2015, 10, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Goto, S.; Fukagawa, M. Role of Uremic Toxins for Kidney, Cardiovascular, and Bone Dysfunction. Toxins 2018, 10, 202. [Google Scholar] [CrossRef]
- Motojima, M.; Hosokawa, A.; Yamato, H.; Muraki, T.; Yoshioka, T. Uremic toxins of organic anions up-regulate PAI-1 expression by induction of NF-kappaB and free radical in proximal tubular cells. Kidney Int. 2003, 63, 1671–1680. [Google Scholar] [CrossRef] [PubMed]
- Arif, E.; Nihalani, D. Glomerular Filtration Barrier Assembly: An insight. Postdoc J. 2013, 1, 33–45. [Google Scholar] [CrossRef]
- Brenner, B.M.; Hostetter, T.H.; Humes, H.D. Molecular basis of proteinuria of glomerular origin. N. Engl. J. Med. 1978, 298, 826–833. [Google Scholar] [CrossRef] [PubMed]
- Faul, C.; Asanuma, K.; Yanagida-Asanuma, E.; Kim, K.; Mundel, P. Actin up: Regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol. 2007, 17, 428–437. [Google Scholar] [CrossRef]
- Schnabel, E.; Anderson, J.M.; Farquhar, M.G. The tight junction protein ZO-1 is concentrated along slit diaphragms of the glomerular epithelium. J. Cell Biol. 1990, 111, 1255–1263. [Google Scholar] [CrossRef]
- Suchy-Dicey, A.M.; Laha, T.; Hoofnagle, A.; Newitt, R.; Sirich, T.L.; Meyer, T.W.; Thummel, K.E.; Yanez, N.D.; Himmelfarb, J.; Weiss, N.S.; et al. Tubular Secretion in CKD. J. Am. Soc. Nephrol. 2016, 27, 2148–2155. [Google Scholar] [CrossRef]
- Masereeuw, R.; Mutsaers, H.A.; Toyohara, T.; Abe, T.; Jhawar, S.; Sweet, D.H.; Lowenstein, J. The kidney and uremic toxin removal: Glomerulus or tubule? Semin. Nephrol. 2014, 34, 191–208. [Google Scholar] [CrossRef] [PubMed]
- Meijers, B.K.; De Loor, H.; Bammens, B.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. p-Cresyl sulfate and indoxyl sulfate in hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1932–1938. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.C.; Rabindranath, K.S.; Craig, J.C.; Roderick, P.J.; Locatelli, F.; Strippoli, G.F. High-flux versus low-flux membranes for end-stage kidney disease. Cochrane Database Syst. Rev. 2012, 2012, CD005016. [Google Scholar] [CrossRef]
- Yamamoto, S.; Kazama, J.J.; Wakamatsu, T.; Takahashi, Y.; Kaneko, Y.; Goto, S.; Narita, I. Removal of uremic toxins by renal replacement therapies: A review of current progress and future perspectives. Ren. Replace. Ther. 2016, 2, 43. [Google Scholar] [CrossRef]
- Itoh, Y.; Ezawa, A.; Kikuchi, K.; Tsuruta, Y.; Niwa, T. Protein-bound uremic toxins in hemodialysis patients measured by liquid chromatography/tandem mass spectrometry and their effects on endothelial ROS production. Anal. Bioanal. Chem. 2012, 403, 1841–1850. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lee, J. Indole as an intercellular signal in microbial communities. FEMS Microbiol. Rev. 2010, 34, 426–444. [Google Scholar] [CrossRef]
- Zhang, L.S.; Davies, S.S. Microbial metabolism of dietary components to bioactive metabolites: Opportunities for new therapeutic interventions. Genome Med. 2016, 8, 46. [Google Scholar] [CrossRef]
- Shimada, Y.; Kinoshita, M.; Harada, K.; Mizutani, M.; Masahata, K.; Kayama, H.; Takeda, K. Commensal bacteria-dependent indole production enhances epithelial barrier function in the colon. PLoS ONE 2013, 8, e80604. [Google Scholar] [CrossRef] [PubMed]
- De Smet, R.; Dhondt, A.; Eloot, S.; Galli, F.; Waterloos, M.A.; Vanholder, R. Effect of the super-flux cellulose triacetate dialyser membrane on the removal of non-protein-bound and protein-bound uraemic solutes. Nephrol. Dial. Transplant. 2007, 22, 2006–2012. [Google Scholar] [CrossRef] [PubMed]
- Lysaght, M.J.; Vonesh, E.F.; Gotch, F.; Ibels, L.; Keen, M.; Lindholm, B.; Nolph, K.D.; Pollock, C.A.; Prowant, B.; Farrell, P.C. The influence of dialysis treatment modality on the decline of remaining renal function. ASAIO Trans. 1991, 37, 598–604. [Google Scholar] [PubMed]
- Miyazaki, T.; Ise, M.; Seo, H.; Niwa, T. Indoxyl sulfate increases the gene expressions of TGF-beta 1, TIMP-1 and pro-alpha 1(I) collagen in uremic rat kidneys. Kidney Int. Suppl. 1997, 62, S15–S22. [Google Scholar]
- Yamamoto, H.; Tsuruoka, S.; Ioka, T.; Ando, H.; Ito, C.; Akimoto, T.; Fujimura, A.; Asano, Y.; Kusano, E. Indoxyl sulfate stimulates proliferation of rat vascular smooth muscle cells. Kidney Int. 2006, 69, 1780–1785. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Jourde-Chiche, N.; Faure, V.; Cerini, C.; Berland, Y.; Dignat-George, F.; Brunet, P. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J. Thromb. Haemost. 2007, 5, 1302–1308. [Google Scholar] [CrossRef]
- Lin, C.-J.; Pan, C.-F.; Liu, H.-L.; Chuang, C.-K.; Jayakumar, T.; Wang, T.-J.; Chen, H.-H.; Wu, C.-J. The role of protein-bound uremic toxins on peripheral artery disease and vascular access failure in patients on hemodialysis. Atherosclerosis 2012, 225, 173–179. [Google Scholar] [CrossRef]
- Lekawanvijit, S.; Adrahtas, A.; Kelly, D.J.; Kompa, A.R.; Wang, B.H.; Krum, H. Does indoxyl sulfate, a uraemic toxin, have direct effects on cardiac fibroblasts and myocytes? Eur. Heart J. 2010, 31, 1771–1779. [Google Scholar] [CrossRef]
- Aoki, K.; Teshima, Y.; Kondo, H.; Saito, S.; Fukui, A.; Fukunaga, N.; Nawata, T.; Shimada, T.; Takahashi, N.; Shibata, H. Role of Indoxyl Sulfate as a Predisposing Factor for Atrial Fibrillation in Renal Dysfunction. J. Am. Heart Assoc. 2015, 4, e002023. [Google Scholar] [CrossRef]
- Mozar, A.; Louvet, L.; Godin, C.; Mentaverri, R.; Brazier, M.; Kamel, S.; Massy, Z.A. Indoxyl sulphate inhibits osteoclast differentiation and function. Nephrol. Dial. Transplant. 2012, 27, 2176–2181. [Google Scholar] [CrossRef]
- Barreto, F.C.; Barreto, D.V.; Canziani, M.E.; Tomiyama, C.; Higa, A.; Mozar, A.; Glorieux, G.; Vanholder, R.; Massy, Z.; de Carvalho, A.B. Association between indoxyl sulfate and bone histomorphometry in pre-dialysis chronic kidney disease patients. J. Bras. Nefrol. 2014, 36, 289–296. [Google Scholar] [CrossRef]
- Nii-Kono, T.; Iwasaki, Y.; Uchida, M.; Fujieda, A.; Hosokawa, A.; Motojima, M.; Yamato, H.; Kurokawa, K.; Fukagawa, M. Indoxyl sulfate induces skeletal resistance to parathyroid hormone in cultured osteoblastic cells. Kidney Int. 2007, 71, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-J.; Pan, C.-F.; Chuang, C.-K.; Liu, H.-L.; Sun, F.-J.; Wang, T.-J.; Chen, H.-H.; Wu, C.-J. Association of Indoxyl Sulfate with Fibroblast Growth Factor 23 in Patients with Advanced Chronic Kidney Disease. Am. J. Med. Sci. 2014, 347, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.-K.; Tanaka, T.; Inagi, R.; Fujita, T.; Nangaku, M. Indoxyl sulfate, a representative uremic toxin, suppresses erythropoietin production in a HIF-dependent manner. Lab. Investig. 2011, 91, 1564–1571. [Google Scholar] [CrossRef]
- Nangaku, M.; Mimura, I.; Yamaguchi, J.; Higashijima, Y.; Wada, T.; Tanaka, T. Role of Uremic Toxins in Erythropoiesis-Stimulating Agent Resistance in Chronic Kidney Disease and Dialysis Patients. J. Ren. Nutr. 2015, 25, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.S.E.; Abed, M.; Voelkl, J.; Lang, F. Triggering of suicidal erythrocyte death by uremic toxin indoxyl sulfate. BMC Nephrol. 2013, 14, 244. [Google Scholar] [CrossRef]
- Cummings, J.H. Fermentation in the human large intestine: Evidence and implications for health. Lancet 1983, 1, 1206–1209. [Google Scholar] [CrossRef]
- Meijers, B.K.; Evenepoel, P. The gut-kidney axis: Indoxyl sulfate, p-cresyl sulfate and CKD progression. Nephrol. Dial. Transplant. 2011, 26, 759–761. [Google Scholar] [CrossRef]
- Mutsaers, H.A.; Wilmer, M.J.; van den Heuvel, L.P.; Hoenderop, J.G.; Masereeuw, R. Basolateral transport of the uraemic toxin p-cresyl sulfate: Role for organic anion transporters? Nephrol. Dial. Transplant. 2011, 26, 4149. [Google Scholar] [CrossRef]
- Poesen, R.; Viaene, L.; Verbeke, K.; Claes, K.; Bammens, B.; Sprangers, B.; Naesens, M.; Vanrenterghem, Y.; Kuypers, D.; Evenepoel, P.; et al. Renal clearance and intestinal generation of p-cresyl sulfate and indoxyl sulfate in CKD. Clin. J. Am. Soc. Nephrol. 2013, 8, 1508–1514. [Google Scholar] [CrossRef]
- Meijers, B.K.; Bammens, B.; De Moor, B.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. Free p-cresol is associated with cardiovascular disease in hemodialysis patients. Kidney Int. 2008, 73, 1174–1180. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.Y.; Chang, S.C.; Wu, M.S. Uremic toxins induce kidney fibrosis by activating intrarenal renin-angiotensin-aldosterone system associated epithelial-to-mesenchymal transition. PLoS ONE 2012, 7, e34026. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Miyamoto, Y.; Honda, D.; Tanaka, H.; Wu, Q.; Endo, M.; Noguchi, T.; Kadowaki, D.; Ishima, Y.; Kotani, S.; et al. p-Cresyl sulfate causes renal tubular cell damage by inducing oxidative stress by activation of NADPH oxidase. Kidney Int. 2013, 83, 582–592. [Google Scholar] [CrossRef]
- Meijers, B.K.; Claes, K.; Bammens, B.; de Loor, H.; Viaene, L.; Verbeke, K.; Kuypers, D.; Vanrenterghem, Y.; Evenepoel, P. p-Cresol and cardiovascular risk in mild-to-moderate kidney disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Sato, E.; Mori, T.; Mishima, E.; Suzuki, A.; Sugawara, S.; Kurasawa, N.; Saigusa, D.; Miura, D.; Morikawa-Ichinose, T.; Saito, R.; et al. Metabolic alterations by indoxyl sulfate in skeletal muscle induce uremic sarcopenia in chronic kidney disease. Sci. Rep. 2016, 6, 36618. [Google Scholar] [CrossRef]
- Vanholder, R.; Schepers, E.; Pletinck, A.; Nagler, E.V.; Glorieux, G. The uremic toxicity of indoxyl sulfate and p-cresyl sulfate: A systematic review. J. Am. Soc. Nephrol. 2014, 25, 1897–1907. [Google Scholar] [CrossRef]
- Mishima, E.; Fukuda, S.; Kanemitsu, Y.; Saigusa, D.; Mukawa, C.; Asaji, K.; Matsumoto, Y.; Tsukamoto, H.; Tachikawa, T.; Tsukimi, T.; et al. Canagliflozin reduces plasma uremic toxins and alters the intestinal microbiota composition in a chronic kidney disease mouse model. Am. J. Physiol. Ren. Physiol. 2018, 315, F824–F833. [Google Scholar] [CrossRef]
- Wu, I.W.; Hsu, K.H.; Hsu, H.J.; Lee, C.C.; Sun, C.Y.; Tsai, C.J.; Wu, M.S. Serum free p-cresyl sulfate levels predict cardiovascular and all-cause mortality in elderly hemodialysis patients--a prospective cohort study. Nephrol. Dial. Transplant. 2012, 27, 1169–1175. [Google Scholar] [CrossRef]
- Rossi, M.; Campbell, K.L.; Johnson, D.W.; Stanton, T.; Vesey, D.A.; Coombes, J.S.; Weston, K.S.; Hawley, C.M.; McWhinney, B.C.; Ungerer, J.P.; et al. Protein-bound uremic toxins, inflammation and oxidative stress: A cross-sectional study in stage 3-4 chronic kidney disease. Arch. Med. Res. 2014, 45, 309–317. [Google Scholar] [CrossRef]
- Shen, W.C.; Liang, C.J.; Huang, T.M.; Liu, C.W.; Wang, S.H.; Young, G.H.; Tsai, J.S.; Tseng, Y.C.; Peng, Y.S.; Wu, V.C.; et al. Indoxyl sulfate enhances IL-1β-induced E-selectin expression in endothelial cells in acute kidney injury by the ROS/MAPKs/NFκB/AP-1 pathway. Arch. Toxicol. 2016, 90, 2779–2792. [Google Scholar] [CrossRef]
- Perna, A.F.; Glorieux, G.; Zacchia, M.; Trepiccione, F.; Capolongo, G.; Vigorito, C.; Anishchenko, E.; Ingrosso, D. The role of the intestinal microbiota in uremic solute accumulation: A focus on sulfur compounds. J. Nephrol. 2019, 32, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, L.; Egestad, B.; Jörnvall, H.; Bergström, J. Identification and determination of phenylacetylglutamine, a major nitrogenous metabolite in plasma of uremic patients. Clin. Nephrol. 1989, 32, 124–128. [Google Scholar] [PubMed]
- Wang, Z.; Roberts, A.B.; Buffa, J.A.; Levison, B.S.; Zhu, W.; Org, E.; Gu, X.; Huang, Y.; Zamanian-Daryoush, M.; Culley, M.K.; et al. Non-lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell 2015, 163, 1585–1595. [Google Scholar] [CrossRef]
- Zeisel, S.H.; Warrier, M. Trimethylamine N-Oxide, the Microbiome, and Heart and Kidney Disease. Annu. Rev. Nutr. 2017, 37, 157–181. [Google Scholar] [CrossRef]
- Tang, W.H.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Wang, Z.; Kennedy, D.J.; Wu, Y.; Buffa, J.A.; Agatisa-Boyle, B.; Li, X.S.; Levison, B.S.; Hazen, S.L. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ. Res. 2015, 116, 448–455. [Google Scholar] [CrossRef]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef]
- Stubbs, J.R.; House, J.A.; Ocque, A.J.; Zhang, S.; Johnson, C.; Kimber, C.; Schmidt, K.; Gupta, A.; Wetmore, J.B.; Nolin, T.D.; et al. Serum Trimethylamine-N-Oxide is Elevated in CKD and Correlates with Coronary Atherosclerosis Burden. J. Am. Soc. Nephrol. 2016, 27, 305–313. [Google Scholar] [CrossRef]
- Missailidis, C.; Hällqvist, J.; Qureshi, A.R.; Barany, P.; Heimbürger, O.; Lindholm, B.; Stenvinkel, P.; Bergman, P. Serum Trimethylamine-N-Oxide Is Strongly Related to Renal Function and Predicts Outcome in Chronic Kidney Disease. PLoS ONE 2016, 11, e0141738. [Google Scholar] [CrossRef]
- Depner, T.A. Uremic toxicity: Urea and beyond. Semin. Dial. 2001, 14, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Pahl, M.V.; Vaziri, N.D. The Chronic Kidney Disease—Colonic Axis. Semin. Dial. 2015, 28, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Armani, R.G.; Ramezani, A.; Yasir, A.; Sharama, S.; Canziani, M.E.F.; Raj, D.S. Gut Microbiome in Chronic Kidney Disease. Curr. Hypertens. Rep. 2017, 19, 29. [Google Scholar] [CrossRef]
- Cummings, J.H.; Hill, M.J.; Bone, E.S.; Branch, W.J.; Jenkins, D.J. The effect of meat protein and dietary fiber on colonic function and metabolism. II. Bacterial metabolites in feces and urine. Am. J. Clin. Nutr. 1979, 32, 2094–2101. [Google Scholar] [CrossRef]
- Poesen, R.; Mutsaers, H.A.; Windey, K.; van den Broek, P.H.; Verweij, V.; Augustijns, P.; Kuypers, D.; Jansen, J.; Evenepoel, P.; Verbeke, K.; et al. The Influence of Dietary Protein Intake on Mammalian Tryptophan and Phenolic Metabolites. PLoS ONE 2015, 10, e0140820. [Google Scholar] [CrossRef]
- Patel, K.P.; Luo, F.J.; Plummer, N.S.; Hostetter, T.H.; Meyer, T.W. The production of p-cresol sulfate and indoxyl sulfate in vegetarians versus omnivores. Clin. J. Am. Soc. Nephrol. 2012, 7, 982–988. [Google Scholar] [CrossRef]
- Sirich, T.L.; Plummer, N.S.; Gardner, C.D.; Hostetter, T.H.; Meyer, T.W. Effect of increasing dietary fiber on plasma levels of colon-derived solutes in hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2014, 9, 1603–1610. [Google Scholar] [CrossRef] [PubMed]
- Guida, B.; Germanò, R.; Trio, R.; Russo, D.; Memoli, B.; Grumetto, L.; Barbato, F.; Cataldi, M. Effect of short-term synbiotic treatment on plasma p-cresol levels in patients with chronic renal failure: A randomized clinical trial. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 1043–1049. [Google Scholar] [CrossRef]
- Poesen, R.; Evenepoel, P.; de Loor, H.; Delcour, J.A.; Courtin, C.M.; Kuypers, D.; Augustijns, P.; Verbeke, K.; Meijers, B. The Influence of Prebiotic Arabinoxylan Oligosaccharides on Microbiota Derived Uremic Retention Solutes in Patients with Chronic Kidney Disease: A Randomized Controlled Trial. PLoS ONE 2016, 11, e0153893. [Google Scholar] [CrossRef]
- Meijers, B.K.; De Preter, V.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. p-Cresyl sulfate serum concentrations in haemodialysis patients are reduced by the prebiotic oligofructose-enriched inulin. Nephrol. Dial. Transplant. 2010, 25, 219–224. [Google Scholar] [CrossRef]
- FAO; WHO. Probiotics in Food: Health and Nutritional Properties and Guidelines for Evaluation: Report of a Joint FAO/WHO Expert Consultation on Evaluation of Health and Nutritional Properties of Probiotics in Food including Powder Milk with Live Lactic Acid Bacteria, Cordoba, Argentina, 1–4 October 2001 [and] Report of a Joint FAO/WHO Working Group on Drafting Guidelines for the Evaluation of Probiotics in Food, London, Ontario, Canada, 30 April–1 May 2002; Food and Agriculture Organization of the United Nations and World Health Organization: Rome, Italy, 2006. [Google Scholar]
- Rastall, R.A.; Gibson, G.R.; Gill, H.S.; Guarner, F.; Klaenhammer, T.R.; Pot, B.; Reid, G.; Rowland, I.R.; Sanders, M.E. Modulation of the microbial ecology of the human colon by probiotics, prebiotics and synbiotics to enhance human health: An overview of enabling science and potential applications. FEMS Microbiol. Ecol. 2005, 52, 145–152. [Google Scholar] [CrossRef]
- Van Baarlen, P.; Troost, F.J.; van Hemert, S.; van der Meer, C.; de Vos, W.M.; de Groot, P.J.; Hooiveld, G.J.E.J.; Brummer, R.-J.M.; Kleerebezem, M. Differential NF-κB pathways induction by Lactobacillus plantarum in the duodenum of healthy humans correlating with immune tolerance. Proc. Natl. Acad. Sci. USA 2009, 106, 2371–2376. [Google Scholar] [CrossRef]
- Takayama, F.; Taki, K.; Niwa, T. Bifidobacterium in gastro-resistant seamless capsule reduces serum levels of indoxyl sulfate in patients on hemodialysis. Am. J. Kidney Dis. 2003, 41, S142–S145. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.-K.; Wu, Y.-Y.; Yang, Y.-F.; Ting, I.-W.; Lin, C.-C.; Yen, T.-H.; Chen, J.-H.; Wang, C.-H.; Huang, C.-C.; Lin, H.-C. The effect of probiotics on serum levels of cytokine and endotoxin in peritoneal dialysis patients: A randomised, double-blind, placebo-controlled trial. Benef. Microbes 2015, 6, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Hutkins, R.W.; Krumbeck, J.A.; Bindels, L.B.; Cani, P.D.; Fahey, G., Jr.; Goh, Y.J.; Hamaker, B.; Martens, E.C.; Mills, D.A.; Rastal, R.A.; et al. Prebiotics: Why definitions matter. Curr. Opin. Biotechnol. 2016, 37, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chiavaroli, L.; Mirrahimi, A.; Sievenpiper, J.L.; Jenkins, D.J.; Darling, P.B. Dietary fiber effects in chronic kidney disease: A systematic review and meta-analysis of controlled feeding trials. Eur. J. Clin. Nutr. 2015, 69, 761–768. [Google Scholar] [CrossRef]
- Rossi, M.; Johnson, D.W.; Xu, H.; Carrero, J.J.; Pascoe, E.; French, C.; Campbell, K.L. Dietary protein-fiber ratio associates with circulating levels of indoxyl sulfate and p-cresyl sulfate in chronic kidney disease patients. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 860–865. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, N.; Ranganathan, P.; Friedman, E.A.; Joseph, A.; Delano, B.; Goldfarb, D.S.; Tam, P.; Rao, A.V.; Anteyi, E.; Musso, C.G. Pilot study of probiotic dietary supplementation for promoting healthy kidney function in patients with chronic kidney disease. Adv. Ther. 2010, 27, 634–647. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, R.; Pechenyak, B.; Vyas, U.; Ranganathan, P.; Weinberg, A.; Liang, P.; Mallappallil, M.C.; Norin, A.J.; Friedman, E.A.; Saggi, S.J. Randomized controlled trial of strain-specific probiotic formulation (Renadyl) in dialysis patients. BioMed Res. Int. 2014, 2014, 568571. [Google Scholar] [CrossRef]
- Rossi, M.; Johnson, D.W.; Morrison, M.; Pascoe, E.M.; Coombes, J.S.; Forbes, J.M.; Szeto, C.-C.; McWhinney, B.C.; Ungerer, J.P.J.; Campbell, K.L. Synbiotics Easing Renal Failure by Improving Gut Microbiology (SYNERGY): A Randomized Trial. Clin. J. Am. Soc. Nephrol. 2016, 11, 223–231. [Google Scholar] [CrossRef]
- Lau, W.L.; Kalantar-Zadeh, K.; Vaziri, N.D. The Gut as a Source of Inflammation in Chronic Kidney Disease. Nephron 2015, 130, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Mishima, E.; Fukuda, S.; Shima, H.; Hirayama, A.; Akiyama, Y.; Takeuchi, Y.; Fukuda, N.N.; Suzuki, T.; Suzuki, C.; Yuri, A.; et al. Alteration of the Intestinal Environment by Lubiprostone Is Associated with Amelioration of Adenine-Induced CKD. J. Am. Soc. Nephrol. 2015, 26, 1787–1794. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.; Desai, M.; Jardine, M.; Balis, D.; Meininger, G.; Perkovic, V. Canagliflozin Slows Progression of Renal Function Decline Independently of Glycemic Effects. J. Am. Soc. Nephrol. 2017, 28, 368–375. [Google Scholar] [CrossRef]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Pan, C.F.; Chuang, C.K.; Sun, F.J.; Wang, D.J.; Chen, H.H.; Liu, H.L.; Wu, C.J. P-cresyl sulfate is a valuable predictor of clinical outcomes in pre-ESRD patients. BioMed Res. Int. 2014, 2014, 526932. [Google Scholar] [CrossRef]
- Schulman, G.; Agarwal, R.; Acharya, M.; Berl, T.; Blumenthal, S.; Kopyt, N. A Multicenter, Randomized, Double-Blind, Placebo-Controlled, Dose-Ranging Study of AST-120 (Kremezin) in Patients With Moderate to Severe CKD. Am. J. Kidney Dis. 2006, 47, 565–577. [Google Scholar] [CrossRef]
- Yamamoto, S.; Kazama, J.J.; Omori, K.; Matsuo, K.; Takahashi, Y.; Kawamura, K.; Matsuto, T.; Watanabe, H.; Maruyama, T.; Narita, I. Continuous Reduction of Protein-Bound Uraemic Toxins with Improved Oxidative Stress by Using the Oral Charcoal Adsorbent AST-120 in Haemodialysis Patients. Sci. Rep. 2015, 5, 14381. [Google Scholar] [CrossRef] [PubMed]
- Wu, I.-W.; Hsu, K.-H.; Sun, C.-Y.; Tsai, C.-J.; Wu, M.-S.; Lee, C.-C. Oral adsorbent AST-120 potentiates the effect of erythropoietin-stimulating agents on Stage 5 chronic kidney disease patients: A randomized crossover study. Nephrol. Dial. Transplant. 2014, 29, 1719–1727. [Google Scholar] [CrossRef]
- Sato, E.; Tanaka, A.; Oyama, J.-I.; Yamasaki, A.; Shimomura, M.; Hiwatashi, A.; Ueda, Y.; Amaha, M.; Nomura, M.; Matsumura, D.; et al. Long-term effects of AST-120 on the progression and prognosis of pre-dialysis chronic kidney disease: A 5-year retrospective study. Heart Vessel. 2016, 31, 1625–1632. [Google Scholar] [CrossRef]


{kind=link}
| Design of Study | Study Group | Adverse Effect | Ref. |
|---|---|---|---|
| ANIMAL STUDIES | |||
| Uremic toxins administration to subtotally nephrectomized rats | HA-treated rats vs. non-HA-treated controls, IAA-treated rats vs. non-IAA-treated controls |
| [66] |
| Analysis of effect of indole on ECs | Germ free mouse model |
| [101] |
| Animal model study | Mouse proximal renal tubular cells (PKSV-PRs) treated with IS or PCS Half-nephrectomized B-6 mice treated with IS or PCS for 4 weeks. |
| [122] |
| Imaging mass spectrometry | Mouse model of CKD |
| [125] |
| CELLS CULTURE | |||
| Incubation of HUVEC with uremic retention solutes at concentrations found in uremic patients | HUVEC culture |
| [68] |
| Incubation of heparinized whole blood with p-cresol, p-cresylsulphate or their respective solvent control | Heparinized whole blood |
| [69] |
| Incubation of J774A.1 macrophages with IS at concentrations observed in uremic patients (1000-62.5 µM) | J774A.1 macrophages |
| [70] |
| Renal cortex of 5/6-nephrectomized uremic rats given indoxyl sulfate | Renal cortex of 5/6-nephrectomized uremic rats |
| [104] |
| Examination of the effect of IS on rat VSMC proliferation | Rat VSMC |
| [105] |
| Analysis of direct effect of IS at uraemic concentrations on OCL differentiation and bone-resorbing activity | Well-established cellular models of monocyte/macrophage (peripheral blood mononuclear cells and the RAW 264.7 cell line). |
| [110] |
| HUMAN STUDIES | |||
| Cross-sectional observational study included a convenience sample of 327 participants with normal kidney function, non-dialysis CKD and end-stage kidney disease (ESKD) patients | 327 participants with kidney function categorized as normal, CKD and ESKD. |
| [72] |
| Prospective observational study | 268 patients with different stages of CKD |
| [128] |
| Observational study | 139 patients with different stages of CKD |
| [69] |
| Observational study | 103 stable CKD patients (stage 3–5 and hemodialysis (HD) patients) |
| [78] |
| Observational study | 120 patients with CKD |
| [77] |
| Observational study | 100 stable and eligible HD patients |
| [107] |
| Cross-sectional observational cohort study | Participants with stage 3–4 CKD |
| [129] |
| Observational study | 4007 patients undergoing elective coronary angiography |
| [135] |
| Observational study | CKD cohort (n = 104) CKD cohort (n = 220) undergoing coronary angiography |
| [139] |
| Longitudinal study | CKD patients ranging from mild-moderate to ESRD |
| [140] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rysz, J.; Franczyk, B.; Ławiński, J.; Olszewski, R.; Ciałkowska-Rysz, A.; Gluba-Brzózka, A. The Impact of CKD on Uremic Toxins and Gut Microbiota. Toxins 2021, 13, 252. https://doi.org/10.3390/toxins13040252
Rysz J, Franczyk B, Ławiński J, Olszewski R, Ciałkowska-Rysz A, Gluba-Brzózka A. The Impact of CKD on Uremic Toxins and Gut Microbiota. Toxins. 2021; 13(4):252. https://doi.org/10.3390/toxins13040252
Chicago/Turabian StyleRysz, Jacek, Beata Franczyk, Janusz Ławiński, Robert Olszewski, Aleksanda Ciałkowska-Rysz, and Anna Gluba-Brzózka. 2021. "The Impact of CKD on Uremic Toxins and Gut Microbiota" Toxins 13, no. 4: 252. https://doi.org/10.3390/toxins13040252
APA StyleRysz, J., Franczyk, B., Ławiński, J., Olszewski, R., Ciałkowska-Rysz, A., & Gluba-Brzózka, A. (2021). The Impact of CKD on Uremic Toxins and Gut Microbiota. Toxins, 13(4), 252. https://doi.org/10.3390/toxins13040252