Ochratoxin A Induces Steatosis via PPARγ-CD36 Axis

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
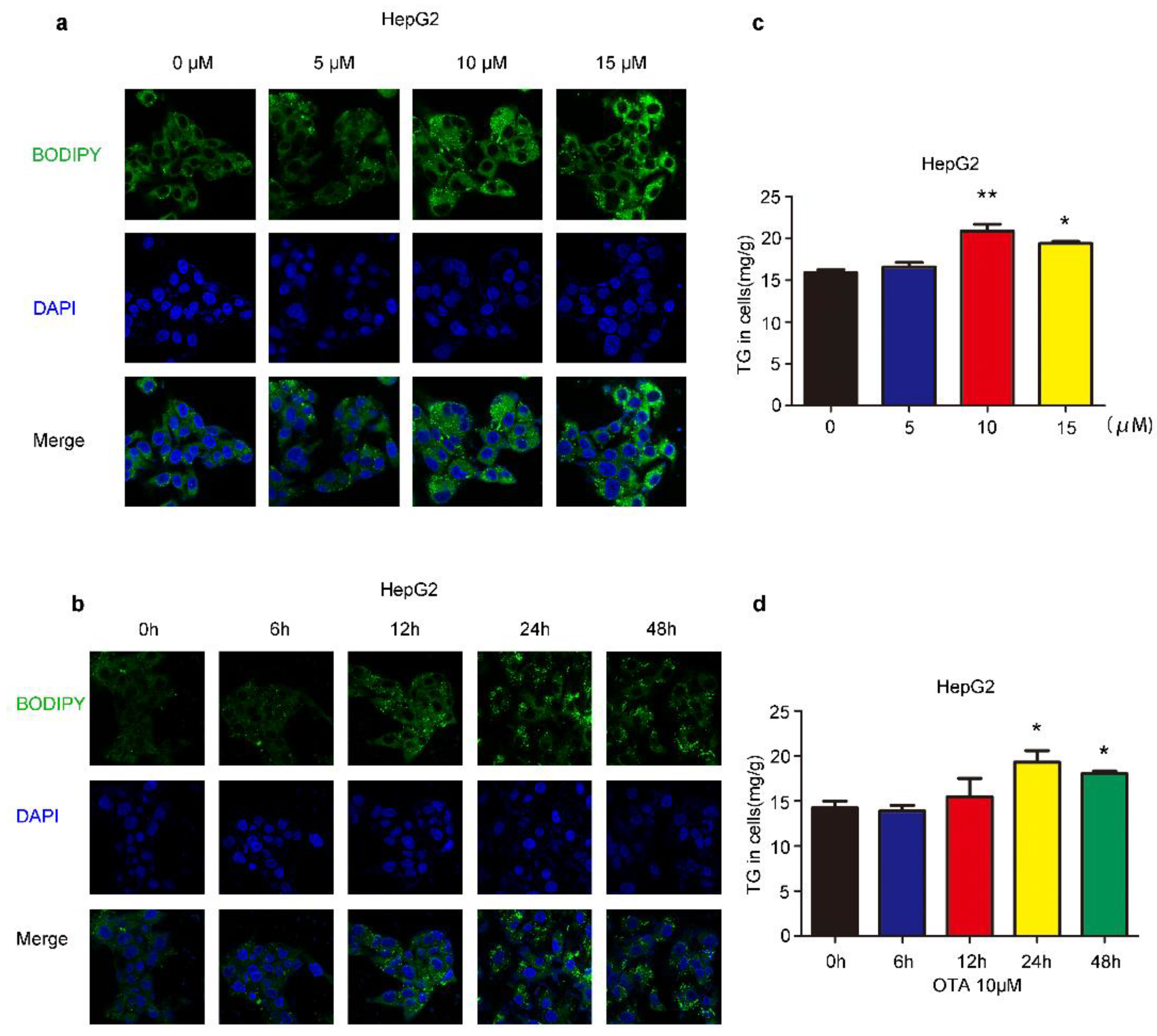
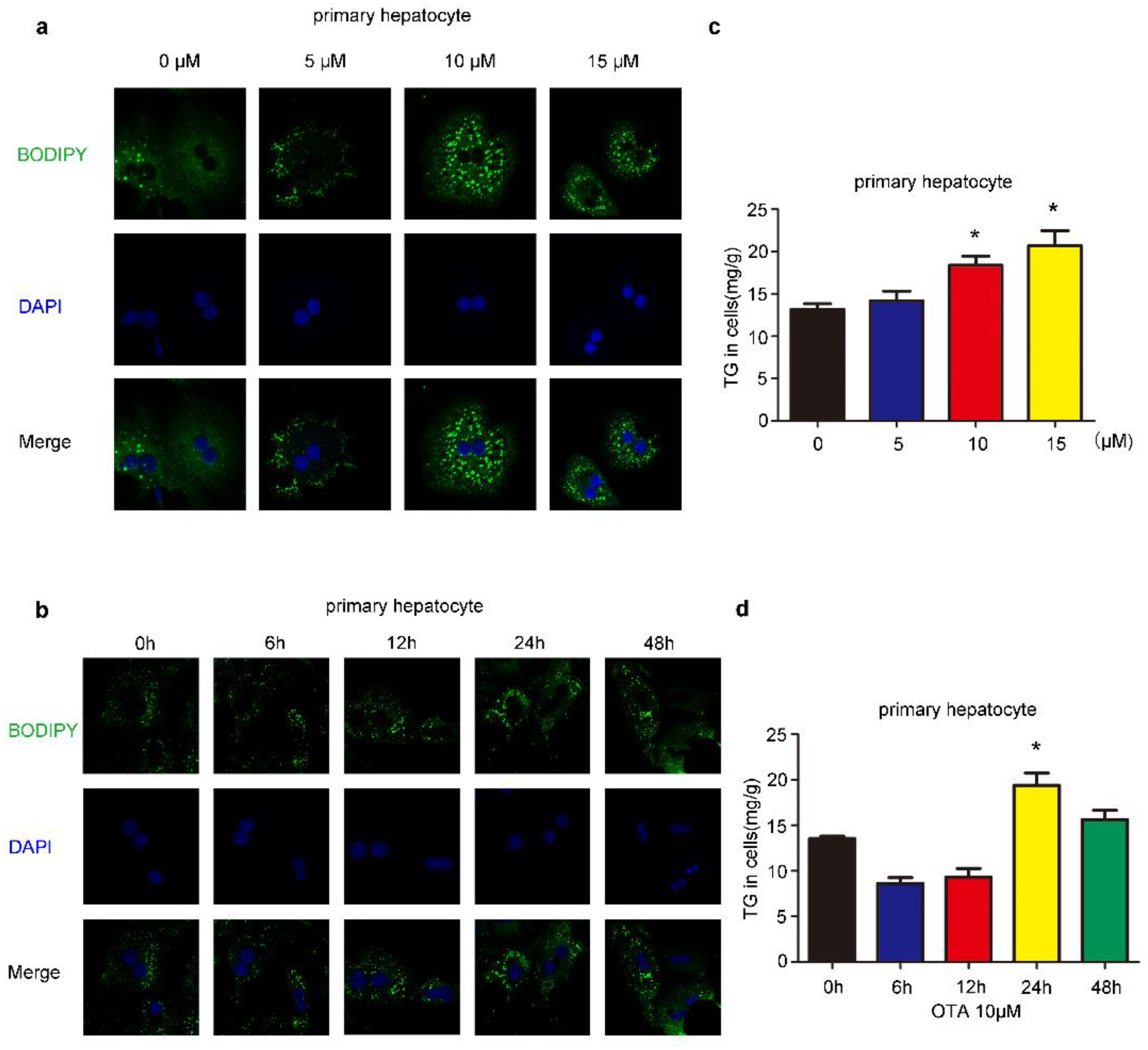
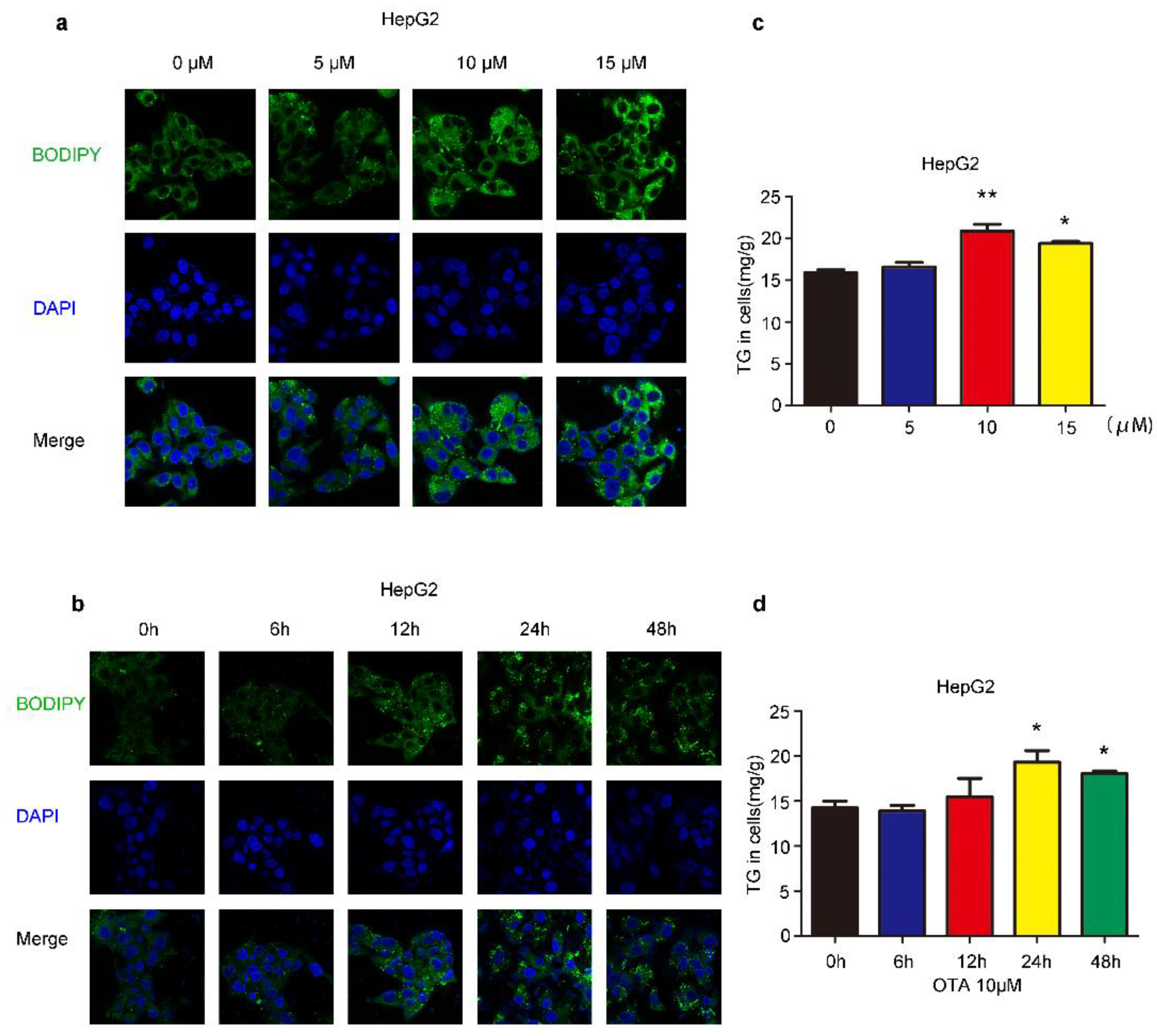
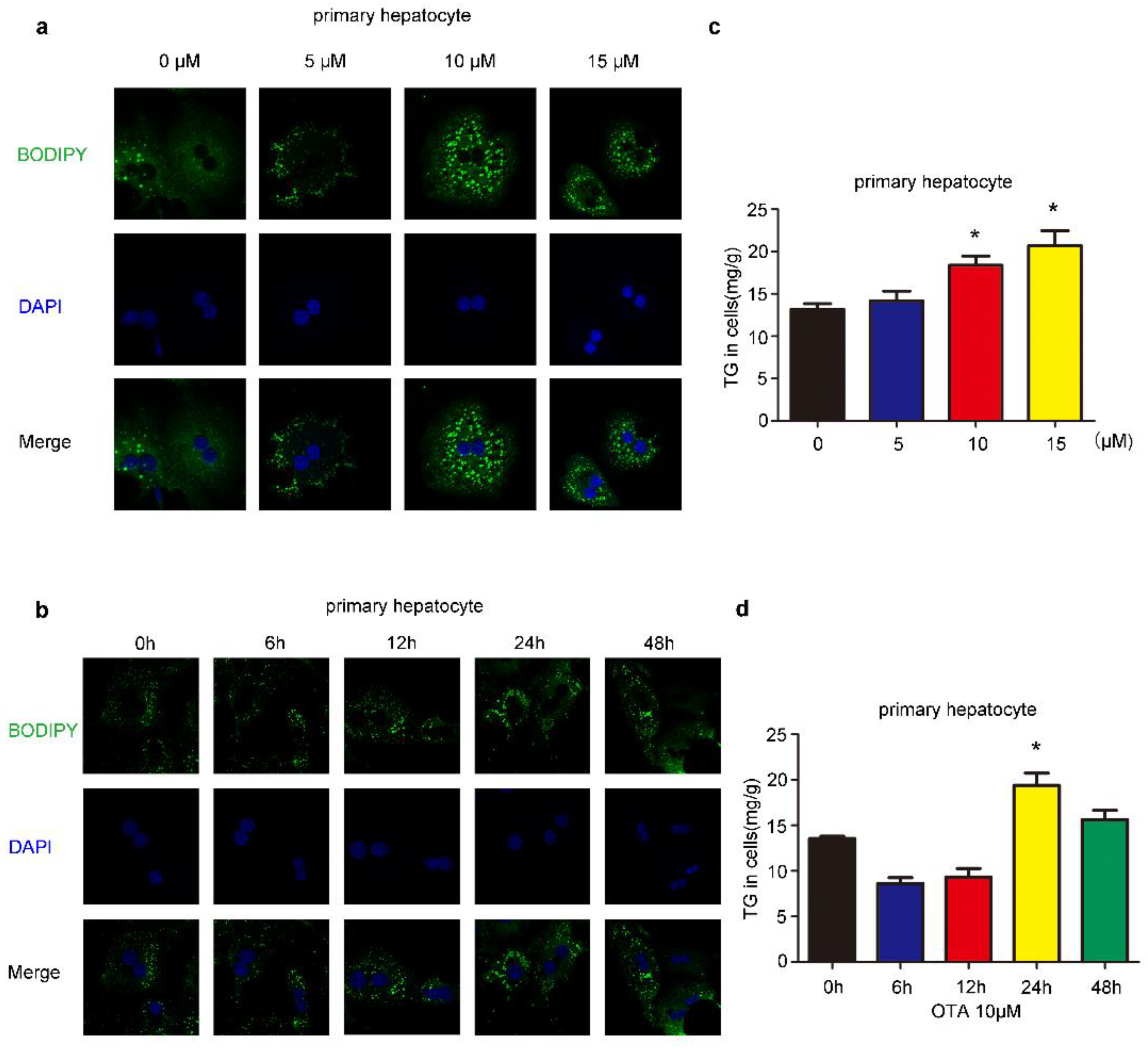
2.1. Effects of OTA on Lipid Accumulation In Vitro
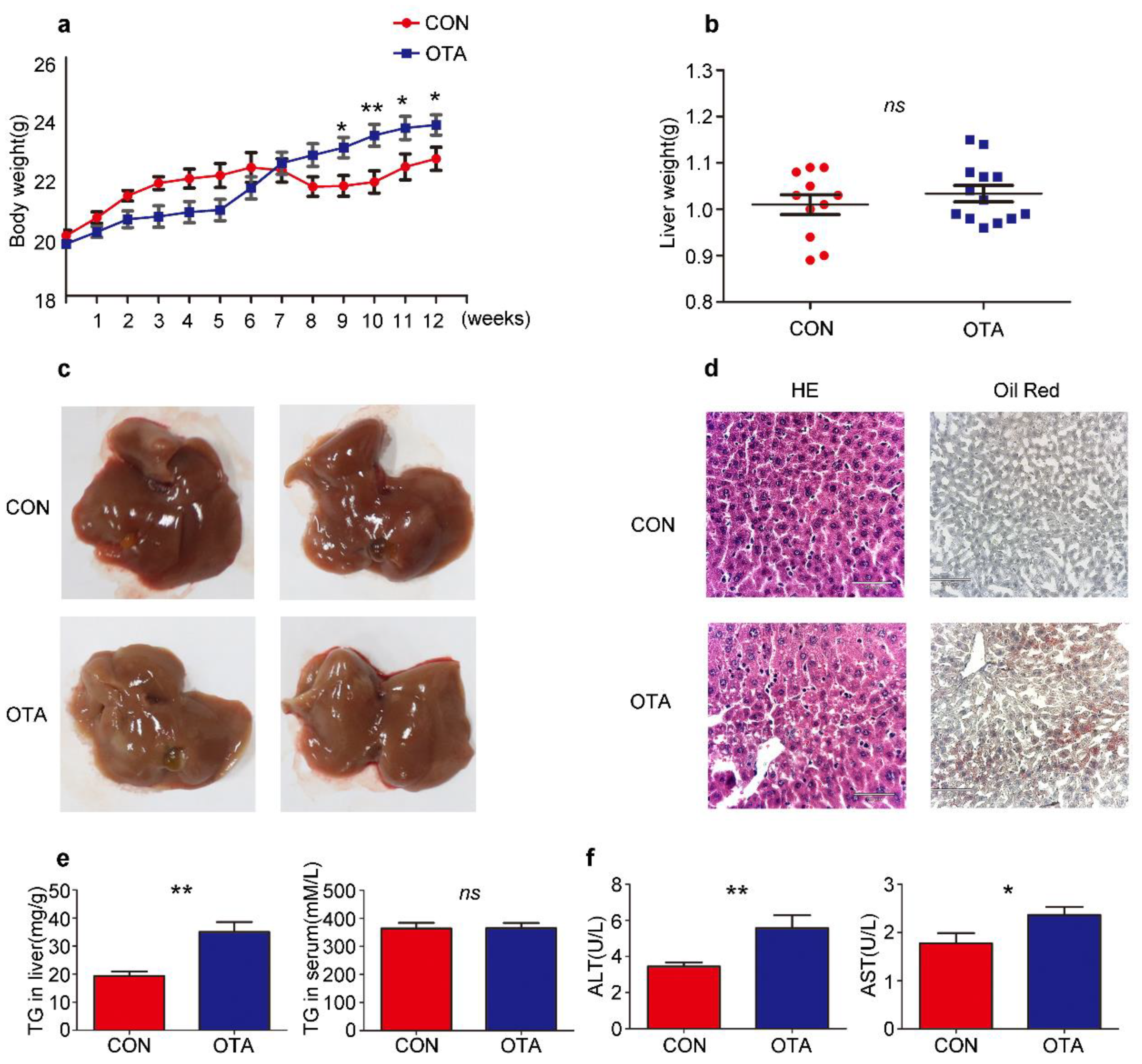
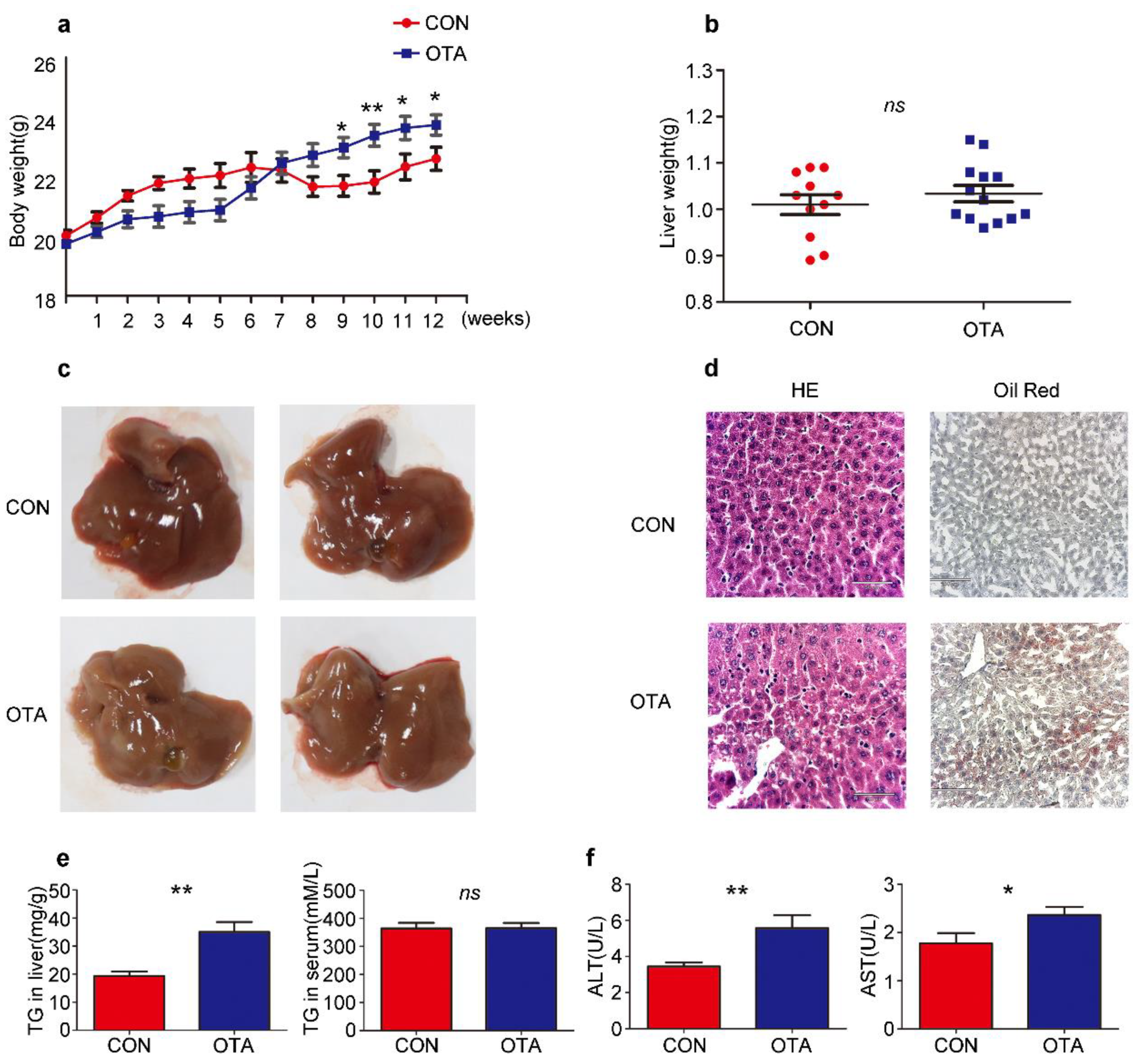
2.2. OTA Promotes Development of Hepatic Steatosis in Mice
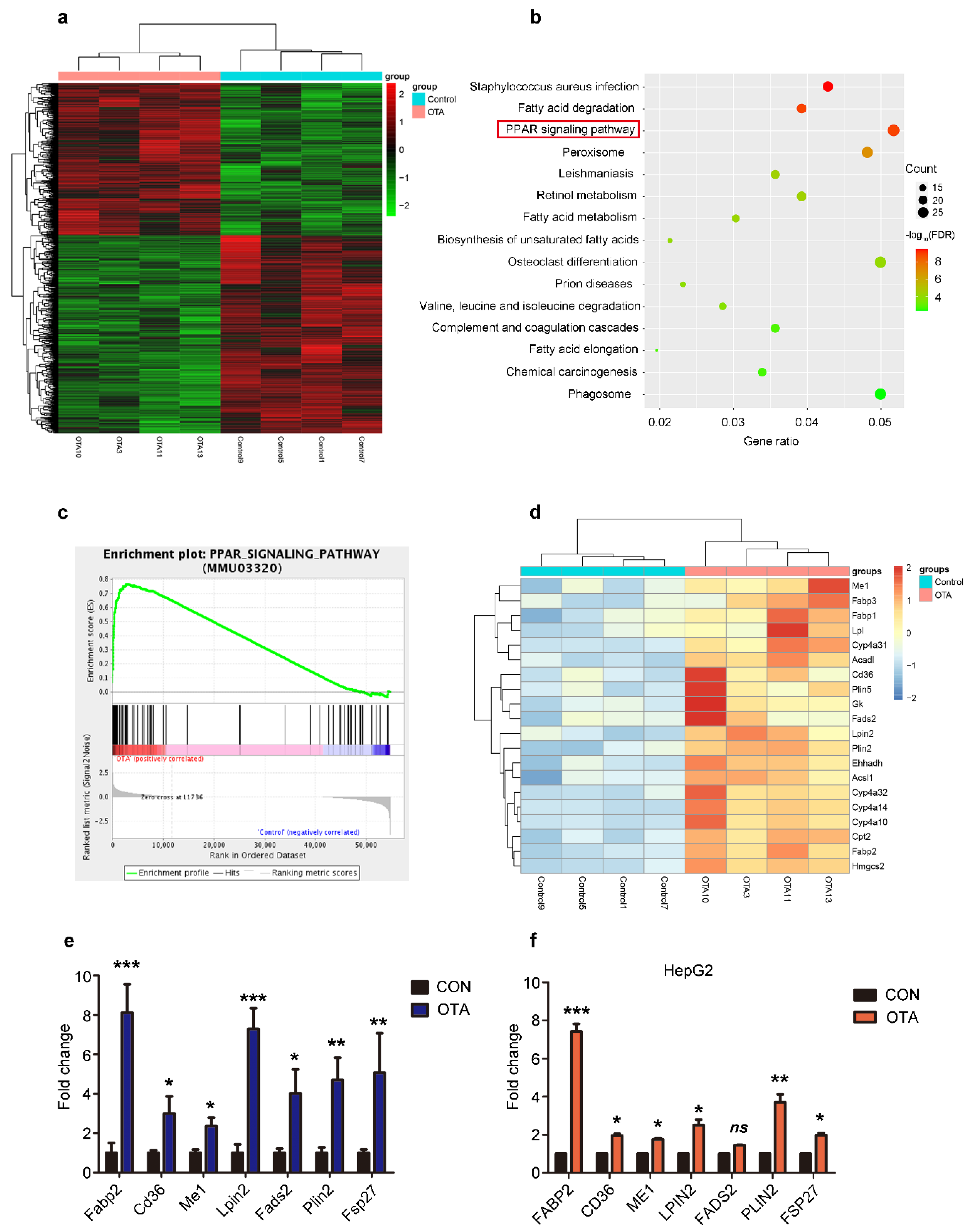
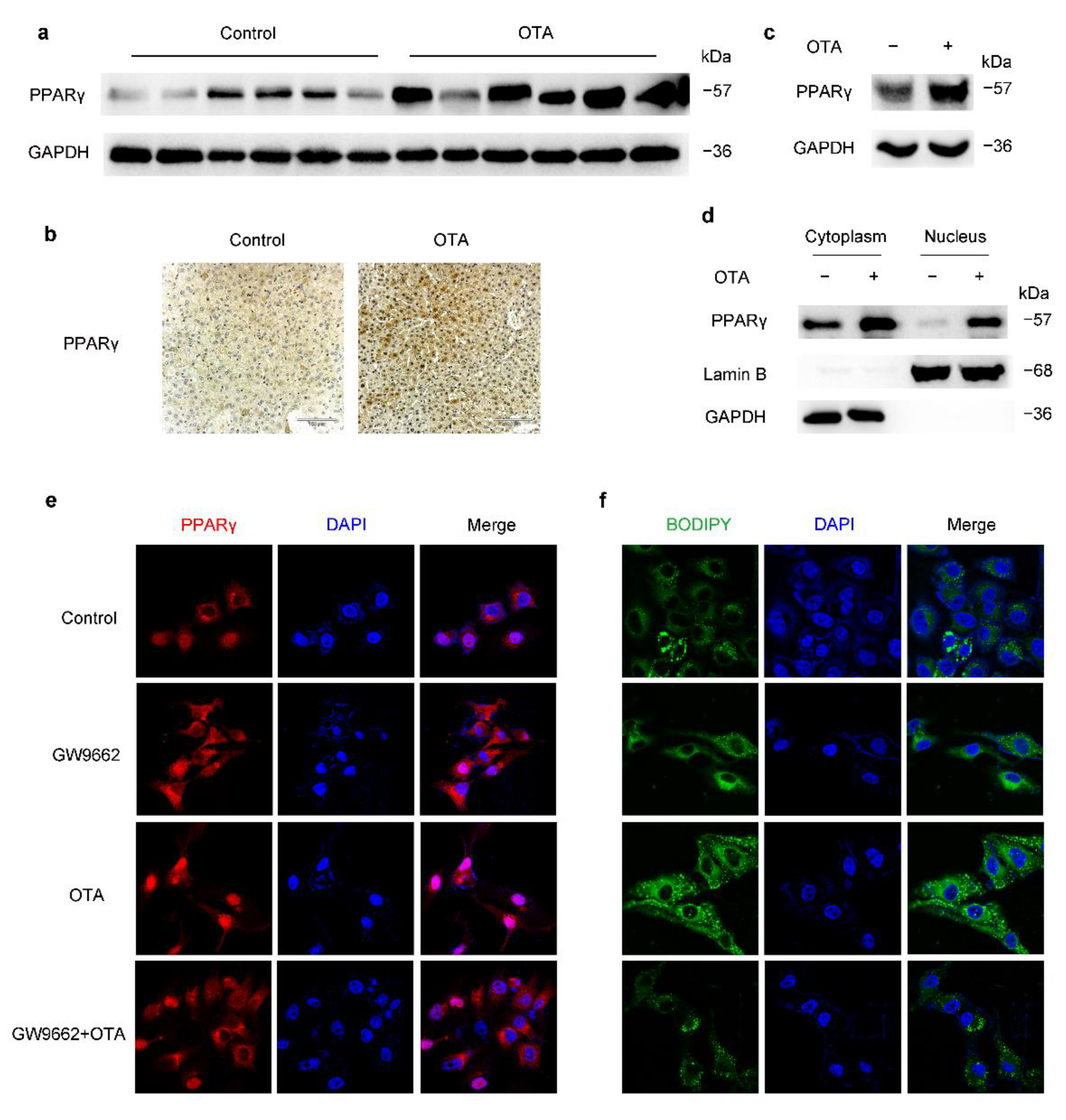
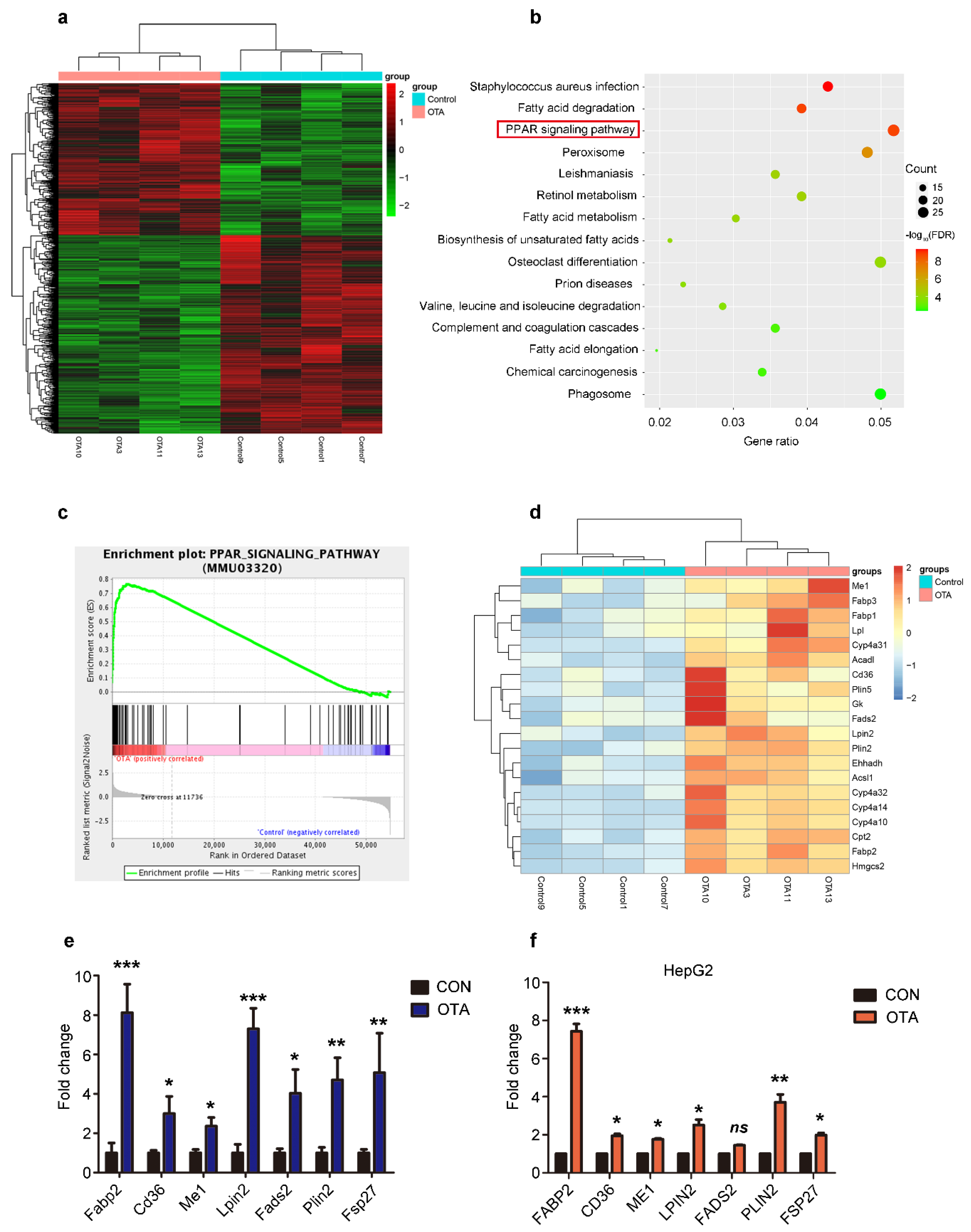
2.3. OTA Induces Hepatic Steatosis through PPARγ Signaling
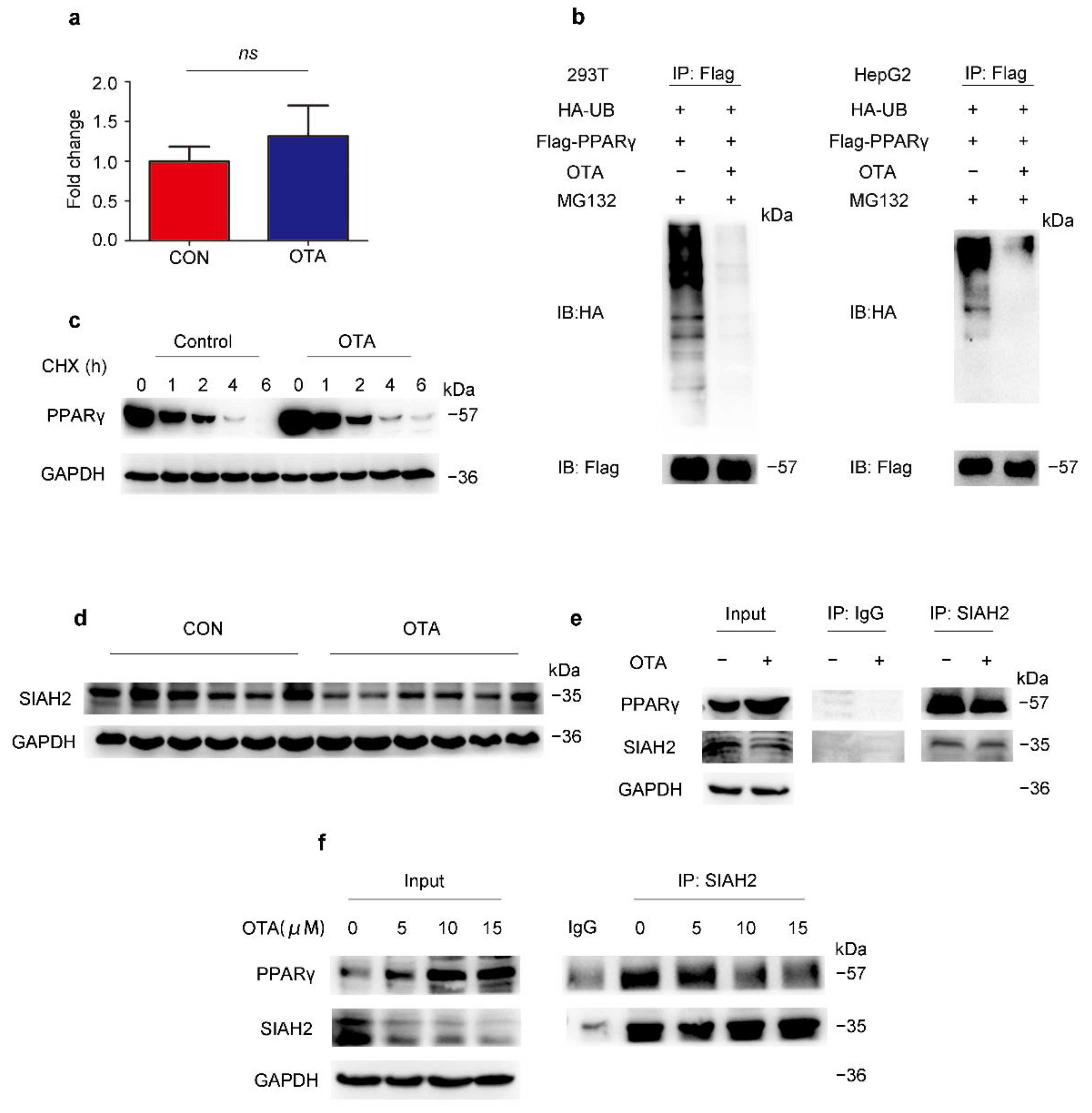
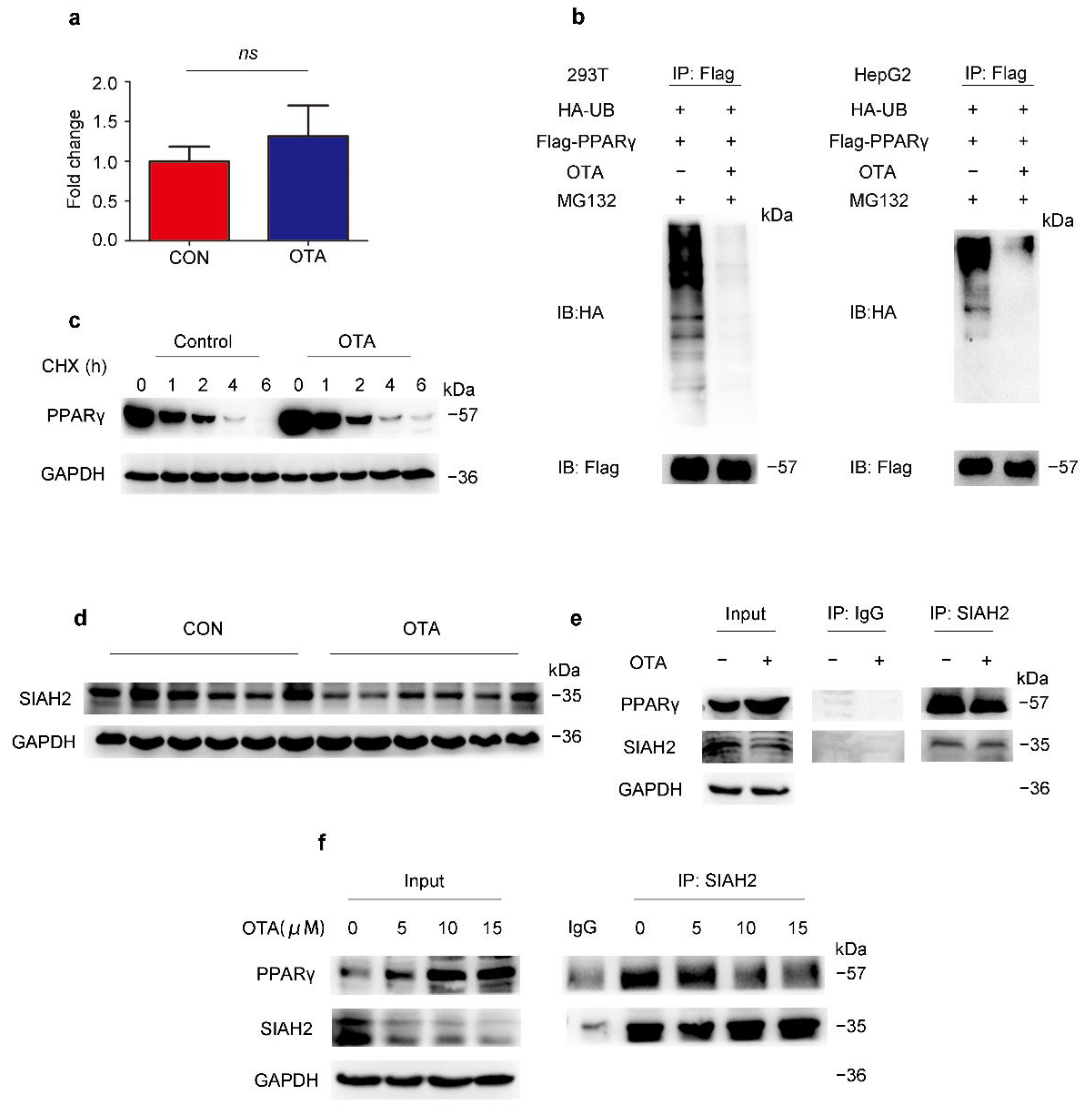
2.4. OTA Regulates PPARγ Protein Stability via Ubiquitin E3 Ligase SIAH2
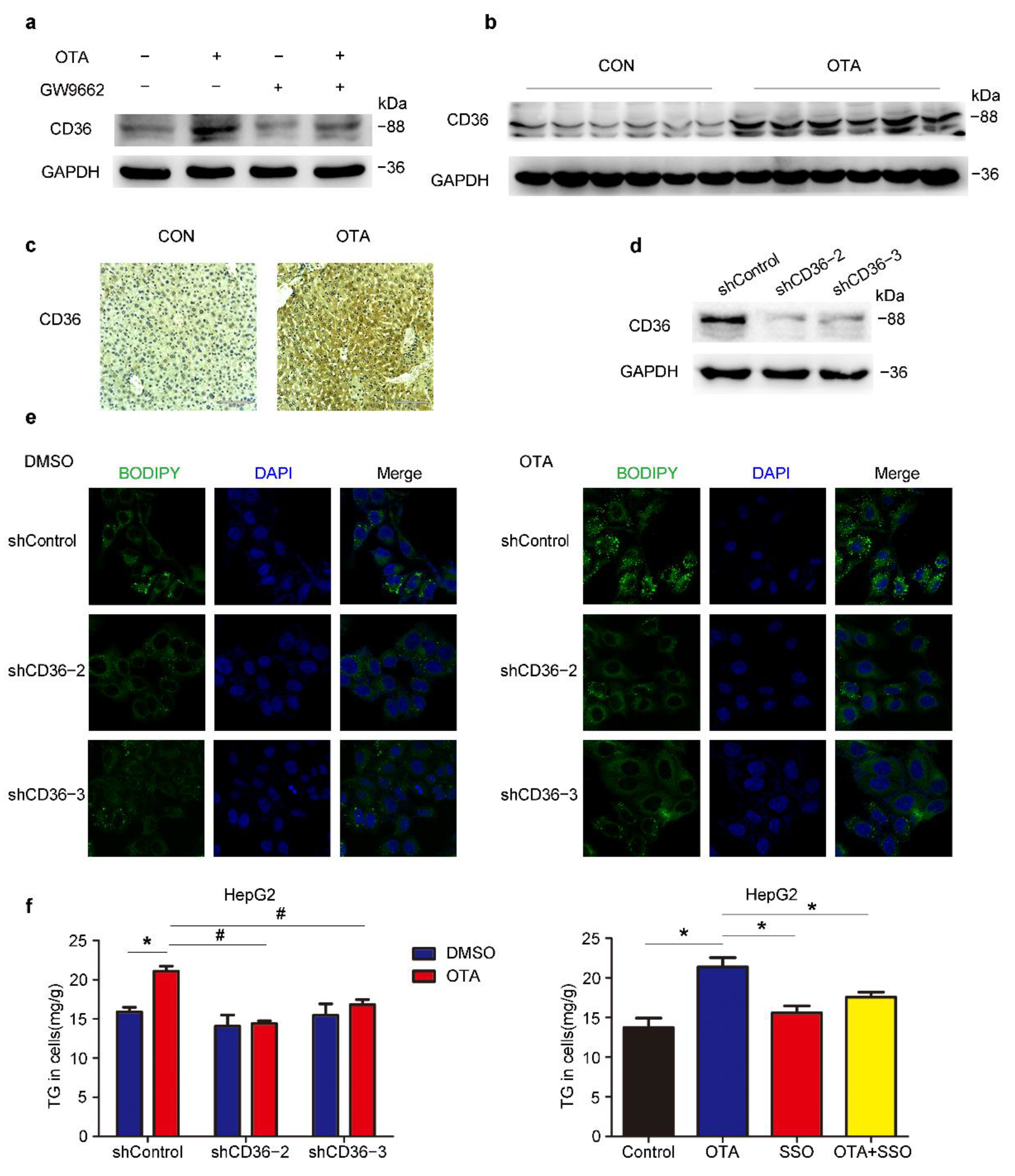
2.5. OTA Induces Hepatic Steatosis in a CD36-Dependent Manner
3. Discussion
4. Materials and Methods
4.1. Experimental Animals
4.2. Cell Lines, Plasmids and Reagents
4.3. Primary Hepatocytes Separation
4.4. Real-Time PCR
4.5. Western Blot Analysis
4.6. Oil Red O Staining
4.7. Mouse Serum and Hepatic Lipid Analyses
4.8. BODIPY Staining
4.9. RNA Sequencing and Processing
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Ringot, D.; Chango, A.; Schneider, Y.-J.; Larondelle, Y. Toxicokinetics and toxicodynamics of ochratoxin A, an update. Chem. Biol. Interact. 2006, 159, 18–46. [Google Scholar] [CrossRef] [PubMed]
- Hajok, I.; Kowalska, A.; Piekut, A.; Ćwieląg-Drabek, M. A risk assessment of dietary exposure to ochratoxin A for the Polish population. Food Chem. 2019, 284, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Connolly, L.; Frizzell, C.; Elliott, C.T. Challenging conventional risk assessment with respect to human exposure to multiple food contaminants in food: A case study using maize. Toxicol. Lett. 2015, 238, 54–64. [Google Scholar] [CrossRef]
- Bui-Klimke, T.R.; Wu, F. Ochratoxin A and Human Health Risk: A Review of the Evidence. Crit. Rev. Food Sci. Nutr. 2015, 55, 1860–1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damiano, S.; Iovane, V.; Squillacioti, C.; Mirabella, N.; Prisco, F.; Ariano, A.; Amenta, M.; Giordano, A.; Florio, S.; Ciarcia, R. Red orange and lemon extract prevents the renal toxicity induced by ochratoxin A in rats. J. Cell. Physiol. 2020, 235, 5386–5393. [Google Scholar] [CrossRef]
- Bendele, A.M.; Carlton, W.W.; Krogh, P.; Lillehoj, E.B. Ochratoxin A carcinogenesis in the (C57BL/6J X C3H)F1 mouse. J. Natl. Cancer Inst. 1985, 75, 733–742. [Google Scholar]
- Rached, E.; Hard, G.; Blumbach, K.; Weber, K.; Draheim, R.; Lutz, W.; Ozden, S.; Steger, U.; Dekant, W.; Mally, A. Ochratoxin A: 13-Week Oral Toxicity and Cell Proliferation in Male F344/N Rats. Toxicol. Sci. Off. J. Soc. Toxicol. 2007, 97, 288–298. [Google Scholar] [CrossRef] [Green Version]
- Vettorazzi, A.; Trocóniz, I.; González-Peñas, E.; Arbillaga, L.; Corcuera, L.; Gil, A.; Cerain, A. Kidney and liver distribution of ochratoxin A in male and female F344 rats. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2011, 49, 1935–1942. [Google Scholar] [CrossRef]
- Al-Eryani, L.; Wahlang, B.; Falkner, K.C.; Guardiola, J.J.; Clair, H.B.; Prough, R.A.; Cave, M. Identification of Environmental Chemicals Associated with the Development of Toxicant-associated Fatty Liver Disease in Rodents. Toxicol. Pathol. 2015, 43, 482–497. [Google Scholar] [CrossRef]
- Shin, H.; Lee, H.J.; Pyo, M.C.; Ryu, D.; Lee, K. Ochratoxin A-Induced Hepatotoxicity through Phase I and Phase II Reactions Regulated by AhR in Liver Cells. Toxins 2019, 11, 377. [Google Scholar] [CrossRef] [Green Version]
- Ramyaa, P.; Krishnaswamy, R.; Padma, V.V. Quercetin modulates OTA-induced oxidative stress and redox signalling in HepG2 cells—Up regulation of Nrf2 expression and down regulation of NF-κB and COX-2. Biochim. Biophys. Acta 2014, 1840, 681–692. [Google Scholar] [CrossRef]
- Wang, W.; Zhai, S.; Xia, Y.; Wang, H.; Ruan, D.; Zhou, T.; Zhu, Y.; Zhang, H.; Zhang, M.; Ye, H.; et al. Ochratoxin A induces liver inflammation: Involvement of intestinal microbiota. Microbiome 2019, 7, 151. [Google Scholar] [CrossRef]
- Wang, H.; Wei, Y.; Xie, Y.; Yan, C.; Du, H.; Li, Z. Ochratoxin A and fumonisin B1 exhibit synergistic cytotoxic effects by inducing apoptosis on rat liver cells. Toxicon 2020, 181, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Atroshi, F.; Biese, I.; Saloniemi, H.; Ali-Vehmas, T.; Saari, S.; Rizzo, A.; Veijalainen, P. Significance of apoptosis and its relationship to antioxidants after ochratoxin A administration in mice. J. Pharm. Pharm. Sci. 2000, 3, 281–291. [Google Scholar]
- Chopra, M.; Link, P.; Michels, C.; Schrenk, D. Characterization of ochratoxin A-induced apoptosis in primary rat hepatocytes. Cell Biol. Toxicol. 2010, 26, 239–254. [Google Scholar] [CrossRef]
- Ehrlich, V.; Darroudi, F.; Uhl, M.; Steinkellner, H.; Gann, M.; Majer, B.; Eisenbauer, M.; Knasmüller, S. Genotoxic effects of ochratoxin A in human-derived hepatoma (HepG2) cells. Food Chem. Toxicol. 2002, 40, 1085–1090. [Google Scholar] [CrossRef]
- Gayathri, L.; Dhivya, R.; Dhanasekaran, D.; Periasamy, V.S.; Alshatwi, A.A.; Akbarsha, M.A. Hepatotoxic effect of ochratoxin A and citrinin, alone and in combination, and protective effect of vitamin E: In vitro study in HepG2 cell. Food Chem. Toxicol. 2015, 83, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Xu, W.-T.; Huang, K.; Zhang, B.; Wang, H.; Zhang, X.; Gong, L.; Luo, Y.; Xiaoyun, H. Precision toxicology shows that troxerutin alleviates ochratoxin A–induced renal lipotoxicity. FASEB J. 2018, 33, fj.201800742R. [Google Scholar] [CrossRef] [Green Version]
- Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptors: Nuclear control of metabolism. Endocr. Rev. 1999, 20, 649–688. [Google Scholar]
- Brocker, C.N.; Patel, D.P.; Velenosi, T.J.; Kim, D.; Yan, T.; Yue, J.; Li, G.; Krausz, K.W.; Gonzalez, F.J. Extrahepatic PPARα modulates fatty acid oxidation and attenuates fasting-induced hepatosteatosis in mice. J. Lipid Res. 2018, 59, 2140–2152. [Google Scholar] [CrossRef] [Green Version]
- Koh, J.H.; Hancock, C.R.; Terada, S.; Higashida, K.; Holloszy, J.O.; Han, D.H. PPARβ Is Essential for Maintaining Normal Levels of PGC-1α and Mitochondria and for the Increase in Muscle Mitochondria Induced by Exercise. Cell Metab. 2017, 25, 1176–1185.e5. [Google Scholar] [CrossRef] [PubMed]
- Georgiadi, A.; Lichtenstein, L.; Degenhardt, T.; Boekschoten, M.V.; van Bilsen, M.; Desvergne, B.; Müller, M.; Kersten, S. Induction of cardiac Angptl4 by dietary fatty acids is mediated by peroxisome proliferator-activated receptor beta/delta and protects against fatty acid-induced oxidative stress. Circ. Res. 2010, 106, 1712–1721. [Google Scholar] [CrossRef] [Green Version]
- Gavrilova, O.; Haluzik, M.; Matsusue, K.; Cutson, J.J.; Johnson, L.; Dietz, K.R.; Nicol, C.J.; Vinson, C.; Gonzalez, F.J.; Reitman, M.L. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J. Biol. Chem. 2003, 278, 34268–34276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilroy, G.; Kirk-Ballard, H.; Carter, L.E.; Floyd, Z.E. The ubiquitin ligase Siah2 regulates PPARγ activity in adipocytes. Endocrinology 2012, 153, 1206–1218. [Google Scholar] [CrossRef] [Green Version]
- Zain, M.E. Impact of mycotoxins on humans and animals. J. Saudi Chem. Soc. 2011, 15, 129–144. [Google Scholar] [CrossRef] [Green Version]
- Bennett, J.W.; Klich, M. Mycotoxins. Encycl. Microbiol. 2009, 16, 559–565. [Google Scholar]
- Hanif, N.Q.; Muhammad, G.; Siddique, M.; Khanum, A.; Ahmed, T.; Gadahai, J.A.; Kaukab, G. Clinico-pathomorphological, serum biochemical and histological studies in broilers fed ochratoxin A and a toxin deactivator (Mycofix Plus). Br. Poult. Sci. 2008, 49, 632–642. [Google Scholar] [CrossRef] [PubMed]
- Damiano, S.; Longobardi, C.; Andretta, E.; Prisco, F.; Piegari, G.; Squillacioti, C.; Montagnaro, S.; Pagnini, F.; Badino, P.; Florio, S.; et al. Antioxidative Effects of Curcumin on the Hepatotoxicity Induced by Ochratoxin A in Rats. Antioxidants 2021, 10, 125. [Google Scholar] [CrossRef] [PubMed]
- Longobardi, C.; Damiano, S.; Andretta, E.; Prisco, F.; Russo, V.; Pagnini, F.; Florio, S.; Ciarcia, R. Curcumin Modulates Nitrosative Stress, Inflammation, and DNA Damage and Protects against Ochratoxin A-Induced Hepatotoxicity and Nephrotoxicity in Rats. Antioxidants 2021, 10, 1239. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Dwivedi, P.; Sharma, A.K.; Sankar, M.; Patil, R.D.; Singh, N.D. Apoptosis and lipid peroxidation in ochratoxin A- and citrinin-induced nephrotoxicity in rabbits. Toxicol. Ind. Health 2014, 30, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Soyöz, M.; Ozçelik, N.; Kilinç, I.; Altuntaş, I. The effects of ochratoxin A on lipid peroxidation and antioxidant enzymes: A protective role of melatonin. Cell Biol. Toxicol. 2004, 20, 213–219. [Google Scholar] [CrossRef]
- Cave, M.C.; Clair, H.B.; Hardesty, J.E.; Falkner, K.C.; Feng, W.; Clark, B.J.; Sidey, J.; Shi, H.; Aqel, B.A.; McClain, C.J.; et al. Nuclear receptors and nonalcoholic fatty liver disease. Biochim. Biophys. Acta 2016, 1859, 1083–1099. [Google Scholar] [CrossRef] [Green Version]
- Hong, F.; Pan, S.; Guo, Y.; Xu, P.; Zhai, Y. PPARs as Nuclear Receptors for Nutrient and Energy Metabolism. Molecules 2019, 24, 2545. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.; Jang, H.J.; Kim, J.K.; Kim, J.M.; Park, E.H.; Yang, J.H.; Kim, Y.H.; Yea, K.; Ryu, S.H.; Suh, P.G. Ochratoxin A inhibits adipogenesis through the extracellular signal-related kinases-peroxisome proliferator-activated receptor-γ pathway in human adipose tissue-derived mesenchymal stem cells. Stem Cells Dev. 2011, 20, 415–426. [Google Scholar] [CrossRef]
- Akpinar, H.A.; Kahraman, H.; Yaman, I. Ochratoxin A Sequentially Activates Autophagy and the Ubiquitin-Proteasome System. Toxins 2019, 11, 615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Febbraio, M.; Wada, T.; Zhai, Y.; Xie, W. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosis. Gastroenterology 2008, 134, 556–567.e1. [Google Scholar] [CrossRef]
- Koonen, D.P.; Jacobs, R.L.; Febbraio, M.; Young, M.E.; Soltys, C.L.; Ong, H.; Vance, D.E.; Dyck, J.R. Increased hepatic CD36 expression contributes to dyslipidemia associated with diet-induced obesity. Diabetes 2007, 56, 2863–2871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storch, J.; McDermott, L. Structural and functional analysis of fatty acid-binding proteins. J. Lipid Res. 2009, 50, S126–S131. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.Z.; Chen, P.P.; Wang, Y.; Yin, D.; Koeffler, H.P.; Li, B.; Tong, X.J.; Xie, D. Connective Tissue Growth Factor Is Overexpressed in Esophageal Squamous Cell Carcinoma and Promotes Tumorigenicity through β-Catenin-T-cell Factor/Lef Signaling. J. Biol. Chem. 2007, 282, 36571–36581. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, Q.-W.; Ding, X.-F.; Cao, H.-J.; Ni, Q.-Z.; Zhu, B.; Ma, N.; Zhang, F.-K.; Wang, Y.-K.; Xu, S.; Chen, T.-W.; et al. Ochratoxin A Induces Steatosis via PPARγ-CD36 Axis. Toxins 2021, 13, 802. https://doi.org/10.3390/toxins13110802
Zheng Q-W, Ding X-F, Cao H-J, Ni Q-Z, Zhu B, Ma N, Zhang F-K, Wang Y-K, Xu S, Chen T-W, et al. Ochratoxin A Induces Steatosis via PPARγ-CD36 Axis. Toxins. 2021; 13(11):802. https://doi.org/10.3390/toxins13110802
Chicago/Turabian StyleZheng, Qian-Wen, Xu-Fen Ding, Hui-Jun Cao, Qian-Zhi Ni, Bing Zhu, Ning Ma, Feng-Kun Zhang, Yi-Kang Wang, Sheng Xu, Tian-Wei Chen, and et al. 2021. "Ochratoxin A Induces Steatosis via PPARγ-CD36 Axis" Toxins 13, no. 11: 802. https://doi.org/10.3390/toxins13110802
APA StyleZheng, Q.-W., Ding, X.-F., Cao, H.-J., Ni, Q.-Z., Zhu, B., Ma, N., Zhang, F.-K., Wang, Y.-K., Xu, S., Chen, T.-W., Xia, J., Qiu, X.-S., Yu, D.-Z., Xie, D., & Li, J.-J. (2021). Ochratoxin A Induces Steatosis via PPARγ-CD36 Axis. Toxins, 13(11), 802. https://doi.org/10.3390/toxins13110802