Nicotinamide Attenuates the Progression of Renal Failure in a Mouse Model of Adenine-Induced Chronic Kidney Disease

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
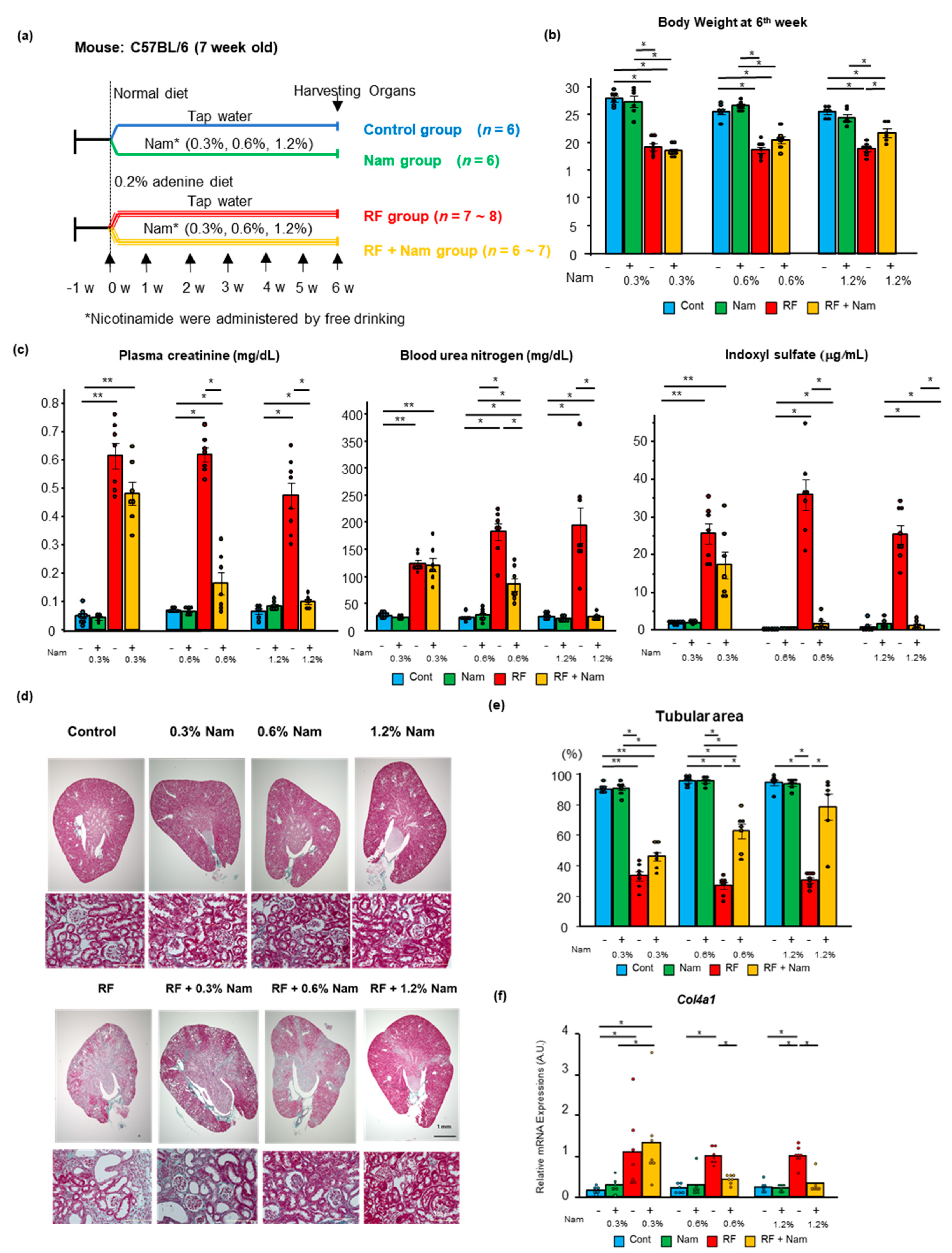
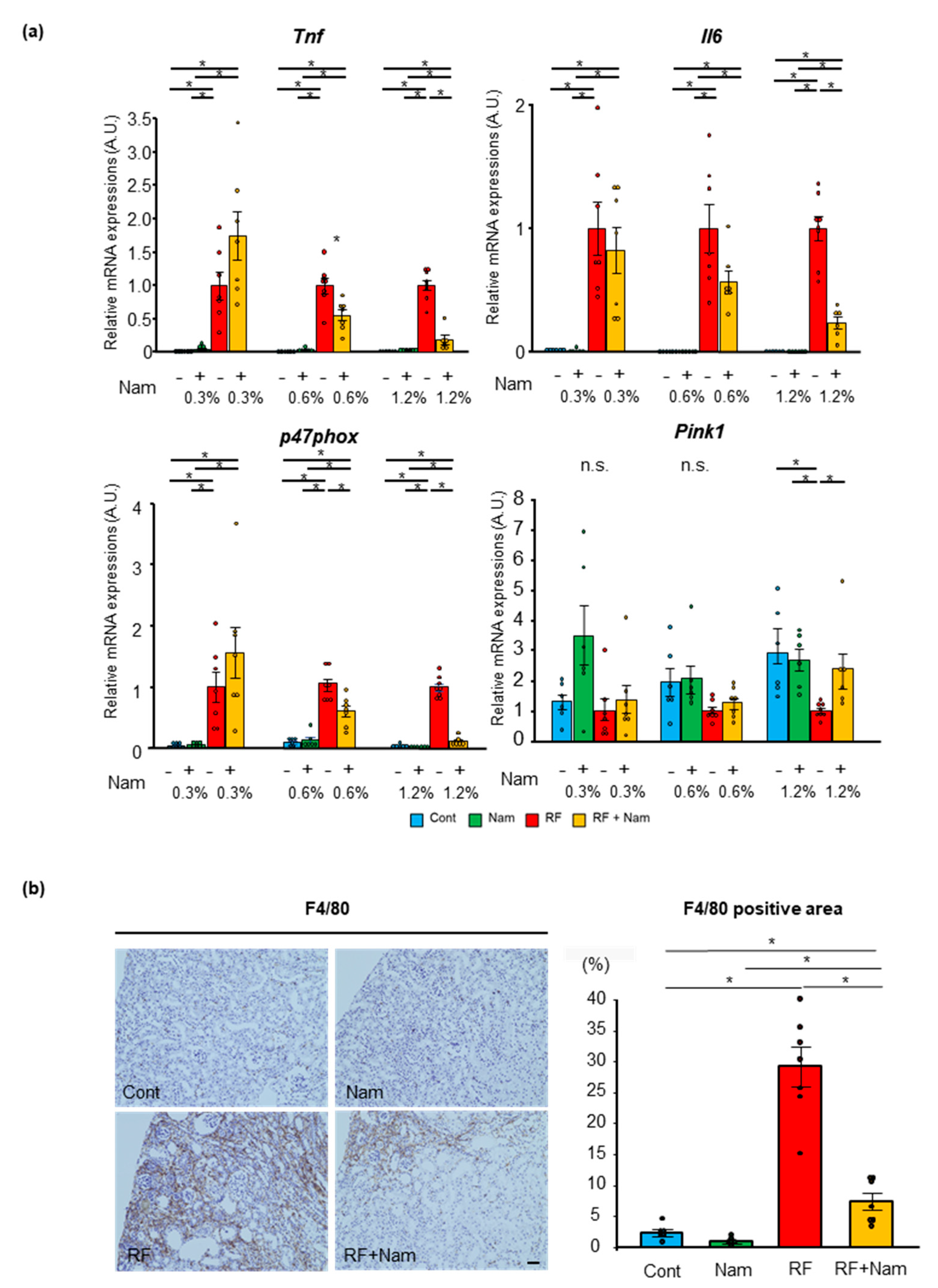
2.1. Prophylactic Nam Supplementation Prevented the Progression of Kidney Disease
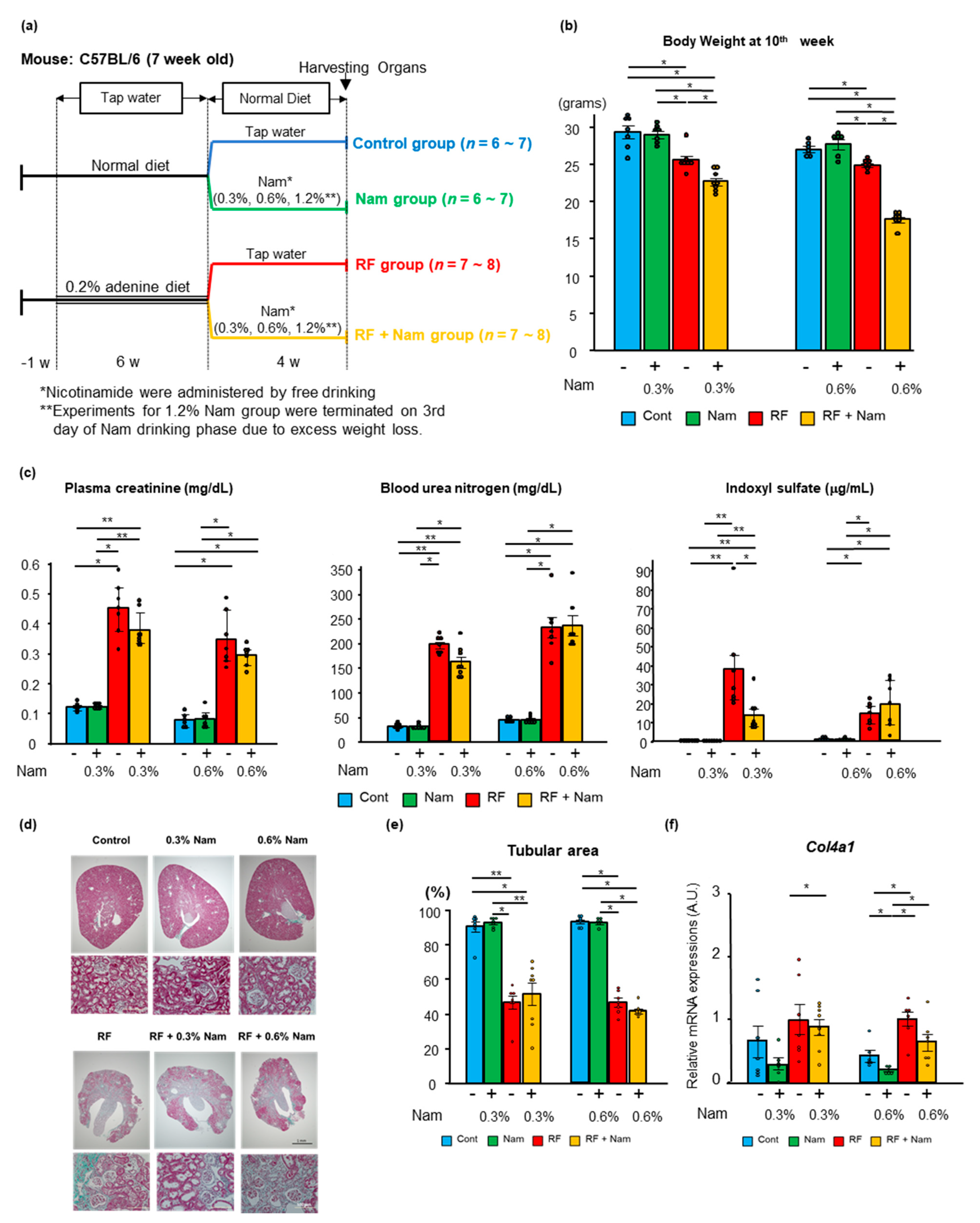
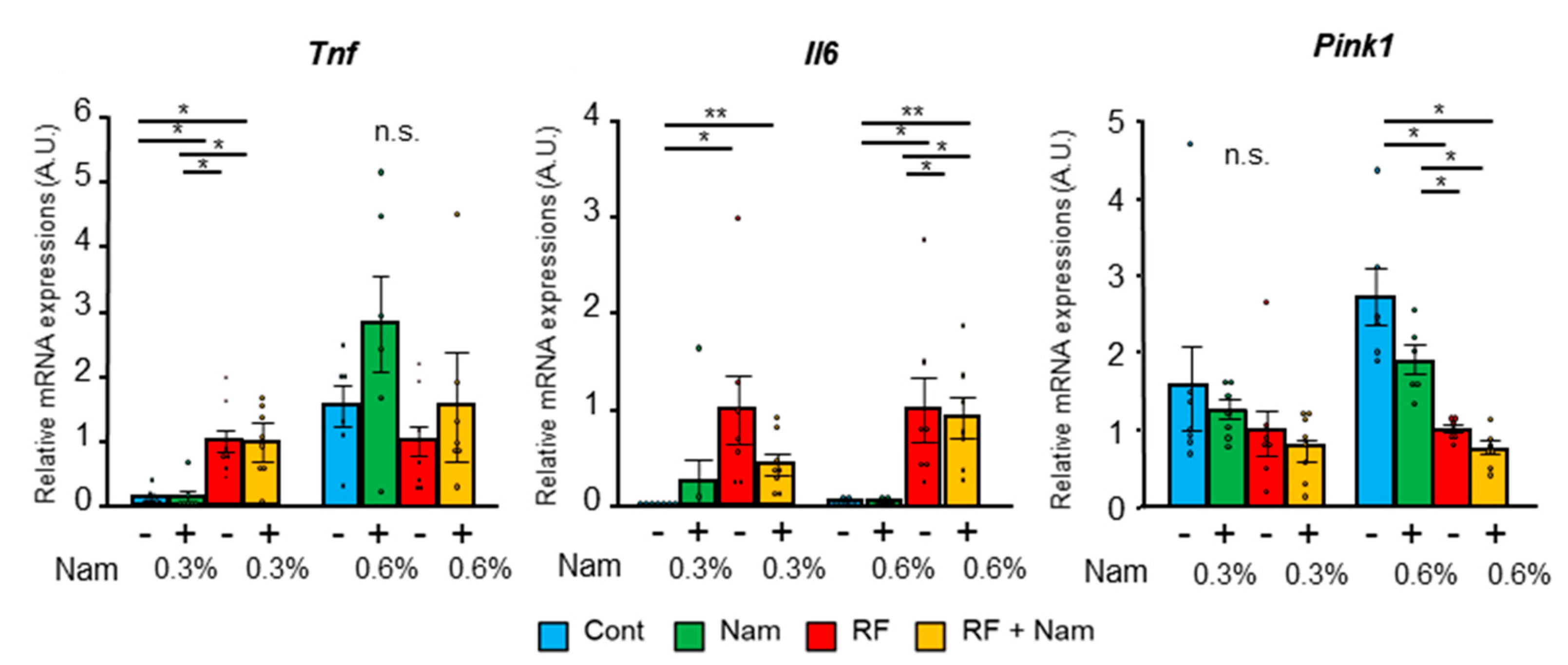
2.2. Nam Supplementation Did Not Attenuate Advanced Kidney Disease
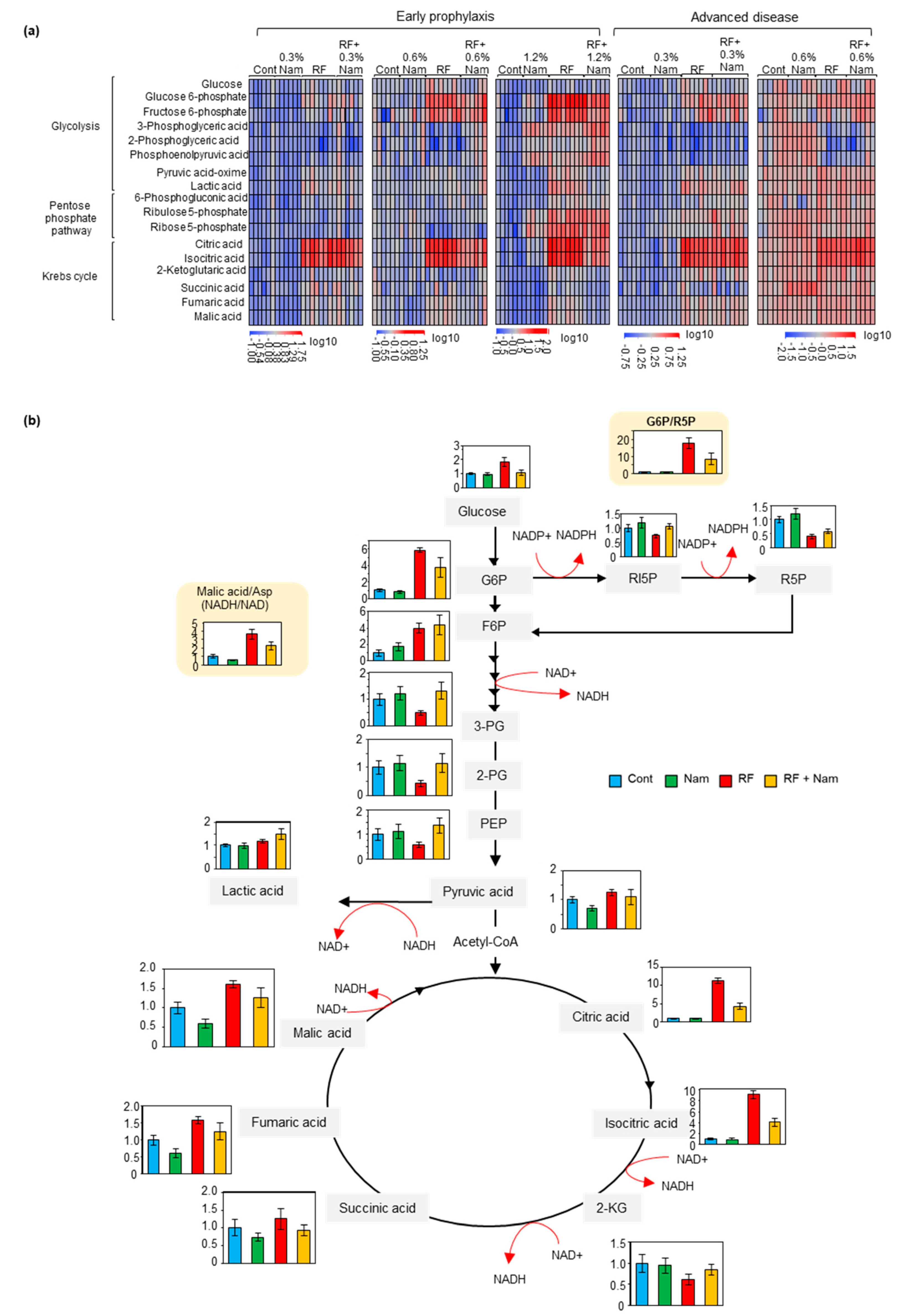
2.3. Nam Administration Affected the Accumulation of Glycolysis, Pentose Phosphate Shuttle, and Krebs Cycle Metabolites at Early, But Not Advanced, Stages of Kidney Disease
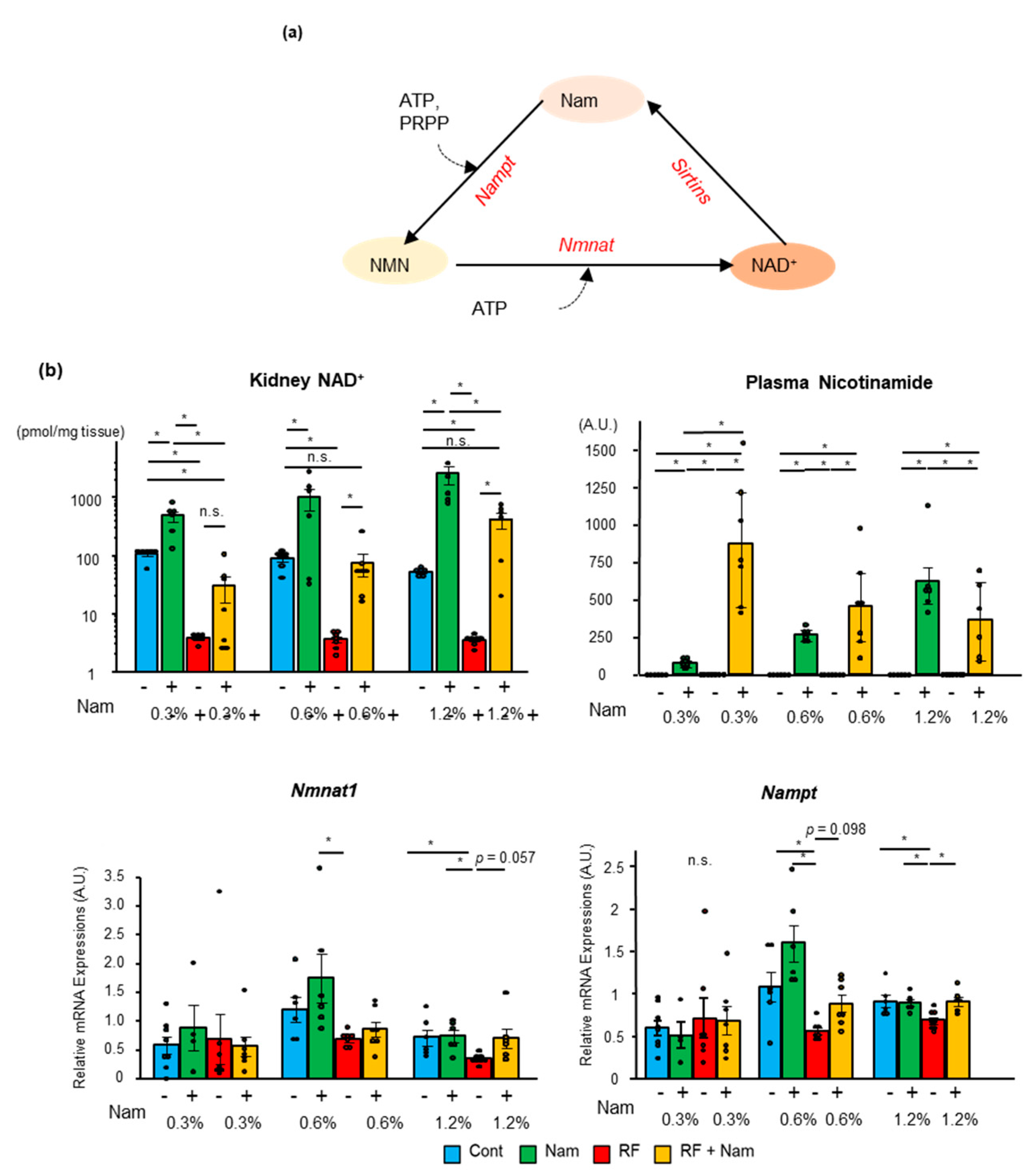
2.4. The Levels of NAD+ Increased in the Kidney with the Concentration of Nam
3. Discussion
3.1. Nam Administration May Be Beneficial in Early Stages of Kidney Disease
3.2. Nam Administration Did Not Exert Beneficial Effects in Mice with Advanced Kidney Disease
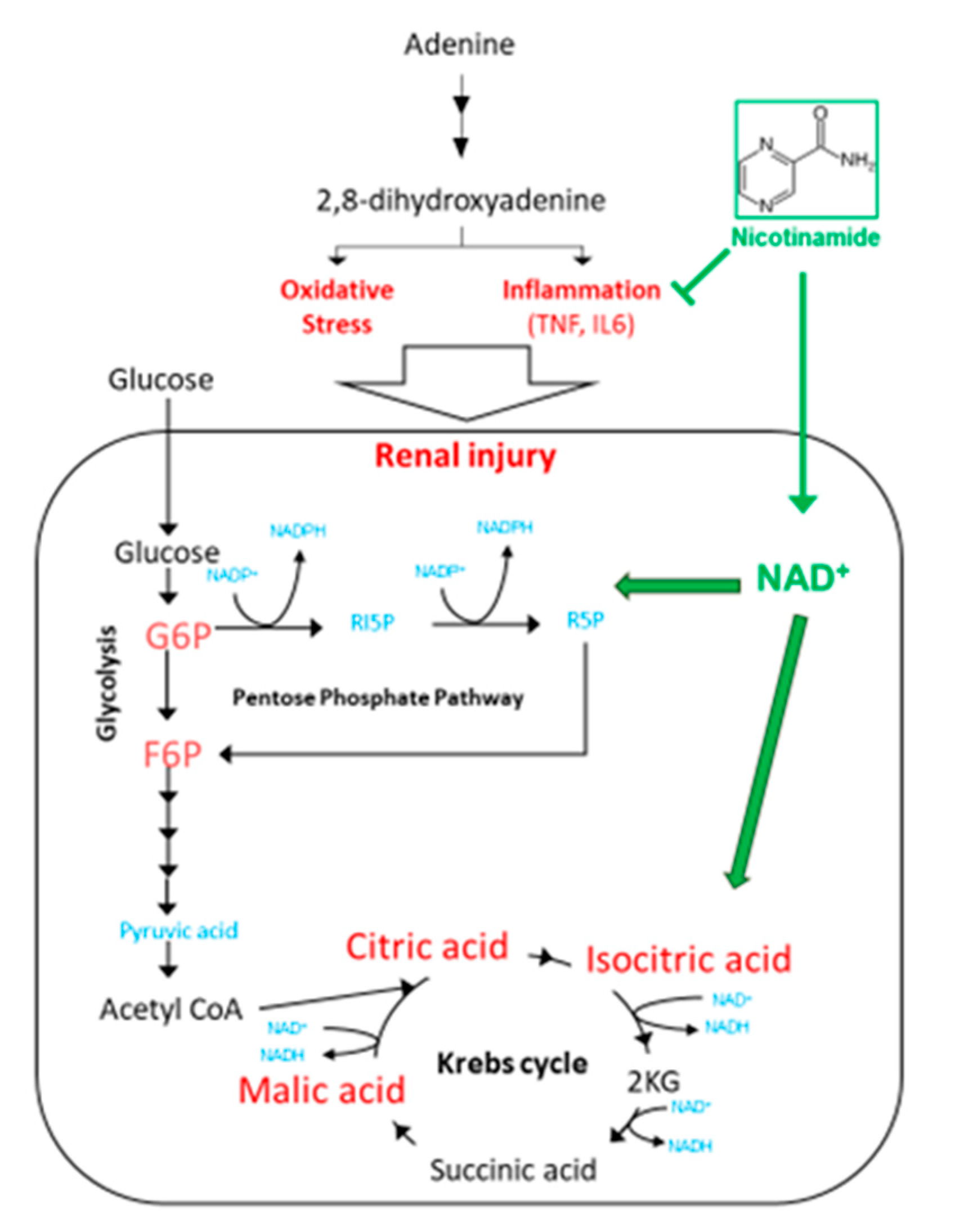
3.3. Nam Supplementation Altered the Levels of Glycolysis, Pentose Phosphate Pathway, and Krebs Cycle Metabolites in the Kidney
3.4. Perspectives in the Use of Nam for the Treatment of CKD
4. Conclusions
5. Materials and Methods
5.1. Animal Experiments
5.2. Histological Analysis
5.3. Quantitative Real-Time PCR Analysis
5.4. LC-MS/MS Measurements for Indoxyl Sulfate and Creatinine
5.5. Sample Preparation for GC-MS Measurements
5.6. GC-MS Measurements
5.7. NAD+ Measurements
5.8. BUN Measurements
5.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mills, K.T.; Xu, Y.; Zhang, W.; Bundy, J.D.; Chen, C.S.; Kelly, T.N.; Chen, J.; He, J. A systematic analysis of worldwide population-based data on the global burden of chronic kidney disease in 2010. Kidney Int. 2015, 88, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.; Yang, C.W. Chronic kidney disease: Global dimension and perspectives. Lancet 2013, 382, 260–272. [Google Scholar] [CrossRef]
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.A.; O’Callaghan, C.A.; Lasserson, D.S.; Hobbs, F.D. Global Prevalence of Chronic Kidney Disease-A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0158765. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Watanabe, T.; Takeuchi, A.; Ohashi, Y.; Nitta, K.; Akizawa, T.; Matsuo, S.; Imai, E.; Makino, H.; Hishida, A.; et al. Cardiovascular events and death in Japanese patients with chronic kidney disease. Kidney Int. 2017, 91, 227–234. [Google Scholar] [CrossRef]
- Kahn, M.R.; Robbins, M.J.; Kim, M.C.; Fuster, V. Management of cardiovascular disease in patients with kidney disease. Nat. Rev. Cardiol. 2013, 10, 261–273. [Google Scholar] [CrossRef]
- Cheikh Hassan, H.I.; Tang, M.; Djurdjev, O.; Langsford, D.; Sood, M.M.; Levin, A. Infection in advanced chronic kidney disease leads to increased risk of cardiovascular events, end-stage kidney disease and mortality. Kidney Int. 2016, 90, 897–904. [Google Scholar] [CrossRef]
- Dalrymple, L.S.; Katz, R.; Kestenbaum, B.; de Boer, I.H.; Fried, L.; Sarnak, M.J.; Shlipak, M.G. The risk of infection-related hospitalization with decreased kidney function. Am. J. Kidney Dis. 2012, 59, 356–363. [Google Scholar] [CrossRef]
- Komaki, Y.; Komaki, F.; Micic, D.; Ido, A.; Sakuraba, A. Risk of Colorectal Cancer in Chronic Kidney Disease: A Systematic Review and Meta-Analysis. J. Clin. Gastroenterol. 2018, 52, 796–804. [Google Scholar] [CrossRef]
- Wimmer, C.D.; Rentsch, M.; Crispin, A.; Illner, W.D.; Arbogast, H.; Graeb, C.; Jauch, K.W.; Guba, M. The janus face of immunosuppression-de novo malignancy after renal transplantation: The experience of the Transplantation Center Munich. Kidney Int. 2007, 71, 1271–1278. [Google Scholar] [CrossRef]
- Nagai, K.; Iseki, C.; Iseki, K.; Kondo, M.; Asahi, K.; Saito, C.; Tsunoda, R.; Okubo, R.; Yamagata, K. Higher medical costs for CKD patients with a rapid decline in eGFR: A cohort study from the Japanese general population. PLoS ONE 2019, 14, e0216432. [Google Scholar] [CrossRef]
- Perlman, R.L.; Finkelstein, F.O.; Liu, L.; Roys, E.; Kiser, M.; Eisele, G.; Burrows-Hudson, S.; Messana, J.M.; Levin, N.; Rajagopalan, S.; et al. Quality of life in chronic kidney disease (CKD): A cross-sectional analysis in the Renal Research Institute-CKD study. Am. J. Kidney Dis. 2005, 45, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.; Howard, K.; Chapman, J.; Pollock, C.; Chadban, S.; Salkeld, G.; Tong, A.; Williams, N.; Webster, A.; Craig, J.C. How do people with chronic kidney disease value cancer-related quality of life? Nephrology (Carlton) 2012, 17, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Zoccali, C.; Vanholder, R.; Massy, Z.A.; Ortiz, A.; Sarafidis, P.; Dekker, F.W.; Fliser, D.; Fouque, D.; Heine, G.H.; Jager, K.J.; et al. Cardiovascular Medicine Working Group of the European Renal Association-European Dialysis Transplantation, A. The systemic nature of CKD. Nat. Rev. Nephrol. 2017, 13, 344–358. [Google Scholar] [CrossRef] [PubMed]
- Yakulov, T.A.; Todkar, A.P.; Slanchev, K.; Wiegel, J.; Bona, A.; Gross, M.; Scholz, A.; Hess, I.; Wurditsch, A.; Grahammer, F.; et al. CXCL12 and MYC control energy metabolism to support adaptive responses after kidney injury. Nat. Commun. 2018, 9, 3660. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Sato, E.; Mori, T.; Mishima, E.; Suzuki, A.; Sugawara, S.; Kurasawa, N.; Saigusa, D.; Miura, D.; Morikawa-Ichinose, T.; Saito, R.; et al. Metabolic alterations by indoxyl sulfate in skeletal muscle induce uremic sarcopenia in chronic kidney disease. Sci. Rep. 2016, 6, 36618. [Google Scholar] [CrossRef]
- Ying, W. NAD+ and NADH in neuronal death. J. Neuroimmune Pharmacol. 2007, 2, 270–275. [Google Scholar] [CrossRef]
- Imai, S.; Yoshino, J. The importance of NAMPT/NAD/SIRT1 in the systemic regulation of metabolism and ageing. Diabetes Obes. Metab. 2013, 15 (Suppl. 3), 26–33. [Google Scholar] [CrossRef]
- Ryall, J.G.; Dell’Orso, S.; Derfoul, A.; Juan, A.; Zare, H.; Feng, X.; Clermont, D.; Koulnis, M.; Gutierrez-Cruz, G.; Fulco, M.; et al. The NAD(+)-dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells. Cell Stem Cell 2015, 16, 171–183. [Google Scholar] [CrossRef]
- Guan, Y.; Wang, S.R.; Huang, X.Z.; Xie, Q.H.; Xu, Y.Y.; Shang, D.; Hao, C.M. Nicotinamide Mononucleotide, an NAD(+) Precursor, Rescues Age-Associated Susceptibility to AKI in a Sirtuin 1-Dependent Manner. J. Am. Soc. Nephrol. 2017, 28, 2337–2352. [Google Scholar] [CrossRef]
- Oh, G.S.; Kim, H.J.; Choi, J.H.; Shen, A.; Choe, S.K.; Karna, A.; Lee, S.H.; Jo, H.J.; Yang, S.H.; Kwak, T.H.; et al. Pharmacological activation of NQO1 increases NAD(+) levels and attenuates cisplatin-mediated acute kidney injury in mice. Kidney Int. 2014, 85, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Poyan Mehr, A.; Tran, M.T.; Ralto, K.M.; Leaf, D.E.; Washco, V.; Messmer, J.; Lerner, A.; Kher, A.; Kim, S.H.; Khoury, C.C.; et al. De novo NAD(+) biosynthetic impairment in acute kidney injury in humans. Nat. Med. 2018, 24, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Cai, J.; Liu, Z.; Shu, S.; Wang, Y.; Tang, C.; Dong, Z. Nicotinamide reduces renal interstitial fibrosis by suppressing tubular injury and inflammation. J. Cell. Mol. Med. 2019, 23, 3995–4004. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E. NAD(+) in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Li, F.; Fushima, T.; Oyanagi, G.; Townley-Tilson, H.W.; Sato, E.; Nakada, H.; Oe, Y.; Hagaman, J.R.; Wilder, J.; Li, M.; et al. Nicotinamide benefits both mothers and pups in two contrasting mouse models of preeclampsia. Proc. Natl. Acad. Sci. USA 2016, 113, 13450–13455. [Google Scholar] [CrossRef]
- Imaruoka, K.; Oe, Y.; Fushima, T.; Sato, E.; Sekimoto, A.; Sato, H.; Sugawara, J.; Ito, S.; Takahashi, N. Nicotinamide alleviates kidney injury and pregnancy outcomes in lupus-prone MRL/lpr mice treated with lipopolysaccharide. Biochem. Biophys. Res. Commun. 2019, 510, 587–593. [Google Scholar] [CrossRef]
- Muraoka, H.; Hasegawa, K.; Sakamaki, Y.; Minakuchi, H.; Kawaguchi, T.; Yasuda, I.; Kanda, T.; Tokuyama, H.; Wakino, S.; Itoh, H. Role of Nampt-Sirt6 Axis in Renal Proximal Tubules in Extracellular Matrix Deposition in Diabetic Nephropathy. Cell Rep. 2019, 27, 199–212.e5. [Google Scholar] [CrossRef]
- Eto, N.; Miyata, Y.; Ohno, H.; Yamashita, T. Nicotinamide prevents the development of hyperphosphataemia by suppressing intestinal sodium-dependent phosphate transporter in rats with adenine-induced renal failure. Nephrol. Dial. Transpl. 2005, 20, 1378–1384. [Google Scholar] [CrossRef]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A.; European Uremic Toxin Work, G. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef]
- Rutkowski, B.; Slominska, E.; Szolkiewicz, M.; Smolenski, R.T.; Striley, C.; Rutkowski, P.; Swierczynski, J. N-methyl-2-pyridone-5-carboxamide: A novel uremic toxin? Kidney Int. Suppl. 2003, 63, S19–S21. [Google Scholar] [CrossRef]
- Cosmetic Ingredient Review Expert Panel. Final report of the safety assessment of niacinamide and niacin. Int. J. Toxicol. 2005, 24 (Suppl. 5), 1–31. [Google Scholar] [CrossRef]
- Sharma, K.; Karl, B.; Mathew, A.V.; Gangoiti, J.A.; Wassel, C.L.; Saito, R.; Pu, M.; Sharma, S.; You, Y.H.; Wang, L.; et al. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. J. Am. Soc. Nephrol. 2013, 24, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Biasioli, S.; Feriani, M.; Bigi, L.; Dell’Aquila, R.; Bragantini, L.; Chiaramonte, S.; Fabris, A.; Brendolan, A.; Ronco, C.; Pradella, M.; et al. Tricarboxylic acid cycle intermediates in chronic renal failure. Nephrol. Dial. Transpl. 1987, 2, 313–315. [Google Scholar]
- Hallan, S.; Afkarian, M.; Zelnick, L.R.; Kestenbaum, B.; Sharma, S.; Saito, R.; Darshi, M.; Barding, G.; Raftery, D.; Ju, W.; et al. Metabolomics and Gene Expression Analysis Reveal Down-regulation of the Citric Acid (TCA) Cycle in Non-diabetic CKD Patients. EBioMedicine 2017, 26, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Glosse, P.; Foller, M. AMP-Activated Protein Kinase (AMPK)-Dependent Regulation of Renal Transport. Int. J. Mol. Sci. 2018, 19, 3481. [Google Scholar] [CrossRef]
- Yamamoto, S.; Yamamoto, M.; Nakamura, J.; Mii, A.; Yamamoto, S.; Takahashi, M.; Kaneko, K.; Uchino, E.; Sato, Y.; Fukuma, S.; et al. Spatiotemporal ATP Dynamics during AKI Predict Renal Prognosis. J. Am. Soc. Nephrol. 2020, 31, 2855–2869. [Google Scholar] [CrossRef]
- Ying, W.; Alano, C.C.; Garnier, P.; Swanson, R.A. NAD+ as a metabolic link between DNA damage and cell death. J. Neurosci. Res. 2005, 79, 216–223. [Google Scholar] [CrossRef]
- Ding, H.; Jiang, L.; Xu, J.; Bai, F.; Zhou, Y.; Yuan, Q.; Luo, J.; Zen, K.; Yang, J. Inhibiting aerobic glycolysis suppresses renal interstitial fibroblast activation and renal fibrosis. Am. J. Physiol. Ren. Physiol. 2017, 313, F561–F575. [Google Scholar] [CrossRef]
- Bollee, G.; Dollinger, C.; Boutaud, L.; Guillemot, D.; Bensman, A.; Harambat, J.; Deteix, P.; Daudon, M.; Knebelmann, B.; Ceballos-Picot, I. Phenotype and genotype characterization of adenine phosphoribosyltransferase deficiency. J. Am. Soc. Nephrol. 2010, 21, 679–688. [Google Scholar] [CrossRef]
- Klinkhammer, B.M.; Djudjaj, S.; Kunter, U.; Palsson, R.; Edvardsson, V.O.; Wiech, T.; Thorsteinsdottir, M.; Hardarson, S.; Foresto-Neto, O.; Mulay, S.R.; et al. Cellular and Molecular Mechanisms of Kidney Injury in 2,8-Dihydroxyadenine Nephropathy. J. Am. Soc. Nephrol. 2020, 31, 799–816. [Google Scholar] [CrossRef]
- Mulay, S.R.; Eberhard, J.N.; Desai, J.; Marschner, J.A.; Kumar, S.V.; Weidenbusch, M.; Grigorescu, M.; Lech, M.; Eltrich, N.; Muller, L.; et al. Hyperoxaluria Requires TNF Receptors to Initiate Crystal Adhesion and Kidney Stone Disease. J. Am. Soc. Nephrol. 2017, 28, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Omote, K.; Gohda, T.; Murakoshi, M.; Sasaki, Y.; Kazuno, S.; Fujimura, T.; Ishizaka, M.; Sonoda, Y.; Tomino, Y. Role of the TNF pathway in the progression of diabetic nephropathy in KK-A(y) mice. Am. J. Physiol. Ren. Physiol. 2014, 306, F1335–F1347. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, Y.; Gohda, T.; Suzuki, Y.; Omote, K.; Ishizaka, M.; Matsuoka, J.; Tomino, Y. Circulating TNF receptors 1 and 2 are associated with the severity of renal interstitial fibrosis in IgA nephropathy. PLoS ONE 2015, 10, e0122212. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, S.; Oe, Y.; Sekimoto, A.; Sato, E.; Hashizume, Y.; Yamakage, S.; Kumakura, S.; Sato, H.; Ito, S.; Takahashi, N. Dual blockade of protease-activated receptor 1 and 2 additively ameliorates diabetic kidney disease. Am. J. Physiol. Ren. Physiol. 2020, 318, F1067–F1073. [Google Scholar] [CrossRef] [PubMed]
- Sato, E.; Saigusa, D.; Mishima, E.; Uchida, T.; Miura, D.; Morikawa-Ichinose, T.; Kisu, K.; Sekimoto, A.; Saito, R.; Oe, Y.; et al. Impact of the Oral Adsorbent AST-120 on Organ-Specific Accumulation of Uremic Toxins: LC-MS/MS and MS Imaging Techniques. Toxins (Basel) 2017, 10, 19. [Google Scholar] [CrossRef]
- Sato, E.; Tsunokuni, Y.; Kaneko, M.; Saigusa, D.; Saito, R.; Shimma, S.; Sekimoto, A.; Kawana, Y.; Oe, Y.; Ito, S.; et al. Metabolomics of a mouse model of preeclampsia induced by overexpressing soluble fms-like tyrosine kinase 1. Biochem. Biophys. Res. Commun. 2020, 527, 1064–1071. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumakura, S.; Sato, E.; Sekimoto, A.; Hashizume, Y.; Yamakage, S.; Miyazaki, M.; Ito, S.; Harigae, H.; Takahashi, N. Nicotinamide Attenuates the Progression of Renal Failure in a Mouse Model of Adenine-Induced Chronic Kidney Disease. Toxins 2021, 13, 50. https://doi.org/10.3390/toxins13010050
Kumakura S, Sato E, Sekimoto A, Hashizume Y, Yamakage S, Miyazaki M, Ito S, Harigae H, Takahashi N. Nicotinamide Attenuates the Progression of Renal Failure in a Mouse Model of Adenine-Induced Chronic Kidney Disease. Toxins. 2021; 13(1):50. https://doi.org/10.3390/toxins13010050
Chicago/Turabian StyleKumakura, Satoshi, Emiko Sato, Akiyo Sekimoto, Yamato Hashizume, Shu Yamakage, Mariko Miyazaki, Sadayoshi Ito, Hideo Harigae, and Nobuyuki Takahashi. 2021. "Nicotinamide Attenuates the Progression of Renal Failure in a Mouse Model of Adenine-Induced Chronic Kidney Disease" Toxins 13, no. 1: 50. https://doi.org/10.3390/toxins13010050
APA StyleKumakura, S., Sato, E., Sekimoto, A., Hashizume, Y., Yamakage, S., Miyazaki, M., Ito, S., Harigae, H., & Takahashi, N. (2021). Nicotinamide Attenuates the Progression of Renal Failure in a Mouse Model of Adenine-Induced Chronic Kidney Disease. Toxins, 13(1), 50. https://doi.org/10.3390/toxins13010050