Role of Uremic Toxins in Early Vascular Ageing and Calcification

 ,
,  , , and
, , and 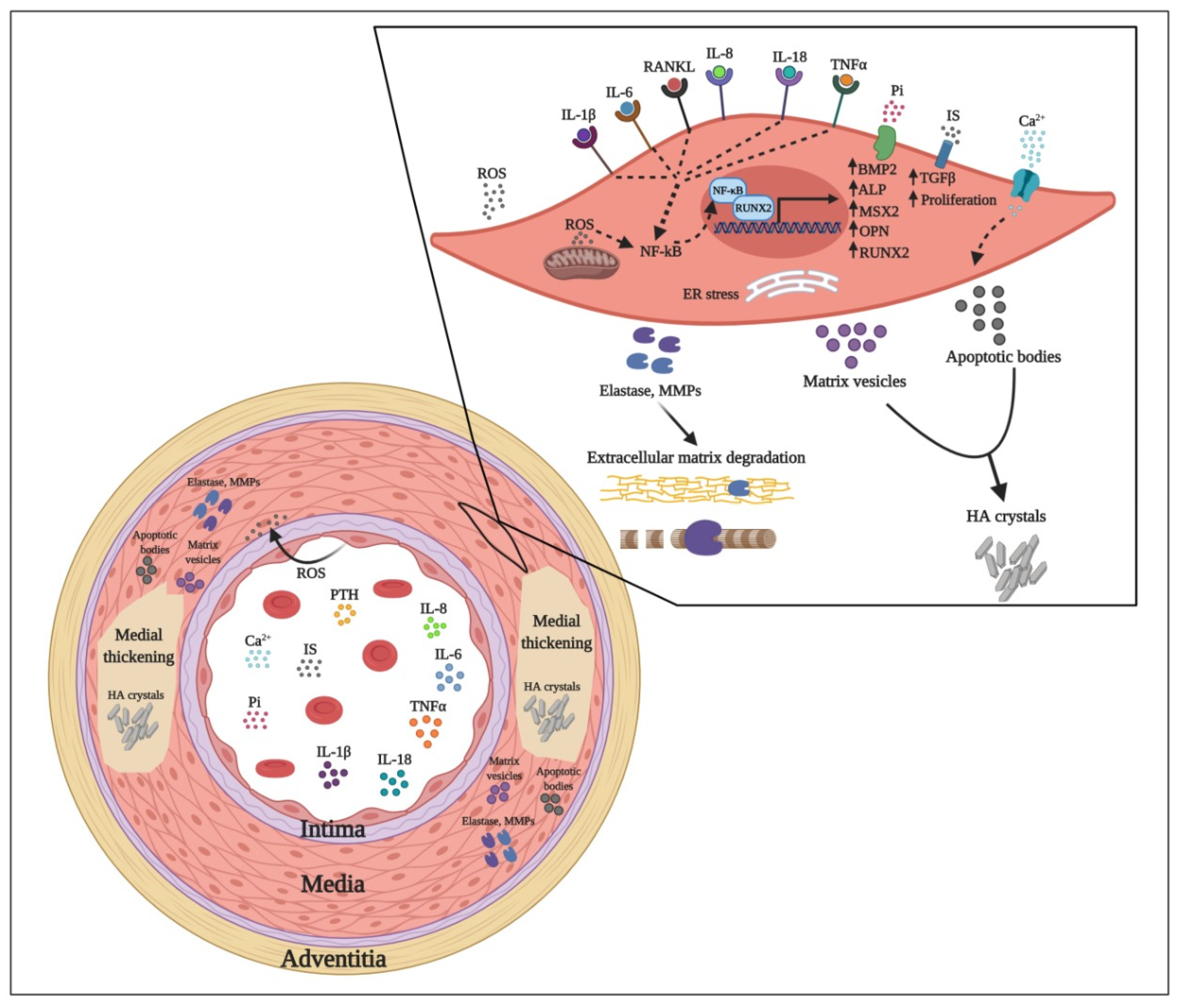{kind=link}
{kind=link}
Abstract
1. Introduction
2. Vascular Calcification and Early Vascular Ageing in CKD Patients
3. Uremic Toxin Mediated Pathways Leading to Vascular Calcification
3.1. Mineral Dysregulation
3.2. Endothelial Dysfunction
3.3. Oxidative Stress
3.4. Pro-Inflammatory Cytokines
3.4.1. Tumor Necrosis Factor
3.4.2. Interleukin-6
3.4.3. Interleukin-1β
3.4.4. Interleukin-8
3.4.5. IL-18
3.5. Other Uremic Toxins
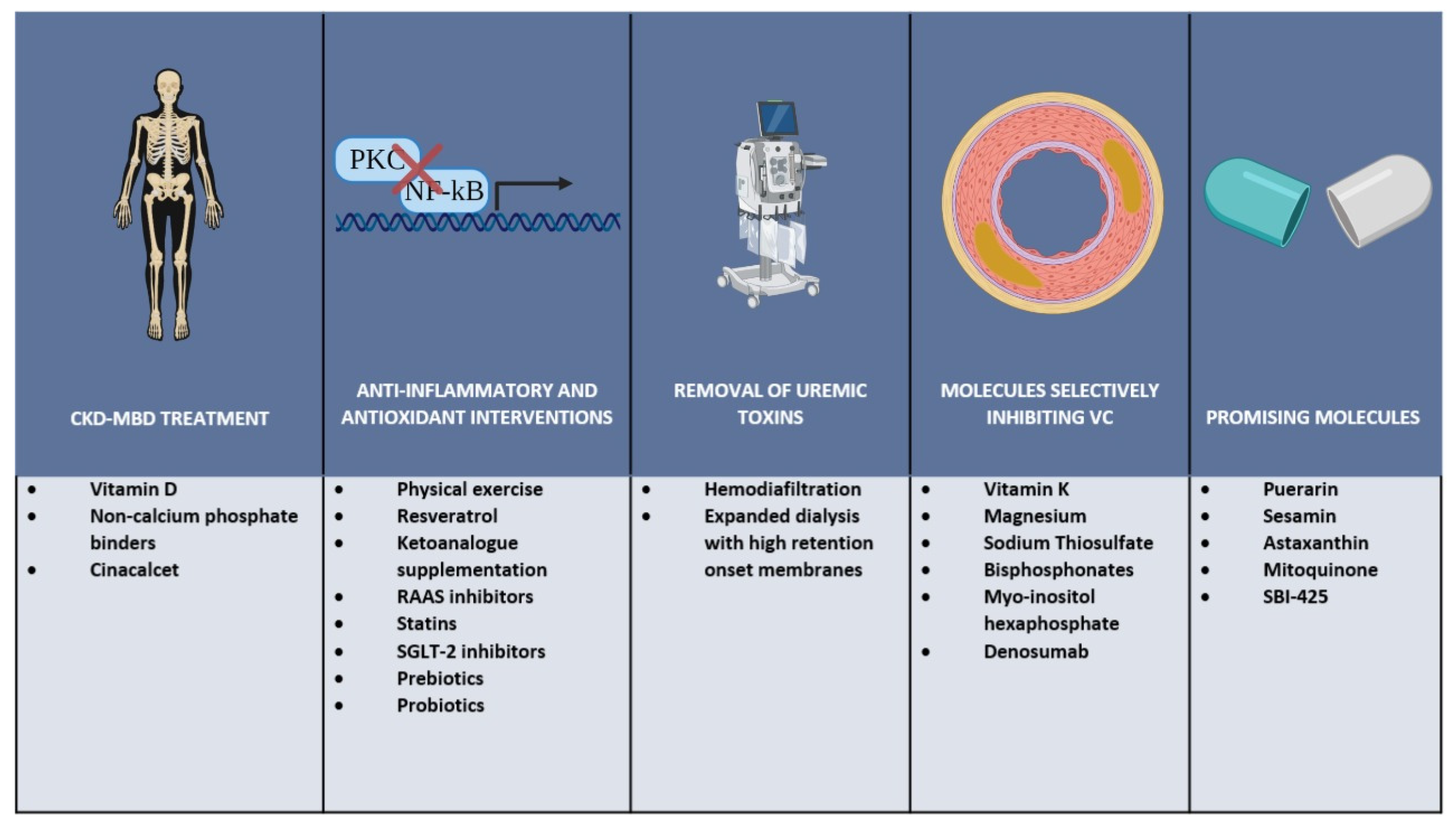
4. Possible Therapeutic Strategies
4.1. CKD-MBD Treatment
4.2. Anti-Inflammatory and Antioxidant Interventions
4.3. Removal of Uremic Toxins
4.4. Molecules Selectively Inhibiting Vascular Calcification
4.4.1. Vitamin K
4.4.2. Magnesium
4.4.3. Sodium Thiosulfate
4.4.4. Bisphosphonates
4.4.5. Myo-Insitol Hexaphosphate
4.4.6. Denosumab
4.5. Promising Novel Molecules
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bikbov, B.; Purcell, C.; Levey, A.S.; Smith, M.; Abdoli, A.; Abebe, M.; Adebayo, O.M.; Afarideh, M.; Agarwal, S.K.; Ahmaidan, E.; et al. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef]
- Vanholder, R.; Henle, T.; De Smet, R.; Glorieux, G.; Argilés, A.; Baurmeister, U.; Brunet, P.; Clark, W.F.; Cohen, G.M.; De Deyn, P.P.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [PubMed]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argilés, À. Normal and Pathologic Concentrations of Uremic Toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Pletinck, A.; Schepers, E.; Glorieux, G. Biochemical and Clinical Impact of Organic Uremic Retention Solutes: A Comprehensive Update. Toxins 2018, 10, 33. [Google Scholar] [CrossRef]
- Castillo-Rodriguez, E.; Pizarro-Sánchez, S.; Sanz, A.B.; Ramos, A.M.; Sanchez-Niño, M.D.; Martin-Cleary, C.; Fernandez, B.; Ortiz, A. Inflammatory Cytokines as Uremic Toxins: “Ni Son Todos Los Que Estan, Ni Estan Todos Los Que Son”. Toxins 2017, 9, 114. [Google Scholar] [CrossRef]
- Wolley, M.; Jardine, M.; Hutchison, C.A. Exploring the Clinical Relevance of Providing Increased Removal of Large Middle Molecules. Clin. J. Am. Soc. Nephrol. 2018, 13, 805–814. [Google Scholar] [CrossRef]
- Dai, L.; Qureshi, A.R.; Witasp, A.; Lindholm, B.; Stenvinkel, P. Early Vascular Ageing and Cellular Senescence in Chronic Kidney Disease. Comput. Struct. Biotechnol. J. 2019, 17, 721–729. [Google Scholar] [CrossRef]
- Cobo, G.; Lindholm, B.; Stenvinkel, P. Chronic inflammation in end-stage renal disease and dialysis. Nephrol. Dial. Transplant. 2018, 33, iii35–iii40. [Google Scholar] [CrossRef]
- Cobo, G.; Qureshi, A.R.; Lindholm, B.; Stenvinkel, P. C-reactive Protein: Repeated Measurements will Improve Dialysis Patient Care. Semin. Dial. 2015, 29, 7–14. [Google Scholar] [CrossRef]
- Foley, R.N.; Parfrey, P.S.; Sarnak, M.J. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am. J. Kidney Dis. 1998, 32, S112–S119. [Google Scholar] [CrossRef]
- Hwang, I.-C.; Park, H.E.; Kim, H.-L.; Kim, H.M.; Park, J.-B.; Yoon, Y.E.; Lee, S.-P.; Kim, H.-K.; Cho, G.-Y.; Sohn, D.-W.; et al. Systemic Inflammation Is Associated with Coronary Artery Calcification and All-Cause Mortality in Chronic Kidney Disease. Circ. J. 2016, 80, 1644–1652. [Google Scholar] [CrossRef] [PubMed]
- Petchey, W.G.; Hawley, C.M.; Johnson, D.W.; Haluska, B.A.; Watkins, T.W.; Isbel, N.M. Multimodality vascular imaging in CKD: Divergence of risk between measured parameters. Nephrol. Dial. Transplant. 2011, 27, 1004–1012. [Google Scholar] [CrossRef] [PubMed]
- Lanzer, P.; Boehm, M.; Sorribas, V.; Thiriet, M.; Janzen, J.; Zeller, T.; Hilaire, C.S.; Shanahan, C. Medial vascular calcification revisited: Review and perspectives. Eur. Heart J. 2014, 35, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Giachelli, C.M. Vascular calcification in CKD-MBD: Roles for phosphate, FGF23, and Klotho. Bone 2017, 100, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, J.C.; Moreno, P.; Hachinski, V.; Nabel, E.G.; Fuster, V. Cellular senescence, vascular disease, and aging: Part 1 of a 2-part review. Circulation 2011, 123, 1650–1660. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, J.C.; Moreno, P.; Nabel, E.G.; Hachinski, V.; Fuster, V. Cellular Senescence, Vascular Disease, and Aging: Part 2 of a 2-Part Review: Clinical Vascular Disease in the Elderly. Circulation 2011, 123, 1900–1910. [Google Scholar] [CrossRef]
- Boström, K.; Watson, K.E.; Horn, S.; Wortham, C.; Herman, I.M.; Demer, L.L. Bone morphogenetic protein expression in human atherosclerotic lesions. J. Clin. Investig. 1993, 91, 1800–1809. [Google Scholar] [CrossRef]
- Iseri, K.; Dai, L.; Chen, Z.; Qureshi, A.R.; Brismar, T.B.; Stenvinkel, P.; Lindholm, B. Bone mineral density and mortality in end-stage renal disease patients. Clin. Kidney J. 2020, 13, 307–321. [Google Scholar] [CrossRef]
- Azpiazu, D.; Gonzalo, S.; Gonzales-Parra, E.; Egido, J.; Villa-Bellosta, R. Role of pyrophosphate in vascular calcification in chronic kidney disease. Nefrologia 2018, 38, 250–257. [Google Scholar] [CrossRef]
- Leopold, J.A. Vascular calcification: Mechanisms of vascular smooth muscle cell calcification. Trends Cardiovasc. Med. 2015, 25, 267–274. [Google Scholar] [CrossRef]
- Hénaut, L.; Chillon, J.-M.; Kamel, S.; Massy, Z.A. Updates on the Mechanisms and the Care of Cardiovascular Calcification in Chronic Kidney Disease. Semin. Nephrol. 2018, 38, 233–250. [Google Scholar] [CrossRef] [PubMed]
- Arefin, S.; Buchanan, S.; Hobson, S.; Steinmetz, J.; AlSalhi, S.; Shiels, P.G.; Kublickiene, K.; Stenvinkel, P. Nrf2 in early vascular ageing: Calcification, senescence and therapy. Clin. Chim. Acta 2020, 505, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Debowska, M.; Lukaszuk, T.; Bobrowski, L.; Barany, P.; Söderberg, M.; Thiagarajan, D.; Frostegård, J.; Wennberg, L.; Lindholm, B.; et al. Phenotypic features of vascular calcification in chronic kidney disease. J. Intern. Med. 2020, 287, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Evenepoel, P.; DeJongh, S.; Verbeke, K.; Meijers, B. The Role of Gut Dysbiosis in the Bone–Vascular Axis in Chronic Kidney Disease. Toxins 2020, 12, 285. [Google Scholar] [CrossRef] [PubMed]
- Chettimada, S.; Joshi, S.R.; Dhagia, V.; Aiezza, A.; Lincoln, T.M.; Gupte, R.; Miano, J.M.; Gupte, S.A. Vascular smooth muscle cell contractile protein expression is increased through protein kinase G-dependent and -independent pathways by glucose-6-phosphate dehydrogenase inhibition and deficiency. Am. J. Physiol. Circ. Physiol. 2016, 311, H904–H912. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.H.; Campbell, G.R. Smooth Muscle Phenotypic Modulation—A Personal Experience. Arter. Thromb. Vasc. Biol. 2012, 32, 1784–1789. [Google Scholar] [CrossRef]
- Cunningham, J.; Locatelli, F.; Rodriguez, M. Secondary Hyperparathyroidism: Pathogenesis, Disease Progression, and Therapeutic Options. Clin. J. Am. Soc. Nephrol. 2011, 6, 913–921. [Google Scholar] [CrossRef]
- Martin, A.; David, V.; Quarles, L.D. Regulation and Function of the FGF23/Klotho Endocrine Pathways. Physiol. Rev. 2012, 92, 131–155. [Google Scholar] [CrossRef]
- Bacchetta, J.; Sea, J.L.; Chun, R.F.; Lisse, T.S.; Wesseling-Perry, K.; Gales, B.; Adams, J.S.; Salusky, I.B.; Hewison, M. Fibroblast growth factor 23 inhibits extrarenal synthesis of 1,25-dihydroxyvitamin D in human monocytes. J. Bone Miner. Res. 2012, 28, 46–55. [Google Scholar] [CrossRef]
- Aghagolzadeh, P.; Bachtler, M.; Bijarnia, R.; Jackson, C.; Smith, E.R.; Odermatt, A.; Radpour, R.; Pasch, A. Calcification of vascular smooth muscle cells is induced by secondary calciprotein particles and enhanced by tumor necrosis factor-α. Atherosclerosis 2016, 251, 404–414. [Google Scholar] [CrossRef]
- Akiyama, K.-I.; Miura, Y.; Hayashi, H.; Sakata, A.; Matsumura, Y.; Kojima, M.; Tsuchiya, K.; Nitta, K.; Shiizaki, K.; Kurosu, H.; et al. Calciprotein particles regulate fibroblast growth factor-23 expression in osteoblasts. Kidney Int. 2020, 97, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Staud, R. Vitamin D: More than just affecting calcium and bone. Curr. Rheumatol. Rep. 2005, 7, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Vahed, S.Z.; Mostafavi, S.; Khatibi, S.M.H.; Shoja, M.M.; Ardalan, M. Vascular Calcification: An Important Understanding in Nephrology. Vasc. Health Risk Manag. 2020, 16, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Stenvinkel, P.; Luttropp, K.; McGuinness, D.; Witasp, A.; Qureshi, A.R.; Wernerson, A.; Nordfors, L.; Schalling, M.; Ripsweden, J.; Wennberg, L.; et al. CDKN2A/p16INK4a expression is associated with vascular progeria in chronic kidney disease. Aging 2017, 9, 494–507. [Google Scholar] [CrossRef] [PubMed]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial Calcification in Chronic Kidney Disease: Key Roles for Calcium and Phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef]
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Bennett, M.R.; Shanahan, C.M.; Weissberg, P.L. Apoptosis regulates human vascular calcification in vitro: Evidence for initiation of vascular calcification by apoptotic bodies. Circ. Res. 2000, 87, 1055–1062. [Google Scholar] [CrossRef]
- Ewence, A.E.; Bootman, M.; Roderick, H.L.; Skepper, J.N.; McCarthy, G.; Epple, M.; Neumann, M.; Shanahan, C.M.; Proudfoot, D. Calcium phosphate crystals induce cell death in human vascular smooth muscle cells: A potential mechanism in atherosclerotic plaque destabilization. Circ. Res. 2008, 103, e28–e34. [Google Scholar] [CrossRef]
- Clarke, M.C.; Littlewood, T.D.; Figg, N.; Maguire, J.J.; Davenport, A.P.; Goddard, M.; Bennett, M.R. Chronic Apoptosis of Vascular Smooth Muscle Cells Accelerates Atherosclerosis and Promotes Calcification and Medial Degeneration. Circ. Res. 2008, 102, 1529–1538. [Google Scholar] [CrossRef]
- Luo, G.; Ducy, P.; McKee, M.D.; Pinero, G.J.; Loyer, E.; Behringer, R.R.; Karsenty, G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nat. Cell Biol. 1997, 386, 78–81. [Google Scholar] [CrossRef]
- Speer, M.Y.; Yang, H.-Y.; Brabb, T.; Leaf, E.; Look, A.; Lin, W.-L.; Frutkin, A.; Dichek, D.; Giachelli, C.M. Smooth Muscle Cells Give Rise to Osteochondrogenic Precursors and Chondrocytes in Calcifying Arteries. Circ. Res. 2009, 104, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.L. Can biological calcification occur in the presence of pyrophosphate? Arch. Biochem. Biophys. 1984, 231, 1–8. [Google Scholar] [CrossRef]
- Schafer, C.; Heiss, A.; Schwarz, A.; Westenfeld, R.; Ketteler, M.; Floege, J.; Muller-Esterl, W.; Schinke, T.; Jahnen-Dechent, W. The serum protein alpha 2-Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcifi-cation. J. Clin. Investig. 2003, 112, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Bucay, N.; Sarosi, I.; Dunstan, C.R.; Morony, S.; Tarpley, J.; Capparelli, C.; Scully, S.; Tan, H.L.; Xu, W.; Lacey, D.L.; et al. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998, 12, 1260–1268. [Google Scholar] [CrossRef]
- Yao, Y.; Bennett, B.J.; Wang, X.; Rosenfeld, M.E.; Giachelli, C.M.; Lusis, A.J.; Bostrom, K.I. Inhibition of Bone Morphogenetic Proteins Protects Against Atherosclerosis and Vascular Calcification. Circ. Res. 2010, 107, 485–494. [Google Scholar] [CrossRef]
- Jaminon, A.M.G.; Dai, L.; Qureshi, A.R.; Evenepoel, P.; Ripsweden, J.; Söderberg, M.; Witasp, A.; Olauson, H.; Schurgers, L.J.; Stenvinkel, P. Matrix Gla protein is an independent predictor of both intimal and medial vascular calcification in chronic kidney disease. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Reynolds, J.L.; Joannides, A.J.; Skepper, J.N.; McNair, R.; Schurgers, L.J.; Proudfoot, D.; Jahnen-Dechent, W.; Weissberg, P.L.; Shanahan, C.M. Human Vascular Smooth Muscle Cells Undergo Vesicle-Mediated Calcification in Response to Changes in Extracellular Calcium and Phosphate Concentrations: A Potential Mechanism for Accelerated Vascular Calcification in ESRD. J. Am. Soc. Nephrol. 2004, 15, 2857–2867. [Google Scholar] [CrossRef]
- Kapustin, A.N.; Davies, J.D.; Reynolds, J.L.; McNair, R.; Jones, G.T.; Sidibe, A.; Schurgers, L.J.; Skepper, J.N.; Proudfoot, D.; Mayr, M.; et al. Calcium Regulates Key Components of Vascular Smooth Muscle Cell–Derived Matrix Vesicles to Enhance Mineralization. Circ. Res. 2011, 109, e1–e12. [Google Scholar] [CrossRef]
- O’Neill, W.C. Pyrophosphate, Alkaline Phosphatase, and Vascular Calcification. Circ. Res. 2006, 99, e2. [Google Scholar] [CrossRef]
- Lomashvili, K.; Garg, P.; Narisawa, S.; Millan, J.; O’Neill, W.C. Upregulation of alkaline phosphatase and pyrophosphate hydrolysis: Potential mechanism for uremic vascular calcification. Kidney Int. 2008, 73, 1024–1030. [Google Scholar] [CrossRef]
- Narisawa, S.; Harmey, D.; Yadav, M.C.; O’Neill, W.C.; Hoylaerts, M.F.; Millán, J.L. Novel Inhibitors of Alkaline Phosphatase Suppress Vascular Smooth Muscle Cell Calcification. J. Bone Miner. Res. 2007, 22, 1700–1710. [Google Scholar] [CrossRef] [PubMed]
- Schinke, T.; Amendt, C.; Trindl, A.; Pöschke, O.; Müller-Esterl, W.; Jahnen-Dechent, W. The serum protein alpha2-HS glycoprotein/fetuin inhibits apatite formation in vitro and in mineralizing calvaria cells. A possible role in mineralization and calcium homeostasis. J. Biol. Chem. 1996, 271, 20789–20796. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.L.; Skepper, J.N.; McNair, R.; Kasama, T.; Gupta, K.; Weissberg, P.L.; Jahnen-Dechent, W.; Shanahan, C.M. Multifunctional Roles for Serum Protein Fetuin-A in Inhibition of Human Vascular Smooth Muscle Cell Calcification. J. Am. Soc. Nephrol. 2005, 16, 2920–2930. [Google Scholar] [CrossRef] [PubMed]
- Hegner, B.; Schaub, T.; Janke, D.; Zickler, D.; Lange, C.; Girndt, M.; Jankowski, J.; Schindler, R.; Dragun, D. Targeting proinflammatory cytokines ameliorates calcifying phenotype conversion of vascular progenitors under uremic conditions in vitro. Sci. Rep. 2018, 8, 12087. [Google Scholar] [CrossRef]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef]
- Tesauro, M.; Mauriello, A.; Rovella, V.; Annicchiarico-Petruzzelli, M.; Cardillo, C.; Melino, G.; Di Daniele, N. Arterial ageing: From endothelial dysfunction to vascular calcification. J. Intern. Med. 2017, 281, 471–482. [Google Scholar] [CrossRef]
- Six, I.; Flissi, N.; Lenglet, G.; Louvet, L.; Kamel, S.; Gallet, M.; Massy, Z.A.; Liabeuf, S. Uremic Toxins and Vascular Dysfunction. Toxins 2020, 12, 404. [Google Scholar] [CrossRef]
- Zafeiropoulou, K.; Bita, T.; Polykratis, A.; Karabina, S.; Vlachojannis, J.; Katsoris, P. Hemodialysis Removes Uremic Toxins That Alter the Biological Actions of Endothelial Cells. PLoS ONE 2012, 7, e30975. [Google Scholar] [CrossRef]
- Maizel, J.; Six, I.; Slama, M.; Tribouilloy, C.; Sevestre, H.; Poirot, S.; Giummelly, P.; Atkinson, J.; Choukroun, G.; Andrejak, M.; et al. Mechanisms of Aortic and Cardiac Dysfunction in Uremic Mice with Aortic Calcification. Circulation 2009, 119, 306–313. [Google Scholar] [CrossRef]
- Six, I.; Maizel, J.; Barreto, F.C.; Rangrez, A.Y.; Dupont, S.; Slama, M.; Tribouilloy, C.; Choukroun, G.; Mazière, J.C.; Bode-Boeger, S.; et al. Effects of phosphate on vascular function under normal conditions and influence of the uraemic state. Cardiovasc. Res. 2012, 96, 130–139. [Google Scholar] [CrossRef]
- Gross, P.; Six, I.; Kamel, S.; Massy, Z.A. Vascular toxicity of phosphate in chronic kidney disease: Beyond vascular calcification. Circ. J. 2014, 78, 2339–2346. [Google Scholar] [CrossRef] [PubMed]
- Luksha, L.; Stenvinkel, P.; Hammarqvist, F.; Carrero, J.J.; Davidge, S.T.; Kublickiene, K. Mechanisms of Endothelial Dysfunction in Resistance Arteries from Patients with End-Stage Renal Disease. PLoS ONE 2012, 7, e36056. [Google Scholar] [CrossRef] [PubMed]
- Novo, E.; Parola, M. The role of redox mechanisms in hepatic chronic wound healing and fibrogenesis. Fibrogenes. Tissue Repair 2012, 5, S4. [Google Scholar] [CrossRef] [PubMed]
- Lassègue, B.; Griendling, K.K. NADPH Oxidases: Functions and Pathologies in the Vasculature. Arter. Thromb. Vasc. Biol. 2010, 30, 653–661. [Google Scholar] [CrossRef]
- Byon, C.H.; Heath, J.M.; Chen, Y. Redox signaling in cardiovascular pathophysiology: A focus on hydrogen peroxide and vascular smooth muscle cells. Redox Biol. 2016, 9, 244–253. [Google Scholar] [CrossRef]
- Tóth, A.; Balogh, E.; Jeney, V. Regulation of Vascular Calcification by Reactive Oxygen Species. Antioxidants 2020, 9, 963. [Google Scholar] [CrossRef]
- Agharazii, M.; St-Louis, R.; Gautier-Bastien, A.; Ung, R.V.; Mokas, S.; Larivière, R.; Richard, D.E. Inflammatory cytokines and reactive oxygen species as mediators of chronic kidney disease-related vascular calcifi-cation. Am. J. Hypertens. 2015, 28, 746–755. [Google Scholar] [CrossRef]
- Liberman, M.; Johnson, R.C.; Handy, D.E.; Loscalzo, J.; Leopold, J.A. Bone morphogenetic protein-2 activates NADPH oxidase to increase endoplasmic reticulum stress and human coronary artery smooth muscle cell calcification. Biochem. Biophys. Res. Commun. 2011, 413, 436–441. [Google Scholar] [CrossRef]
- Furmanik, M.; Chatrou, M.; Van Gorp, R.H.; Akbulut, A.; Willems, B.; Schmidt, H.H.; Van Eys, G.; Bochaton-Piallat, M.-L.; Proudfoot, D.; Al Biessen, E.; et al. Reactive Oxygen-Forming Nox5 Links Vascular Smooth Muscle Cell Phenotypic Switching and Extracellular Vesicle-Mediated Vascular Calcification. Circ. Res. 2020, 127, 911–927. [Google Scholar] [CrossRef]
- Cui, L.; Li, Z.; Chang, X.; Cong, G.; Hao, L. Quercetin attenuates vascular calcification by inhibiting oxidative stress and mitochondrial fission. Vasc. Pharmacol. 2017, 88, 21–29. [Google Scholar] [CrossRef]
- Zhao, M.-M.; Xu, M.-J.; Cai, Y.; Zhao, G.; Guan, Y.; Kong, W.; Tang, C.; Wang, X. Mitochondrial reactive oxygen species promote p65 nuclear translocation mediating high-phosphate-induced vascular calcification in vitro and in vivo. Kidney Int. 2011, 79, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Paddenberg, R.; Ishaq, B.; Goldenberg, A.; Faulhammer, P.; Rose, F.; Weissmann, N.; Braun-Dullaeus, R.C.; Kummer, W. Essential role of complex II of the respiratory chain in hypoxia-induced ROS generation in the pulmonary vasculature. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L710–L719. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate hypoxic signaling. Curr. Opin. Cell Biol. 2009, 21, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Kanai, H.; Uchiyama, T.; Iso, T.; Ohyama, Y.; Sakamoto, H.; Tamura, J.; Nagai, R.; Kurabayashi, M. Mitochondrial reactive oxygen species and c-Src play a critical role in hypoxic response in vascular smooth muscle cells. Cardiovasc. Res. 2005, 67, 714–722. [Google Scholar] [CrossRef]
- Waypa, G.B.; Smith, K.A.; Schumacker, P.T. O2 sensing, mitochondria and ROS signaling: The fog is lifting. Mol. Asp. Med. 2016, 47, 76–89. [Google Scholar] [CrossRef]
- Balogh, E.; Tóth, A.; Méhes, G.; Trencsényi, G.; Paragh, G.; Jeney, V. Hypoxia Triggers Osteochondrogenic Differentiation of Vascular Smooth Muscle Cells in an HIF-1 (Hypoxia-Inducible Factor 1)–Dependent and Reactive Oxygen Species–Dependent Manner. Arter. Thromb. Vasc. Biol. 2019, 39, 1088–1099. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Meyer, C.J.; Block, G.A.; Chertow, G.M.; Shiels, P.G. Understanding the role of the cytoprotective transcription factor nuclear factor erythroid 2-related factor 2—Lessons from evolution, the animal kingdom and rare progeroid syndromes. Nephrol. Dial. Transplant. 2020, 35, 2036–2045. [Google Scholar] [CrossRef]
- Fulop, G.A.; Kiss, T.; Tarantini, S.; Balasubramanian, P.; Yabluchanskiy, A.; Farkas, E.; Bari, F.; Ungvari, Z.; Csiszar, A. Nrf2 deficiency in aged mice exacerbates cellular senescence promoting cerebrovascular inflammation. GeroScience 2018, 40, 513–521. [Google Scholar] [CrossRef]
- Yao, L.; Wang, J.; Tian, B.-Y.; Xu, T.-H.; Sheng, Z.-T. Activation of the Nrf2-ARE Signaling Pathway Prevents Hyperphosphatemia-Induced Vascular Calcification by Inducing Autophagy in Renal Vascular Smooth Muscle Cells. J. Cell. Biochem. 2017, 118, 4708–4715. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Ketteler, M.; Johnson, R.J.; Lindholm, B.; Pecoits-Filho, R.; Riella, M.; Heimbürger, O.; Cederholm, T.; Girndt, M. IL-10, IL-6, and TNF-α: Central factors in the altered cytokine network of uremia—The good, the bad, and the ugly. Kidney Int. 2005, 67, 1216–1233. [Google Scholar] [CrossRef] [PubMed]
- Rapp, N.; Evenepoel, P.; Stenvinkel, P.; Schurgers, L.J. Uremic Toxins and Vascular Calcification—Missing the Forest for All the Trees. Toxins 2020, 12, 624. [Google Scholar] [CrossRef] [PubMed]
- Kopf, M.; Bachmann, M.F.; Marsland, B.J. Averting inflammation by targeting the cytokine environment. Nat. Rev. Drug Discov. 2010, 9, 703–718. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.A.; Izquierdo, M.C.; Sánchez-Niño, M.D.; Suárez-Alvarez, B.; Lopez-Larrea, C.; Jakubowski, A.; Blanco-Parra, J.A.; Ramirez, R.; Selgas, R.; Ruiz-Ortega, M.; et al. The Inflammatory Cytokines TWEAK and TNFα Reduce Renal Klotho Expression through NFκB. J. Am. Soc. Nephrol. 2011, 22, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Souma, Y.; Akakabe, Y.; Kitamura, Y.; Matsuo, K.; Shimoda, Y.; Ueyama, T.; Matoba, S.; Yamada, H.; Okigaki, M.; et al. Macrophages play a unique role in the plaque calcification by enhancing the osteogenic signals exerted by vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2012, 425, 39–44. [Google Scholar] [CrossRef]
- García-Miguel, M.; Riquelme, J.A.; Norambuena-Soto, I.; Morales, P.E.; Sanhueza-Olivares, F.; Núñez-Soto, C.; Mondaca-Ruff, D.; Cancino-Arenas, N.; Martín, A.S.; Chiong, M. Autophagy mediates tumor necrosis factor-α-induced phenotype switching in vascular smooth muscle A7r5 cell line. PLoS ONE 2018, 13, e0197210. [Google Scholar] [CrossRef]
- Shioi, A.; Katagi, M.; Okuno, Y.; Mori, K.; Jono, S.; Koyama, H.; Nishizawa, Y. Induction of bone-type alkaline phosphatase in human vascular smooth muscle cells: Roles of tumor necrosis factor-alpha and oncostatin M derived from macrophages. Circ. Res. 2002, 91, 9–16. [Google Scholar] [CrossRef]
- Freise, C.; Querfeld, U. The lignan (+)-episesamin interferes with TNF-α-induced activation of VSMC via diminished activation of NF-ĸB, ERK1/2 and AKT and decreased activity of gelatinases. Acta Physiol. 2015, 213, 642–652. [Google Scholar] [CrossRef]
- Lee, H.-L.; Woo, K.M.; Ryoo, H.-M.; Baek, J.-H. Tumor necrosis factor-α increases alkaline phosphatase expression in vascular smooth muscle cells via MSX2 induction. Biochem. Biophys. Res. Commun. 2010, 391, 1087–1092. [Google Scholar] [CrossRef]
- Zickler, D.; Luecht, C.; Willy, K.; Chen, L.; Witowski, J.; Girndt, M.; Fiedler, R.; Storr, M.; Kamhieh-Milz, J.; Schoon, J.; et al. Tumour necrosis factor-alpha in uraemic serum promotes osteoblastic transition and calcification of vascular smooth muscle cells via extracellular signal-regulated kinases and activator protein 1/c-FOS-mediated induction of interleukin 6 expression. Nephrol. Dial. Transplant. 2018, 33, 574–585. [Google Scholar] [CrossRef]
- Bundy, J.D.; Chen, J.; Yang, W.; Budoff, M.; Go, A.S.; Grunwald, J.E.; Kallem, R.R.; Post, W.S.; Reilly, M.P.; Ricardo, A.C.; et al. Risk factors for progression of coronary artery calcification in patients with chronic kidney disease: The CRIC study. Atherosclerosis 2018, 271, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Stenvinkel, P.; Heimburger, O.; Jogestrand, T. Elevated interleukin-6 predicts progressive carotid artery atherosclerosis in dialysis patients: Association with Chlamydia pneumoniae seropositivity. Am. J. Kidney Dis. 2002, 39, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zheng, S.-G. Hall of Fame among Pro-inflammatory Cytokines: Interleukin-6 Gene and Its Transcriptional Regulation Mechanisms. Front. Immunol. 2016, 7, 604. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Axelsson, J.; Machowska, A.; Heimbürger, O.; Bárány, P.; Lindholm, B.; Lindström, K.; Stenvinkel, P.; Qureshi, A.R. Biomarkers of Cardiovascular Disease and Mortality Risk in Patients with Advanced CKD. Clin. J. Am. Soc. Nephrol. 2016, 11, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Kurozumi, A.; Nakano, K.; Yamagata, K.; Okada, Y.; Nakayamada, S.; Tanaka, Y. IL-6 and sIL-6R induces STAT3-dependent differentiation of human VSMCs into osteoblast-like cells through JMJD2B-mediated histone demethylation of RUNX2. Bone 2019, 124, 53–61. [Google Scholar] [CrossRef]
- Deuell, K.A.; Callegari, A.; Giachelli, C.M.; Rosenfeld, M.E.; Scatena, M. RANKL Enhances Macrophage Paracrine Pro-Calcific Activity in High Phosphate-Treated Smooth Muscle Cells: Dependence on IL-6 and TNF-α. J. Vasc. Res. 2012, 49, 510–521. [Google Scholar] [CrossRef]
- Callegari, A.; Coons, M.L.; Ricks, J.L.; Rosenfeld, M.E.; Scatena, M. Increased calcification in osteoprotegerin-deficient smooth muscle cells: Dependence on receptor activator of NF-κB ligand and interleukin 6. J. Vasc. Res. 2014, 51, 118–131. [Google Scholar] [CrossRef]
- Sun, M.; Chang, Q.; Xin, M.; Wang, Q.; Li, H.; Qian, J. Endogenous bone morphogenetic protein 2 plays a role in vascular smooth muscle cell calcification induced by interleukin 6 in vitro. Int. J. Immunopathol. Pharmacol. 2017, 30, 227–237. [Google Scholar] [CrossRef]
- Xu, D.; Zeng, F.; Han, L.; Wang, J.; Yin, Z.; Lv, L.; Guo, L.; Wang, D.-G.; Xu, Y.; Zhou, H.-S. The synergistic action of phosphate and interleukin-6 enhances senescence-associated calcification in vascular smooth muscle cells depending on p53. Mech. Ageing Dev. 2019, 182, 111124. [Google Scholar] [CrossRef]
- Shavelle, D.M.; Katz, R.; Takasu, J.; Lima, J.A.; Jenny, N.S.; Budoff, M.J.; O’Brien, K.D. Soluble intercellular adhesion molecule-1 (sICAM-1) and aortic valve calcification in the multi-ethnic study of atherosclerosis (MESA). J. Heart Valve Dis. 2008, 17, 388–395. [Google Scholar]
- Takasu, J.; Katz, R.; Shavelle, D.M.; O’Brien, K.; Mao, S.; Carr, J.J.; Cushman, M.; Budoff, M.J. Inflammation and descending thoracic aortic calcification as detected by computed tomography: The Multi-Ethnic Study of Atherosclerosis. Atherosclerosis 2008, 199, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Barreto, D.V.; Barreto, F.C.; Liabeuf, S.; Temmar, M.; Lemke, H.-D.; Tribouilloy, C.; Choukroun, G.; Vanholder, R.; Massy, Z. Plasma interleukin-6 is independently associated with mortality in both hemodialysis and pre-dialysis patients with chronic kidney disease. Kidney Int. 2010, 77, 550–556. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Reilly, M.; Yang, W.; Chen, J.; Go, A.S.; Lash, J.P.; Rahman, M.; Defilippi, C.; Gadegbeku, C.; Kanthety, R.; et al. Risk Factors for Coronary Artery Calcium Among Patients with Chronic Kidney Disease (from the Chronic Renal Insufficiency Cohort Study). Am. J. Cardiol. 2012, 110, 1735–1741. [Google Scholar] [CrossRef] [PubMed]
- Kraśniak, A.; Drozdz, M.; Pasowicz, M.; Chmiel, G.; Michałek, M.; Szumilak, D.; Podolec, P.; Klimeczek, P.; Konieczyńska, M.; Wicher-Muniak, E.; et al. Factors involved in vascular calcification and atherosclerosis in maintenance haemodialysis patients. Nephrol. Dial. Transplant. 2006, 22, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Larsen, B.A.; Laughlin, G.A.; Cummins, K.; Barrett-Connor, E.; Wassel, C.L. Adipokines and severity and progression of coronary artery calcium: Findings from the Rancho Bernardo Study. Atherosclerosis 2017, 265, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Pecoits-Filho, R. Interleukin-6 is an independent predictor of mortality in patients starting dialysis treatment. Nephrol. Dial. Transplant. 2002, 17, 1684–1688. [Google Scholar] [CrossRef] [PubMed]
- Perrier, S.; Darakhshan, F.; Hajduch, E. IL-1 receptor antagonist in metabolic diseases: Dr Jekyll or Mr Hyde? FEBS Lett. 2006, 580, 6289–6294. [Google Scholar] [CrossRef]
- Hung, A.M.; Ellis, C.D.; Shintani, A.; Booker, C.; Ikizler, T.A. IL-1β Receptor Antagonist Reduces Inflammation in Hemodialysis Patients. J. Am. Soc. Nephrol. 2011, 22, 437–442. [Google Scholar] [CrossRef]
- Gupta, J.; Mitra, N.; Kanetsky, P.A.; Devaney, J.; Wing, M.R.; Reilly, M.; Shah, V.O.; Balakrishnan, V.S.; Guzman, N.J.; Girndt, M.; et al. Association between Albuminuria, Kidney Function, and Inflammatory Biomarker Profile in CKD in CRIC. Clin. J. Am. Soc. Nephrol. 2012, 7, 1938–1946. [Google Scholar] [CrossRef]
- Awan, Z.; Denis, M.; Roubtsova, A.; Essalmani, R.; Marcinkiewicz, J.; Awan, A.; Gram, H.; Seidah, N.G.; Genest, J. Reducing Vascular Calcification by Anti-IL-1β Monoclonal Antibody in a Mouse Model of Familial Hypercholesterolemia. Angiology 2015, 67, 157–167. [Google Scholar] [CrossRef]
- Dautova, Y.; Kapustin, A.N.; Pappert, K.; Epple, M.; Okkenhaug, H.; Cook, S.J.; Shanahan, C.M.; Bootman, M.D.; Proudfoot, D. Calcium phosphate particles stimulate interleukin-1β release from human vascular smooth muscle cells: A role for spleen tyrosine kinase and exosome release. J. Mol. Cell. Cardiol. 2018, 115, 82–93. [Google Scholar] [CrossRef] [PubMed]
- David, J.M.; Dominguez, C.; Hamilton, D.H.; Palena, C. The IL-8/IL-8R Axis: A Double Agent in Tumor Immune Resistance. Vaccines 2016, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Bouabdallah, J.; Zibara, K.; Issa, H.; Lenglet, G.; Kchour, G.; Caus, T.; Six, I.; Choukroun, G.; Kamel, S.; Bennis, Y. Endothelial cells exposed to phosphate and indoxyl sulphate promote vascular calcification through interleukin-8 secretion. Nephrol. Dial. Transplant. 2019, 34, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Raaz-Schrauder, D.; Klinghammer, L.; Baum, C.; Frank, T.; Lewczuk, P.; Achenbach, S.; Cicha, I.; Stumpf, C.; Wiltfang, J.; Kornhuber, J.; et al. Association of systemic inflammation markers with the presence and extent of coronary artery calcification. Cytokine 2012, 57, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Gauss, S.; Klinghammer, L.; Steinhoff, A.; Raaz-Schrauder, R.; Marwan, M.; Achenbach, S.; Garlichs, C.D. Association of systemic inflammation with epicardial fat and coronary artery calcification. Inflamm. Res. 2015, 64, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Panichi, V.; Paoletti, S.; Mantuano, E.; Manca-Rizza, G.; Filippi, C.; Santi, S.; Taccola, D.; Donadio, C.; Tramonti, G.; Innocenti, M.; et al. In vivo and in vitro effects of simvastatin on inflammatory markers in pre-dialysis patients. Nephrol. Dial. Transplant. 2005, 21, 337–344. [Google Scholar] [CrossRef]
- Yu, C.; Zhang, C.; Kuang, Z.; Zheng, Q. The Role of NLRP3 Inflammasome Activities in Bone Diseases and Vascular Calcification. Inflamm. 2020, 1–16. [Google Scholar] [CrossRef]
- Sahar, S.; Dwarakanath, R.S.; Reddy, M.A.; Lanting, L.; Todorov, I.; Natarajan, R. Angiotensin II enhances interleukin-18 mediated inflammatory gene expression in vascular smooth muscle cells: A novel cross-talk in the pathogenesis of atherosclerosis. Circ. Res. 2005, 96, 1064–1071. [Google Scholar] [CrossRef]
- Formanowicz, D.; Wanic-Kossowska, M.; Pawliczak, E.; Radom, M.; Formanowicz, P. Usefulness of serum interleukin-18 in predicting cardiovascular mortality in patients with chronic kidney disease—Systems and clinical approach. Sci. Rep. 2015, 5, 18332. [Google Scholar] [CrossRef]
- Al-Suhaimi, E.A.; Shehzad, A. Leptin, resistin and visfatin: The missing link between endocrine metabolic disorders and immunity. Eur. J. Med. Res. 2013, 18, 12. [Google Scholar] [CrossRef]
- Li, L.; Mamputu, M.; Wiernsperger, N.; Renier, G. Signaling Pathways Involved in Human Vascular Smooth Muscle Cell Proliferation and Matrix Metalloproteinase-2 Expression Induced by Leptin: Inhibitory Effect of Metformin. Diabetes 2005, 54, 2227–2234. [Google Scholar] [CrossRef] [PubMed]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, K.; Maeda, T.; Kawane, T.; Matsunuma, A.; Horiuchi, N. Leptin stimulates fibroblast growth factor 23 expression in bone and suppresses renal 1α,25-dihydroxyvitamin D3 synthesis in leptin-deficient ob/ob Mice. J. Bone Miner. Res. 2010, 25, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.X.; O’Neill, K.; Akl, N.K.; Moe, S.M. Adipocyte induced arterial calcification is prevented with sodium thiosulfate. Biochem. Biophys. Res. Commun. 2014, 449, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Parhami, F.; Tintut, Y.; Ballard, A.; Fogelman, A.M.; Demer, L.L. Leptin enhances the calcification of vascular cells: Artery wall as a target of leptin. Circ. Res. 2001, 88, 954–960. [Google Scholar] [CrossRef]
- Zeadin, M.G.; Butcher, M.K.; Shaughnessy, S.G.; Werstuck, G.H. Leptin promotes osteoblast differentiation and mineralization of primary cultures of vascular smooth muscle cells by inhibiting glycogen synthase kinase (GSK)-3β. Biochem. Biophys. Res. Commun. 2012, 425, 924–930. [Google Scholar] [CrossRef]
- Zeadin, M.; Butcher, M.; Werstuck, G.; Khan, M.; Yee, C.K.; Shaughnessy, S.G. Effect of Leptin on Vascular Calcification in Apolipoprotein E–Deficient Mice. Arter. Thromb. Vasc. Biol. 2009, 29, 2069–2075. [Google Scholar] [CrossRef]
- Iribarren, C.; Husson, G.; Go, A.S.; Lo, J.C.; Fair, J.M.; Rubin, G.D.; Hlatky, M.A.; Fortmann, S.P. Plasma Leptin Levels and Coronary Artery Calcification in Older Adults. J. Clin. Endocrinol. Metab. 2006, 92, 729–732. [Google Scholar] [CrossRef]
- Szulc, P.; Amri, E.-Z.; Varennes, A.; Panaia-Ferrari, P.; Fontas, E.; Goudable, J.; Chapurlat, R.; Breuil, V. Positive Association of High Leptin Level and Abdominal Aortic Calcification in Men—The Prospective MINOS Study. Circ. J. 2018, 82, 2954–2961. [Google Scholar] [CrossRef]
- de Oliveira, R.B.; Liabeuf, S.; Okazaki, H.; Lenglet, A.; Desjardins, L.; Lemke, H.D.; Vanholder, R.; Choukroun, G.; Massy, Z.A.; European Uremic Toxin Work Group. The clinical impact of plasma leptin levels in a cohort of chronic kidney disease patients. Clin. Kidney J. 2013, 6, 63–70. [Google Scholar] [CrossRef]
- Aoqui, C.; Cuppari, L.; Kamimura, M.A.; Canziani, M.E.F. Increased visceral adiposity is associated with coronary artery calcification in male patients with chronic kidney disease. Eur. J. Clin. Nutr. 2013, 67, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Hyun, Y.Y.; Kim, H.; Oh, Y.K.; Oh, K.-H.; Ahn, C.; Sung, S.; Choi, K.H.; Kim, S.; Lee, K.-B. High fibroblast growth factor 23 is associated with coronary calcification in patients with high adiponectin: Analysis from the KoreaN cohort study for Outcome in patients With Chronic Kidney Disease (KNOW-CKD) study. Nephrol. Dial. Transplant. 2018, 34, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Neirynck, N.; Glorieux, G.; Schepers, E.; Pletinck, A.; Dhondt, A.; Vanholder, R. Review of Protein-Bound Toxins, Possibility for Blood Purification Therapy. Blood Purif. 2013, 35, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Aronov, P.A.; Luo, F.J.-G.; Plummer, N.S.; Quan, Z.; Holmes, S.; Hostetter, T.H.; Meyer, T.W. Colonic Contribution to Uremic Solutes. J. Am. Soc. Nephrol. 2011, 22, 1769–1776. [Google Scholar] [CrossRef] [PubMed]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.; on behalf of the European Uremic Toxin Work Group (EUTox). Serum Indoxyl Sulfate Is Associated with Vascular Disease and Mortality in Chronic Kidney Disease Patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Schepers, E.; Pletinck, A.; Nagler, E.V.; Glorieux, G. The Uremic Toxicity of Indoxyl Sulfate and p-Cresyl Sulfate: A Systematic Review. J. Am. Soc. Nephrol. 2014, 25, 1897–1907. [Google Scholar] [CrossRef]
- Matsumoto, T.; Takayanagi, K.; Kojima, M.; Katome, T.; Taguchi, K.; Kobayashi, T. Direct Impairment of the Endothelial Function by Acute Indoxyl Sulfate through Declined Nitric Oxide and Not Endothelium-Derived Hyperpolarizing Factor or Vasodilator Prostaglandins in the Rat Superior Mesenteric Artery. Biol. Pharm. Bull. 2019, 42, 1236–1242. [Google Scholar] [CrossRef]
- Lano, G.; Burtey, S.; Sallée, M. Indoxyl Sulfate, a Uremic Endotheliotoxin. Toxins 2020, 12, 229. [Google Scholar] [CrossRef]
- Tumur, Z.; Shimizu, H.; Enomoto, A.; Miyazaki, H.; Niwa, T. Indoxyl sulfate upregulates expression of ICAM-1 and MCP-1 by oxidative stress-induced NF-kappaB activation. Am. J. Nephrol. 2010, 31, 435–441. [Google Scholar] [CrossRef]
- Stinghen, A.E.M.; Chillon, J.-M.; Massy, Z.; Boullier, A. Differential Effects of Indoxyl Sulfate and Inorganic Phosphate in a Murine Cerebral Endothelial Cell Line (bEnd.3). Toxins 2014, 6, 1742–1760. [Google Scholar] [CrossRef]
- Kharait, S.; Haddad, D.J.; Springer, M.L. Nitric oxide counters the inhibitory effects of uremic toxin indoxyl sulfate on endothelial cells by governing ERK MAP kinase and myosin light chain activation. Biochem. Biophys. Res. Commun. 2011, 409, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Bertrand, E.; Cerini, C.; Faure, V.; Sampol, J.; Vanholder, R.; Berland, Y.; Brunet, P. The uremic solutes p-cresol and indoxyl sulfate inhibit endothelial proliferation and wound repair. Kidney Int. 2004, 65, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.-C.; Kuo, K.-L.; Huang, H.-L.; Lin, C.-C.; Tsai, T.-H.; Wang, C.-H.; Chen, J.-W.; Lin, S.-J.; Huang, P.-H.; Tarng, D.-C. Indoxyl sulfate suppresses endothelial progenitor cell–mediated neovascularization. Kidney Int. 2016, 89, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Faure, V.; Dou, L.; Sabatier, F.; Cerini, C.; Sampol, J.; Berland, Y.; Brunet, P.; Dignat-George, F. Elevation of circulating endothelial microparticles in patients with chronic renal failure. J. Thromb. Haemost. 2006, 4, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.-H.; Jeon, E.-Y.; Kim, S.-J. Indoxyl Sulfate-Induced Extracellular Vesicles Released from Endothelial Cells Stimulate Vascular Smooth Muscle Cell Proliferation by Inducing Transforming Growth Factor-Beta Production. J. Vasc. Res. 2019, 56, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Boenink, R.; Stel, V.S.; Waldum-Grevbo, B.E.; Collart, F.; Kerschbaum, J.; Heaf, J.G.; De Meester, J.; Finne, P.; García-Marcos, S.A.; Evans, M.; et al. Data from the ERA-EDTA Registry were examined for trends in excess mortality in European adults on kidney replacement therapy. Kidney Int. 2020, 98, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Ruospo, M.; Palmer, S.C.; Natale, P.; Craig, J.C.; Vecchio, M.; Elder, G.J.; Strippoli, G.F. Phosphate binders for preventing and treating chronic kidney disease-mineral and bone disorder (CKD-MBD). Cochrane Database Syst. Rev. 2018, 8, CD006023. [Google Scholar] [CrossRef]
- Greeviroj, P.; Kitrungphaiboon, T.; Katavetin, P.; Praditpornsilpa, K.; Eiam-Ong, S.; Jaber, B.L.; Susantitaphong, P. Cinacalcet for Treatment of Chronic Kidney Disease-Mineral and Bone Disorder: A Meta-Analysis of Randomized Controlled Trials. Nephron 2018, 139, 197–210. [Google Scholar] [CrossRef]
- Ureña-Torres, P.A.; Floege, J.; Hawley, C.M.; Pedagogos, E.; Goodman, W.G.; Pétavy, F.; Reiner, M.; Raggi, P. Protocol adherence and the progression of cardiovascular calcification in the ADVANCE study. Nephrol. Dial. Transplant. 2013, 28, 146–152. [Google Scholar] [CrossRef][Green Version]
- Raggi, P.; Chertow, G.M.; Torres, P.U.; Csiky, B.; Naso, A.; Nossuli, K.; Moustafa, M.; Goodman, W.G.; Lopez, N.; Downey, G.; et al. The ADVANCE study: A randomized study to evaluate the effects of cinacalcet plus low-dose vitamin D on vascular calcification in patients on hemodialysis. Nephrol. Dial. Transplant. 2011, 26, 1327–1339. [Google Scholar] [CrossRef]
- Barcellos, F.C.; Del Vecchio, F.B.; Reges, A.; Mielke, G.I.; Santos, I.S.; Umpierre, D.; Bohlke, M.; Hallal, P.C. Exercise in patients with hypertension and chronic kidney disease: A randomized controlled trial. J. Hum. Hypertens. 2018, 32, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Ikizler, T.A.; Robinson-Cohen, C.; Ellis, C.; Headley, S.A.; Tuttle, K.; Wood, R.J.; Evans, E.E.; Milch, C.M.; Moody, K.A.; Germain, M.; et al. Metabolic Effects of Diet and Exercise in Patients with Moderate to Severe CKD: A Randomized Clinical Trial. J. Am. Soc. Nephrol. 2017, 29, 250–259. [Google Scholar] [CrossRef] [PubMed]
- E Silva, V.R.O.; Stringuetta Belik, F.; Hueb, J.C.; de Souza Gonçalves, R.; Teixeira Caramori, J.C.; Perez Vogt, B.; Barretti, P.; Zanati Bazan, S.G.; De Stefano, G.M.M.F.; Martin, L.C. Aerobic Exercise Training and Nontraditional Cardiovascular Risk Factors in Hemodialysis Patients: Results from a Prospective Randomized Trial. Cardiorenal Med. 2019, 9, 391–399. [Google Scholar]
- Abreu, C.C.; Cardozo, L.F.M.F.; Stockler-Pinto, M.B.; Esgalhado, M.; Barboza, J.E.; Frauches, R.; Mafra, D. Does resistance exercise performed during dialysis modulate Nrf2 and NF-κB in patients with chronic kidney disease? Life Sci. 2017, 188, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Mafra, D.; Borges, N.A.; Lindholm, B.; Shiels, P.G.; Evenepoel, P.; Stenvinkel, P. Food as medicine: Targeting the uraemic phenotype in chronic kidney disease. Nat. Rev. Nephrol. 2020, 1–19. [Google Scholar] [CrossRef]
- Mafra, D.; Borges, N.A.; Alvarenga, L.; Esgalhado, M.; Cardozo, L.F.; Lindholm, B.; Stenvinkel, P. Dietary Components That May Influence the Disturbed Gut Microbiota in Chronic Kidney Disease. Nutrients 2019, 11, 496. [Google Scholar] [CrossRef]
- Chudzińska, M.; Rogowicz, D.; Wołowiec, Ł.; Banach, J.; Sielski, S.; Bujak, R.; Sinkiewicz, A.; Grześk, G. Resveratrol and cardiovascular system—The unfulfilled hopes. Ir. J. Med. Sci. 2020, 1–6. [Google Scholar]
- Milovanova, L.; Fomin, V.; Moiseev, S.; Taranova, M.; Milovanov, Y.; Lysenko, L.; Kozlovskaya, L.L.; Kozevnikova, E.; Milovanova, S.; Lebedeva, M.; et al. Effect of essential amino acid кetoanalogues and protein restriction diet on morphogenetic proteins (FGF-23 and Кlotho) in 3b–4 stages chronic кidney disease patients: A randomized pilot study. Clin. Exp. Nephrol. 2018, 22, 1351–1359. [Google Scholar] [CrossRef]
- Merino, A.; Alvarez-Lara, M.A.; Ramirez, R.; Carracedo, J.; Martin-Malo, A.; Aljama, P. Losartan prevents the development of the pro-inflammatory monocytes CD14+CD16+ in haemodialysis patients. Nephrol. Dial. Transplant. 2012, 27, 2907–2912. [Google Scholar] [CrossRef][Green Version]
- Makówka, A.; Olejniczak-Fortak, M.; Nowicki, M. A Comparison of the Antihypertensive and Anti-Inflammatory Effects of Aliskiren and Ramipril Add-On Therapy in Peritoneal Dialysis Patients—A Pilot Open Label Study. Kidney Blood Press. Res. 2012, 36, 18–25. [Google Scholar] [CrossRef]
- Iijima, K.; Ito, Y.; Son, B.-K.; Akishita, M.; Ouchi, Y. Pravastatin and Olmesartan Synergistically Ameliorate Renal Failure-Induced Vascular Calcification. J. Atheroscler. Thromb. 2014, 21, 917–929. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Heimbürger, O.; Stenvinkel, P. Statins to treat chronic inflammation in dialysis patients—Is this feasible? Perit. Dial. Int. 2007, 27, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Qureshi, A.R.; Parini, P.; Hurt-Camejo, E.; Ripsweden, J.; Brismar, T.B.; Barany, P.; Jaminon, A.M.; Schurgers, L.J.; Heimburger, O.; et al. Does statins promote vascular calcification in chronic kidney disease? Eur. J. Clin. Investig. 2017, 47, 137–148. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.; Wheeler, D.C.; Stefánsson, B.V.; Jongs, N.; Postmus, D.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Held, C.; Hou, F.F.; et al. Effect of Dapagliflozin on Clinical Outcomes in Patients with Chronic Kidney Disease, With and Without Cardiovascular Disease. Circulation 2020. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.-F.; Mann, J.F.; McMurray, J.J.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef]
- Filippatos, T.D.; Liontos, A.; Papakitsou, I.; Moses, E.S. SGLT2 inhibitors and cardioprotection: A matter of debate and multiple hypotheses. Postgrad. Med. 2019, 131, 82–88. [Google Scholar] [CrossRef]
- Ojima, A.; Matsui, T.; Nishino, Y.; Nakamura, N.; Yamagishi, S. Empagliflozin, an Inhibitor of Sodium-Glucose Cotransporter 2 Exerts Anti-Inflammatory and Antifibrotic Effects on Experimental Diabetic Nephropathy Partly by Suppressing AGEs-Receptor Axis. Horm. Metab. Res. 2015, 47, 686–692. [Google Scholar] [CrossRef]
- Lau, W.L.; Savoj, J.; Nakata, M.B.; Vaziri, N.D. Altered microbiome in chronic kidney disease: Systemic effects of gut-derived uremic toxins. Clin. Sci. 2018, 132, 509–522. [Google Scholar] [CrossRef]
- Niwa, T. Role of Indoxyl Sulfate in the Progression of Chronic Kidney Disease and Cardiovascular Disease: Experimental and Clinical Effects of Oral Sorbent AST-120. Ther. Apher. Dial. 2011, 15, 120–124. [Google Scholar] [CrossRef]
- Sakaguchi, Y.; Hamano, T.; Obi, Y.; Monden, C.; Oka, T.; Yamaguchi, S.; Matsui, I.; Hashimoto, N.; Matsumoto, A.; Shimada, K.; et al. A Randomized Trial of Magnesium Oxide and Oral Carbon Adsorbent for Coronary Artery Calcification in Predialysis CKD. J. Am. Soc. Nephrol. 2019, 30, 1073–1085. [Google Scholar] [CrossRef]
- Zheng, H.J.; Guo, J.; Wang, Q.; Wang, L.; Wang, Y.; Zhang, F.; Huang, W.-J.; Zhang, W.; Liu, W.J.; Wang, Y. Probiotics, prebiotics, and synbiotics for the improvement of metabolic profiles in patients with chronic kidney disease: A systematic review and meta-analysis of randomized controlled trials. Crit. Rev. Food Sci. Nutr. 2020, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Cavallari, C.; Dellepiane, S.; Fonsato, V.; Medica, D.; Marengo, M.; Migliori, M.; Quercia, A.D.; Pitino, A.; Formica, M.; Panichi, V.; et al. Online Hemodiafiltration Inhibits Inflammation-Related Endothelial Dysfunction and Vascular Calcification of Uremic Patients Modulating miR-223 Expression in Plasma Extracellular Vesicles. J. Immunol. 2019, 202, 2372–2383. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C. The Rise of Expanded Hemodialysis. Blood Purif. 2017, 44, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zickler, D.; Schindler, R.; Willy, K.; Martus, P.; Pawlak, M.; Storr, M.; Hulko, M.; Boehler, T.; Glomb, M.A.; Liehr, K.; et al. Medium Cut-Off (MCO) Membranes Reduce Inflammation in Chronic Dialysis Patients—A Randomized Controlled Clinical Trial. PLoS ONE 2017, 12, e0169024. [Google Scholar] [CrossRef] [PubMed]
- Willy, K.; Girndt, M.; Voelkl, J.; Fiedler, R.; Martus, P.; Storr, M.; Schindler, R.; Zickler, D. Expanded Haemodialysis Therapy of Chronic Haemodialysis Patients Prevents Calcification and Apoptosis of Vascular Smooth Muscle Cells in vitro. Blood Purif. 2017, 45, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Murshed, M.; Schinke, T.; McKee, M.D.; Karsenty, G. Extracellular matrix mineralization is regulated locally; different roles of two gla-containing proteins. J. Cell Biol. 2004, 165, 625–630. [Google Scholar] [CrossRef]
- Shearer, M.J. Vitamin K. Lancet 1995, 345, 229–234. [Google Scholar] [CrossRef]
- Schurgers, L.J.; Spronk, H.M.H.; Soute, B.A.M.; Schiffers, P.M.; Demey, J.G.R.; Vermeer, C. Regression of warfarin-induced medial elastocalcinosis by high intake of vitamin K in rats. Blood 2006, 109, 2823–2831. [Google Scholar] [CrossRef]
- O’Donnell, C.J.; Shea, M.K.; Price, P.A.; Gagnon, D.R.; Wilson, P.W.F.; Larson, M.G.; Kiel, D.P.; Hoffmann, U.; Ferencik, M.; Clouse, M.E.; et al. Matrix Gla Protein Is Associated With Risk Factors for Atherosclerosis but not With Coronary Artery Calcification. Arter. Thromb. Vasc. Biol. 2006, 26, 2769–2774. [Google Scholar] [CrossRef]
- Delanaye, P.; Krzesinski, J.-M.; Warling, X.; Moonen, M.; Smelten, N.; Médart, L.; Pottel, H.; Cavalier, E. Dephosphorylated-uncarboxylated Matrix Gla protein concentration is predictive of vitamin K status and is correlated with vascular calcification in a cohort of hemodialysis patients. BMC Nephrol. 2014, 15, 1–8. [Google Scholar] [CrossRef]
- Shroff, R.; Shah, V.; Hiorns, M.P.; Schoppet, M.; Hofbauer, L.C.; Hawa, G.; Schurgers, L.J.; Singhal, A.; Merryweather, I.; Brogan, P.; et al. The circulating calcification inhibitors, fetuin-A and osteoprotegerin, but not Matrix Gla protein, are associated with vascular stiffness and calcification in children on dialysis. Nephrol. Dial. Transplant. 2008, 23, 3263–3271. [Google Scholar] [CrossRef] [PubMed]
- Witham, M.D.; Lees, J.S.; White, M.; Band, M.; Bell, S.; Chantler, D.J.; Ford, I.; Fulton, R.L.; Kennedy, G.; Littleford, R.C.; et al. Vitamin K Supplementation to Improve Vascular Stiffness in CKD: The K4Kidneys Randomized Controlled Trial. J. Am. Soc. Nephrol. 2020, 31, 2434–2445. [Google Scholar] [CrossRef] [PubMed]
- Bressendorff, I.; Hansen, D.; Schou, M.; Kragelund, C.; Brandi, L. The effect of magnesium supplementation on vascular calcification in chronic kidney disease—A randomised clinical trial (MAGiCAL-CKD): Essential study design and rationale. BMJ Open 2017, 7, e016795. [Google Scholar] [CrossRef] [PubMed]
- Raymond, C.B.; Wazny, L.D. Sodium thiosulfate, bisphosphonates, and cinacalcet for treatment of calciphylaxis. Am. J. Health Pharm. 2008, 65, 1419–1429. [Google Scholar] [CrossRef] [PubMed]
- Djuric, P.; Dimkovic, N.; Schlieper, G.; Djuric, Z.; Pantelic, M.; Mitrovic, M.; Jankovic, A.; Milanov, M.; Pficer, J.K.; Floege, J. Sodium thiosulphate and progression of vascular calcification in end-stage renal disease patients: A double-blind, randomized, placebo-controlled study. Nephrol. Dial. Transplant. 2019, 35, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Bi, Z.M.; Wang, Y.; Chen, Z.Q.; Xu, S.W. Effect of sodium thiosulfate on coronary artery calcification in maintenance hemodialysis patients. Zhonghua Yi Xue Za Zhi 2016, 96, 3724–3728. [Google Scholar] [PubMed]
- Hildebrand, S.; Cunningham, J. Is there a role for bisphosphonates in vascular calcification in chronic kidney disease? Bone 2020, 115751. [Google Scholar] [CrossRef]
- Nitta, K.; Akiba, T.; Suzuki, K.; Uchida, K.; Watanabe, R.-I.; Majima, K.; Aoki, T.; Nihei, H. Effects of cyclic intermittent etidronate therapy on coronary artery calcification in patients receiving long-term hemodialysis. Am. J. Kidney Dis. 2004, 44, 680–688. [Google Scholar] [CrossRef]
- Raggi, P.; Bellasi, A.; Bushinsky, D.; Bover, J.; Rodriguez, M.; Ketteler, M.; Sinha, S.; Salcedo, C.; Gillotti, K.; Padgett, C.; et al. Slowing Progression of Cardiovascular Calcification with SNF472 in Patients on Hemodialysis: Results of a Randomized Phase 2b Study. Circulation 2020, 141, 728–739. [Google Scholar] [CrossRef]
- Ferrer, M.D.; Ketteler, M.; Tur, F.; Tur, E.; Isern, B.; Salcedo, C.; Joubert, P.H.; Behets, G.J.; Neven, E.; D’Haese, P.C.; et al. Characterization of SNF472 pharmacokinetics and efficacy in uremic and non-uremic rats models of cardiovascular calcification. PLoS ONE 2018, 13, e0197061. [Google Scholar] [CrossRef]
- Chen, C.-L.; Chen, N.-C.; Wu, F.-Z.; Wu, M.-T. Impact of denosumab on cardiovascular calcification in patients with secondary hyperparathyroidism undergoing dialysis: A pilot study. Osteoporos. Int. 2020, 31, 1507–1516. [Google Scholar] [CrossRef] [PubMed]
- Festuccia, F.; Jafari, M.T.; Moioli, A.; Fofi, C.; Barberi, S.; Amendola, S.; Sciacchitano, S.; Punzo, G.; Menè, P. Safety and efficacy of denosumab in osteoporotic hemodialysed patients. J. Nephrol. 2017, 30, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Marlow, C.F.; Sharma, S.; Babar, F.; Lin, J. Severe Hypocalcemia and Hypomagnesemia with Denosumab in Advanced Chronic Kidney Disease: Case Report and Literature Review. Case Rep. Oncol. Med. 2018, 2018, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.-X.; Zhang, H.; Peng, C. Puerarin: A Review of Pharmacological Effects. Phytother. Res. 2014, 28, 961–975. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, X.; Zhong, X.; Li, Z.; Cai, S.; Yang, P.; Ou, C.; Chen, M. Puerarin inhibits vascular calcification of uremic rats. Eur. J. Pharmacol. 2019, 855, 235–243. [Google Scholar] [CrossRef]
- Dalibalta, S.; Majdalawieh, A.; Manjikian, H. Health benefits of sesamin on cardiovascular disease and its associated risk factors. Saudi Pharm. J. 2020, 28, 1276–1289. [Google Scholar] [CrossRef]
- Han, J.-H.; Lee, S.-G.; Jung, S.-H.; Lee, J.-J.; Park, H.-S.; Kim, Y.H.; Myung, C.-S. Sesamin Inhibits PDGF-Mediated Proliferation of Vascular Smooth Muscle Cells by Upregulating p21 and p27. J. Agric. Food Chem. 2015, 63, 7317–7325. [Google Scholar] [CrossRef]
- Kugo, H.; Miyamoto, C.; Sawaragi, A.; Hoshino, K.; Hamatani, Y.; Matsumura, S.; Yoshioka, Y.; Moriyama, T.; Zaima, N. Sesame Extract Attenuates the Degradation of Collagen and Elastin Fibers in the Vascular Walls of Nicotine-administered Mice. J. Oleo Sci. 2018, 68, 79–85. [Google Scholar] [CrossRef]
- Chao, C.-T.; Yeh, H.-Y.; Tsai, Y.-T.; Yuan, T.-H.; Liao, M.-T.; Huang, J.-W.; Chen, H.-W. Astaxanthin Counteracts Vascular Calcification In Vitro Through an Early Up-Regulation of SOD2 Based on a Transcriptomic Approach. Int. J. Mol. Sci. 2020, 21, 8530. [Google Scholar] [CrossRef]
- Cui, L.; Zhou, Q.; Zheng, X.; Sun, B.; Zhao, S. Mitoquinone attenuates vascular calcification by suppressing oxidative stress and reducing apoptosis of vascular smooth muscle cells via the Keap1/Nrf2 pathway. Free. Radic. Biol. Med. 2020, 161, 23–31. [Google Scholar] [CrossRef]
- Tani, T.; Fujiwara, M.; Orimo, H.; Shimizu, A.; Narisawa, S.; Pinkerton, A.B.; Millán, J.L.; Tsuruoka, S. Inhibition of tissue-nonspecific alkaline phosphatase protects against medial arterial calcification and improves survival probability in the CKD-MBD mouse model. J. Pathol. 2020, 250, 30–41. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kyriakidis, N.C.; Cobo, G.; Dai, L.; Lindholm, B.; Stenvinkel, P. Role of Uremic Toxins in Early Vascular Ageing and Calcification. Toxins 2021, 13, 26. https://doi.org/10.3390/toxins13010026
Kyriakidis NC, Cobo G, Dai L, Lindholm B, Stenvinkel P. Role of Uremic Toxins in Early Vascular Ageing and Calcification. Toxins. 2021; 13(1):26. https://doi.org/10.3390/toxins13010026
Chicago/Turabian StyleKyriakidis, Nikolaos C., Gabriela Cobo, Lu Dai, Bengt Lindholm, and Peter Stenvinkel. 2021. "Role of Uremic Toxins in Early Vascular Ageing and Calcification" Toxins 13, no. 1: 26. https://doi.org/10.3390/toxins13010026
APA StyleKyriakidis, N. C., Cobo, G., Dai, L., Lindholm, B., & Stenvinkel, P. (2021). Role of Uremic Toxins in Early Vascular Ageing and Calcification. Toxins, 13(1), 26. https://doi.org/10.3390/toxins13010026