Gut-Derived Protein-Bound Uremic Toxins


Abstract
1. Introduction
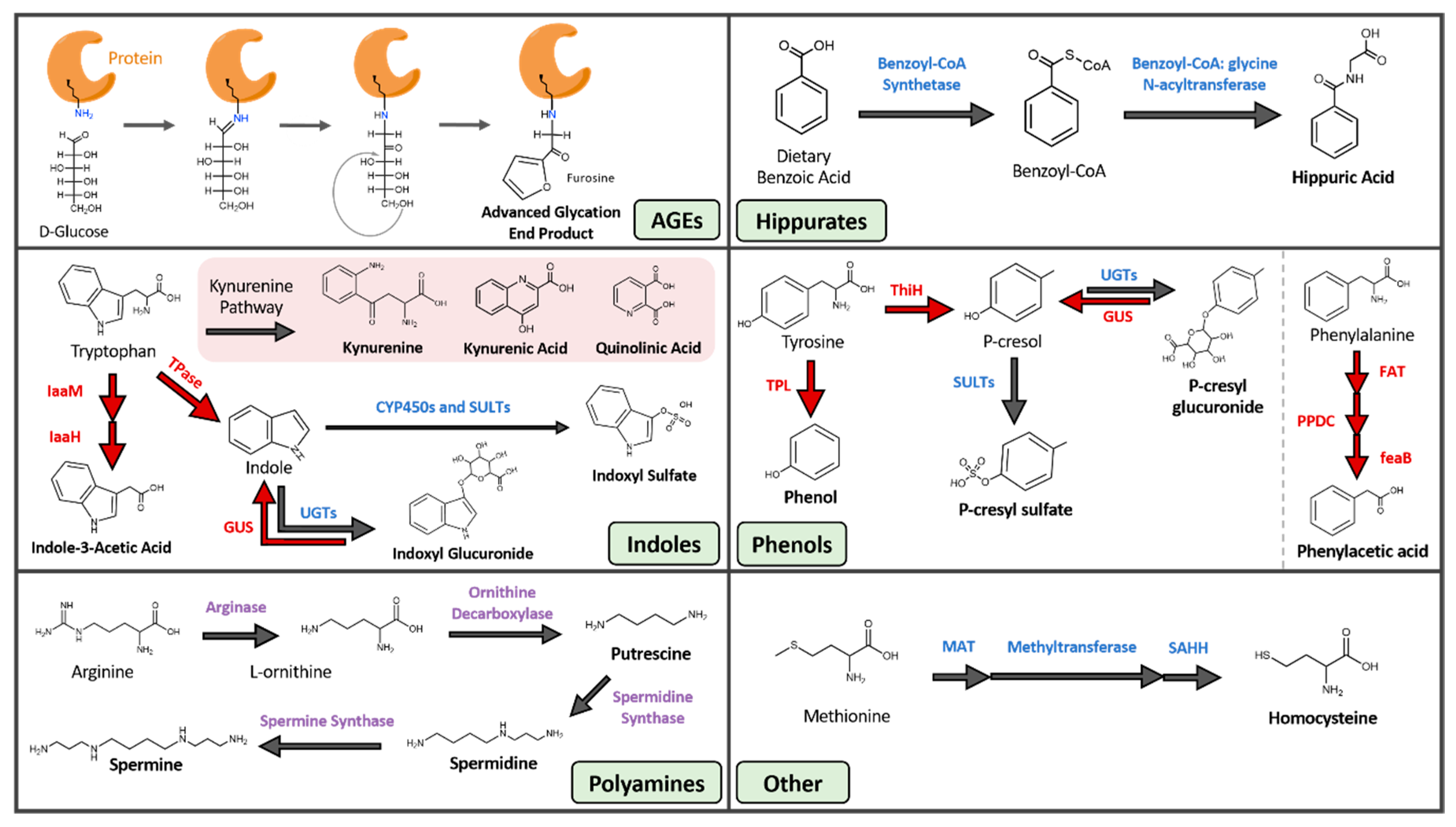
2. PBUT Derivation and Pathological Mechanisms
2.1. Advanced Glycation End Products (AGEs)
2.2. Hippurates
2.3. Indoles
2.4. Phenols
2.5. Polyamines
2.6. Other
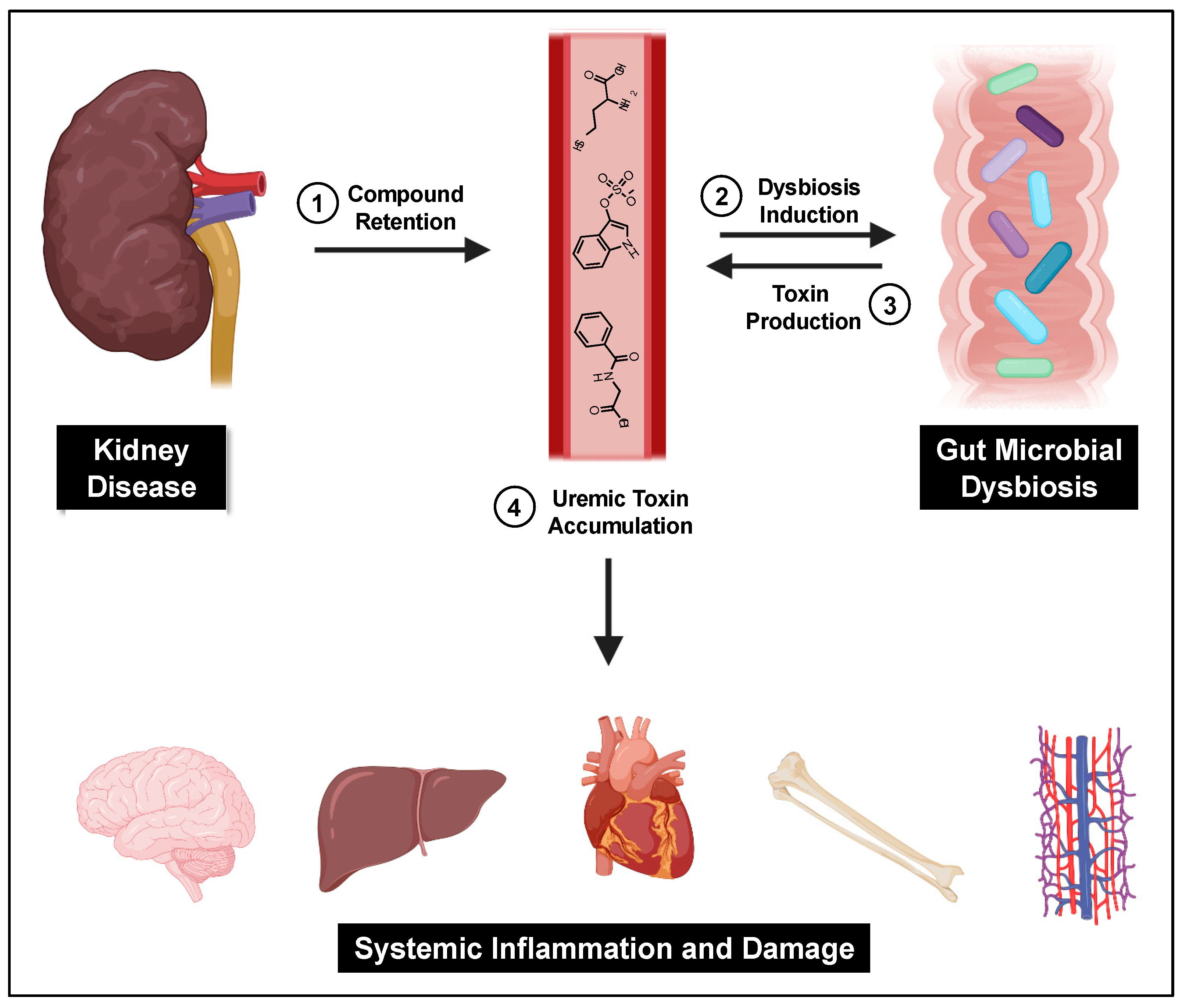
3. PBUTs and the Gut Microbiome
3.1. Gut Microbial Dysbiosis
3.2. Reducing Gut-Derived PBUTs
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mills, K.T.; Xu, Y.; Zhang, W.; Bundy, J.D.; Chen, C.-S.; Kelly, T.N.; Chen, J.; He, J. A systematic analysis of world-wide population-based data on the global burden of chronic kidney disease in 2010. Kidney Int. 2016, 88, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Anothaisintawee, T.; Rattanasiri, S.; Ingsathit, A.; Attia, J.; Thakkinstian, A. Prevalence of chronic kidney disease: A systematic review and meta-analysis. Clin. Nephrol. 2009, 71, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argilés, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Pletinck, A.; Schepers, E.; Glorieux, G. Biochemical and Clinical Impact of Organic Uremic Retention Solutes: A Comprehensive Update. Toxins 2018, 10, 33. [Google Scholar] [CrossRef]
- Vanholder, R.; Baurmeister, U.; Brunet, P.; Cohen, G.; Glorieux, G.; Jankowski, J. A bench to bedside view of uremic toxins. J. Am. Soc. Nephrol. 2008, 19, 863–870. [Google Scholar] [CrossRef]
- Aronov, P.A.; Luo, F.J.G.; Plummer, N.S.; Quan, Z.; Holmes, S.; Hostetter, T.H.; Meyer, T.W. Colonic contribution to uremic solutes. J. Am. Soc. Nephrol. 2011, 22, 1769–1776. [Google Scholar] [CrossRef] [PubMed]
- Mair, R.D.; Sirich, T.L.; Plummer, N.S.; Meyer, T.W. Characteristics of colon-derived uremic solutes. Clin. J. Am. Soc. Nephrol. 2018, 13, 1398–1404. [Google Scholar] [CrossRef]
- Jourde-Chiche, N.; Dou, L.; Cerini, C.; Dignat-George, F.; Vanholder, R.; Brunet, P. Protein-Bound Toxins-Update 2009. Semin. Dial. 2009, 22, 334–339. [Google Scholar] [CrossRef]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Wong, J.; Pahl, M.; Piceno, Y.M.; Yuan, J.; DeSantis, T.Z.; Ni, Z.; Nguyen, T.-H.; Andersen, G.L. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013, 83, 308–315. [Google Scholar] [CrossRef]
- Ramezani, A.; Raj, D.S. The Gut Microbiome, Kidney Disease, and Targeted Interventions. J. Am. Soc. Nephrol. 2013, 25, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D. CKD impairs barrier function and alters microbial flora of the intestine: A major link to inflammation and uremic toxicity. Curr. Opin. Nephrol. Hypertens. 2012, 21, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Barrios, C.; Beaumont, M.; Pallister, T.; Villar, J.; Goodrich, J.K.; Clark, A.; Pascual, J.; E Ley, R.; Spector, T.D.; Bell, J.T.; et al. Gut-Microbiota-Metabolite Axis in Early Renal Function Decline. PLoS ONE 2015, 10, e0134311. [Google Scholar] [CrossRef]
- Fukuuchi, F.; Hida, M.; Aiba, Y.; Koga, Y.; Endoh, M.; Kurokawa, K.; Sakai, H. Intestinal bacteria-derived putrefactants in chronic renal failure. Clin. Exp. Nephrol. 2002, 6, 99–104. [Google Scholar] [CrossRef]
- Strid, H.; Simrén, M.; Stotzer, P.O.; Ringström, G.; Abrahamsson, H.; Björnsson, E.S. Patients with chronic renal failure have abnormal small intestinal motility and a high prevalence of small intestinal bacterial overgrowth. Digestion 2003, 67, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, P.; Jiang, H.; Cheng, S. Gut bacterial translocation contributes to microinflammation in experimental uremia. Dig. Dis. Sci. 2012, 57, 2856–2862. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Y.; Chen, D.Q.; Chen, L.; Liu, J.-R.; Vaziri, N.D.; Guo, Y.; Zhao, Y.-Y. Microbiome-metabolome reveals the contribution of gut–kidney axis on kidney disease. J. Transl. Med. 2019, 17, 1–11. [Google Scholar] [CrossRef]
- Cigarrán-Guldrís, S.; Parra, E.G.; Amenós, A.C. Gut microbiota in chronic kidney disease. Nefrologia 2017, 37, 9–19. [Google Scholar] [CrossRef]
- Liu, Y.; Li, J.; Yu, J.; Wang, Y.; Lu, J.; Shang, E.-X.; Zhu, Z.; Guo, J.; Duan, J. Disorder of gut amino acids metabolism during CKD progression is related with gut microbiota dysbiosis and metagenome change. J. Pharm. Biomed. Anal. 2018, 149, 425–435. [Google Scholar] [CrossRef]
- Gouroju, S.; Srinivasa Rao, P.V.L.N.; Bitla, A.R.; Vinapamula, K.S.; Manohar, S.M.; Vishnubhotla, S. Role of gut-derived uremic toxins on oxidative stress and inflammation in patients with chronic kidney disease. Indian J. Nephrol. 2017, 27, 359–364. [Google Scholar] [CrossRef]
- Stinghen, A.E.M.; Massy, Z.A.; Vlassara, H.; Striker, G.E.; Boullier, A. Uremic toxicity of advanced glycation end products in CKD. J. Am. Soc. Nephrol. 2016, 27, 354–370. [Google Scholar] [CrossRef]
- Miyata, T.; Ueda, Y.; Yamada, Y.; Izuhara, Y.; Wada, T.; Jadoul, M.; Saito, A.; Kurokawa, K.; De Strihou, C.V.Y. Accumulation of Carbonyls an Advanced Stress in Uremia Accelerates the Formation of Glycation End Product: Carbonyl Stress in Uremia. J. Am. Soc. Nephrol. 1998, 9, 2349–2356. [Google Scholar]
- Mallipattu, S.K.; He, J.C.; Uribarri, J. Role of advanced glycation endproducts and potential therapeutic interventions in dialysis patients. Semin. Dial. 2012, 25, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Snelson, M.; Coughlan, M.T. Dietary Advanced Glycation End Products: Digestion, Metabolism and Modulation of Gut Microbial Ecology. Nutrients 2019, 11, 215. [Google Scholar] [CrossRef]
- Abordo, E.A.; Minhas, H.S.; Thornalley, P.J. Accumulation of α-oxoaldehydes during oxidative stress: A role in cytotoxicity. Biochem. Pharmacol. 1999, 58, 641–648. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. Advanced glycation end products in the pathogenesis of chronic kidney disease. Kidney Int. 2018, 93, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Perrone, A.; Giovino, A.; Benny, J.; Martinelli, F. Advanced Glycation End Products (AGEs): Biochemistry, Signaling, Analytical Methods, and Epigenetic Effects. Oxidative Med. Cell. Longev. 2020, 2020, 1–18. [Google Scholar] [CrossRef]
- Xie, J.; Méndez, J.D.; Méndez-Valenzuela, V.; Aguilar-Hernández, M.M. Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell Signal. 2013, 25, 2185–2197. [Google Scholar] [CrossRef]
- Franko, B.; Brault, J.; Jouve, T.; Beaumel, S.; Benhamou, P.-Y.; Zaoui, P.; Stasia, M.J. Differential impact of glucose levels and advanced glycation end-products on tubular cell viability and pro-inflammatory/profibrotic functions. Biochem. Biophys. Res. Commun. 2014, 451, 627–631. [Google Scholar] [CrossRef]
- Ding, Q.; Keller, J.N. Evaluation of rage isoforms, ligands, and signaling in the brain. Biochim. Biophys. Acta-Mol. Cell Res. 2005, 1746, 18–27. [Google Scholar] [CrossRef]
- Ramasamy, R.; Yan, S.F.; Schmidt, A.M. Advanced glycation endproducts: From precursors to RAGE: Round and round we go. Amino Acids 2010, 42, 1151–1161. [Google Scholar] [CrossRef]
- Grossin, N.; Auger, F.; Niquet-Léridon, C.; Durieux, N.; Montaigne, D.; Schmidt, A.M.; Susen, S.; Jacolot, P.; Beuscart, J.-B.; Tessier, F.J.; et al. Dietary CML-enriched protein induces functional arterial aging in a RAGE-dependent manner in mice. Mol. Nutr. Food Res. 2015, 59, 927–938. [Google Scholar] [CrossRef]
- Ueno, H.; Koyama, H.; Fukumoto, S.; Tanaka, S.; Shoji, T.; Shoji, T.; Emoto, M.; Tahara, H.; Inaba, M.; Kakiya, R.; et al. Advanced glycation end products, carotid atherosclerosis, and circulating endothelial progenitor cells in patients with end-stage renal disease. Metabolism 2011, 60, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced glycation end products: Sparking the development of diabetic vascular injury. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef]
- Vlassara, H.; Fuh, H.; Makita, Z.; Krungkrai, S.; Cerami, A.; Bucala, R. Exogenous advanced glycosylation end products induce complex vascular dysfunction in normal animals: A model for diabetic and aging complications. Proc. Natl. Acad. Sci. USA 1992, 89, 12043–12047. [Google Scholar] [CrossRef] [PubMed]
- Park, L.; Raman, K.G.; Lee, K.J.; Lu, Y.; Ferran, L.J., Jr.; Chow, W.S.; Stern, D.; Schmidt, A.M. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat. Med. 1998, 4, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Ingersoll, A.W.; Babcock, S.H. Hippuric Acid. Org. Synth. 2003, 2003, 40. [Google Scholar] [CrossRef]
- Lees, H.J.; Swann, J.R.; Wilson, I.D.; Nicholson, J.K.; Holmes, E. Hippurate: The Natural History of a Mammalian–Microbial Cometabolite. J. Proteome Res. 2013, 12, 1527–1546. [Google Scholar] [CrossRef]
- Toromanović, J.; Kovac-Besović, E.; Šapčanin, A.; Tahirovic, I.; Rimpapa, Z.; Kroyer, G.; Sofić, E. Urinary Hippuric Acid after Ingestion of Edible Fruits. Bosn. J. Basic Med. Sci. 2008, 8, 38–43. [Google Scholar] [CrossRef]
- Williams, H.; Cox, I.J.; Walker, D.G.; Cobbold, J.F.L.; Taylor-Robinson, S.D.; E Marshall, S.; Orchard, T.R. Differences in gut microbial metabolism are responsible for reduced hippurate synthesis in Crohn’s disease. BMC Gastroenterol. 2010, 10, 108. [Google Scholar] [CrossRef]
- Decharat, S. Hippuric Acid levels in paint workers at steel furniture manufacturers in Thailand. Saf. Health Work. 2014, 5, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Duydu, Y.; Süzen, S.; Erdem, N.; Uysal, H.; Vural, N. Validation of hippuric acid as a biomarker of toluene exposure. Bull. Environ. Contam. Toxicol. 1999, 63, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Angerer, J. Occupational chronic exposure to organic solvents. Int. Arch. Occup. Environ. Health 1985, 50, 323–328. [Google Scholar] [CrossRef]
- Satoh, M.; Hayashi, H.; Watanabe, M.; Ueda, K.; Yamato, H.; Yoshioka, T.; Motojima, M. Uremic toxins overload accelerates renal damage in a rat model of chronic renal failure. Nephron Exp. Nephrol. 2003, 95, e111–e118. [Google Scholar] [CrossRef] [PubMed]
- Edamatsu, T.; Fujieda, A.; Ezawa, A.; Itoh, Y. Classification of five uremic solutes according to their effects on renal tubular cells. Int. J. Nephrol. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Wei, R.; Wang, Y.; Su, T.; Li, P.; Chen, X. The uremic toxin hippurate promotes endothelial dysfunction via the activation of Drp1-mediated mitochondrial fission. Redox Biol. 2018, 16, 303–313. [Google Scholar] [CrossRef]
- Zaidi, N.; Ajmal, M.R.; Rabbani, G.; Ahmad, E.; Khan, R.H. A Comprehensive Insight into Binding of Hippuric Acid to Human Serum Albumin: A Study to Uncover Its Impaired Elimination through Hemodialysis. PLoS ONE 2013, 8, e71422. [Google Scholar] [CrossRef]
- Höglund, E.; Øverli, Ø.; Winberg, S. Tryptophan Metabolic Pathways and Brain Serotonergic Activity: A Comparative Review. Front. Endocrinol. 2019, 10, 158. [Google Scholar] [CrossRef]
- Fernandez-Prado, R.; Esteras, R.; Perez-Gomez, M.V.; Gracia-Iguacel, C.; Gonzalez-Parra, E.; Sanz, A.B.; Ortiz, A.; Sanchez-Niño, M.D. Nutrients Turned into Toxins: Microbiota Modulation of Nutrient Properties in Chronic Kidney Disease. Nutrients 2017, 9, 489. [Google Scholar] [CrossRef]
- Mahony, S.M.O.; Clarke, G.; Borre, Y.E.; Dinan, T.G.; Cryan, J.F. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav. Brain Res. 2015, 277, 32–48. [Google Scholar] [CrossRef]
- Farrell, K.O.; Harkin, A. Stress-related regulation of the kynurenine pathway: Relevance to neuropsychiatric and degenerative disorders. Neuropharmacology 2017, 112, 307–323. [Google Scholar] [CrossRef] [PubMed]
- Savitz, J. The kynurenine pathway: A finger in every pie. Mol. Psychiatry 2020, 25, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Xu, K.; Liu, H.; Liu, G.; Bai, M.; Peng, C.; Li, T.; Yin, J. Impact of the Gut Microbiota on Intestinal Immunity Mediated by Tryptophan Metabolism. Front. Cell Infect. Microbiol. 2018, 8, 1–22. [Google Scholar] [CrossRef]
- Hubbard, T.D.; Murray, I.A.; Perdew, G.H. Indole and Tryptophan Metabolism: Endogenous and Dietary Routes to Ah Receptor Activation. Drug Metab. Dispos. 2015, 43, 1522–1535. [Google Scholar] [CrossRef]
- Banoglu, E.; Jha, G.G.; King, R. Hepatic microsomal metabolism of indole to indoxyl, a precursor of indoxyl sulfate. Eur. J. Drug Metab. Pharmacokinet. 2001, 26, 235–240. [Google Scholar] [CrossRef]
- Asai, H.; Hirata, J.; Watanabe-Akanuma, M. Indoxyl glucuronide, a protein-bound uremic toxin, inhibits hypoxia-inducible factor-dependent erythropoietin expression through activation of aryl hydrocarbon receptor. Biochem. Biophys. Res. Commun. 2018, 504, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Vázquez, C.; Quintana, F.J. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity 2018, 48, 19–33. [Google Scholar] [CrossRef]
- De Brito, J.S.; Borges, N.A.; Dos Anjos, J.S.; Nakao, L.S.; Stockler-Pinto, M.B.; Paiva, B.R.; Cardoso-Weide, L.D.C.; Cardozo, L.F.M.D.F.; Mafra, D. Aryl Hydrocarbon Receptor and Uremic Toxins from the Gut Microbiota in Chronic Kidney Disease Patients: Is There a Relationship between Them? Biochemistry 2019, 58, 2054–2060. [Google Scholar] [CrossRef]
- Choi, W.; Eum, S.Y.; Lee, Y.W.; Hennig, B.; Robertson, L.W.; Toborek, M. PCB 104-Induced Proinflammatory Reactions in Human Vascular Endothelial Cells: Relationship to Cancer Metastasis and Atherogenesis. Toxicol. Sci. 2003, 75, 47–56. [Google Scholar] [CrossRef]
- Thome, T.; Salyers, Z.R.; Kumar, R.A.; Hahn, D.; Berru, F.N.; Ferreira, L.F.; Scali, S.T.; Ryan, T.E. Uremic metabolites impair skeletal muscle mitochondrial energetics through disruption of the electron transport system and matrix dehydrogenase activity. Am. J. Physiol. Cell Physiol. 2019, 317, 701–713. [Google Scholar] [CrossRef]
- Guillemin, G.J. Quinolinic acid, the inescapable neurotoxin. FEBS J. 2012, 279, 1356–1365. [Google Scholar] [CrossRef] [PubMed]
- Lekawanvijit, S.; Adrahtas, A.; Kelly, D.J.; Kompa, A.R.; Wang, B.H.; Krum, H. Does indoxyl sulfate, a uraemic toxin, have direct effects on cardiac fibroblasts and myocytes? Eur. Heart J. 2010, 31, 1771–1779. [Google Scholar] [CrossRef] [PubMed]
- Lano, G.; Burtey, S.; Sallée, M. Indoxyl Sulfate, a Uremic Endotheliotoxin. Toxins 2020, 12, 229. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Wang, C.; Chung, F.; Huang, L.L.H.; Yu, T. Uremic Retention Solute Indoxyl Sulfate Level Is Associated with Prolonged QTc Interval in Early CKD Patients. PLoS ONE 2015, 10, e0119545. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Chen, Y.; Hsieh, M.-H.; Huang, S.-Y.; Kao, Y.-H.; Chen, Y.-A.; Lin, Y.-K.; Chen, S.-A.; Chen, Y.-J. The Uremic Toxin Indoxyl Sulfate Increases Pulmonary Vein and Atrial Arrhythmogenesis. J. Cardiovasc. Electrophysiol. 2014, 26, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Adijiang, A.; Goto, S.; Uramoto, S.; Nishijima, F.; Niwa, T. Indoxyl sulphate promotes aortic calcification with expression of osteoblast-specific proteins in hypertensive rats. Nephrol. Dial. Transplant. 2008, 23, 1892–1901. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Osaka, M.; Higuchi, Y.; Nishijima, F.; Ishii, H.; Yoshida, M. Indoxyl sulfate induces leukocyte-endothelial interactions through up-regulation of E-selectin. J. Biol. Chem. 2010, 285, 38869–38875. [Google Scholar] [CrossRef] [PubMed]
- Tumur, Z.; Niwa, T. Indoxyl Sulfate Inhibits Nitric Oxide Production and Cell Viability by Inducing Oxidative Stress in Vascular Endothelial Cells. Am. J. Nephrol. 2009, 29, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.; Kuo, K.; Huang, H.; Lin, C.-C.; Tsai, T.-H.; Wang, C.-H.; Chen, J.-W.; Lin, S.-J.; Huang, P.-H.; Tarng, D.-C. Indoxyl sulfate suppresses endothelial progenitor cell–mediated neovascularization. Kidney Int. 2016, 89, 574–585. [Google Scholar] [CrossRef]
- Chitalia, V.C.; Shivanna, S.; Martorell, J.; Balcells, M.; Bosch, I. Uremic serum and solutes increase post-vascular interventional thrombotic risk through altered stability of smooth muscle cell tissue factor. Circulation 2012, 127, 365–376. [Google Scholar] [CrossRef]
- Nii-Kono, T.; Iwasaki, Y.; Uchida, M.; Fujieda, A.; Hosokawa, A.; Motojima, M.; Yamato, H.; Kurokawa, K.; Fukagawa, M. Indoxyl sulfate induces skeletal resistance to parathyroid hormone in cultured osteoblastic cells. Kidney Int. 2007, 71, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Hirata, J.; Hirai, K.; Asai, H.; Matsumoto, C.; Inada, M.; Miyaura, C. Indoxyl sulfate exacerbates low bone turnover induced by parathyroidectomy in young adult rats. Bone 2015, 79, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Leong, S.C.; Sirich, T.L. Indoxyl Sulfate—Review of Toxicity and Therapeutic Strategies. Toxins 2016, 8, 358. [Google Scholar] [CrossRef]
- Windey, K.; de Preter, V.; Verbeke, K. Relevance of protein fermentation to gut health. Mol. Nutr. Food Res. 2011, 56, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhao, Y. Gut microbiota derived metabolites in cardiovascular health and disease. Protein Cell 2018, 9, 416–431. [Google Scholar] [CrossRef]
- Aklujkar, M.; Risso, C.; Smith, J.A.; Beaulieu, D.; Dubay, R.; Giloteaux, L.; Diburro, K.; Holmes, D.E. Anaerobic degradation of aromatic amino acids by the hyperthermophilic archaeon Ferroglobus placidus. Microbiology 2014, 160, 2694–2709. [Google Scholar] [CrossRef]
- McGregor, D. Hydroquinone: An Evaluation of the Human Risks from its Carcinogenic and Mutagenic Properties. J. Allergy Clin. Immunol. 2007, 37, 887–914. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, J.; Van Der Giet, M.; Jankowski, V.; Schmidt, S.; Hemeier, M.; Mahn, B.; Giebing, G.; Tolle, M.; Luftmann, H.; Sculuter, H.; et al. Increased plasma phenylacetic acid in patients with end-stage renal failure inhibits iNOS expression. J. Clin. Investig. 2003, 112, 256–264. [Google Scholar] [CrossRef]
- Schmidt, S.; Westhoff, T.H.; Krauser, P.; Zidek, W.; Van Der Giet, M. The uraemic toxin phenylacetic acid increases the formation of reactive oxygen species in vascular smooth muscle cells. Nephrol. Dial. Transplant. 2007, 23, 65–71. [Google Scholar] [CrossRef]
- Cohen, G.; Raupachova, J.; Hörl, W.H. The uraemic toxin phenylacetic acid contributes to inflammation by priming polymorphonuclear leucocytes. Nephrol. Dial. Transplant. 2012, 28, 421–429. [Google Scholar] [CrossRef]
- Saito, Y.; Sato, T.; Nomoto, K.; Tsuji, H. Identification of phenol- and p-cresol-producing intestinal bacteria by using media supplemented with tyrosine and its metabolites. FEMS Microbiol. Ecol. 2018, 94, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gryp, T.; Vanholder, R.; Vaneechoutte, M.; Glorieux, G. p-Cresyl Sulfate. Toxins 2017, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Wu, I.W.; Hsu, K.H.; Hsu, H.J.; Lee, C.-C.; Sun, C.-Y.; Tsai, C.-J.; Wu, M.-S.; Kim, Y.C.; Oh, S.-W.; Koo, H.; et al. Serum free p-cresyl sulfate levels predict cardiovascular and all-cause mortality in elderly hemodialysis patients--a prospective cohort study. Nephrol. Dial. Transplant. 2011, 27, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Zhu, J.; Zhu, Z.; Ni, J.; Du, R.; Dai, Y.; Chen, Y.; Wu, Z.; Lu, L.; Zhang, R. p -Cresyl Sulfate Aggravates Cardiac Dysfunction Associated with Chronic Kidney Disease by Enhancing Apoptosis of Cardiomyocytes. J. Am. Heart Assoc. 2015, 4, e001852. [Google Scholar] [CrossRef]
- Guerrero, F.; Carmona, A.; Obrero, T.; Jiménez, M.J.; Soriano, S.; Moreno, J.A.; Martín-Malo, A.; Aljama, P. Role of endothelial microvesicles released by p-cresol on endothelial dysfunction. Sci. Rep. 2020, 10, 10657. [Google Scholar] [CrossRef]
- Gross, P.; Massy, Z.A.; Hénaut, L.; Boudot, C.; Cagnard, J.; March, C.; Kamel, S.; Drüeke, T.B.; Six, I. Para-cresyl sulfate acutely impairs vascular reactivity and induces vascular remodeling. J. Cell. Physiol. 2015, 230, 2927–2935. [Google Scholar] [CrossRef]
- Poveda, J.; Sanchez-Niño, M.D.; Glorieux, G.; Sanz, A.B.; Egido, J.; Vanholder, R.; Ortiz, A.; Ortiz, A. p-Cresyl sulphate has pro-inflammatory and cytotoxic actions on human proximal tubular epithelial cells. Nephrol. Dial. Transplant. 2013, 29, 56–64. [Google Scholar] [CrossRef]
- Sun, C.Y.; Chang, S.C.; Wu, M.S. Uremic Toxins Induce Kidney Fibrosis by Activating Intrarenal Renin–Angiotensin–Aldosterone System Associated Epithelial-to-Mesenchymal Transition. PLoS ONE 2012, 7, e34026. [Google Scholar] [CrossRef]
- Liabeuf, S.; Glorieux, G.; Lenglet, A.; Diouf, M.; Schepers, E.; Desjardins, L.; Choukroun, G.; Vanholder, R.; Massy, Z.A. Does P-Cresylglucuronide Have the Same Impact on Mortality as Other Protein-Bound Uremic Toxins? PLoS ONE 2013, 8, e67168. [Google Scholar] [CrossRef]
- Mutsaers, H.A.M.; Caetano-Pinto, P.; Seegers, A.E.M.; Dankers, A.C.; Broek, P.H.V.D.; Wetzels, J.F.; Brand, J.V.D.; Heuvel, L.P.V.D.; Hoenderop, J.G.; Wilmer, M.J.; et al. Proximal tubular efflux transporters involved in renal excretion of p-cresyl sulfate and p-cresyl glucuronide: Implications for chronic kidney disease pathophysiology. Toxicol. Vitr. 2015, 29, 1868–1877. [Google Scholar] [CrossRef]
- Ramos-Molina, B.; Queipo-Ortuño, M.I.; Lambertos, A.; Tinahones, F.J.; Peñafiel, R. Dietary and Gut Microbiota Polyamines in Obesity- and Age-Related Diseases. Front. Nutr. 2019, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Tofalo, R.; Cocchi, S.; Suzzi, G. Polyamines and Gut Microbiota. Front. Nutr. 2019, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Pegg, A.E. Introduction to the Thematic Minireview Series: Sixty plus years of polyamine research. J. Biol. Chem. 2018, 293, 18681–18692. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.A.; Poortvliet, E.; Stromberg, R.; Yngve, A. Polyamines in foods: Development of a food database. Food Nutr. Res. 2011, 55, 5572. [Google Scholar] [CrossRef]
- Igarashi, K.; Ueda, S.; Yoshida, K.; Kashiwagi, K. Polyamines in renal failure. Amino Acids 2006, 31, 477–483. [Google Scholar] [CrossRef]
- Sindhu, K.K. Uremic toxins: Some thoughts on acrolein and spermine. Ren. Fail. 2016, 38, 1755–1758. [Google Scholar] [CrossRef]
- Yoshida, K.; Yoneda, T.; Kimura, S.; Fujimoto, K.; Okajima, E.; Hirao, Y. Polyamines as an Inhibitor on Erythropoiesis of Hemodialysis Patients by In Vitro Bioassay Using the Fetal Mouse Liver Assay. Ther. Apher. Dial. 2006, 10, 267–272. [Google Scholar] [CrossRef]
- Dou, L.; Bertrand, E.; Cerini, C.; Faure, V.; Sampol, J.; Vanholder, R.; Berland, Y.; Brunet, P. The uremic solutes p-cresol and indoxyl sulfate inhibit endothelial proliferation and wound repair. Kidney Int. 2004, 65, 442–451. [Google Scholar] [CrossRef]
- Hoet, P.H.M.; Nemery, B. Polyamines in the lung: Polyamine uptake and polyamine-linked pathological or toxicological conditions. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 138, 377. [Google Scholar] [CrossRef]
- Conklin, D.J.; Barski, O.A.; Lesgards, J.F.; Juvan, P.; Režen, T.; Rozman, D.; Prough, R.A.; Vladykovskaya, E.; Liu, S.; Srivastava, S.; et al. Acrolein consumption induces systemic dyslipidemia and lipoprotein modification. Toxicol. Appl. Pharmacol. 2010, 243, 1–12. [Google Scholar] [CrossRef]
- Sakata, K.; Kashiwagi, K.; Sharmin, S.; Ueda, S.; Igarashi, K. Acrolein produced from polyamines as one of the uraemic toxins. Biochem. Soc. Trans. 2003, 31, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.H.; Lee, P.A.H.; Lu, Y.C.; Huang, C.-Y.; Chen, C.-H.; Chiang, C.-H.; Chow, P.-M.; Jaw, F.-S.; Wang, C.-C.; Huang, C.-Y.; et al. Acrolein contributes to urothelial carcinomas in patients with chronic kidney disease. Urol. Oncol. Semin. Orig. Investig. 2020, 38, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Luce, M.; Bouchara, A.; Pastural, M.; Granjon, S.; Szelag, J.C.; Laville, M.; Arkouche, W.; Fouque, D.; Soulage, C.O.; Koppe, L. Is 3-Carboxy-4-methyl-5-propyl-2-furanpropionate (CMPF) a clinically relevant uremic toxin in haemodialysis patients? Toxins 2018, 10, 205. [Google Scholar] [CrossRef] [PubMed]
- Prentice, K.J.; Wendell, S.G.; Liu, Y.; Eversley, J.A.; Salvatore, S.R.; Mohan, H.; Brandt, S.L.; Adams, A.C.; Wang, X.S.; Wei, D.; et al. CMPF, a Metabolite Formed Upon Prescription Omega-3-Acid Ethyl Ester Supplementation, Prevents and Reverses Steatosis. EBioMedicine 2018, 27, 200–213. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, P.; Alam, S.F. Role of homocysteine in the development of cardiovascular disease. Nutr. J. 2015, 14, 6. [Google Scholar] [CrossRef]
- Miller, A.L. The methionine-homocysteine cycle and its effects on cognitive diseases. Altern. Med. Rev. 2003, 8, 7–19. [Google Scholar]
- Chinnappa, S.; Tu, Y.K.; Yeh, Y.C.; Glorieux, G.; Vanholder, R.; Mooney, A. Association between Protein-Bound Uremic Toxins and Asymptomatic Cardiac Dysfunction in Patients with Chronic Kidney Disease. Toxins 2018, 10, 520. [Google Scholar] [CrossRef]
- Tsutsumi, Y.; Maruyama, T.; Takadate, A.; Shimada, H.; Otagiri, M. Decreased bilirubin-binding capacity in uremic serum caused by an accumulation of furan dicarboxylic acid. Nephron 2000, 85, 60–64. [Google Scholar] [CrossRef]
- Lim, C.F.; Stockigt, J.R.; Curtis, A.J.; Wynne, K.N.; Barlow, J.W.; Topliss, D.J. A naturally occurring furan fatty acid enhances drug inhibition of thyroxine binding in serum. Metabolism 1993, 42, 1468–1474. [Google Scholar] [CrossRef]
- Deguchi, T.; Kusuhara, H.; Takadate, A.; Endou, H.; Otagiri, M.; Sugiyama, Y. Characterization of uremic toxin transport by organic anion transporters in the kidney. Kidney Int. 2004, 65, 162–174. [Google Scholar] [CrossRef]
- Costigan, M.G.; O’callaghan, C.A.; Lindup, W.E. Hypothesis: Is accumulation of a furan dicarboxylic acid (3-carboxy-4-methyl-5-propyl-2-furanpropanoic acid) related to the neurological abnormalities in patients with renal failure? Nephron 1996, 73, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Mabuchi, H.; Nakahashi, H. Inhibition of Hepatic Glutathione S-Transferases by a Major Endogenous Ligand Substance Present in Uremic Serum. Nephron 1988, 49, 281–283. [Google Scholar] [CrossRef]
- Lim, C.F.; Bernard, B.F.; de Jong, M.; Docter, R.; Krenning, E.P.; Hennenmann, G. A Furan Fatty Acid and Indoxyl Sulfate Are the Putative Inhibitors of Thyroxine Hepatocyte Transport in Uremia. J. Clin. Endocrinol. Metab. 1993, 76, 318–324. [Google Scholar] [PubMed]
- Tsujimoto, M.; Kinoshita, Y.; Hirata, S.; Otagiri, M.; Ohtani, H.; Sawada, Y. Effects of Uremic Serum and Uremic Toxins on Hepatic Uptake of Digoxin. Ther. Drug Monit. 2008, 30, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, Y.; Iwao, Y.; Mera, K.; Watanabe, H.; Kadowaki, D.; Ishima, Y.; Chuang, V.T.G.; Sato, K.; Otagiri, M.; Maruyama, T. A uremic toxin, 3-carboxy-4-methyl-5-propyl-2-furanpropionate induces cell damage to proximal tubular cells via the generation of a radical intermediate. Biochem. Pharmacol. 2012, 84, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.C.; Perrella, M.A.; Yoshizumi, M.; Hsieh, C.M.; Haber, E.; Schlegel, R.; Lee, M.E. Promotion of vascular smooth muscle cell growth by homocysteine: A link to atherosclerosis. Proc. Natl. Acad. Sci. USA 1994, 91, 6369–6373. [Google Scholar] [CrossRef]
- Harpel, P.C.; Zhang, X.; Borth, W. Homocysteine and Hemostasis: Pathogenetic Mechanisms Predisposing to Thrombosis. Am. Inst. Nutr. 1996, 126, 1285–1289. [Google Scholar] [CrossRef]
- Liang, S.; Liu, S.; Liu, H.; He, X.; Sun, L.; Chen, L.; Wei, M.; Gao, F.; Jiang, H. Homocysteine Aggravates Intestinal Epithelial Barrier Dysfunction in Rats with Experimental Uremia. Kidney Blood Press. Res. 2018, 43, 1516–1528. [Google Scholar] [CrossRef]
- Marcus, J.; Sarnak, M.J.; Menon, V. Homocysteine lowering and cardiovascular disease risk: Lost in translation. Can. J. Cardiol. 2007, 23, 707–710. [Google Scholar] [CrossRef]
- Long, Y.; Nie, J. Homocysteine in Renal Injury. Kidney Dis. 2016, 2, 80–87. [Google Scholar] [CrossRef]
- Wong, J.; Piceno, Y.M.; DeSantis, T.Z.; Pahl, M.; Andersen, G.L.; Vaziri, N.D. Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am. J. Nephrol. 2015, 39, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Han, T.H.; Lee, J.-H.; Cho, M.H.; Wood, T.K.; Lee, J. Environmental Factor Affecting Indole Production in Escherichia coli. Res. Microbiol. 2011, 162, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Young, K.D. Indole production by the tryptophanase TnaA in Escherichia coli is determined by the amount of exogenous tryptophan. Microbiology 2013, 159, 402–410. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, J. Indole as an intercellular signal in microbial communities. FEMS Microbiol. Rev. 2010, 34, 426–444. [Google Scholar] [CrossRef]
- Camilleri, M. Serotonin in the gastrointestinal tract. Curr. Opin. Endocrinol. Diabetes Obes. 2009, 16, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Bammens, B.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. Evidence for impaired assimilation of protein in chronic renal failure. Kidney Int. 2003, 64, 2196–2203. [Google Scholar] [CrossRef]
- Bammens, B.; Evenepoel, P.; Verbeke, K.; Vanrenterghem, Y. Impairment of small intestinal protein assimilation in patients with end-stage renal disease: Extending the malnutrition-inflammation-atherosclerosis concept. Am. J. Clin. Nutr. 2004, 80, 1536–1543. [Google Scholar] [CrossRef]
- Wu, I.W.; Hsu, K.H.; Lee, C.C.; Sun, C.-Y.; Hsu, H.-J.; Tsai, C.-J.; Tzen, C.-Y.; Wang, Y.-C.; Lin, C.-Y.; Wu, M.-S. P-cresyl sulphate and indoxyl sulphate predict progression of chronic kidney disease. Nephrol. Dial. Transplant. 2011, 26, 938–947. [Google Scholar] [CrossRef]
- Lin, C.N.; Wu, I.W.; Huang, Y.F.; Peng, S.Y.; Huang, Y.C.; Ning, H.C. Measuring serum total and free indoxyl sulfate and p-cresyl sulfate in chronic kidney disease using UPLC-MS/MS. J. Food Drug Anal. 2019, 27, 502–509. [Google Scholar] [CrossRef]
- Gryp, T.; De Paepe, K.; Vanholder, R.; Kerckhof, F.-M.; Van Biesen, W.; Van De Wiele, T.; Verbeke, F.; Speeckaert, M.; Joossens, M.; Couttenye, M.M.; et al. Gut microbiota generation of protein-bound uremic toxins and related metabolites is not altered at different stages of chronic kidney disease. Kidney Int. 2020, 97, 1230–1242. [Google Scholar] [CrossRef]
- Poesen, R.; Windey, K.; Neven, E.; Kuypers, D.R.; De Preter, V.; Augustijns, P.; D’Haese, P.; Evenepoel, P.; Verbeke, K.; Meijers, B. The Influence of CKD on Colonic Microbial Metabolism. J. Am. Soc. Nephrol. 2016, 27, 1389–1399. [Google Scholar] [CrossRef] [PubMed]
- Joossens, M.; Faust, K.; Gryp, T.; Nguyen, A.T.L.; Wang, J.; Eloot, S.; Schepers, E.; Dhondt, A.; Pletinck, A.; Vieira-Silva, S.; et al. Gut microbiota dynamics and uraemic toxins: One size does not fit all. Gut 2019, 68, 2257–2260. [Google Scholar] [CrossRef] [PubMed]
- Russell, W.R.; Duncan, S.H.; Scobbie, L.; Duncan, G.; Cantlay, L.; Calder, A.G.; Anderson, S.E.; Flint, H.J. Major phenylpropanoid-derived metabolites in the human gut can arise from microbial fermentation of protein. Mol. Nutr. Food Res. 2013, 57, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Devlin, A.S.; Marcobal, A.; Dodd, D.; Nayfach, S.; Plummer, N.; Meyer, T.; Pollard, K.S.; Sonnenburg, J.L.; Fischbach, M.A. Modulation of a circulating uremic solute via rational genetic manipulation of the gut microbiota. Cell Host Microbe 2016, 20, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, S.; Li, S.; Zhao, L.; Hao, Y.; Qin, J.; Zhang, L.; Zhang, C.; Bian, W.; I Zuo, L.; et al. Aberrant gut microbiota alters host metabolome and impacts renal failure in humans and rodents. Gut 2020, 2020, 1–12. [Google Scholar] [CrossRef]
- Kim, J.E.; Kim, H.E.; Park, J.I.; Cho, H.; Kwak, M.-J.; Kim, B.-Y.; Yang, S.H.; Lee, J.P.; Kim, D.K.; Joo, K.W.; et al. The Association between Gut Microbiota and Uremia of Chronic Kidney Disease. Microorganisms 2020, 8, 907. [Google Scholar] [CrossRef]
- Jazani, N.; Savoj, J.; Lustgarten, M.; Lau, W.; Vaziri, N. Impact of Gut Dysbiosis on Neurohormonal Pathways in Chronic Kidney Disease. Diseases 2019, 7, 21. [Google Scholar] [CrossRef]
- Smith, S.M.; Vale, W.W. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialog. Clin. Neurosci. 2006, 8, 383–395. [Google Scholar]
- Mudd, A.T.; Berding, K.; Wang, M.; Donovan, S.M.; Dilger, R.N. Serum cortisol mediates the relationship between fecal Ruminococcus and brain N-acetylaspartate in the young pig. Gut Microbes 2017, 8, 589–600. [Google Scholar] [CrossRef]
- Sudo, N.; Chida, Y.; Aiba, Y.; Sonoda, J.; Oyama, N.; Yu, X.-N.; Kubo, C.; Koga, Y. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J. Physiol. 2004, 558, 263–275. [Google Scholar] [CrossRef]
- Larraufie, P.; Martin-Gallausiaux, C.; Lapaque, N.; Dore, J.; Gribble, F.M.; Reimann, F.; Blottière, H.M. SCFAs strongly stimulate PYY production in human enteroendocrine cells. Sci. Rep. 2018, 8, 74. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Yun, M.; Oh, Y.J.; Choi, H.J. Mind-altering with the gut: Modulation of the gut-brain axis with probiotics. J. Microbiol. 2018, 56, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Takayama, F.; Taki, K.; Niwa, T. Bifidobacterium in gastro-resistant seamless capsule reduces serum levels of indoxyl sulfate in patients on hemodialysis. Am. J. Kidney Dis. 2003, 41, 142–145. [Google Scholar] [CrossRef] [PubMed]
- Hida, M.; Aiba, Y.; Sawamura, S.; Suzuki, N.; Satoh, T.; Koga, Y. Inhibition of the Accumulation of Uremic Toxins in the Blood and Their Precursors in the Feces after Oral Administration of Lebenin®, a Lactic Acid Bacteria Preparation, to Uremic Patients Undergoing Hemodialysis? Nephron 1996, 74, 349–355. [Google Scholar] [CrossRef]
- Borges, N.A.; Carmo, F.L.; Stockler-Pinto, M.B.; De Brito, J.S.; Dolenga, C.J.; Ferreira, D.D.C.; Nakao, L.S.; Rosado, A.S.; Fouque, D.; Mafra, D. Probiotic Supplementation in Chronic Kidney Disease: A Double-blind, Randomized, Placebo-controlled Trial. J. Ren. Nutr. 2018, 28, 28–36. [Google Scholar] [CrossRef]
- Davani-Davari, D.; Negahdaripour, M.; Karimzadeh, I.; Seifan, M.; Mohkam, M.; Masoumi, S.J.; Berenjian, A.; Ghasemi, Y. Prebiotics: Definition, Types, Sources, Mechanisms, and Clinical Applications. Foods 2019, 8, 92. [Google Scholar] [CrossRef]
- Salmean, Y.A.; Segal, M.S.; Palii, S.P.; Dahl, W.J. Fiber supplementation lowers plasma p-cresol in chronic kidney disease patients. J. Ren. Nutr. 2014, 25, 316–320. [Google Scholar] [CrossRef]
- Sirich, T.L.; Plummer, N.S.; Gardner, C.D.; Hostetter, T.H.; Meyer, T.W. Effect of Increasing Dietary Fiber on Plasma Levels of Colon-Derived Solutes in Hemodialysis Patients. Clin. J. Am. Soc. Nephrol. 2014, 9, 1603–1610. [Google Scholar] [CrossRef]
- Mafra, D.; Borges, N.; Alvarenga, L.; Esgalhado, M.; Cardozo, L.F.; Lindholm, B.; Stenvinkel, P. Dietary Components That May Influence the Disturbed Gut Microbiota in Chronic Kidney Disease. Nutrients 2019, 11, 496. [Google Scholar] [CrossRef]
- Dehghani, H.; Heidari, F.; Mozaffari-Khosravi, H.; Nouri-Majelan, N.; Dehghani, A. Synbiotic Supplementations for Azotemia in Patients with Chronic Kidney Disease: A Randomized Controlled Trial. Iran. J. Kidney Dis. 2016, 10, 351–357. [Google Scholar]
- Nakabayashi, I.; Nakamura, M.; Kawakami, K.; Ohta, T.; Kato, I.; Uchida, K.; Yoshida, M. Effects of synbiotic treatment on serum level of p-cresol in haemodialysis patients: A preliminary study. Nephrol. Dial. Transplant. 2010, 26, 1094–1098. [Google Scholar] [CrossRef]
- Rossi, M.; Johnson, D.W.; Morrison, M.; Pascoe, E.M.; Coombes, J.S.; Forbes, J.M.; Szeto, C.-C.; McWhinney, B.C.; Ungerer, J.; Campbell, K.L. Synbiotics Easing Renal Failure by Improving Gut Microbiology (SYNERGY): A Randomized Trial. Clin. J. Am. Soc. Nephrol. 2016, 11, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Marzocco, S.; Piaz, F.D.; Di Micco, L.; Torraca, S.; Sirico, M.L.; Tartaglia, D.; Autore, G.; Di Iorio, B.R. Very Low Protein Diet Reduces Indoxyl Sulfate Levels in Chronic Kidney Disease. Blood Purif. 2013, 35, 196–201. [Google Scholar] [CrossRef]
- Black, A.P.; Anjos, J.S.; Cardozo, L.; Carmo, F.L.; Dolenga, C.J.; Nakao, L.S.; Ferreira, D.D.C.; Rosado, A.S.; Eduardo, J.C.C.; Mafra, D. Does Low-Protein Diet Influence the Uremic Toxin Serum Levels From the Gut Microbiota in Nondialysis Chronic Kidney Disease Patients? J. Ren. Nutr. 2018, 28, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Mafra, D.; Barros, A.F.; Fouque, D. Dietary protein metabolism by gut microbiota and its consequences for chronic kidney disease patients. Futur. Microbiol. 2013, 8, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Kandouz, S.; Mohamed, A.M.S.; Zheng, Y.; Sandeman, S.; Davenport, A. Reduced protein bound uraemic toxins in vegetarian kidney failure patients treated by haemodiafiltration. Hemodial. Int. 2016, 20, 610–617. [Google Scholar] [CrossRef]
- Montemurno, E.; Cosola, C.; Dalfino, G.; Gesualdo, L.; Daidone, G.; De Angelis, M.; Gobbetti, M. What Would You Like to Eat, Mr CKD Microbiota? A Mediterranean Diet, please! Kidney Blood Press. Res. 2014, 39, 114–123. [Google Scholar] [CrossRef]
- Del Chierico, F.; Vernocchi, P.; Dallapiccola, B.; Putignani, L. Mediterranean Diet and Health: Food Effects on Gut Microbiota and Disease Control. Int. J. Mol. Sci. 2014, 15, 11678–11699. [Google Scholar] [CrossRef]
- Schulman, G.; Agarwal, R.; Acharya, M.; Berl, T.; Blumenthal, S.; Kopyt, N. A Multicenter, Randomized, Double-Blind, Placebo-Controlled, Dose-Ranging Study of AST-120 (Kremezin) in Patients with Moderate to Severe CKD. Am. J. Kidney Dis. 2006, 47, 565–577. [Google Scholar] [CrossRef]
- Yamamoto, S.; Kazama, J.J.; Omori, K.; Matsuo, K.; Takahashi, Y.; Kawamura, K.; Matsuto, T.; Watanabe, H.; Maruyama, T.; Narita, I. Continuous Reduction of Protein-Bound Uraemic Toxins with Improved Oxidative Stress by Using the Oral Charcoal Adsorbent AST-120 in Haemodialysis Patients. Sci. Rep. 2015, 5, 14381. [Google Scholar] [CrossRef]
- Asai, M.; Kumakura, S.; Kikuchi, M. Review of the efficacy of AST-120 (KREMEZIN®) on renal function in chronic kidney disease patients. Ren. Fail. 2019, 41, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Schulman, G.; Berl, T.; Beck, G.J.; Remuzzi, G.; Ritz, E.; Arita, K.; Kato, A.; Shimizu, M. Randomized Placebo-Controlled EPPIC Trials of AST-120 in CKD. J. Am. Soc. Nephrol. 2015, 26, 1732–1746. [Google Scholar] [CrossRef] [PubMed]
- Cha, R.H.; Kang, S.W.; Park, C.W.; Cha, D.R.; Na, K.Y.; Kim, S.G.; Yoon, S.A.; Han, S.Y.; Chang, J.H.; Park, S.K.; et al. A Randomized, Controlled Trial of Oral Intestinal Sorbent AST-120 on Renal Function Deterioration in Patients with Advanced Renal Dysfunction. Clin. J. Am. Soc. Nephrol. 2016, 11, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Lenglet, A.; Fabresse, N.; Taupin, M.; Gomila, C.; Liabeuf, S.; Kamel, S.; Alvarez, J.C.; Drueke, T.B.; Massy, Z.A. Does the Administration of Sevelamer or Nicotinamide Modify Uremic Toxins or Endotoxemia in Chronic Hemodialysis Patients? Drugs 2019, 79, 855–862. [Google Scholar] [CrossRef]
- Guida, B.; Cataldi, M.; Riccio, E.; Grumetto, L.; Pota, A.; Borrelli, S.; Memoli, A.; Barbato, F.; Argentino, G.; Salerno, G.; et al. Plasma p-Cresol Lowering Effect of Sevelamer in Peritoneal Dialysis Patients: Evidence from a Cross-Sectional Observational Study. PLoS ONE 2013, 8, e73558. [Google Scholar] [CrossRef]
- Lin, C.-J.; Pan, C.-F.; Chuang, C.-K.; Liu, H.-L.; Huang, S.-F.; Chen, H.-H.; Wu, C.-J. Effects of Sevelamer Hydrochloride on Uremic Toxins Serum Indoxyl Sulfate and P-Cresyl Sulfate in Hemodialysis Patients. J. Clin. Med. Res. 2017, 9, 765–770. [Google Scholar] [CrossRef]
- Li, F.; Fu, T.; Tong, W.D.; Liu, B.; Li, C.-X.; Gao, Y.; Wu, J.-S.; Wang, X.-F.; Zhang, A.-P. Lubiprostone Is Effective in the Treatment of Chronic Idiopathic Constipation and Irritable Bowel Syndrome. Mayo Clin. Proc. 2016, 91, 456–468. [Google Scholar] [CrossRef]
- Mishima, E.; Fukuda, S.; Shima, H.; Hirayama, A.; Akiyama, Y.; Takeuchi, Y.; Fukuda, N.N.; Suzuki, T.; Suzuki, C.; Yuri, A.; et al. Alteration of the Intestinal Environment by Lubiprostone Is Associated with Amelioration of Adenine-Induced CKD. J. Am. Soc. Nephrol. 2015, 26, 1787–1794. [Google Scholar] [CrossRef]
- Plata, C.; Cruz, C.; Cervantes, L.G.; Ramírez, V. The gut microbiota and its relationship with chronic kidney disease. Int. Urol. Nephrol. 2019, 51, 2209–2226. [Google Scholar] [CrossRef]
- Castillo-Rodríguez, E.; Fernandez-Prado, R.; Esteras, R.; Perez-Gomez, M.V.; Gracia-Iguacel, C.; Fernandez-Fernandez, B.; Kanbay, M.; Tejedor, A.; Lázaro, A.; Ruiz-Ortega, M.; et al. Impact of Altered Intestinal Microbiota on Chronic Kidney Disease Progression. Toxins 2018, 10, 300. [Google Scholar] [CrossRef]
- Yamamoto, S.; Kazama, J.J.; Wakamatsu, T.; Takahashi, Y.; Kaneko, Y.; Goto, S.; Narita, I. Removal of uremic toxins by renal replacement therapies: A review of current progress and future perspectives. Ren. Replace. Ther. 2016, 2, 43. [Google Scholar] [CrossRef]
- Jula, K.I.M.D.; Robert, M.C.M.D.; Asba, T.; Radha, K.V.M.D.; Christopher, M.B.S.; John, W.S.M.D.; Karen, C.; Uptal, D.P.M.D. The Landscape of Clinical Trials in Nephrology: A Systematic Review of ClinicalTrials.gov. Bone 2012, 12, 276–285. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Gut-Derived PBUT Class | Toxin | Derivation | Pathological Mechanisms | Associated Comorbidities |
|---|---|---|---|---|
| AGEs | 3-Deoxyglucosone Fructoselysine Glyoxal Methylglyoxal N(6)-Carboxymethyllysine Pentosidine | Diet | ECM crosslink formation Impaired endothelial progenitor cell function NF- kB/MAPK/JNK signaling RAGE signaling | Arterial stiffness Diabetic nephropathy Endothelial dysfunction Immune system dysregulation |
| Hippurates | Hippuric acid Hydroxyhippuric acid | Diet | Activation of mitochondrial fission Albumin binding Free radical production NF- kB signaling | Altered drug pharmacokinetics Endothelial dysfunction Renal tubule damage |
| Indoles | Indole-3-acetic acid Indoxyl glucuronide Indoxyl sulfate Kynurenine Kynurenic acid Melatonin Quinolinic acid | Microbial metabolism | AhR activation Excessive glutamate release Impaired mitochondrial OXPHOS NF- kB/MAPK signaling NMDA receptor activation Reduced PTH expression | Bone disease Cardiovascular disease Endothelial dysfunction Inflammation Muscle weakness/atrophy Neurotoxicity Oxidative stress |
| Phenols | Hydroquinone p-cresyl glucuronide p-cresyl sulfate Phenol Phenylacetic acid | Microbial metabolism | Apoptosis Chromosomal aberrations Inhibition of iNOS expression NADPH oxidase activation ROS production Stimulates Rho-associated protein kinase | All-cause mortality Cardiovascular disease Inflammation Oxidative stress Renal fibrosis Vascular remodeling |
| Polyamines | Putrescine Spermidine Spermine | Microbial metabolism/Diet | Inhibition of erythropoietin | Anemia |
| Other | CMPF Homocysteine | Diet | Albumin binding Altered hepatic metabolism CMPF radical adducts Competitive reabsorption by OAT Degradation of gut epithelial TJ VSMC proliferation | Altered drug pharmacokinetics Atherosclerosis Increased intestinal permeability Neurological abnormalities Renal tubule damage |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Graboski, A.L.; Redinbo, M.R. Gut-Derived Protein-Bound Uremic Toxins. Toxins 2020, 12, 590. https://doi.org/10.3390/toxins12090590
Graboski AL, Redinbo MR. Gut-Derived Protein-Bound Uremic Toxins. Toxins. 2020; 12(9):590. https://doi.org/10.3390/toxins12090590
Chicago/Turabian StyleGraboski, Amanda L., and Matthew R. Redinbo. 2020. "Gut-Derived Protein-Bound Uremic Toxins" Toxins 12, no. 9: 590. https://doi.org/10.3390/toxins12090590
APA StyleGraboski, A. L., & Redinbo, M. R. (2020). Gut-Derived Protein-Bound Uremic Toxins. Toxins, 12(9), 590. https://doi.org/10.3390/toxins12090590