Interaction of Macrophages and Cholesterol-Dependent Cytolysins: The Impact on Immune Response and Cellular Survival


Abstract
1. Introduction
2. Cholesterol-Dependent Cytolysins
2.1. CDC-Producing Bacteria and Tissues They Invade
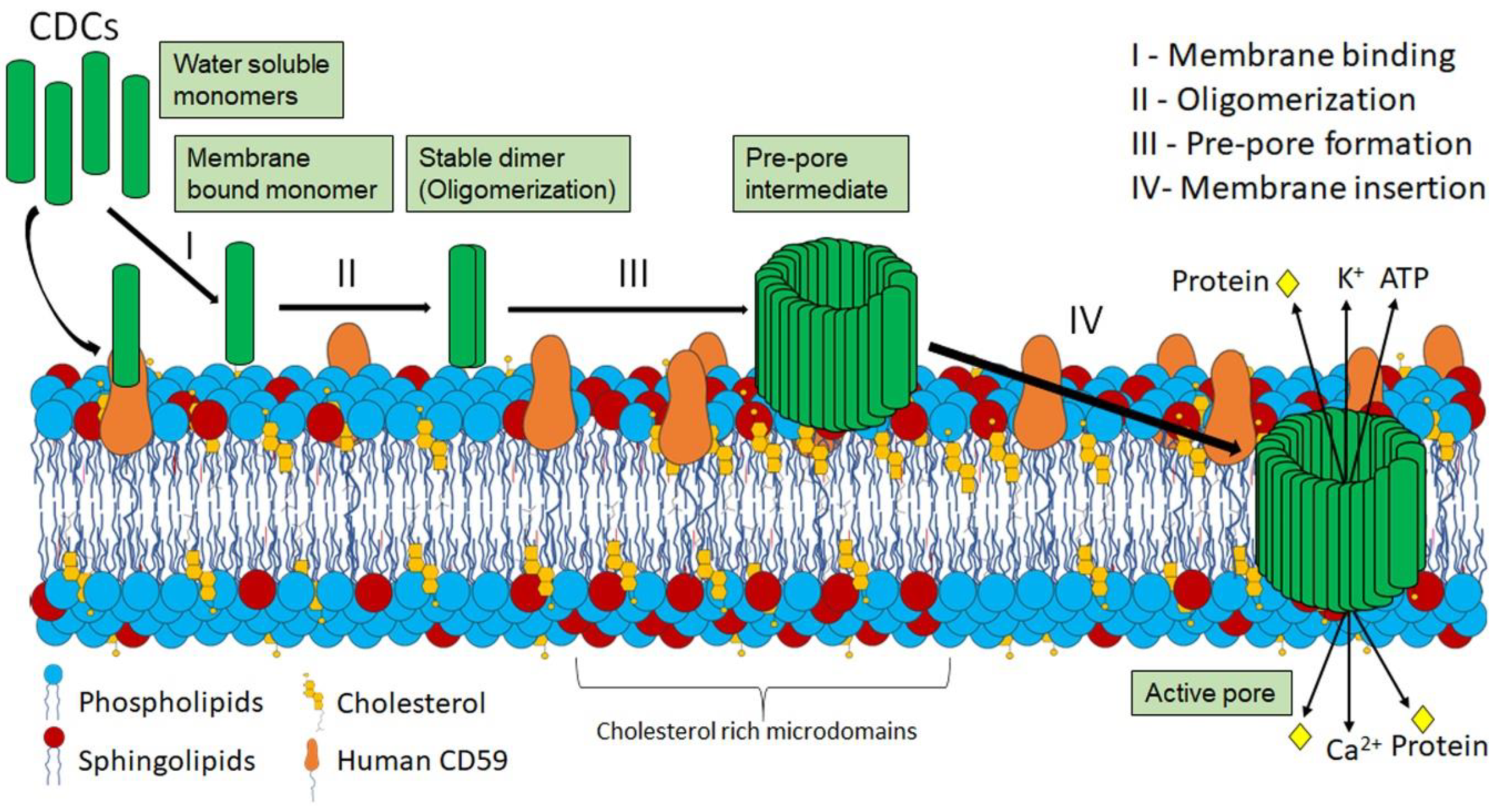
2.2. CDC Structure and Pore-Formation
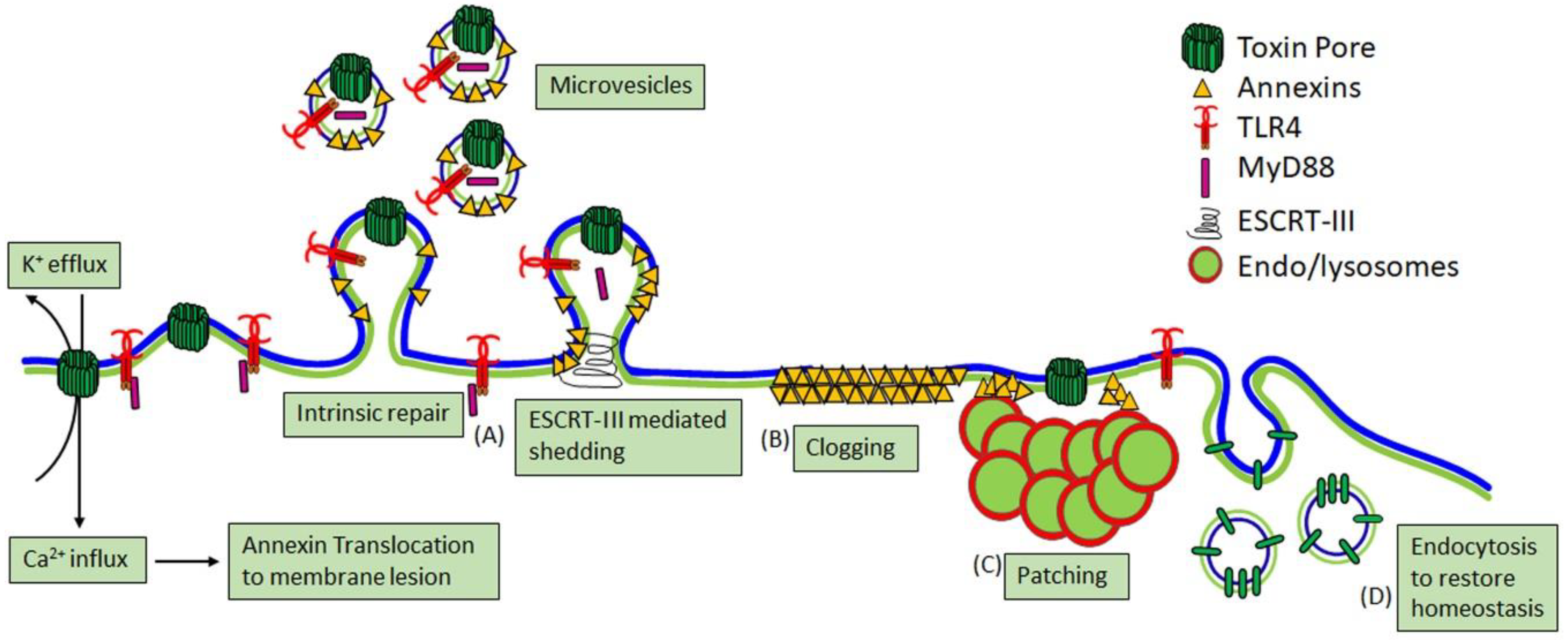
2.3. Cellular Consequences of CDC Pore Formation
3. CDC Interactions with Macrophages
3.1. Cytokine Production in Response to CDCs
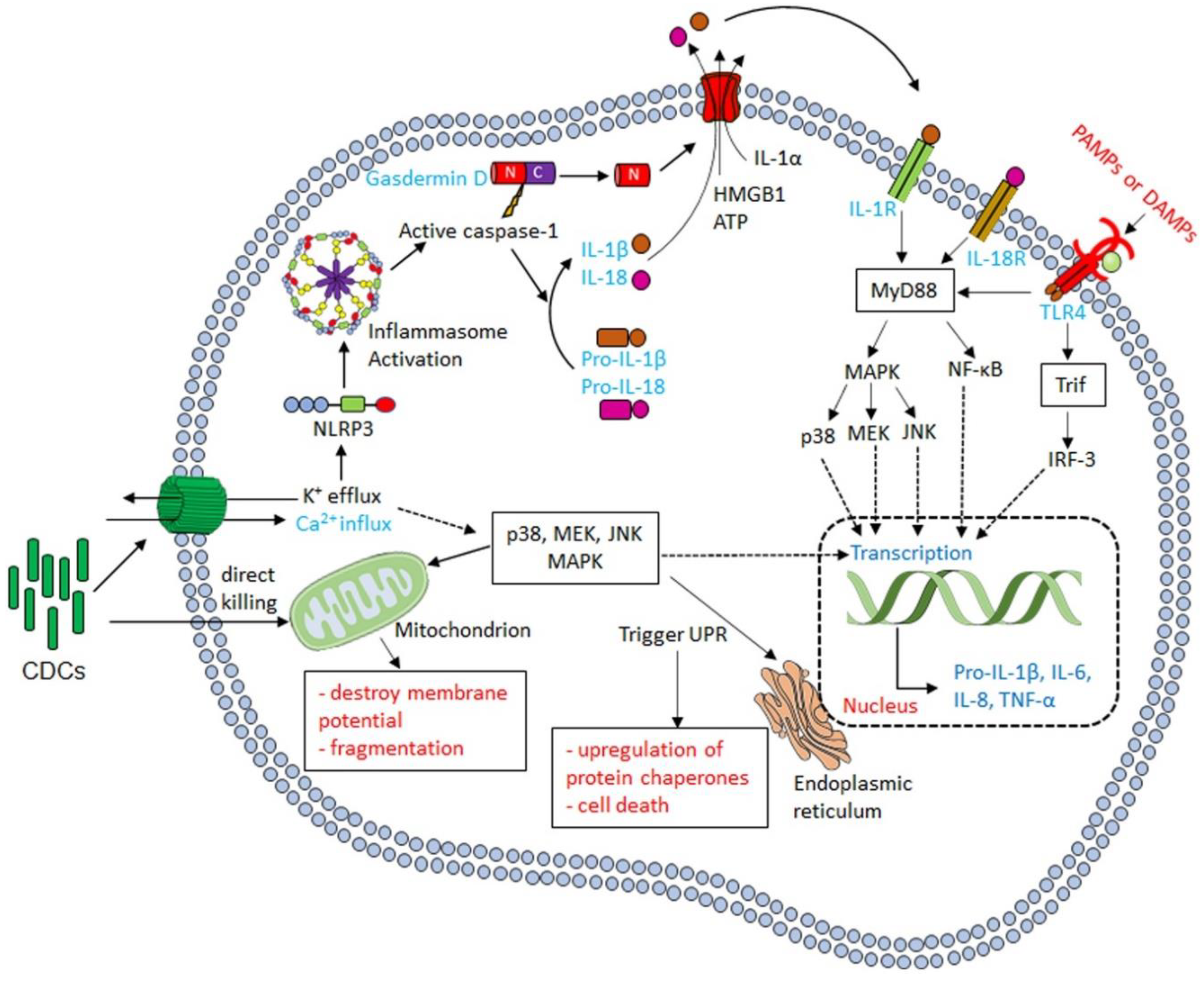
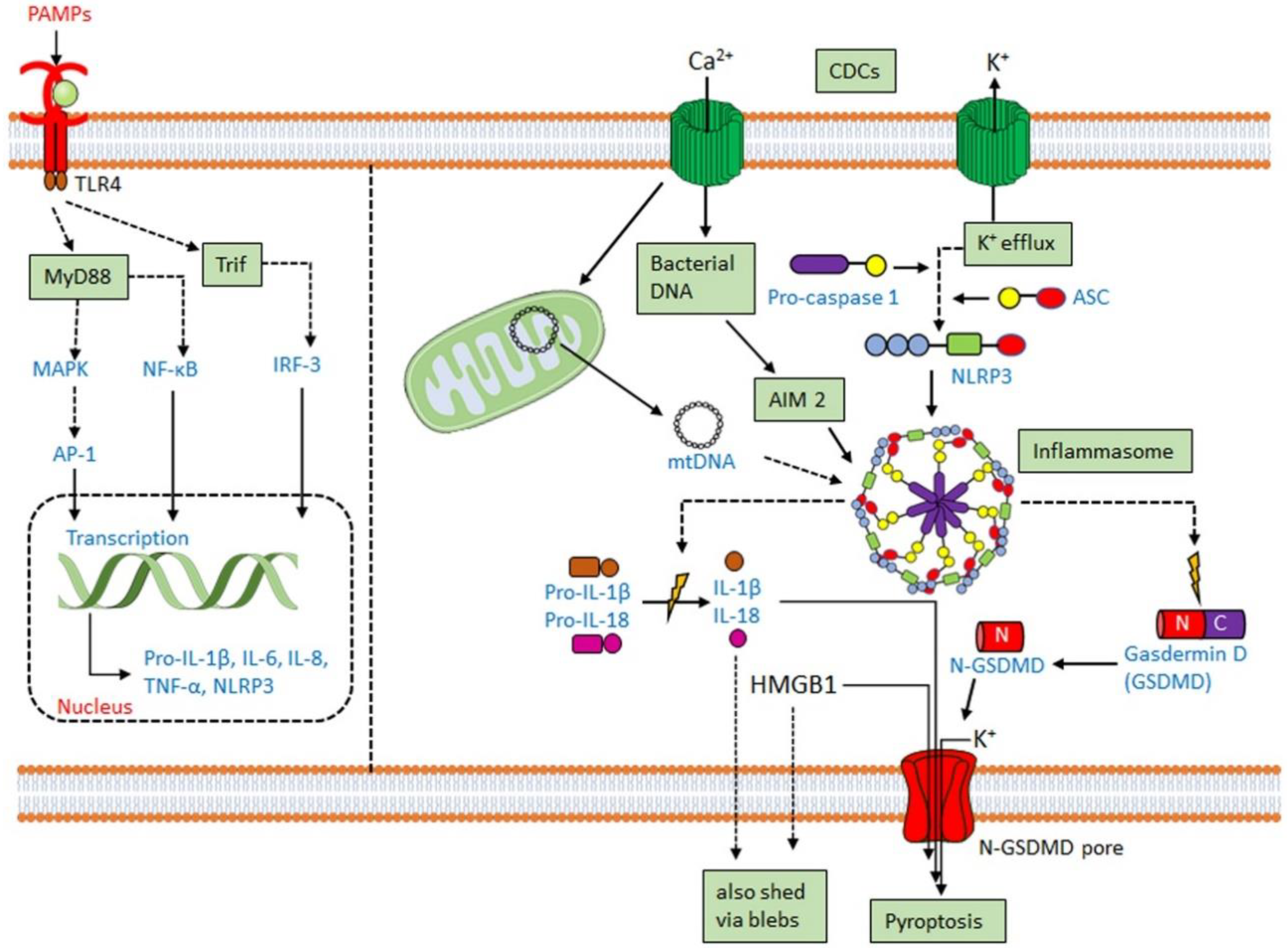
3.2. CDCs Activate the Inflammasome
3.3. CDCs Damage Phagosomes and Permit Phagolysosomal Escape
3.4. CDC-Mediated Innate Immune Evasion
3.5. CDCs as an Adaptive Immune Target
4. Individual CDCs
4.1. Streptolysin O (SLO)
4.2. Pneumolysin (PLY)
4.3. Perfringolysin O (PFO)
4.4. Listeriolysin O (LLO)
4.5. Anthrolysin O (ALO), Tetanolysin O (TLO) and Suilysin (SLY)
4.6. Intermedilysin (ILY), and Vaginolysin (VLY)
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Savinov, S.N.; Heuck, A.P. Interaction of Cholesterol with Perfringolysin O: What Have We Learned from Functional Analysis? Toxins 2017, 9, 381. [Google Scholar] [CrossRef] [PubMed]
- Tweten, R.K. Cholesterol-dependent cytolysins, a family of versatile pore-forming toxins. Infect. Immun. 2005, 73, 6199–6209. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, M.W. Pathogenesis of group A streptococcal infections. Clin. Microbiol. Rev. 2000, 13, 470–511. [Google Scholar] [CrossRef] [PubMed]
- Bolz, D.D.; Li, Z.; McIndoo, E.R.; Tweten, R.K.; Bryant, A.E.; Stevens, D.L. Cardiac myocyte dysfunction induced by streptolysin O is membrane pore and calcium dependent. Shock 2015, 43, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Limbago, B.; Penumalli, V.; Weinrick, B.; Scott, J.R. Role of streptolysin O in a mouse model of invasive group A streptococcal disease. Infect. Immun. 2000, 68, 6384–6390. [Google Scholar] [CrossRef]
- Shiseki, M.; Miwa, K.; Nemoto, Y.; Kato, H.; Suzuki, J.; Sekiya, K.; Murai, T.; Kikuchi, T.; Yamashita, N.; Totsuka, K.; et al. Comparison of pathogenic factors expressed by group A Streptococci isolated from patients with streptococcal toxic shock syndrome and scarlet fever. Microb. Pathog. 1999, 27, 243–252. [Google Scholar] [CrossRef]
- Thigpen, M.C.; Whitney, C.G.; Messonnier, N.E.; Zell, E.R.; Lynfield, R.; Hadler, J.L.; Harrison, L.H.; Farley, M.M.; Reingold, A.; Bennett, N.M.; et al. Bacterial meningitis in the United States, 1998–2007. N. Engl. J. Med. 2011, 364, 2016–2025. [Google Scholar] [CrossRef]
- Prina, E.; Ranzani, O.T.; Torres, A. Community-acquired pneumonia. Lancet 2015, 386, 1097–1108. [Google Scholar] [CrossRef]
- Toltzis, P.; Jacobs, M.R. The epidemiology of childhood pneumococcal disease in the United States in the era of conjugate vaccine use. Infect. Dis. Clin. N. Am. 2005, 19, 629–645. [Google Scholar] [CrossRef]
- Berry, A.M.; Yother, J.; Briles, D.E.; Hansman, D.; Paton, J.C. Reduced virulence of a defined pneumolysin-negative mutant of Streptococcus pneumoniae. Infect. Immun. 1989, 57, 2037–2042. [Google Scholar] [CrossRef]
- Nagamune, H.; Ohnishi, C.; Katsuura, A.; Fushitani, K.; Whiley, R.A.; Tsuji, A.; Matsuda, Y. Intermedilysin, a novel cytotoxin specific for human cells secreted by Streptococcus intermedius UNS46 isolated from a human liver abscess. Infect. Immun. 1996, 64, 3093–3100. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, A.A.; Loeffen, P.L.; van den Berg, A.J.; Storm, P.K. Identification, purification, and characterization of a thiol-activated hemolysin (suilysin) of Streptococcus suis. Infect. Immun. 1994, 62, 1742–1748. [Google Scholar] [CrossRef] [PubMed]
- King, S.J.; Heath, P.J.; Luque, I.; Tarradas, C.; Dowson, C.G.; Whatmore, A.M. Distribution and genetic diversity of suilysin in Streptococcus suis isolated from different diseases of pigs and characterization of the genetic basis of suilysin absence. Infect. Immun. 2001, 69, 7572–7582. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy, C.; Gaillard, J.L.; Alouf, J.E.; Berche, P. Purification, characterization, and toxicity of the sulfhydryl-activated hemolysin listeriolysin O from Listeria monocytogenes. Infect. Immun. 1987, 55, 1641–1646. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Boland, J.A.; Kuhn, M.; Berche, P.; Chakraborty, T.; Dominguez-Bernal, G.; Goebel, W.; Gonzalez-Zorn, B.; Wehland, J.; Kreft, J. Listeria pathogenesis and molecular virulence determinants. Clin. Microbiol. Rev. 2001, 14, 584–640. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Ellemor, D.M.; Boyd, R.L.; Emmins, J.J.; Rood, J.I. Synergistic effects of alpha-toxin and perfringolysin O in Clostridium perfringens-mediated gas gangrene. Infect. Immun. 2001, 69, 7904–7910. [Google Scholar] [CrossRef]
- Stevens, D.L.; Bryant, A.E. The role of clostridial toxins in the pathogenesis of gas gangrene. Clin. Infect. Dis. 2002, 35, S93–S100. [Google Scholar] [CrossRef]
- Hatheway, C.L. Toxigenic clostridia. Clin. Microbiol. Rev. 1990, 3, 66–98. [Google Scholar] [CrossRef]
- Shannon, J.G.; Ross, C.L.; Koehler, T.M.; Rest, R.F. Characterization of anthrolysin O, the Bacillus anthracis cholesterol-dependent cytolysin. Infect. Immun. 2003, 71, 3183–3189. [Google Scholar] [CrossRef]
- Knupp de Souza, D.M.; Diniz, C.G.; Filho, D.S.; Andrade de Oliveira, L.M.; Coelho, D.M.; Talha, L.S.; Nascimento, T.C.; Ferreira-Machado, A.B.; Silva, V.L. Antimicrobial susceptibility and vaginolysin in Gardnerella vaginalis from healthy and bacterial vaginosis diagnosed women. J. Infect. Dev. Ctries. 2016, 10, 913–919. [Google Scholar] [CrossRef]
- Cauci, S.; Monte, R.; Ropele, M.; Missero, C.; Not, T.; Quadrifoglio, F.; Menestrina, G. Pore-forming and haemolytic properties of the Gardnerella vaginalis cytolysin. Mol. Microbiol. 1993, 9, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Gelber, S.E.; Aguilar, J.L.; Lewis, K.L.; Ratner, A.J. Functional and phylogenetic characterization of Vaginolysin, the human-specific cytolysin from Gardnerella vaginalis. J. Bacteriol. 2008, 190, 3896–3903. [Google Scholar] [CrossRef] [PubMed]
- Los, F.C.; Randis, T.M.; Aroian, R.V.; Ratner, A.J. Role of pore-forming toxins in bacterial infectious diseases. Microbiol. Mol. Biol. Rev. 2013, 77, 173–207. [Google Scholar] [CrossRef]
- Heuck, A.P.; Moe, P.C.; Johnson, B.B. The cholesterol-dependent cytolysin family of gram-positive bacterial toxins. Subcell. Biochem. 2010, 51, 551–577. [Google Scholar] [PubMed]
- Christie, M.P.; Johnstone, B.A.; Tweten, R.K.; Parker, M.W.; Morton, C.J. Cholesterol-dependent cytolysins: From water-soluble state to membrane pore. Biophys. Rev. 2018, 10, 1337–1348. [Google Scholar] [CrossRef]
- Farrand, A.J.; LaChapelle, S.; Hotze, E.M.; Johnson, A.E.; Tweten, R.K. Only two amino acids are essential for cytolytic toxin recognition of cholesterol at the membrane surface. Proc. Natl. Acad. Sci. USA 2010, 107, 4341–4346. [Google Scholar] [CrossRef]
- Soltani, C.E.; Hotze, E.M.; Johnson, A.E.; Tweten, R.K. Structural elements of the cholesterol-dependent cytolysins that are responsible for their cholesterol-sensitive membrane interactions. Proc. Natl. Acad. Sci. USA 2007, 104, 20226–20231. [Google Scholar] [CrossRef]
- Giddings, K.S.; Zhao, J.; Sims, P.J.; Tweten, R.K. Human CD59 is a receptor for the cholesterol-dependent cytolysin intermedilysin. Nat. Struct. Mol. Biol. 2004, 11, 1173–1178. [Google Scholar] [CrossRef]
- Shewell, L.K.; Day, C.J.; Jen, F.E.; Haselhorst, T.; Atack, J.M.; Reijneveld, J.F.; Everest-Dass, A.; James, D.B.A.; Boguslawski, K.M.; Brouwer, S.; et al. All major cholesterol-dependent cytolysins use glycans as cellular receptors. Sci. Adv. 2020, 6, eaaz4926. [Google Scholar] [CrossRef]
- Garcia-Suarez Mdel, M.; Florez, N.; Astudillo, A.; Vazquez, F.; Villaverde, R.; Fabrizio, K.; Pirofski, L.A.; Mendez, F.J. The role of pneumolysin in mediating lung damage in a lethal pneumococcal pneumonia murine model. Respir. Res. 2007, 8, 3. [Google Scholar] [CrossRef][Green Version]
- Ato, M.; Ikebe, T.; Kawabata, H.; Takemori, T.; Watanabe, H. Incompetence of neutrophils to invasive group A streptococcus is attributed to induction of plural virulence factors by dysfunction of a regulator. PLoS ONE 2008, 3, e3455. [Google Scholar] [CrossRef] [PubMed]
- Flexner, S.; Noguchi, H. The Influence of Colloids upon the Diffusion of Haemolysins. J. Exp. Med. 1906, 8, 547–563. [Google Scholar] [CrossRef]
- McNeil, P.L.; Terasaki, M. Coping with the inevitable: How cells repair a torn surface membrane. Nat. Cell Biol. 2001, 3, E124–E129. [Google Scholar] [CrossRef] [PubMed]
- Leal, J.K.F.; Adjobo-Hermans, M.J.W.; Bosman, G. Red Blood Cell Homeostasis: Mechanisms and Effects of Microvesicle Generation in Health and Disease. Front. Physiol. 2018, 9, 703. [Google Scholar] [CrossRef] [PubMed]
- Keyel, P.A.; Loultcheva, L.; Roth, R.; Salter, R.D.; Watkins, S.C.; Yokoyama, W.M.; Heuser, J.E. Streptolysin O clearance through sequestration into blebs that bud passively from the plasma membrane. J. Cell Sci. 2011, 124, 2414–2423. [Google Scholar] [CrossRef]
- Romero, M.; Keyel, M.; Shi, G.; Bhattacharjee, P.; Roth, R.; Heuser, J.E.; Keyel, P.A. Intrinsic repair protects cells from pore-forming toxins by microvesicle shedding. Cell Death Differ. 2017, 24, 798–808. [Google Scholar] [CrossRef]
- Keyel, P.A.; Roth, R.; Yokoyama, W.M.; Heuser, J.E.; Salter, R.D. Reduction of streptolysin O (SLO) pore-forming activity enhances inflammasome activation. Toxins 2013, 5, 1105–1118. [Google Scholar] [CrossRef]
- Tanigawa, T.; Suzuki, J.; Ueta, T.; Katsumoto, T.; Tanaka, Y. Different sensitivity to streptolysin-O of cells in macrophage lineage. Microbiol. Immunol. 1996, 40, 81–84. [Google Scholar] [CrossRef]
- Mosser, E.M.; Rest, R.F. The Bacillus anthracis cholesterol-dependent cytolysin, Anthrolysin O, kills human neutrophils, monocytes and macrophages. BMC Microbiol. 2006, 6, 56. [Google Scholar] [CrossRef]
- Hirst, R.A.; Yesilkaya, H.; Clitheroe, E.; Rutman, A.; Dufty, N.; Mitchell, T.J.; O’Callaghan, C.; Andrew, P.W. Sensitivities of human monocytes and epithelial cells to pneumolysin are different. Infect. Immun. 2002, 70, 1017–1022. [Google Scholar] [CrossRef][Green Version]
- Larpin, Y.; Besancon, H.; Iacovache, M.I.; Babiychuk, V.S.; Babiychuk, E.B.; Zuber, B.; Draeger, A.; Koffel, R. Bacterial pore-forming toxin pneumolysin: Cell membrane structure and microvesicle shedding capacity determines differential survival of cell types. FASEB J. 2020, 34, 1665–1678. [Google Scholar] [CrossRef] [PubMed]
- Domon, H.; Oda, M.; Maekawa, T.; Nagai, K.; Takeda, W.; Terao, Y. Streptococcus pneumoniae disrupts pulmonary immune defence via elastase release following pneumolysin-dependent neutrophil lysis. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Babiychuk, E.B.; Draeger, A. Defying death: Cellular survival strategies following plasmalemmal injury by bacterial toxins. Semin. Cell Dev. Biol. 2015, 45, 39–47. [Google Scholar] [CrossRef]
- Brito, C.; Cabanes, D.; Sarmento Mesquita, F.; Sousa, S. Mechanisms protecting host cells against bacterial pore-forming toxins. Cell. Mol. Life Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.T.; McNeil, P.L. Membrane Repair: Mechanisms and Pathophysiology. Physiol. Rev. 2015, 95, 1205–1240. [Google Scholar] [CrossRef]
- Idone, V.; Tam, C.; Goss, J.W.; Toomre, D.; Pypaert, M.; Andrews, N.W. Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J. Cell Biol. 2008, 180, 905–914. [Google Scholar] [CrossRef]
- Wolfmeier, H.; Schoenauer, R.; Atanassoff, A.P.; Neill, D.R.; Kadioglu, A.; Draeger, A.; Babiychuk, E.B. Ca2+-dependent repair of pneumolysin pores: A new paradigm for host cellular defense against bacterial pore-forming toxins. Biochim. Biophys. Acta 2015, 1853, 2045–2054. [Google Scholar] [CrossRef]
- Babiychuk, E.B.; Monastyrskaya, K.; Draeger, A. Fluorescent annexin A1 reveals dynamics of ceramide platforms in living cells. Traffic 2008, 9, 1757–1775. [Google Scholar] [CrossRef]
- Roostalu, U.; Strahle, U. In Vivo imaging of molecular interactions at damaged sarcolemma. Dev. Cell 2012, 22, 515–529. [Google Scholar] [CrossRef]
- Bouter, A.; Gounou, C.; Berat, R.; Tan, S.; Gallois, B.; Granier, T.; d’Estaintot, B.L.; Poschl, E.; Brachvogel, B.; Brisson, A.R. Annexin-A5 assembled into two-dimensional arrays promotes cell membrane repair. Nat. Commun. 2011, 2, 270. [Google Scholar] [CrossRef]
- Demonbreun, A.R.; Quattrocelli, M.; Barefield, D.Y.; Allen, M.V.; Swanson, K.E.; McNally, E.M. An actin-dependent annexin complex mediates plasma membrane repair in muscle. J. Cell Biol. 2016, 213, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Babiychuk, E.B.; Monastyrskaya, K.; Potez, S.; Draeger, A. Blebbing confers resistance against cell lysis. Cell Death Differ. 2011, 18, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, A.J.; Maiuri, P.; Lafaurie-Janvore, J.; Divoux, S.; Piel, M.; Perez, F. ESCRT machinery is required for plasma membrane repair. Science 2014, 343, 1247136. [Google Scholar] [CrossRef] [PubMed]
- Wolfmeier, H.; Radecke, J.; Schoenauer, R.; Koeffel, R.; Babiychuk, V.S.; Drucker, P.; Hathaway, L.J.; Mitchell, T.J.; Zuber, B.; Draeger, A.; et al. Active release of pneumolysin prepores and pores by mammalian cells undergoing a Streptococcus pneumoniae attack. Biochim. Biophys. Acta 2016, 1860, 2498–2509. [Google Scholar] [CrossRef]
- Ray, S.; Thapa, R.; Keyel, P.A. Multiple Parameters Beyond Lipid Binding Affinity Drive Cytotoxicity of Cholesterol-Dependent Cytolysins. Toxins 2018, 11, 1. [Google Scholar] [CrossRef]
- Babiychuk, E.B.; Monastyrskaya, K.; Potez, S.; Draeger, A. Intracellular Ca(2+) operates a switch between repair and lysis of streptolysin O-perforated cells. Cell Death Differ. 2009, 16, 1126–1134. [Google Scholar] [CrossRef]
- Xie, M.; Low, M.G. Streptolysin-O induces release of glycosylphosphatidylinositol-anchored alkaline phosphatase from ROS cells by vesiculation independently of phospholipase action. Biochem. J. 1995, 305 (Pt 2), 529–537. [Google Scholar] [CrossRef]
- Schoenauer, R.; Atanassoff, A.P.; Wolfmeier, H.; Pelegrin, P.; Babiychuk, E.B.; Draeger, A. P2X7 receptors mediate resistance to toxin-induced cell lysis. Biochim. Biophys. Acta 2014, 1843, 915–922. [Google Scholar] [CrossRef][Green Version]
- Sun, C.; Heid, M.E.; Keyel, P.A.; Salter, R.D. The second transmembrane domain of P2X7 contributes to dilated pore formation. PLoS ONE 2013, 8, e61886. [Google Scholar] [CrossRef]
- Browne, L.E.; Compan, V.; Bragg, L.; North, R.A. P2X7 Receptor Channels Allow Direct Permeation of Nanometer-Sized Dyes. J. Neurosci. 2013, 33, 3557–3566. [Google Scholar] [CrossRef]
- Pelegrin, P.; Surprenant, A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 2006, 25, 5071–5082. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.D.; Kao, C.Y.; Liu, B.Y.; Huang, S.W.; Kuo, C.J.; Ruan, J.W.; Lin, Y.H.; Huang, C.R.; Chen, Y.H.; Wang, H.D.; et al. HLH-30/TFEB-mediated autophagy functions in a cell-autonomous manner for epithelium intrinsic cellular defense against bacterial pore-forming toxin in C. elegans. Autophagy 2017, 13, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Morse, N.; Robbins, J.R.; Rae, C.S.; Mochegova, S.N.; Swanson, M.S.; Zhao, Z.; Virgin, H.W.; Portnoy, D. Listeriolysin O is necessary and sufficient to induce autophagy during Listeria monocytogenes infection. PLoS ONE 2010, 5, e8610. [Google Scholar] [CrossRef] [PubMed]
- Birmingham, C.L.; Canadien, V.; Gouin, E.; Troy, E.B.; Yoshimori, T.; Cossart, P.; Higgins, D.E.; Brumell, J.H. Listeria monocytogenes evades killing by autophagy during colonization of host cells. Autophagy 2007, 3, 442–451. [Google Scholar] [CrossRef]
- Kloft, N.; Neukirch, C.; Bobkiewicz, W.; Veerachato, G.; Busch, T.; von Hoven, G.; Boller, K.; Husmann, M. Pro-autophagic signal induction by bacterial pore-forming toxins. Med. Microbiol. Immunol. 2010, 199, 299–309. [Google Scholar] [CrossRef]
- Carrero, J.A.; Calderon, B.; Unanue, E.R. Listeriolysin O from Listeria monocytogenes is a lymphocyte apoptogenic molecule. J. Immunol. 2004, 172, 4866–4874. [Google Scholar] [CrossRef]
- Braun, J.S.; Sublett, J.E.; Freyer, D.; Mitchell, T.J.; Cleveland, J.L.; Tuomanen, E.I.; Weber, J.R. Pneumococcal pneumolysin and H2O2 mediate brain cell apoptosis during meningitis. J. Clin. Investig. 2002, 109, 19–27. [Google Scholar] [CrossRef]
- Rayner, C.F.; Jackson, A.D.; Rutman, A.; Dewar, A.; Mitchell, T.J.; Andrew, P.W.; Cole, P.J.; Wilson, R. Interaction of pneumolysin-sufficient and -deficient isogenic variants of Streptococcus pneumoniae with human respiratory mucosa. Infect. Immun. 1995, 63, 442–447. [Google Scholar] [CrossRef]
- Timmer, A.M.; Timmer, J.C.; Pence, M.A.; Hsu, L.C.; Ghochani, M.; Frey, T.G.; Karin, M.; Salvesen, G.S.; Nizet, V. Streptolysin O promotes group A Streptococcus immune evasion by accelerated macrophage apoptosis. J. Biol. Chem. 2009, 284, 862–871. [Google Scholar] [CrossRef]
- Braun, J.S.; Hoffmann, O.; Schickhaus, M.; Freyer, D.; Dagand, E.; Bermpohl, D.; Mitchell, T.J.; Bechmann, I.; Weber, J.R. Pneumolysin causes neuronal cell death through mitochondrial damage. Infect. Immun. 2007, 75, 4245–4254. [Google Scholar] [CrossRef]
- Goldmann, O.; Sastalla, I.; Wos-Oxley, M.; Rohde, M.; Medina, E. Streptococcus pyogenes induces oncosis in macrophages through the activation of an inflammatory programmed cell death pathway. Cell. Microbiol. 2009, 11, 138–155. [Google Scholar] [CrossRef] [PubMed]
- Stavru, F.; Bouillaud, F.; Sartori, A.; Ricquier, D.; Cossart, P. Listeria monocytogenes transiently alters mitochondrial dynamics during infection. Proc. Natl. Acad. Sci. USA 2011, 108, 3612–3617. [Google Scholar] [CrossRef] [PubMed]
- Ratner, A.J.; Hippe, K.R.; Aguilar, J.L.; Bender, M.H.; Nelson, A.L.; Weiser, J.N. Epithelial cells are sensitive detectors of bacterial pore-forming toxins. J. Biol. Chem. 2006, 281, 12994–12998. [Google Scholar] [CrossRef] [PubMed]
- Husmann, M.; Dersch, K.; Bobkiewicz, W.; Beckmann, E.; Veerachato, G.; Bhakdi, S. Differential role of p38 mitogen activated protein kinase for cellular recovery from attack by pore-forming S. aureus alpha-toxin or streptolysin O. Biochem. Biophys. Res. Commun. 2006, 344, 1128–1134. [Google Scholar] [CrossRef] [PubMed]
- Kloft, N.; Busch, T.; Neukirch, C.; Weis, S.; Boukhallouk, F.; Bobkiewicz, W.; Cibis, I.; Bhakdi, S.; Husmann, M. Pore-forming toxins activate MAPK p38 by causing loss of cellular potassium. Biochem. Biophys. Res. Commun. 2009. [Google Scholar] [CrossRef]
- Stringaris, A.K.; Geisenhainer, J.; Bergmann, F.; Balshusemann, C.; Lee, U.; Zysk, G.; Mitchell, T.J.; Keller, B.U.; Kuhnt, U.; Gerber, J.; et al. Neurotoxicity of pneumolysin, a major pneumococcal virulence factor, involves calcium influx and depends on activation of p38 mitogen-activated protein kinase. Neurobiol. Dis. 2002, 11, 355–368. [Google Scholar] [CrossRef]
- Aguilar, J.L.; Kulkarni, R.; Randis, T.M.; Soman, S.; Kikuchi, A.; Yin, Y.; Ratner, A.J. Phosphatase-dependent regulation of epithelial mitogen-activated protein kinase responses to toxin-induced membrane pores. PLoS ONE 2009, 4, e8076. [Google Scholar] [CrossRef]
- Gonzalez, M.R.; Bischofberger, M.; Freche, B.; Ho, S.; Parton, R.G.; van der Goot, F.G. Pore-forming toxins induce multiple cellular responses promoting survival. Cell. Microbiol. 2011, 13, 1026–1043. [Google Scholar] [CrossRef]
- Cabezas, S.; Ho, S.; Ros, U.; Lanio, M.E.; Alvarez, C.; van der Goot, F.G. Damage of eukaryotic cells by the pore-forming toxin sticholysin II: Consequences of the potassium efflux. Biochim. Biophys. Acta 2017, 1859, 982–992. [Google Scholar] [CrossRef]
- Popova, T.G.; Millis, B.; Bradburne, C.; Nazarenko, S.; Bailey, C.; Chandhoke, V.; Popov, S.G. Acceleration of epithelial cell syndecan-1 shedding by anthrax hemolytic virulence factors. BMC Microbiol. 2006, 6, 8. [Google Scholar] [CrossRef]
- Rampersaud, R.; Planet, P.J.; Randis, T.M.; Kulkarni, R.; Aguilar, J.L.; Lehrer, R.I.; Ratner, A.J. Inerolysin, a cholesterol-dependent cytolysin produced by Lactobacillus iners. J. Bacteriol. 2011, 193, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Stirling, B.; Derry, J.; Koga, T.; Jono, H.; Woo, C.H.; Xu, H.; Bourne, P.; Ha, U.H.; Ishinaga, H.; et al. Tumor suppressor CYLD regulates acute lung injury in lethal Streptococcus pneumoniae infections. Immunity 2007, 27, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; LaRose, M.I.; Hergott, C.B.; Leng, L.; Bucala, R.; Weiser, J.N. Macrophage migration inhibitory factor promotes clearance of pneumococcal colonization. J. Immunol. 2014, 193, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Lemon, J.K.; Weiser, J.N. Degradation products of the extracellular pathogen Streptococcus pneumoniae access the cytosol via its pore-forming toxin. mBio 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Ha, U.H.; Lim, J.H.; Kim, H.J.; Wu, W.; Jin, S.; Xu, H.; Li, J.D. MKP1 regulates the induction of MUC5AC mucin by Streptococcus pneumoniae pneumolysin by inhibiting the PAK4-JNK signaling pathway. J. Biol. Chem. 2008, 283, 30624–30631. [Google Scholar] [CrossRef] [PubMed]
- Weiglein, I.; Goebel, W.; Troppmair, J.; Rapp, U.R.; Demuth, A.; Kuhn, M. Listeria monocytogenes infection of HeLa cells results in listeriolysin O-mediated transient activation of the Raf-MEK-MAP kinase pathway. FEMS Microbiol. Lett. 1997, 148, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.; Sutherland, C.L.; Gold, M.R.; Finlay, B.B. Listeria monocytogenes invasion of epithelial cells requires the MEK-1/ERK-2 mitogen-activated protein kinase pathway. Infect. Immun. 1998, 66, 1106–1112. [Google Scholar] [CrossRef]
- Grootjans, J.; Kaser, A.; Kaufman, R.J.; Blumberg, R.S. The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 2016, 16, 469–484. [Google Scholar] [CrossRef]
- Alhamdi, Y.; Neill, D.R.; Abrams, S.T.; Malak, H.A.; Yahya, R.; Barrett-Jolley, R.; Wang, G.; Kadioglu, A.; Toh, C.H. Circulating Pneumolysin Is a Potent Inducer of Cardiac Injury during Pneumococcal Infection. PLoS Pathog. 2015, 11, e1004836. [Google Scholar] [CrossRef]
- Akazawa, Y.; Isomoto, H.; Matsushima, K.; Kanda, T.; Minami, H.; Yamaghchi, N.; Taura, N.; Shiozawa, K.; Ohnita, K.; Takeshima, F.; et al. Endoplasmic reticulum stress contributes to Helicobacter pylori VacA-induced apoptosis. PLoS ONE 2013, 8, e82322. [Google Scholar] [CrossRef]
- Baruch, M.; Belotserkovsky, I.; Hertzog, B.B.; Ravins, M.; Dov, E.; McIver, K.S.; Le Breton, Y.S.; Zhou, Y.; Cheng, C.Y.; Hanski, E. An extracellular bacterial pathogen modulates host metabolism to regulate its own sensing and proliferation. Cell 2014, 156, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Pillich, H.; Loose, M.; Zimmer, K.P.; Chakraborty, T. Activation of the unfolded protein response by Listeria monocytogenes. Cell. Microbiol. 2012, 14, 949–964. [Google Scholar] [CrossRef] [PubMed]
- Mesquita, F.S.; Brito, C.; Mazon Moya, M.J.; Pinheiro, J.C.; Mostowy, S.; Cabanes, D.; Sousa, S. Endoplasmic reticulum chaperone Gp96 controls actomyosin dynamics and protects against pore-forming toxins. EMBO Rep. 2017, 18, 303–318. [Google Scholar] [CrossRef]
- Sokolov, A.; Radhakrishnan, A. Accessibility of cholesterol in endoplasmic reticulum membranes and activation of SREBP-2 switch abruptly at a common cholesterol threshold. J. Biol. Chem. 2010, 285, 29480–29490. [Google Scholar] [CrossRef]
- Gekara, N.O.; Westphal, K.; Ma, B.; Rohde, M.; Groebe, L.; Weiss, S. The multiple mechanisms of Ca2+ signalling by listeriolysin O, the cholesterol-dependent cytolysin of Listeria monocytogenes. Cell. Microbiol. 2007, 9, 2008–2021. [Google Scholar] [CrossRef] [PubMed]
- Malet, J.K.; Impens, F.; Carvalho, F.; Hamon, M.A.; Cossart, P.; Ribet, D. Rapid Remodeling of the Host Epithelial Cell Proteome by the Listeriolysin O (LLO) Pore-forming Toxin. Mol. Cell. Proteom. 2018, 17, 1627–1636. [Google Scholar] [CrossRef] [PubMed]
- Ribet, D.; Hamon, M.; Gouin, E.; Nahori, M.A.; Impens, F.; Neyret-Kahn, H.; Gevaert, K.; Vandekerckhove, J.; Dejean, A.; Cossart, P. Listeria monocytogenes impairs SUMOylation for efficient infection. Nature 2010, 464, 1192–1195. [Google Scholar] [CrossRef]
- Li, J.; Lam, W.W.; Lai, T.W.; Au, S.W. Degradation of nuclear Ubc9 induced by listeriolysin O is dependent on K+ efflux. Biochem. Biophys. Res. Commun. 2017, 493, 1115–1121. [Google Scholar] [CrossRef]
- Hamon, M.A.; Batsche, E.; Regnault, B.; Tham, T.N.; Seveau, S.; Muchardt, C.; Cossart, P. Histone modifications induced by a family of bacterial toxins. Proc. Natl. Acad. Sci. USA 2007, 104, 13467–13472. [Google Scholar] [CrossRef]
- Hamon, M.A.; Cossart, P. K+ efflux is required for histone H3 dephosphorylation by Listeria monocytogenes listeriolysin O and other pore-forming toxins. Infect. Immun. 2011, 79, 2839–2846. [Google Scholar] [CrossRef]
- Bishop, B.L.; Lodolce, J.P.; Kolodziej, L.E.; Boone, D.L.; Tang, W.J. The role of anthrolysin O in gut epithelial barrier disruption during Bacillus anthracis infection. Biochem. Biophys. Res. Commun. 2010, 394, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Witzenrath, M.; Pache, F.; Lorenz, D.; Koppe, U.; Gutbier, B.; Tabeling, C.; Reppe, K.; Meixenberger, K.; Dorhoi, A.; Ma, J.; et al. The NLRP3 inflammasome is differentially activated by pneumolysin variants and contributes to host defense in pneumococcal pneumonia. J. Immunol. 2011, 187, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D.L.; Troyer, B.E.; Merrick, D.T.; Mitten, J.E.; Olson, R.D. Lethal effects and cardiovascular effects of purified alpha- and theta-toxins from Clostridium perfringens. J. Infect. Dis. 1988, 157, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Asmuth, D.M.; Olson, R.D.; Hackett, S.P.; Bryant, A.E.; Tweten, R.K.; Tso, J.Y.; Zollman, T.; Stevens, D.L. Effects of Clostridium perfringens recombinant and crude phospholipase C and theta-toxin on rabbit hemodynamic parameters. J. Infect. Dis. 1995, 172, 1317–1323. [Google Scholar] [CrossRef]
- Bryant, A.E.; Bayer, C.R.; Chen, R.Y.; Guth, P.H.; Wallace, R.J.; Stevens, D.L. Vascular dysfunction and ischemic destruction of tissue in Streptococcus pyogenes infection: The role of streptolysin O-induced platelet/neutrophil complexes. J. Infect. Dis. 2005, 192, 1014–1022. [Google Scholar] [CrossRef]
- Paton, J.C.; Ferrante, A. Inhibition of human polymorphonuclear leukocyte respiratory burst, bactericidal activity, and migration by pneumolysin. Infect. Immun. 1983, 41, 1212–1216. [Google Scholar] [CrossRef]
- Andersen, B.R.; Van Epps, D.E. Suppression of chemotatic activity of human neutrophils by streptolysin O. J. Infect. Dis. 1972, 125, 353–359. [Google Scholar] [CrossRef]
- Ofek, I.; Bergner-Rabinowitz, S.; Ginsburg, I. Oxygen-stable hemolysins of group A streptococci. 8. Leukotoxic and antiphagocytic effects of streptolysins S and O. Infect. Immun. 1972, 6, 459–464. [Google Scholar] [CrossRef]
- Bryant, A.E.; Bergstrom, R.; Zimmerman, G.A.; Salyer, J.L.; Hill, H.R.; Tweten, R.K.; Sato, H.; Stevens, D.L. Clostridium perfringens invasiveness is enhanced by effects of theta toxin upon PMNL structure and function: The roles of leukocytotoxicity and expression of CD11/CD18 adherence glycoprotein. FEMS Immunol. Med. Microbiol. 1993, 7, 321–336. [Google Scholar] [CrossRef]
- Pinho-Ribeiro, F.A.; Baddal, B.; Haarsma, R.; O’Seaghdha, M.; Yang, N.J.; Blake, K.J.; Portley, M.; Verri, W.A.; Dale, J.B.; Wessels, M.R.; et al. Blocking Neuronal Signaling to Immune Cells Treats Streptococcal Invasive Infection. Cell 2018, 173, 1083–1097.e1022. [Google Scholar] [CrossRef]
- Keyel, P.A. How is inflammation initiated? Individual influences of IL-1, IL-18 and HMGB1. Cytokine 2014, 69, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Abbott, K.N.; Wu, W.; Salter, R.D.; Keyel, P.A. Dnase1L3 Regulates Inflammasome-Dependent Cytokine Secretion. Front. Immunol. 2017, 8, 522. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.; Wu, R.; Du, H.; Jin, M.; Liu, Y.; Lei, G.; Jiang, B.; Lei, Z.; Peng, Y.; Nie, K.; et al. Pneumolysin-Dependent Calpain Activation and Interleukin-1alpha Secretion in Macrophages Infected with Streptococcus pneumoniae. Infect. Immun. 2017, 85. [Google Scholar] [CrossRef]
- Dewamitta, S.R.; Nomura, T.; Kawamura, I.; Hara, H.; Tsuchiya, K.; Kurenuma, T.; Shen, Y.; Daim, S.; Yamamoto, T.; Qu, H.; et al. Listeriolysin O-dependent bacterial entry into the cytoplasm is required for calpain activation and interleukin-1 alpha secretion in macrophages infected with Listeria monocytogenes. Infect. Immun. 2010, 78, 1884–1894. [Google Scholar] [CrossRef] [PubMed]
- Stassen, M.; Muller, C.; Richter, C.; Neudorfl, C.; Hultner, L.; Bhakdi, S.; Walev, I.; Schmitt, E. The streptococcal exotoxin streptolysin O activates mast cells to produce tumor necrosis factor alpha by p38 mitogen-activated protein kinase- and protein kinase C-dependent pathways. Infect. Immun. 2003, 71, 6171–6177. [Google Scholar] [CrossRef]
- Mishalian, I.; Ordan, M.; Peled, A.; Maly, A.; Eichenbaum, M.B.; Ravins, M.; Aychek, T.; Jung, S.; Hanski, E. Recruited macrophages control dissemination of group A Streptococcus from infected soft tissues. J. Immunol. 2011, 187, 6022–6031. [Google Scholar] [CrossRef]
- Tsuchiya, K.; Kawamura, I.; Takahashi, A.; Nomura, T.; Kohda, C.; Mitsuyama, M. Listeriolysin O-induced membrane permeation mediates persistent interleukin-6 production in Caco-2 cells during Listeria monocytogenes infection in vitro. Infect. Immun. 2005, 73, 3869–3877. [Google Scholar] [CrossRef]
- Lun, S.; Perez-Casal, J.; Connor, W.; Willson, P.J. Role of suilysin in pathogenesis of Streptococcus suis capsular serotype 2. Microb. Pathog. 2003, 34, 27–37. [Google Scholar] [CrossRef]
- Rijneveld, A.W.; van den Dobbelsteen, G.P.; Florquin, S.; Standiford, T.J.; Speelman, P.; van Alphen, L.; van der Poll, T. Roles of interleukin-6 and macrophage inflammatory protein-2 in pneumolysin-induced lung inflammation in mice. J. Infect. Dis. 2002, 185, 123–126. [Google Scholar] [CrossRef]
- Rose, F.; Zeller, S.A.; Chakraborty, T.; Domann, E.; Machleidt, T.; Kronke, M.; Seeger, W.; Grimminger, F.; Sibelius, U. Human endothelial cell activation and mediator release in response to Listeria monocytogenes virulence factors. Infect. Immun. 2001, 69, 897–905. [Google Scholar] [CrossRef]
- Malley, R.; Henneke, P.; Morse, S.C.; Cieslewicz, M.J.; Lipsitch, M.; Thompson, C.M.; Kurt-Jones, E.; Paton, J.C.; Wessels, M.R.; Golenbock, D.T. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc. Natl. Acad. Sci. USA 2003, 100, 1966–1971. [Google Scholar] [CrossRef] [PubMed]
- Dessing, M.C.; Hirst, R.A.; de Vos, A.F.; van der Poll, T. Role of Toll-like receptors 2 and 4 in pulmonary inflammation and injury induced by pneumolysin in mice. PLoS ONE 2009, 4, e7993. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Ng, V.H.; Maeda, S.; Rest, R.F.; Karin, M. Anthrolysin O and other gram-positive cytolysins are toll-like receptor 4 agonists. J. Exp. Med. 2004, 200, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, P.; Keyel, P.A. Cholesterol-dependent cytolysins impair pro-inflammatory macrophage responses. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Chu, J.; Thomas, L.M.; Watkins, S.C.; Franchi, L.; Nunez, G.; Salter, R.D. Cholesterol-dependent cytolysins induce rapid release of mature IL-1beta from murine macrophages in a NLRP3 inflammasome and cathepsin B-dependent manner. J. Leukoc. Biol. 2009, 86, 1227–1238. [Google Scholar] [CrossRef]
- Fickl, H.; Cockeran, R.; Steel, H.C.; Feldman, C.; Cowan, G.; Mitchell, T.J.; Anderson, R. Pneumolysin-mediated activation of NFkappaB in human neutrophils is antagonized by docosahexaenoic acid. Clin. Exp. Immunol. 2005, 140, 274–281. [Google Scholar] [CrossRef]
- Kayal, S.; Lilienbaum, A.; Poyart, C.; Memet, S.; Israel, A.; Berche, P. Listeriolysin O-dependent activation of endothelial cells during infection with Listeria monocytogenes: Activation of NF-kappa B and upregulation of adhesion molecules and chemokines. Mol. Microbiol. 1999, 31, 1709–1722. [Google Scholar] [CrossRef]
- Kayal, S.; Lilienbaum, A.; Join-Lambert, O.; Li, X.; Israel, A.; Berche, P. Listeriolysin O secreted by Listeria monocytogenes induces NF-kappaB signalling by activating the IkappaB kinase complex. Mol. Microbiol. 2002, 44, 1407–1419. [Google Scholar] [CrossRef]
- Shoma, S.; Tsuchiya, K.; Kawamura, I.; Nomura, T.; Hara, H.; Uchiyama, R.; Daim, S.; Mitsuyama, M. Critical involvement of pneumolysin in production of interleukin-1alpha and caspase-1-dependent cytokines in infection with Streptococcus pneumoniae In Vitro: A novel function of pneumolysin in caspase-1 activation. Infect. Immun. 2008, 76, 1547–1557. [Google Scholar] [CrossRef]
- Houldsworth, S.; Andrew, P.W.; Mitchell, T.J. Pneumolysin stimulates production of tumor necrosis factor alpha and interleukin-1 beta by human mononuclear phagocytes. Infect. Immun. 1994, 62, 1501–1503. [Google Scholar] [CrossRef]
- Subramanian, K.; Neill, D.R.; Malak, H.A.; Spelmink, L.; Khandaker, S.; Dalla Libera Marchiori, G.; Dearing, E.; Kirby, A.; Yang, M.; Achour, A.; et al. Pneumolysin binds to the mannose receptor C type 1 (MRC-1) leading to anti-inflammatory responses and enhanced pneumococcal survival. Nat. Microbiol. 2019, 4, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Bernatoniene, J.; Zhang, Q.; Dogan, S.; Mitchell, T.J.; Paton, J.C.; Finn, A. Induction of CC and CXC chemokines in human antigen-presenting dendritic cells by the pneumococcal proteins pneumolysin and CbpA, and the role played by toll-like receptor 4, NF-kappaB, and mitogen-activated protein kinases. J. Infect. Dis. 2008, 198, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Harvey, R.M.; Hughes, C.E.; Paton, A.W.; Trappetti, C.; Tweten, R.K.; Paton, J.C. The impact of pneumolysin on the macrophage response to Streptococcus pneumoniae is strain-dependent. PLoS ONE 2014, 9, e103625. [Google Scholar] [CrossRef] [PubMed]
- Parker, D.; Martin, F.J.; Soong, G.; Harfenist, B.S.; Aguilar, J.L.; Ratner, A.J.; Fitzgerald, K.A.; Schindler, C.; Prince, A. Streptococcus pneumoniae DNA initiates type I interferon signaling in the respiratory tract. mBio 2011, 2, e00016-11. [Google Scholar] [CrossRef]
- Koppe, U.; Hogner, K.; Doehn, J.M.; Muller, H.C.; Witzenrath, M.; Gutbier, B.; Bauer, S.; Pribyl, T.; Hammerschmidt, S.; Lohmeyer, J.; et al. Streptococcus pneumoniae stimulates a STING- and IFN regulatory factor 3-dependent type I IFN production in macrophages, which regulates RANTES production in macrophages, cocultured alveolar epithelial cells, and mouse lungs. J. Immunol. 2012, 188, 811–817. [Google Scholar] [CrossRef]
- Mitsui, K.; Takano, K.; Nakatani, S.; Nambu, H.; Shibata, F.; Nakagawa, H. Chemokine production by rat macrophages stimulated with streptolysin O from Streptococcus pyogenes. Microbiol. Immunol. 2002, 46, 37–45. [Google Scholar] [CrossRef]
- Braun, J.S.; Novak, R.; Gao, G.; Murray, P.J.; Shenep, J.L. Pneumolysin, a protein toxin of Streptococcus pneumoniae, induces nitric oxide production from macrophages. Infect. Immun. 1999, 67, 3750–3756. [Google Scholar] [CrossRef]
- Nishibori, T.; Xiong, H.; Kawamura, I.; Arakawa, M.; Mitsuyama, M. Induction of cytokine gene expression by listeriolysin O and roles of macrophages and NK cells. Infect. Immun. 1996, 64, 3188–3195. [Google Scholar] [CrossRef]
- Hackett, S.P.; Stevens, D.L. Streptococcal toxic shock syndrome: Synthesis of tumor necrosis factor and interleukin-1 by monocytes stimulated with pyrogenic exotoxin A and streptolysin O. J. Infect. Dis. 1992, 165, 879–885. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Munoz-Planillo, R.; Kuffa, P.; Martinez-Colon, G.; Smith, B.L.; Rajendiran, T.M.; Nunez, G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, K.; Ashida, H.; Okano, T.; Kinoshita-Daitoku, R.; Suzuki, S.; Ohtani, K.; Hamagaki, M.; Ikeda, T.; Suzuki, T. Inflammasome Activation Induced by Perfringolysin O of Clostridium perfringens and Its Involvement in the Progression of Gas Gangrene. Front. Microbiol. 2019, 10, 2406. [Google Scholar] [CrossRef] [PubMed]
- Harder, J.; Franchi, L.; Munoz-Planillo, R.; Park, J.H.; Reimer, T.; Nunez, G. Activation of the Nlrp3 inflammasome by Streptococcus pyogenes requires streptolysin O and NF-kappa B activation but proceeds independently of TLR signaling and P2X7 receptor. J. Immunol. 2009, 183, 5823–5829. [Google Scholar] [CrossRef]
- McNeela, E.A.; Burke, A.; Neill, D.R.; Baxter, C.; Fernandes, V.E.; Ferreira, D.; Smeaton, S.; El-Rachkidy, R.; McLoughlin, R.M.; Mori, A.; et al. Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS Pathog. 2010, 6, e1001191. [Google Scholar] [CrossRef]
- Warren, S.E.; Mao, D.P.; Rodriguez, A.E.; Miao, E.A.; Aderem, A. Multiple Nod-like receptors activate caspase 1 during Listeria monocytogenes infection. J. Immunol. 2008, 180, 7558–7564. [Google Scholar] [CrossRef]
- Meixenberger, K.; Pache, F.; Eitel, J.; Schmeck, B.; Hippenstiel, S.; Slevogt, H.; N’Guessan, P.; Witzenrath, M.; Netea, M.G.; Chakraborty, T.; et al. Listeria monocytogenes-infected human peripheral blood mononuclear cells produce IL-1beta, depending on listeriolysin O and NLRP3. J. Immunol. 2010, 184, 922–930. [Google Scholar] [CrossRef]
- Hara, H.; Tsuchiya, K.; Nomura, T.; Kawamura, I.; Shoma, S.; Mitsuyama, M. Dependency of caspase-1 activation induced in macrophages by Listeria monocytogenes on cytolysin, listeriolysin O, after evasion from phagosome into the cytoplasm. J. Immunol. 2008, 180, 7859–7868. [Google Scholar] [CrossRef]
- Lavagna, A.; Auger, J.P.; Dumesnil, A.; Roy, D.; Girardin, S.E.; Gisch, N.; Segura, M.; Gottschalk, M. Interleukin-1 signaling induced by Streptococcus suis serotype 2 is strain-dependent and contributes to bacterial clearance and inflammation during systemic disease in a mouse model of infection. Vet. Res. 2019, 50, 52. [Google Scholar] [CrossRef]
- Ozoren, N.; Masumoto, J.; Franchi, L.; Kanneganti, T.D.; Body-Malapel, M.; Erturk, I.; Jagirdar, R.; Zhu, L.; Inohara, N.; Bertin, J.; et al. Distinct roles of TLR2 and the adaptor ASC in IL-1beta/IL-18 secretion in response to Listeria monocytogenes. J. Immunol. 2006, 176, 4337–4342. [Google Scholar] [CrossRef]
- Fang, R.; Tsuchiya, K.; Kawamura, I.; Shen, Y.; Hara, H.; Sakai, S.; Yamamoto, T.; Fernandes-Alnemri, T.; Yang, R.; Hernandez-Cuellar, E.; et al. Critical roles of ASC inflammasomes in caspase-1 activation and host innate resistance to Streptococcus pneumoniae infection. J. Immunol. 2011, 187, 4890–4899. [Google Scholar] [CrossRef]
- Rathinam, V.A.; Jiang, Z.; Waggoner, S.N.; Sharma, S.; Cole, L.E.; Waggoner, L.; Vanaja, S.K.; Monks, B.G.; Ganesan, S.; Latz, E.; et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 2010, 11, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Bauernfeind, F.; Ablasser, A.; Hartmann, G.; Fitzgerald, K.A.; Latz, E.; Hornung, V. Listeria monocytogenes is sensed by the NLRP3 and AIM2 inflammasome. Eur. J. Immunol. 2010, 40, 1545–1551. [Google Scholar] [CrossRef] [PubMed]
- Dang, E.V.; McDonald, J.G.; Russell, D.W.; Cyster, J.G. Oxysterol Restraint of Cholesterol Synthesis Prevents AIM2 Inflammasome Activation. Cell 2017, 171, 1057–1071.e1011. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Scott, A.L. Cholesterol 25-hydroxylase production by dendritic cells and macrophages is regulated by type I interferons. J. Leukoc. Biol. 2010, 88, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, A.; Ikeda, Y.; Kwon, H.J.; Brown, M.S.; Goldstein, J.L. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Oxysterols block transport by binding to Insig. Proc. Natl. Acad. Sci. USA 2007, 104, 6511–6518. [Google Scholar] [CrossRef]
- Chang, T.Y.; Chang, C.C.; Ohgami, N.; Yamauchi, Y. Cholesterol sensing, trafficking, and esterification. Annu. Rev. Cell Dev. Biol. 2006, 22, 129–157. [Google Scholar] [CrossRef]
- Reboldi, A.; Dang, E.V.; McDonald, J.G.; Liang, G.; Russell, D.W.; Cyster, J.G. Inflammation. 25-Hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon. Science 2014, 345, 679–684. [Google Scholar] [CrossRef]
- Abrams, M.E.; Johnson, K.A.; Perelman, S.S.; Zhang, L.S.; Endapally, S.; Mar, K.B.; Thompson, B.M.; McDonald, J.G.; Schoggins, J.W.; Radhakrishnan, A.; et al. Oxysterols provide innate immunity to bacterial infection by mobilizing cell surface accessible cholesterol. Nat. Microbiol. 2020, 5, 929–942. [Google Scholar] [CrossRef]
- Zhou, Q.D.; Chi, X.; Lee, M.S.; Hsieh, W.Y.; Mkrtchyan, J.J.; Feng, A.C.; He, C.; York, A.G.; Bui, V.L.; Kronenberger, E.B.; et al. Interferon-mediated reprogramming of membrane cholesterol to evade bacterial toxins. Nat. Immunol. 2020, 21, 746–755. [Google Scholar] [CrossRef]
- Miller, L.S.; Pietras, E.M.; Uricchio, L.H.; Hirano, K.; Rao, S.; Lin, H.; O’Connell, R.M.; Iwakura, Y.; Cheung, A.L.; Cheng, G.; et al. Inflammasome-mediated production of IL-1beta is required for neutrophil recruitment against Staphylococcus aureus in vivo. J. Immunol. 2007, 179, 6933–6942. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Kono, H.; Golenbock, D.; Reed, G.; Akira, S.; Rock, K.L. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat. Med. 2007, 13, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.C.; Enzler, T.; Seita, J.; Timmer, A.M.; Lee, C.Y.; Lai, T.Y.; Yu, G.Y.; Lai, L.C.; Temkin, V.; Sinzig, U.; et al. IL-1beta-driven neutrophilia preserves antibacterial defense in the absence of the kinase IKKbeta. Nat. Immunol. 2011, 12, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Kafka, D.; Ling, E.; Feldman, G.; Benharroch, D.; Voronov, E.; Givon-Lavi, N.; Iwakura, Y.; Dagan, R.; Apte, R.N.; Mizrachi-Nebenzahl, Y. Contribution of IL-1 to resistance to Streptococcus pneumoniae infection. Int. Immunol. 2008, 20, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Zwijnenburg, P.J.; van der Poll, T.; Florquin, S.; Akira, S.; Takeda, K.; Roord, J.J.; van Furth, A.M. Interleukin-18 gene-deficient mice show enhanced defense and reduced inflammation during pneumococcal meningitis. J. Neuroimmunol. 2003, 138, 31–37. [Google Scholar] [CrossRef]
- Tsuji, N.M.; Tsutsui, H.; Seki, E.; Kuida, K.; Okamura, H.; Nakanishi, K.; Flavell, R.A. Roles of caspase-1 in Listeria infection in mice. Int. Immunol. 2004, 16, 335–343. [Google Scholar] [CrossRef]
- Flannagan, R.S.; Jaumouille, V.; Grinstein, S. The cell biology of phagocytosis. Annu. Rev. Pathol. 2012, 7, 61–98. [Google Scholar] [CrossRef]
- Portnoy, D.A.; Jacks, P.S.; Hinrichs, D.J. Role of hemolysin for the intracellular growth of Listeria monocytogenes. J. Exp. Med. 1988, 167, 1459–1471. [Google Scholar] [CrossRef]
- Malet, J.K.; Cossart, P.; Ribet, D. Alteration of epithelial cell lysosomal integrity induced by bacterial cholesterol-dependent cytolysins. Cell. Microbiol. 2017, 19, e12682. [Google Scholar] [CrossRef]
- Shaughnessy, L.M.; Hoppe, A.D.; Christensen, K.A.; Swanson, J.A. Membrane perforations inhibit lysosome fusion by altering pH and calcium in Listeria monocytogenes vacuoles. Cell. Microbiol. 2006, 8, 781–792. [Google Scholar] [CrossRef]
- Jones, S.; Portnoy, D.A. Characterization of Listeria monocytogenes pathogenesis in a strain expressing perfringolysin O in place of listeriolysin O. Infect. Immun. 1994, 62, 5608–5613. [Google Scholar] [CrossRef] [PubMed]
- Portnoy, D.A.; Tweten, R.K.; Kehoe, M.; Bielecki, J. Capacity of listeriolysin O, streptolysin O, and perfringolysin O to mediate growth of Bacillus subtilis within mammalian cells. Infect. Immun. 1992, 60, 2710–2717. [Google Scholar] [CrossRef] [PubMed]
- Bielecki, J.; Youngman, P.; Connelly, P.; Portnoy, D.A. Bacillus subtilis expressing a haemolysin gene from Listeria monocytogenes can grow in mammalian cells. Nature 1990, 345, 175–176. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, D.K.; Melville, S.B. Effects of Clostridium perfringens alpha-toxin (PLC) and perfringolysin O (PFO) on cytotoxicity to macrophages, on escape from the phagosomes of macrophages, and on persistence of C. perfringens in host tissues. Infect. Immun. 2004, 72, 5204–5215. [Google Scholar] [CrossRef] [PubMed]
- Bastiat-Sempe, B.; Love, J.F.; Lomayesva, N.; Wessels, M.R. Streptolysin O and NAD-glycohydrolase prevent phagolysosome acidification and promote group A Streptococcus survival in macrophages. mBio 2014, 5, e01690-14. [Google Scholar] [CrossRef] [PubMed]
- Hickey, M.J.; Kwan, R.Y.; Awad, M.M.; Kennedy, C.L.; Young, L.F.; Hall, P.; Cordner, L.M.; Lyras, D.; Emmins, J.J.; Rood, J.I. Molecular and cellular basis of microvascular perfusion deficits induced by Clostridium perfringens and Clostridium septicum. PLoS Pathog. 2008, 4, e1000045. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, A.M.; Thurston, T.L.; Holden, D.W. Cytosolic Replication of Group A Streptococcus in Human Macrophages. mBio 2016, 7, e00020-16. [Google Scholar] [CrossRef]
- De Chastellier, C.; Berche, P. Fate of Listeria monocytogenes in murine macrophages: Evidence for simultaneous killing and survival of intracellular bacteria. Infect. Immun. 1994, 62, 543–553. [Google Scholar] [CrossRef]
- Vadia, S.; Arnett, E.; Haghighat, A.C.; Wilson-Kubalek, E.M.; Tweten, R.K.; Seveau, S. The pore-forming toxin listeriolysin O mediates a novel entry pathway of L. monocytogenes into human hepatocytes. PLoS Pathog. 2011, 7, e1002356. [Google Scholar] [CrossRef]
- Dramsi, S.; Cossart, P. Listeriolysin O-mediated calcium influx potentiates entry of Listeria monocytogenes into the human Hep-2 epithelial cell line. Infect. Immun. 2003, 71, 3614–3618. [Google Scholar] [CrossRef]
- Sierig, G.; Cywes, C.; Wessels, M.R.; Ashbaugh, C.D. Cytotoxic effects of streptolysin o and streptolysin s enhance the virulence of poorly encapsulated group a streptococci. Infect. Immun. 2003, 71, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Corrotte, M.; Almeida, P.E.; Tam, C.; Castro-Gomes, T.; Fernandes, M.C.; Millis, B.A.; Cortez, M.; Miller, H.; Song, W.; Maugel, T.K.; et al. Caveolae internalization repairs wounded cells and muscle fibers. eLife 2013, 2, e00926. [Google Scholar] [CrossRef]
- Heid, M.E.; Keyel, P.A.; Kamga, C.; Shiva, S.; Watkins, S.C.; Salter, R.D. Mitochondrial Reactive Oxygen Species Induces NLRP3-Dependent Lysosomal Damage and Inflammasome Activation. J. Immunol. 2013, 191, 5230–5238. [Google Scholar] [CrossRef]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Bewley, M.A.; Naughton, M.; Preston, J.; Mitchell, A.; Holmes, A.; Marriott, H.M.; Read, R.C.; Mitchell, T.J.; Whyte, M.K.; Dockrell, D.H. Pneumolysin activates macrophage lysosomal membrane permeabilization and executes apoptosis by distinct mechanisms without membrane pore formation. mBio 2014, 5, e01710–e01714. [Google Scholar] [CrossRef]
- Skowyra, M.L.; Schlesinger, P.H.; Naismith, T.V.; Hanson, P.I. Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science 2018, 360. [Google Scholar] [CrossRef]
- Schuerch, D.W.; Wilson-Kubalek, E.M.; Tweten, R.K. Molecular basis of listeriolysin O pH dependence. Proc. Natl. Acad. Sci. USA 2005, 102, 12537–12542. [Google Scholar] [CrossRef]
- Singh, R.; Jamieson, A.; Cresswell, P. GILT is a critical host factor for Listeria monocytogenes infection. Nature 2008, 455, 1244–1247. [Google Scholar] [CrossRef]
- Hancz, D.; Westerlund, E.; Valfridsson, C.; Aemero, G.M.; Bastiat-Sempe, B.; Orning, P.; Lien, E.; Wessels, M.R.; Persson, J.J. Streptolysin O Induces the Ubiquitination and Degradation of Pro-IL-1beta. J. Innate Immun. 2019, 11, 457–468. [Google Scholar] [CrossRef]
- Littmann, M.; Albiger, B.; Frentzen, A.; Normark, S.; Henriques-Normark, B.; Plant, L. Streptococcus pneumoniae evades human dendritic cell surveillance by pneumolysin expression. EMBO Mol. Med. 2009, 1, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Lam, G.Y.; Fattouh, R.; Muise, A.M.; Grinstein, S.; Higgins, D.E.; Brumell, J.H. Listeriolysin O suppresses phospholipase C-mediated activation of the microbicidal NADPH oxidase to promote Listeria monocytogenes infection. Cell Host Microbe 2011, 10, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, S.; Dohrmann, S.; Timmer, A.M.; Dixit, N.; Ghochani, M.; Bhandari, T.; Timmer, J.C.; Sprague, K.; Bubeck-Wardenburg, J.; Simon, S.I.; et al. Streptolysin O Rapidly Impairs Neutrophil Oxidative Burst and Antibacterial Responses to Group A Streptococcus. Front. Immunol. 2015, 6, 581. [Google Scholar] [CrossRef] [PubMed]
- Keyel, P.A.; Heid, M.E.; Salter, R.D. Macrophage responses to bacterial toxins: A balance between activation and suppression. Immunol. Res. 2011, 50, 118–123. [Google Scholar] [CrossRef]
- Keyel, P.A.; Tkacheva, O.A.; Larregina, A.T.; Salter, R.D. Coordinate stimulation of macrophages by microparticles and TLR ligands induces foam cell formation. J. Immunol. 2012, 189, 4621–4629. [Google Scholar] [CrossRef]
- Panousis, C.G.; Zuckerman, S.H. Regulation of cholesterol distribution in macrophage-derived foam cells by interferon-gamma. J. Lipid Res. 2000, 41, 75–83. [Google Scholar]
- Koffel, R.; Wolfmeier, H.; Larpin, Y.; Besancon, H.; Schoenauer, R.; Babiychuk, V.S.; Drucker, P.; Pabst, T.; Mitchell, T.J.; Babiychuk, E.B.; et al. Host-Derived Microvesicles Carrying Bacterial Pore-Forming Toxins Deliver Signals to Macrophages: A Novel Mechanism of Shaping Immune Responses. Front. Immunol. 2018, 9, 1688. [Google Scholar] [CrossRef]
- Whyte, C.S.; Bishop, E.T.; Ruckerl, D.; Gaspar-Pereira, S.; Barker, R.N.; Allen, J.E.; Rees, A.J.; Wilson, H.M. Suppressor of cytokine signaling (SOCS)1 is a key determinant of differential macrophage activation and function. J. Leukoc. Biol. 2011, 90, 845–854. [Google Scholar] [CrossRef]
- Wolf, A.I.; Strauman, M.C.; Mozdzanowska, K.; Williams, K.L.; Osborne, L.C.; Shen, H.; Liu, Q.; Garlick, D.; Artis, D.; Hensley, S.E.; et al. Pneumolysin expression by streptococcus pneumoniae protects colonized mice from influenza virus-induced disease. Virology 2014, 462–463, 254–265. [Google Scholar] [CrossRef]
- Edelson, B.T.; Cossart, P.; Unanue, E.R. Cutting edge: Paradigm revisited: Antibody provides resistance to Listeria infection. J. Immunol. 1999, 163, 4087–4090. [Google Scholar]
- Pamer, E.G.; Harty, J.T.; Bevan, M.J. Precise prediction of a dominant class I MHC-restricted epitope of Listeria monocytogenes. Nature 1991, 353, 852–855. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.Q.; Tuma-Warrino, R.J.; Bryan, M.A.; Mitchell, K.G.; Higgins, D.E.; Watkins, S.C.; Salter, R.D. Escherichia coli expressing recombinant antigen and listeriolysin O stimulate class I-restricted CD8+ T cells following uptake by human APC. J. Immunol. 2004, 172, 1595–1601. [Google Scholar] [CrossRef] [PubMed]
- Bouwer, H.G.; Nelson, C.S.; Gibbins, B.L.; Portnoy, D.A.; Hinrichs, D.J. Listeriolysin O is a target of the immune response to Listeria monocytogenes. J. Exp. Med. 1992, 175, 1467–1471. [Google Scholar] [CrossRef] [PubMed]
- Mureithi, M.W.; Finn, A.; Ota, M.O.; Zhang, Q.; Davenport, V.; Mitchell, T.J.; Williams, N.A.; Adegbola, R.A.; Heyderman, R.S. T cell memory response to pneumococcal protein antigens in an area of high pneumococcal carriage and disease. J. Infect. Dis. 2009, 200, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bagrade, L.; Bernatoniene, J.; Clarke, E.; Paton, J.C.; Mitchell, T.J.; Nunez, D.A.; Finn, A. Low CD4 T cell immunity to pneumolysin is associated with nasopharyngeal carriage of pneumococci in children. J. Infect. Dis. 2007, 195, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Nakouzi, A.; Rivera, J.; Rest, R.F.; Casadevall, A. Passive administration of monoclonal antibodies to anthrolysin O prolong survival in mice lethally infected with Bacillus anthracis. BMC Microbiol. 2008, 8, 159. [Google Scholar] [CrossRef]
- Stevens, D.L.; Tweten, R.K.; Awad, M.M.; Rood, J.I.; Bryant, A.E. Clostridial gas gangrene: Evidence that alpha and theta toxins differentially modulate the immune response and induce acute tissue necrosis. J. Infect. Dis. 1997, 176, 189–195. [Google Scholar] [CrossRef]
- Chiarot, E.; Faralla, C.; Chiappini, N.; Tuscano, G.; Falugi, F.; Gambellini, G.; Taddei, A.; Capo, S.; Cartocci, E.; Veggi, D.; et al. Targeted amino acid substitutions impair streptolysin O toxicity and group A Streptococcus virulence. mBio 2013, 4, e00387-12. [Google Scholar] [CrossRef]
- Jacobs, A.A.; van den Berg, A.J.; Loeffen, P.L. Protection of experimentally infected pigs by suilysin, the thiol-activated haemolysin of Streptococcus suis. Vet. Rec. 1996, 139, 225–228. [Google Scholar] [CrossRef]
- Musher, D.M.; Phan, H.M.; Baughn, R.E. Protection against bacteremic pneumococcal infection by antibody to pneumolysin. J. Infect. Dis. 2001, 183, 827–830. [Google Scholar] [CrossRef][Green Version]
- Wade, K.R.; Hotze, E.M.; Briles, D.E.; Tweten, R.K. Mouse, but not human, ApoB-100 lipoprotein cholesterol is a potent innate inhibitor of Streptococcus pneumoniae pneumolysin. PLoS Pathog. 2014, 10, e1004353. [Google Scholar] [CrossRef]
- Carrero, J.A.; Vivanco-Cid, H.; Unanue, E.R. Listeriolysin o is strongly immunogenic independently of its cytotoxic activity. PLoS ONE 2012, 7, e32310. [Google Scholar] [CrossRef] [PubMed]
- Kamtchoua, T.; Bologa, M.; Hopfer, R.; Neveu, D.; Hu, B.; Sheng, X.; Corde, N.; Pouzet, C.; Zimmermann, G.; Gurunathan, S. Safety and immunogenicity of the pneumococcal pneumolysin derivative PlyD1 in a single-antigen protein vaccine candidate in adults. Vaccine 2013, 31, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Odutola, A.; Ota, M.O.C.; Antonio, M.; Ogundare, E.O.; Saidu, Y.; Owiafe, P.K.; Worwui, A.; Idoko, O.T.; Owolabi, O.; Kampmann, B.; et al. Immunogenicity of pneumococcal conjugate vaccine formulations containing pneumococcal proteins, and immunogenicity and reactogenicity of co-administered routine vaccines—A phase II, randomised, observer-blind study in Gambian infants. Vaccine 2019, 37, 2586–2599. [Google Scholar] [CrossRef] [PubMed]
- Hammitt, L.L.; Campbell, J.C.; Borys, D.; Weatherholtz, R.C.; Reid, R.; Goklish, N.; Moulton, L.H.; Traskine, M.; Song, Y.; Swinnen, K.; et al. Efficacy, safety and immunogenicity of a pneumococcal protein-based vaccine co-administered with 13-valent pneumococcal conjugate vaccine against acute otitis media in young children: A phase IIb randomized study. Vaccine 2019, 37, 7482–7492. [Google Scholar] [CrossRef]
- Sumby, P.; Porcella, S.F.; Madrigal, A.G.; Barbian, K.D.; Virtaneva, K.; Ricklefs, S.M.; Sturdevant, D.E.; Graham, M.R.; Vuopio-Varkila, J.; Hoe, N.P.; et al. Evolutionary origin and emergence of a highly successful clone of serotype M1 group a Streptococcus involved multiple horizontal gene transfer events. J. Infect. Dis. 2005, 192, 771–782. [Google Scholar] [CrossRef]
- Zhu, L.; Olsen, R.J.; Nasser, W.; Beres, S.B.; Vuopio, J.; Kristinsson, K.G.; Gottfredsson, M.; Porter, A.R.; DeLeo, F.R.; Musser, J.M. A molecular trigger for intercontinental epidemics of group A Streptococcus. J. Clin. Investig. 2015, 125, 3545–3559. [Google Scholar] [CrossRef]
- Escajadillo, T.; Olson, J.; Luk, B.T.; Zhang, L.; Nizet, V. A Red Blood Cell Membrane-Camouflaged Nanoparticle Counteracts Streptolysin O-Mediated Virulence Phenotypes of Invasive Group A Streptococcus. Front. Pharmacol. 2017, 8, 477. [Google Scholar] [CrossRef]
- Farrand, A.J.; Hotze, E.M.; Sato, T.K.; Wade, K.R.; Wimley, W.C.; Johnson, A.E.; Tweten, R.K. The Cholesterol-dependent Cytolysin Membrane-binding Interface Discriminates Lipid Environments of Cholesterol to Support beta-Barrel Pore Insertion. J. Biol. Chem. 2015, 290, 17733–17744. [Google Scholar] [CrossRef]
- Johnson, B.B.; Brena, M.; Anguita, J.; Heuck, A.P. Mechanistic Insights into the Cholesterol-dependent Binding of Perfringolysin O-based Probes and Cell Membranes. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Bricker, A.L.; Cywes, C.; Ashbaugh, C.D.; Wessels, M.R. NAD+-glycohydrolase acts as an intracellular toxin to enhance the extracellular survival of group A streptococci. Mol. Microbiol. 2002, 44, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Madden, J.C.; Ruiz, N.; Caparon, M. Cytolysin-mediated translocation (CMT): A functional equivalent of type III secretion in gram-positive bacteria. Cell 2001, 104, 143–152. [Google Scholar] [CrossRef]
- Magassa, N.; Chandrasekaran, S.; Caparon, M.G. Streptococcus pyogenes cytolysin-mediated translocation does not require pore formation by streptolysin O. EMBO Rep. 2010, 11, 400–405. [Google Scholar] [CrossRef]
- Michos, A.; Gryllos, I.; Hakansson, A.; Srivastava, A.; Kokkotou, E.; Wessels, M.R. Enhancement of streptolysin O activity and intrinsic cytotoxic effects of the group A streptococcal toxin, NAD-glycohydrolase. J. Biol. Chem. 2006, 281, 8216–8223. [Google Scholar] [CrossRef] [PubMed]
- Kimoto, H.; Fujii, Y.; Yokota, Y.; Taketo, A. Molecular characterization of NADase-streptolysin O operon of hemolytic streptococci. Biochim. Biophys. Acta 2005, 1681, 134–149. [Google Scholar] [CrossRef] [PubMed]
- Mozola, C.C.; Caparon, M.G. Dual modes of membrane binding direct pore formation by Streptolysin O. Mol. Microbiol. 2015, 97, 1036–1050. [Google Scholar] [CrossRef]
- O’Seaghdha, M.; Wessels, M.R. Streptolysin O and its co-toxin NAD-glycohydrolase protect group A Streptococcus from Xenophagic killing. PLoS Pathog. 2013, 9, e1003394. [Google Scholar] [CrossRef]
- Hancz, D.; Westerlund, E.; Bastiat-Sempe, B.; Sharma, O.; Valfridsson, C.; Meyer, L.; Love, J.F.; O’Seaghdha, M.; Wessels, M.R.; Persson, J.J. Inhibition of Inflammasome-Dependent Interleukin 1beta Production by Streptococcal NAD+-Glycohydrolase: Evidence for Extracellular Activity. mBio 2017, 8. [Google Scholar] [CrossRef]
- Fontaine, M.C.; Lee, J.J.; Kehoe, M.A. Combined contributions of streptolysin O and streptolysin S to virulence of serotype M5 Streptococcus pyogenes strain Manfredo. Infect. Immun. 2003, 71, 3857–3865. [Google Scholar] [CrossRef]
- Miyoshi-Akiyama, T.; Takamatsu, D.; Koyanagi, M.; Zhao, J.; Imanishi, K.; Uchiyama, T. Cytocidal effect of Streptococcus pyogenes on mouse neutrophils in vivo and the critical role of streptolysin S. J. Infect. Dis. 2005, 192, 107–116. [Google Scholar] [CrossRef]
- Lin, A.; Loughman, J.A.; Zinselmeyer, B.H.; Miller, M.J.; Caparon, M.G. Streptolysin S inhibits neutrophil recruitment during the early stages of Streptococcus pyogenes infection. Infect. Immun. 2009, 77, 5190–5201. [Google Scholar] [CrossRef] [PubMed]
- Benton, K.A.; Everson, M.P.; Briles, D.E. A pneumolysin-negative mutant of Streptococcus pneumoniae causes chronic bacteremia rather than acute sepsis in mice. Infect. Immun. 1995, 63, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Tsuprun, V.; Cureoglu, S.; Schachern, P.A.; Ferrieri, P.; Briles, D.E.; Paparella, M.M.; Juhn, S.K. Role of pneumococcal proteins in sensorineural hearing loss due to otitis media. Otol. Neurotol. 2008, 29, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Wippel, C.; Maurer, J.; Fortsch, C.; Hupp, S.; Bohl, A.; Ma, J.; Mitchell, T.J.; Bunkowski, S.; Bruck, W.; Nau, R.; et al. Bacterial cytolysin during meningitis disrupts the regulation of glutamate in the brain, leading to synaptic damage. PLoS Pathog. 2013, 9, e1003380. [Google Scholar] [CrossRef] [PubMed]
- Gilley, R.P.; Gonzalez-Juarbe, N.; Shenoy, A.T.; Reyes, L.F.; Dube, P.H.; Restrepo, M.I.; Orihuela, C.J. Infiltrated Macrophages Die of Pneumolysin-Mediated Necroptosis following Pneumococcal Myocardial Invasion. Infect. Immun. 2016, 84, 1457–1469. [Google Scholar] [CrossRef]
- Henry, B.D.; Neill, D.R.; Becker, K.A.; Gore, S.; Bricio-Moreno, L.; Ziobro, R.; Edwards, M.J.; Muhlemann, K.; Steinmann, J.; Kleuser, B.; et al. Engineered liposomes sequester bacterial exotoxins and protect from severe invasive infections in mice. Nat. Biotechnol. 2015, 33, 81–88. [Google Scholar] [CrossRef]
- Van der Poll, T.; Keogh, C.V.; Buurman, W.A.; Lowry, S.F. Passive immunization against tumor necrosis factor-alpha impairs host defense during pneumococcal pneumonia in mice. Am. J. Respir. Crit. Care Med. 1997, 155, 603–608. [Google Scholar] [CrossRef]
- Orman, K.L.; Shenep, J.L.; English, B.K. Pneumococci stimulate the production of the inducible nitric oxide synthase and nitric oxide by murine macrophages. J. Infect. Dis. 1998, 178, 1649–1657. [Google Scholar] [CrossRef]
- Cockeran, R.; Steel, H.C.; Mitchell, T.J.; Feldman, C.; Anderson, R. Pneumolysin potentiates production of prostaglandin E2 and leukotriene B4 by human neutrophils. Infect. Immun. 2001, 69, 3494–3496. [Google Scholar] [CrossRef]
- Paton, J.C.; Rowan-Kelly, B.; Ferrante, A. Activation of human complement by the pneumococcal toxin pneumolysin. Infect. Immun. 1984, 43, 1085–1087. [Google Scholar] [CrossRef]
- Jounblat, R.; Kadioglu, A.; Mitchell, T.J.; Andrew, P.W. Pneumococcal behavior and host responses during bronchopneumonia are affected differently by the cytolytic and complement-activating activities of pneumolysin. Infect. Immun. 2003, 71, 1813–1819. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, T.J.; Andrew, P.W.; Saunders, F.K.; Smith, A.N.; Boulnois, G.J. Complement activation and antibody binding by pneumolysin via a region of the toxin homologous to a human acute-phase protein. Mol. Microbiol. 1991, 5, 1883–1888. [Google Scholar] [CrossRef] [PubMed]
- Rubins, J.B.; Charboneau, D.; Paton, J.C.; Mitchell, T.J.; Andrew, P.W.; Janoff, E.N. Dual function of pneumolysin in the early pathogenesis of murine pneumococcal pneumonia. J. Clin. Investig. 1995, 95, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Yuste, J.; Botto, M.; Paton, J.C.; Holden, D.W.; Brown, J.S. Additive inhibition of complement deposition by pneumolysin and PspA facilitates Streptococcus pneumoniae septicemia. J. Immunol. 2005, 175, 1813–1819. [Google Scholar] [CrossRef]
- Cockeran, R.; Theron, A.J.; Steel, H.C.; Matlola, N.M.; Mitchell, T.J.; Feldman, C.; Anderson, R. Proinflammatory interactions of pneumolysin with human neutrophils. J. Infect. Dis. 2001, 183, 604–611. [Google Scholar] [CrossRef]
- Rubins, J.B.; Mitchell, T.J.; Andrew, P.W.; Niewoehner, D.E. Pneumolysin activates phospholipase A in pulmonary artery endothelial cells. Infect. Immun. 1994, 62, 3829–3836. [Google Scholar] [CrossRef]
- Ellemor, D.M.; Baird, R.N.; Awad, M.M.; Boyd, R.L.; Rood, J.I.; Emmins, J.J. Use of genetically manipulated strains of Clostridium perfringens reveals that both alpha-toxin and theta-toxin are required for vascular leukostasis to occur in experimental gas gangrene. Infect. Immun. 1999, 67, 4902–4907. [Google Scholar] [CrossRef]
- Flanagan, J.J.; Tweten, R.K.; Johnson, A.E.; Heuck, A.P. Cholesterol exposure at the membrane surface is necessary and sufficient to trigger perfringolysin O binding. Biochemistry 2009, 48, 3977–3987. [Google Scholar]
- Das, A.; Brown, M.S.; Anderson, D.D.; Goldstein, J.L.; Radhakrishnan, A. Three pools of plasma membrane cholesterol and their relation to cholesterol homeostasis. eLife 2014, 3, e02882. [Google Scholar] [CrossRef]
- Nelson, L.D.; Chiantia, S.; London, E. Perfringolysin O association with ordered lipid domains: Implications for transmembrane protein raft affinity. Biophys. J. 2010, 99, 3255–3263. [Google Scholar] [CrossRef]
- Moe, P.C.; Heuck, A.P. Phospholipid hydrolysis caused by Clostridium perfringens alpha-toxin facilitates the targeting of perfringolysin O to membrane bilayers. Biochemistry 2010, 49, 9498–9507. [Google Scholar] [PubMed]
- Bunting, M.; Lorant, D.E.; Bryant, A.E.; Zimmerman, G.A.; McIntyre, T.M.; Stevens, D.L.; Prescott, S.M. Alpha toxin from Clostridium perfringens induces proinflammatory changes in endothelial cells. J. Clin. Investig. 1997, 100, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Bryant, A.E.; Bayer, C.R.; Aldape, M.J.; Wallace, R.J.; Titball, R.W.; Stevens, D.L. Clostridium perfringens phospholipase C-induced platelet/leukocyte interactions impede neutrophil diapedesis. J. Med. Microbiol. 2006, 55, 495–504. [Google Scholar] [CrossRef]
- Bryant, A.E.; Chen, R.Y.; Nagata, Y.; Wang, Y.; Lee, C.H.; Finegold, S.; Guth, P.H.; Stevens, D.L. Clostridial gas gangrene. I. Cellular and molecular mechanisms of microvascular dysfunction induced by exotoxins of Clostridium perfringens. J. Infect. Dis. 2000, 182, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Xin, L.; Ertelt, J.M.; Rowe, J.H.; Jiang, T.T.; Kinder, J.M.; Chaturvedi, V.; Elahi, S.; Way, S.S. Cutting edge: Committed Th1 CD4+ T cell differentiation blocks pregnancy-induced Foxp3 expression with antigen-specific fetal loss. J. Immunol. 2014, 192, 2970–2974. [Google Scholar] [CrossRef]
- Schluter, D.; Domann, E.; Buck, C.; Hain, T.; Hof, H.; Chakraborty, T.; Deckert-Schluter, M. Phosphatidylcholine-specific phospholipase C from Listeria monocytogenes is an important virulence factor in murine cerebral listeriosis. Infect. Immun. 1998, 66, 5930–5938. [Google Scholar] [CrossRef] [PubMed]
- Beauregard, K.E.; Lee, K.D.; Collier, R.J.; Swanson, J.A. pH-dependent perforation of macrophage phagosomes by listeriolysin O from Listeria monocytogenes. J. Exp. Med. 1997, 186, 1159–1163. [Google Scholar] [CrossRef]
- Bavdek, A.; Gekara, N.O.; Priselac, D.; Gutierrez Aguirre, I.; Darji, A.; Chakraborty, T.; Macek, P.; Lakey, J.H.; Weiss, S.; Anderluh, G. Sterol and pH interdependence in the binding, oligomerization, and pore formation of Listeriolysin O. Biochemistry 2007, 46, 4425–4437. [Google Scholar]
- Chen, C.; Nguyen, B.N.; Mitchell, G.; Margolis, S.R.; Ma, D.; Portnoy, D.A. The Listeriolysin O PEST-like Sequence Co-opts AP-2-Mediated Endocytosis to Prevent Plasma Membrane Damage during Listeria Infection. Cell Host Microbe 2018, 23, 786–795.e785. [Google Scholar] [CrossRef]
- Czuczman, M.A.; Fattouh, R.; van Rijn, J.M.; Canadien, V.; Osborne, S.; Muise, A.M.; Kuchroo, V.K.; Higgins, D.E.; Brumell, J.H. Listeria monocytogenes exploits efferocytosis to promote cell-to-cell spread. Nature 2014, 509, 230–234. [Google Scholar] [CrossRef]
- Freitag, N.E.; Port, G.C.; Miner, M.D. Listeria monocytogenes—From saprophyte to intracellular pathogen. Nat. Rev. Microbiol. 2009, 7, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Sibelius, U.; Chakraborty, T.; Krogel, B.; Wolf, J.; Rose, F.; Schmidt, R.; Wehland, J.; Seeger, W.; Grimminger, F. The listerial exotoxins listeriolysin and phosphatidylinositol-specific phospholipase C synergize to elicit endothelial cell phosphoinositide metabolism. J. Immunol. 1996, 157, 4055–4060. [Google Scholar] [PubMed]
- Smith, G.A.; Marquis, H.; Jones, S.; Johnston, N.C.; Portnoy, D.A.; Goldfine, H. The two distinct phospholipases C of Listeria monocytogenes have overlapping roles in escape from a vacuole and cell-to-cell spread. Infect. Immun. 1995, 63, 4231–4237. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, G.; Ge, L.; Huang, Q.; Chen, C.; Kianian, S.; Roberts, M.F.; Schekman, R.; Portnoy, D.A. Avoidance of autophagy mediated by PlcA or ActA is required for Listeria monocytogenes growth in macrophages. Infect. Immun. 2015, 83, 2175–2184. [Google Scholar] [CrossRef] [PubMed]
- Tattoli, I.; Sorbara, M.T.; Yang, C.; Tooze, S.A.; Philpott, D.J.; Girardin, S.E. Listeria phospholipases subvert host autophagic defenses by stalling pre-autophagosomal structures. EMBO J. 2013, 32, 3066–3078. [Google Scholar] [CrossRef]
- Wadsworth, S.J.; Goldfine, H. Listeria monocytogenes phospholipase C-dependent calcium signaling modulates bacterial entry into J774 macrophage-like cells. Infect. Immun. 1999, 67, 1770–1778. [Google Scholar] [CrossRef]
- La Pietra, L.; Hudel, M.; Pillich, H.; Abu Mraheil, M.; Berisha, B.; Aden, S.; Hodnik, V.; Lochnit, G.; Rafiq, A.; Perniss, A.; et al. Phosphocholine antagonizes listeriolysin O-induced host cell responses of Listeria monocytogenes. J. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Walev, I.; Bhakdi, S.C.; Hofmann, F.; Djonder, N.; Valeva, A.; Aktories, K.; Bhakdi, S. Delivery of proteins into living cells by reversible membrane permeabilization with streptolysin-O. Proc. Natl. Acad. Sci. USA 2001, 98, 3185–3190. [Google Scholar] [CrossRef]
- Wippel, C.; Fortsch, C.; Hupp, S.; Maier, E.; Benz, R.; Ma, J.; Mitchell, T.J.; Iliev, A.I. Extracellular calcium reduction strongly increases the lytic capacity of pneumolysin from streptococcus pneumoniae in brain tissue. J. Infect. Dis. 2011, 204, 930–936. [Google Scholar] [CrossRef]
- Maurer, J.; Hupp, S.; Pillich, H.; Mitchell, T.J.; Chakraborty, T.; Iliev, A.I. Missing elimination via membrane vesicle shedding contributes to the diminished calcium sensitivity of listeriolysin O. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Radtke, A.L.; Anderson, K.L.; Davis, M.J.; DiMagno, M.J.; Swanson, J.A.; O’Riordan, M.X. Listeria monocytogenes exploits cystic fibrosis transmembrane conductance regulator (CFTR) to escape the phagosome. Proc. Natl. Acad. Sci. USA 2011, 108, 1633–1638. [Google Scholar] [CrossRef] [PubMed]
- Heffernan, B.J.; Thomason, B.; Herring-Palmer, A.; Hanna, P. Bacillus anthracis anthrolysin O and three phospholipases C are functionally redundant in a murine model of inhalation anthrax. FEMS Microbiol. Lett. 2007, 271, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Gay, A.; Rye, D.; Radhakrishnan, A. Switch-like responses of two cholesterol sensors do not require protein oligomerization in membranes. Biophys. J. 2015, 108, 1459–1469. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Weston, T.A.; He, C.; Jung, R.S.; Heizer, P.J.; Young, B.D.; Tu, Y.; Tontonoz, P.; Wohlschlegel, J.A.; Jiang, H.; et al. Release of cholesterol-rich particles from the macrophage plasma membrane during movement of filopodia and lamellipodia. eLife 2019, 8. [Google Scholar] [CrossRef]
- Hardegree, M.C.; Palmer, A.E.; Duffin, N. Tetanolysin: In-Vivo effects in animals. J. Infect. Dis. 1971, 123, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.B.; Hardegree, C.; Fornwald, R. Effect of tetanolysin on platelets and lysosomes. Infect. Immun. 1974, 9, 696–701. [Google Scholar] [CrossRef]
- Rottem, S.; Cole, R.M.; Habig, W.H.; Barile, M.F.; Hardegree, M.C. Structural characteristics of tetanolysin and its binding to lipid vesicles. J. Bacteriol. 1982, 152, 888–892. [Google Scholar]
- Allen, A.G.; Bolitho, S.; Lindsay, H.; Khan, S.; Bryant, C.; Norton, P.; Ward, P.; Leigh, J.; Morgan, J.; Riches, H.; et al. Generation and characterization of a defined mutant of Streptococcus suis lacking suilysin. Infect. Immun. 2001, 69, 2732–2735. [Google Scholar] [CrossRef]
- Lecours, M.P.; Gottschalk, M.; Houde, M.; Lemire, P.; Fittipaldi, N.; Segura, M. Critical role for Streptococcus suis cell wall modifications and suilysin in resistance to complement-dependent killing by dendritic cells. J. Infect. Dis. 2011, 204, 919–929. [Google Scholar] [CrossRef]
- Chabot-Roy, G.; Willson, P.; Segura, M.; Lacouture, S.; Gottschalk, M. Phagocytosis and killing of Streptococcus suis by porcine neutrophils. Microb. Pathog. 2006, 41, 21–32. [Google Scholar] [CrossRef]
- Segura, M.; Gottschalk, M. Streptococcus suis interactions with the murine macrophage cell line J774: Adhesion and cytotoxicity. Infect. Immun. 2002, 70, 4312–4322. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.; Hao, H.; Bi, L.; Zheng, Y.; Zhou, X.; Jiang, Y. Suilysin remodels the cytoskeletons of human brain microvascular endothelial cells by activating RhoA and Rac1 GTPase. Protein Cell 2014, 5, 261–264. [Google Scholar] [CrossRef]
- Zhang, S.; Zheng, Y.; Chen, S.; Huang, S.; Liu, K.; Lv, Q.; Jiang, Y.; Yuan, Y. Suilysin-induced Platelet-Neutrophil Complexes Formation is Triggered by Pore Formation-dependent Calcium Influx. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Feil, S.C.; Lawrence, S.; Mulhern, T.D.; Holien, J.K.; Hotze, E.M.; Farrand, S.; Tweten, R.K.; Parker, M.W. Structure of the lectin regulatory domain of the cholesterol-dependent cytolysin lectinolysin reveals the basis for its lewis antigen specificity. Structure 2012, 20, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Farrand, S.; Hotze, E.; Friese, P.; Hollingshead, S.K.; Smith, D.F.; Cummings, R.D.; Dale, G.L.; Tweten, R.K. Characterization of a streptococcal cholesterol-dependent cytolysin with a lewis y and b specific lectin domain. Biochemistry 2008, 47, 7097–7107. [Google Scholar] [CrossRef]
- Zilnyte, M.; Venclovas, C.; Zvirbliene, A.; Pleckaityte, M. The cytolytic activity of vaginolysin strictly depends on cholesterol and is potentiated by human CD59. Toxins 2015, 7, 110–128. [Google Scholar] [CrossRef]
- Lawrence, S.L.; Feil, S.C.; Holien, J.K.; Kuiper, M.J.; Doughty, L.; Dolezal, O.; Mulhern, T.D.; Tweten, R.K.; Parker, M.W. Manipulating the Lewis antigen specificity of the cholesterol-dependent cytolysin lectinolysin. Front. Immunol. 2012, 3, 330. [Google Scholar] [CrossRef]
- Feng, D.; Dai, S.; Liu, F.; Ohtake, Y.; Zhou, Z.; Wang, H.; Zhang, Y.; Kearns, A.; Peng, X.; Zhu, F.; et al. Cre-inducible human CD59 mediates rapid cell ablation after intermedilysin administration. J. Clin. Investig. 2016, 126, 2321–2333. [Google Scholar] [CrossRef][Green Version]
- Nagamune, H.; Ohkura, K.; Umezu, K.; Shouji, H.; Kourai, H. A cell membrane modification technique using domain 4 of intermedilysin for immunotherapy against cancer. Anticancer Res. 2004, 24, 3367–3372. [Google Scholar]
- Issa, E.; Salloum, T.; Tokajian, S. From Normal Flora to Brain Abscesses: A Review of Streptococcus intermedius. Front. Microbiol. 2020, 11, 826. [Google Scholar] [CrossRef]
- Mitchell, J. Streptococcus mitis: Walking the line between commensalism and pathogenesis. Mol. Oral Microbiol. 2011, 26, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Morrill, S.; Gilbert, N.M.; Lewis, A.L. Gardnerella vaginalis as a Cause of Bacterial Vaginosis: Appraisal of the Evidence From in vivo Models. Front. Cell. Infect. Microbiol. 2020, 10, 168. [Google Scholar] [CrossRef] [PubMed]
- Garcia, E.M.; Kraskauskiene, V.; Koblinski, J.E.; Jefferson, K.K. Interaction of Gardnerella vaginalis and Vaginolysin with the Apical versus Basolateral Face of a Three-Dimensional Model of Vaginal Epithelium. Infect. Immun. 2019, 87. [Google Scholar] [CrossRef]
- Randis, T.M.; Zaklama, J.; LaRocca, T.J.; Los, F.C.; Lewis, E.L.; Desai, P.; Rampersaud, R.; Amaral, F.E.; Ratner, A.J. Vaginolysin drives epithelial ultrastructural responses to Gardnerella vaginalis. Infect. Immun. 2013, 81, 4544–4550. [Google Scholar] [CrossRef] [PubMed][Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Toxin Name | Abbreviation | Organism | Diseases |
|---|---|---|---|
| Streptolysin O | SLO | Streptococcus pyogenes | Necrotizing fasciitis, septic shock, cardiomyopathy, pharyngitis |
| Pneumolysin | PLY | S. pneumoniae | Pneumonia, meningitis, otitis media |
| Perfringolysin O | PFO | Clostridium perfringens | Gas gangrene |
| Listeriolysin O | LLO | Listeria monocytogenes | Meningitis/Miscarriage |
| Suilysin | SLY | S. suis | Meningitis/septicemia |
| Anthrolysin O | ALO | Bacillus anthracis | Anthrax |
| Tetanolysin O | TLO | C. tetani | Tetanus |
| Intermedilysin | ILY | S. intermedius | Brain/liver abscess |
| Vaginolysin | VLY | Gardnerella vaginalis | Bacterial vaginosis |
| Lectinolysin | LLY | S. mitis and S. pseudopneumoniae | Endocarditis/septicemia |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thapa, R.; Ray, S.; Keyel, P.A. Interaction of Macrophages and Cholesterol-Dependent Cytolysins: The Impact on Immune Response and Cellular Survival. Toxins 2020, 12, 531. https://doi.org/10.3390/toxins12090531
Thapa R, Ray S, Keyel PA. Interaction of Macrophages and Cholesterol-Dependent Cytolysins: The Impact on Immune Response and Cellular Survival. Toxins. 2020; 12(9):531. https://doi.org/10.3390/toxins12090531
Chicago/Turabian StyleThapa, Roshan, Sucharit Ray, and Peter A. Keyel. 2020. "Interaction of Macrophages and Cholesterol-Dependent Cytolysins: The Impact on Immune Response and Cellular Survival" Toxins 12, no. 9: 531. https://doi.org/10.3390/toxins12090531
APA StyleThapa, R., Ray, S., & Keyel, P. A. (2020). Interaction of Macrophages and Cholesterol-Dependent Cytolysins: The Impact on Immune Response and Cellular Survival. Toxins, 12(9), 531. https://doi.org/10.3390/toxins12090531