α-Conotoxin Peptidomimetics: Probing the Minimal Binding Motif for Effective Analgesia

 ,
,  and
and
Abstract
1. Introduction
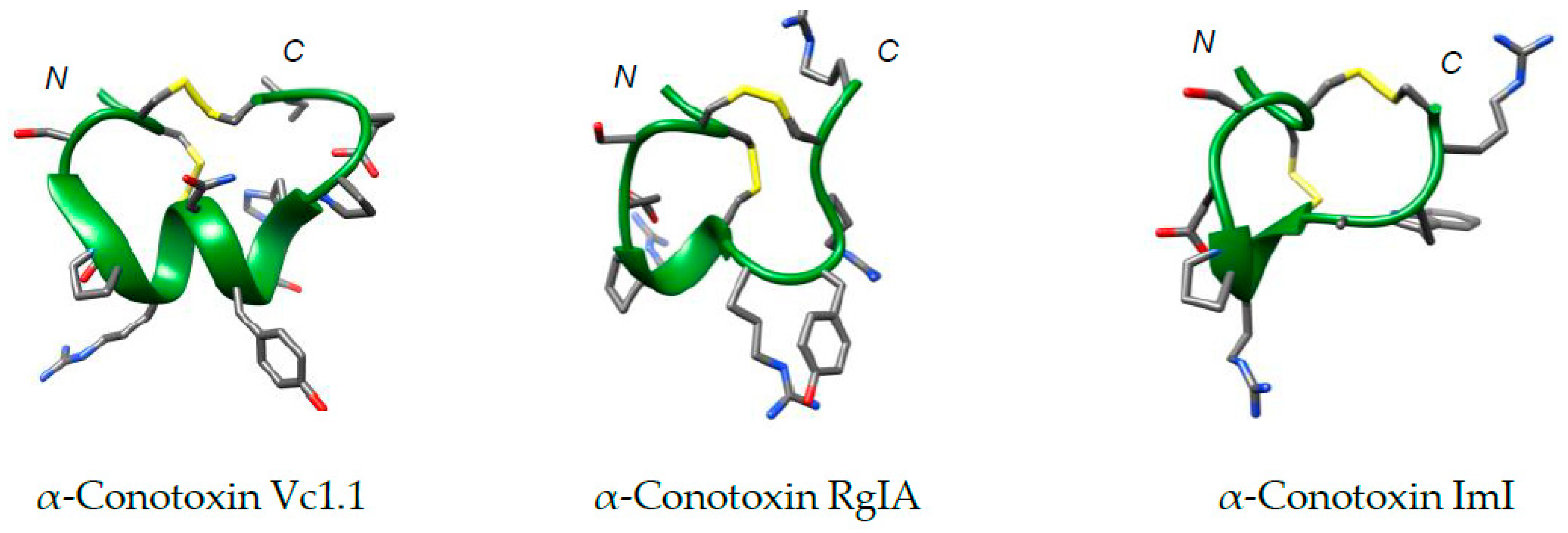
2. Therapeutic α-Conotoxin Development
3. Disulfide Replacement Strategies
3.1. The Importance of the Disulfide Bridge
3.2. Diselenide Bridges
3.3. Triazole

{kind=link}
{kind=link}
{kind=link}
| Replacement Strategy | Conotoxin (Targeted Bridge) | Primary Receptor Target | Improved In Vitro Activity? | Retained Native Structure? | Reduced Disulfide Scrambling? | Improved Plasma Stability? | Ref |
|---|---|---|---|---|---|---|---|
| Diselenide | MI (3–13) | (α1)2β1δγ nAChR | ✓ | n.r. | n.r. | n.r. | [83] |
| AuIB (2–8) | α3β4 nAChR | ✓ | ✓ a,b | ✓ | ≈ | [83] | |
| AuIB (3–15) | α3β4 nAChR | ✓ | ✓ a,b | ✓ | ≈ | [83] | |
| ImI (2–8) | α7 nAChR | ✓ | ✓ a,c | ✓ | ✓ | [79,83] | |
| ImI (3–12) | α7 nAChR | ✓ | ✓ a,c | ✓ | ✓ | [79,83] | |
| ImI (2–8,3–12) | α7 nAChR | ✓ | ✓ a,c | ✓ | ✓ | [79] | |
| Vc1.1 (2–8) | α3β4 nAChR | ✓ | n.r. | n.r. | n.r. | [83] | |
| (A10L)-PnIA (2–8) | α7 nAChR | ≈ | n.r. | n.r. | n.r. | [83] | |
| (A10L)-PnIA (3–16) | α7 nAChR | n.r. | ✓ d | n.r. | n.r. | [83] | |
| Triazole | GI (2–7) | muscle nAChR | ✗ | n.r. | ✓ i | n.r. | [98] |
| GI (3–16) | muscle nAChR | ✓ | n.r. | ✓ i | n.r. | [98] | |
| Thioether | GI (2–7,3–16) | muscle nAChR | ✗ | n.r. | ✓ i | n.r. | [101] |
| ImI (2–8) | α7 nAChR | ✗ | ✓ c | ✓ i | n.r. | [102] | |
| ImI (3–12) | α7 nAChR | ≈ | ✓ c | ✓ i | n.r. | [102] | |
| ImI (2–8,3–12) | α7 nAChR | ✗ | ✓ b | ✓ i | n.r. | [102] | |
| Lactam | (des-Glu1)-GI (2–7) | undefined | ✗ | n.r. | ✓ i | n.r | [103] |
| (des-Glu1)-GI (3–16) | undefined | ≈ | n.r. | ✓ i | n.r. | [103] | |
| SI (2–7) e | α2βγδ nAChR | ✗ | n.r. | ✓ i | n.r. | [104] | |
| SI (2–7) f | α2βγδ nAChR | ✗ | n.r. | ✓ i | n.r. | [104] | |
| SI (3–13) e | α2βγδ nAChR | ✓ | n.r. | ✓ i | n.r. | [104] | |
| SI (3–13) f | α2βγδ nAChR | ✗ | n.r. | ✓ i | n.r. | [104] | |
| Dicarba | ImI cis-(2–8) | α7 nAChR | ≈ | ✓ c | ✓ i | n.r. | [105] |
| ImI trans-(2–8) | α7 nAChR | ✗ | ✓ c | ✓ i | n.r. | [105] | |
| Vc1.1 cis-(2–8) | GABAB | ✓ g | ✓ c | ✓ i | n.r. | [84] | |
| Vc1.1 trans-(2–8) | GABAB | ✓ g | ✗ c | ✓ i | n.r. | [84] | |
| Vc1.1 cis-(3–16) | GABAB | ✗ h | ✓ b | ✓ i | n.r. | [84] | |
| Vc1.1 trans-(3–16) | GABAB | ✗ h | ✓ c | ✓ i | n.r. | [84] | |
| RgIA cis-(2–8) | GABAB | ✓ g | ✓ c | ✓ i | ✓ | [106] | |
| RgIA trans-(2–8) | GABAB | ✓ g | ✗ b | ✓ i | ✓ i | [106] | |
| RgIA cis-(3–16) | GABAB | ✗ h | ✓ c | ✓ i | ✓ i | [106] | |
| RgIA trans-(3–16) | GABAB | ✗ h | ✓ c | ✓ i | ✓ i | [106] |
3.4. Thioether
3.5. Lactam Bridge
3.6. Dicarba Bridges
3.7. Disulfide-Based Target Tunability
4. Loop I Residue Analysis
4.1. N-Terminus
4.2. Gly1
4.3. Cys2 (and Cys8)
4.4. Cys3
4.5. Ser4
4.6. Asp5
4.7. Pro6
4.8. Arg7
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lewis, R.J.; Dutertre, S.; Vetter, I.; Christie, M.J. Conus venom peptide pharmacology. Pharmacol. Rev. 2012, 64, 259–298. [Google Scholar] [CrossRef] [PubMed]
- Olivera, B.M.; Hillyard, D.R.; Marsh, M.; Yoshikami, D. Combinatorial peptide libraries in drug design: Lessons from venomous cone snails. Trends Biotechnol. 1995, 13, 422–426. [Google Scholar] [CrossRef]
- Bruce, G.L.; Ken, R.G.; Zeinab, K. Drugs from the sea: Conopeptides as potential therapeutics. Curr. Med. Chem. 2004, 11, 1715–1723. [Google Scholar] [CrossRef]
- Robinson, S.D.; Norton, R.S. Conotoxin gene superfamilies. Mar. Drugs 2014, 12, 6058–6101. [Google Scholar] [CrossRef]
- Norton, R.S.; Olivera, B.M. Conotoxins down under. Toxicon 2006, 48, 780–798. [Google Scholar] [CrossRef]
- Jin, A.-H.; Muttenthaler, M.; Dutertre, S.; Himaya, S.W.A.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Conotoxins: Chemistry and biology. Chem. Rev. 2019, 119, 11510–11549. [Google Scholar] [CrossRef]
- Akondi, K.B.; Muttenthaler, M.; Dutertre, S.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Discovery, synthesis, and structure–activity relationships of conotoxins. Chem. Rev. 2014, 114, 5815–5847. [Google Scholar] [CrossRef]
- Muttenthaler, M.; Akondi, K.; Alewood, P.F. Structure-activity studies on alpha-conotoxins. Curr. Pharm. Des. 2011, 17, 4226–4241. [Google Scholar] [CrossRef]
- Lewis, R.J.; Garcia, M.L. Therapeutic potential of venom peptides. Nat. Rev. Drug Discov. 2003, 2, 790–802. [Google Scholar] [CrossRef]
- Essack, M.; Bajic, V.B.; Archer, J.A. Conotoxins that confer therapeutic possibilities. Mar. Drugs 2012, 10, 1244–1265. [Google Scholar] [CrossRef]
- Bodil, B.C.; Richard, J.C.; Norelle, L.D.; Peta, J.H.; Quentin, K.; David, J.C. Engineering of conotoxins for the treatment of pain. Curr. Pharm. Des. 2011, 17, 4242–4253. [Google Scholar] [CrossRef]
- Fedosov, A.; Moshkovskii, S.; Kuznetsova, K.; Olivera, B. Conotoxins: From the biodiversity of gastropods to new drugs. Biochem. Mosc. Suppl. B Biomed. Chem. 2012, 6, 107–122. [Google Scholar] [CrossRef]
- Olivera, B.M.; Teichert, R.W. Diversity of the neurotoxic Conus peptides. Mol. Interv. 2007, 7, 251. [Google Scholar] [CrossRef] [PubMed]
- Han, T.S.; Teichert, R.W.; Olivera, B.M.; Bulaj, G. Conus venoms—A rich source of peptide-based therapeutics. Curr. Pharm. Des. 2008, 14, 2462–2479. [Google Scholar] [CrossRef]
- Romero, H.K.; Christensen, S.B.; Di Cesare Mannelli, L.; Gajewiak, J.; Ramachandra, R.; Elmslie, K.S.; Vetter, D.E.; Ghelardini, C.; Iadonato, S.P.; Mercado, J.L.; et al. Inhibition of α9α10 nicotinic acetylcholine receptors prevents chemotherapy-induced neuropathic pain. Proc. Natl. Acad. Sci. USA 2017, 114, E1825–E1832. [Google Scholar] [CrossRef]
- Callaghan, B.; Adams, D.J. Analgesic α-conotoxins Vc1.1 and Rg1A inhibit N-type calcium channels in sensory neurons of α9 nicotinic receptor knockout mice. Channels 2010, 4, 51–54. [Google Scholar] [CrossRef]
- Callaghan, B.; Haythornthwaite, A.; Berecki, G.; Clark, R.J.; Craik, D.J.; Adams, D.J. Analgesic α-conotoxins Vc1.1 and Rg1A inhibit N-type calcium channels in rat sensory neurons via GABAB receptor activation. J. Neurosci. 2008, 28, 10943–10951. [Google Scholar] [CrossRef]
- Cuny, H.; de Faoite, A.; Huynh, T.G.; Yasuda, T.; Berecki, G.; Adams, D.J. γ-Aminobutyric acid type B (GABAB) receptor expression is needed for inhibition of N-type (Cav2.2) calcium channels by analgesic α-conotoxins. J. Biol. Chem. 2012, 287, 23948–23957. [Google Scholar] [CrossRef]
- Adams, D.J.; Callaghan, B.; Berecki, G. Analgesic conotoxins: Block and G protein-coupled receptor modulation of N-type (CaV2.2) calcium channels. Br. J. Pharmacol. 2012, 166, 486–500. [Google Scholar] [CrossRef]
- Calver, A.R.; Medhurst, A.D.; Robbins, M.J.; Charles, K.J.; Evans, M.L.; Harrison, D.C.; Stammers, M.; Hughes, S.A.; Hervieu, G.; Couve, A.; et al. The expression of GABAB1 and GABAB2 receptor subunits in the CNS differs from that in peripheral tissues. Neuroscience 2000, 100, 155–170. [Google Scholar] [CrossRef]
- Yang, K.; Wang, D.; Li, Y.-Q. Distribution and depression of the GABAB receptor in the spinal dorsal horn of adult rat. Brain Res. Bull. 2001, 55, 479–485. [Google Scholar] [CrossRef]
- Price, G.W.; Kelly, J.S.; Bowery, N.G. The location of GABAB receptor binding sites in mammalian spinal cord. Synapse 1987, 1, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Castro, A.R.; Pinto, M.; Lima, D.; Tavares, I. Nociceptive spinal neurons expressing NK1 and GABAB receptors are located in lamina I. Brain Res. 2004, 1003, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Goudet, C.; Magnaghi, V.; Landry, M.; Nagy, F.; Gereau, R.W.; Pin, J.-P. Metabotropic receptors for glutamate and GABA in pain. Brain Res. Rev. 2009, 60, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Salio, C.; Merighi, A.; Bardoni, R. GABAB receptors-mediated tonic inhibition of glutamate release from Aβ fibers in rat laminae III/IV of the spinal cord dorsal horn. Mol. Pain 2017, 13, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Désarmenien, M.; Feltz, P.; Occhipinti, G.; Santangelo, F.; Schlichter, R. Coexistence of GABAA and GABAB receptors on Aδ and C primary afferents. Br. J. Pharmacol. 1984, 81, 327–333. [Google Scholar] [CrossRef]
- Yang, K.; Ma, H. Blockade of GABAB receptors facilitates evoked neurotransmitter release at spinal dorsal horn synapse. Neuroscience 2011, 193, 411–420. [Google Scholar] [CrossRef]
- Schmidtko, A.; Lötsch, J.; Freynhagen, R.; Geisslinger, G. Ziconotide for treatment of severe chronic pain. Lancet 2010, 375, 1569–1577. [Google Scholar] [CrossRef]
- Scott, D.A.; Wright, C.E.; Angus, J.A. Actions of intrathecal ω-conotoxins CVID, GVIA, MVIIA, and morphine in acute and neuropathic pain in the rat. Eur. J. Pharamcol. 2002, 451, 279–286. [Google Scholar] [CrossRef]
- Wang, Y.-X.; Gao, D.; Pettus, M.; Phillips, C.; Bowersox, S.S. Interactions of intrathecally administered ziconotide, a selective blocker of neuronal N-type voltage-sensitive calcium channels, with morphine on nociception in rats. Pain 2000, 84, 271–281. [Google Scholar] [CrossRef]
- Halai, R.; Craik, D.J. Conotoxins: Natural product drug leads. Nat. Prod. Rep. 2009, 26, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Norton, R.S. Enhancing the therapeutic potential of peptide toxins. Expert Opin. Drug Discov. 2017, 12, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Sandall, D.W.; Satkunanathan, N.; Keays, D.A.; Polidano, M.A.; Liping, X.; Pham, V.; Down, J.G.; Khalil, Z.; Livett, B.G.; Gayler, K.R. A novel α-conotoxin identified by gene sequencing is active in suppressing the vascular response to selective stimulation of sensory nerves in vivo. Biochemistry 2003, 42, 6904–6911. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.J.; Fischer, H.; Nevin, S.T.; Adams, D.J.; Craik, D.J. The synthesis, structural characterization, and receptor specificity of the α-conotoxin Vc1.1. J. Biol. Chem. 2006, 281, 23254–23263. [Google Scholar] [CrossRef]
- Satkunanathan, N.; Livett, B.; Gayler, K.; Sandall, D.; Down, J.; Khalil, Z. Alpha-conotoxin Vc1.1 alleviates neuropathic pain and accelerates functional recovery of injured neurones. Brain Res. 2005, 1059, 149–158. [Google Scholar] [CrossRef]
- Vincler, M.; Wittenauer, S.; Parker, R.; Ellison, M.; Olivera, B.M.; McIntosh, J.M. Molecular mechanism for analgesia involving specific antagonism of α9α10 nicotinic acetylcholine receptors. Proc. Natl. Acad. Sci. USA 2006, 103, 17880. [Google Scholar] [CrossRef]
- Metabolic. Metabolic Discontinues Clinical Trial Programme for Neuropathic Pain Drug, ACV1; Metabolic Pharmaceuticals Ltd.: Melbourne, Australia, 2007. [Google Scholar]
- Clark, R.J.; Jensen, J.; Nevin, S.T.; Callaghan, B.P.; Adams, D.J.; Craik, D.J. The engineering of an orally active conotoxin for the treatment of neuropathic pain. Angew. Chem. Int. Ed. 2010, 49, 6545–6548. [Google Scholar] [CrossRef]
- Castro, J.; Harrington, A.M.; Garcia-Caraballo, S.; Maddern, J.; Grundy, L.; Zhang, J.; Page, G.; Miller, P.E.; Craik, D.J.; Adams, D.J.; et al. α-Conotoxin Vc1.1 inhibits human dorsal root ganglion neuroexcitability and mouse colonic nociception via GABAB receptors. Gut 2017, 66, 1083. [Google Scholar] [CrossRef]
- Castro, J.; Grundy, L.; Deiteren, A.; Harrington, A.M.; O’Donnell, T.; Maddern, J.; Moore, J.; Garcia-Caraballo, S.; Rychkov, G.Y.; Yu, R.; et al. Cyclic analogues of α-conotoxin Vc1.1 inhibit colonic nociceptors and provide analgesia in a mouse model of chronic abdominal pain. Br. J. Pharmacol. 2018, 175, 2384–2398. [Google Scholar] [CrossRef]
- Ellison, M.; Haberlandt, C.; Gomez-Casati, M.E.; Watkins, M.; Elgoyhen, A.B.; McIntosh, J.M.; Olivera, B.M. α-RgIA: A novel conotoxin that specifically and potently blocks the α9α10 nAChR. Biochemistry 2006, 45, 1511–1517. [Google Scholar] [CrossRef]
- Clark, R.J.; Daly, N.L.; Halai, R.; Nevin, S.T.; Adams, D.J.; Craik, D.J. The three-dimensional structure of the analgesic α-conotoxin, RgIA. FEBS Lett. 2008, 582, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Ellison, M.; Feng, Z.-P.; Park, A.J.; Zhang, X.; Olivera, B.M.; McIntosh, J.M.; Norton, R.S. α-RgIA, a novel conotoxin that blocks the α9α10 nAChR: Structure and identification of key receptor-binding residues. J. Mol. Biol. 2008, 377, 1216–1227. [Google Scholar] [CrossRef] [PubMed]
- Halai, R.; Callaghan, B.; Daly, N.L.; Clark, R.J.; Adams, D.J.; Craik, D.J. Effects of cyclization on stability, structure, and activity of α-conotoxin RgIA at the α9α10 nicotinic acetylcholine receptor and GABAB receptor. J. Med. Chem. 2011, 54, 6984–6992. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.P.; Luginbühl, P.; Shen, G.S.; McCabe, R.T.; Stevens, R.C.; Wemmer, D.E. NMR solution structure of α-conotoxin ImI and comparison to other conotoxins specific for neuronal nicotinic acetylcholine receptors. Biochemistry 1999, 38, 3874–3882. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Christensen, S.B.; Hone, A.J.; Roux, I.; Kniazeff, J.; Pin, J.-P.; Upert, G.; Servent, D.; Glowatzki, E.; McIntosh, J.M. RgIA4 potently blocks mouse α9α10 nAChRs and provides long lasting protection against oxaliplatin-induced cold allodynia. Front. Cell. Neurosci. 2017, 11, 219. [Google Scholar] [CrossRef]
- Armishaw, C.J.; Jensen, A.A.; Balle, L.D.; Scott, K.C.M.; Sorensen, L.; Stromgaard, K. Improving the stability of α-conotoxin AuIB through N-to-C cyclization: The effect of linker length on stability and activity at nicotinic acetylcholine receptors. Antioxid. Redox Signal 2011, 14, 65–76. [Google Scholar] [CrossRef]
- Lovelace, E.S.; Gunasekera, S.; Alvarmo, C.; Clark, R.J.; Nevin, S.T.; Grishin, A.A.; Adams, D.J.; Craik, D.J.; Daly, N.L. Stabilization of α-conotoxin AuIB: Influences of disulfide connectivity and backbone cyclization. Antioxid. Redox Signal 2011, 14, 87–95. [Google Scholar] [CrossRef]
- Luo, S.; Kulak, J.M.; Cartier, G.E.; Jacobsen, R.B.; Yoshikami, D.; Olivera, B.M.; McIntosh, J.M. α-Conotoxin AuIB selectively blocks α3β4 nicotinic acetylcholine receptors and nicotine-evoked norepinephrine release. J. Neurosci. 1998, 18, 8571–8579. [Google Scholar] [CrossRef]
- Klimis, H.; Adams, D.J.; Callaghan, B.; Nevin, S.; Alewood, P.F.; Vaughan, C.W.; Mozar, C.A.; Christie, M.J. A novel mechanism of inhibition of high-voltage activated calcium channels by α-conotoxins contributes to relief of nerve injury-induced neuropathic pain. Pain 2011, 152, 259–266. [Google Scholar] [CrossRef]
- Dutton, J.L.; Bansal, P.S.; Hogg, R.C.; Adams, D.J.; Alewood, P.F.; Craik, D.J. A new level of conotoxin diversity, a non-native disulfide bond connectivity in α-conotoxin AuIB reduces structural definition but increases biological activity. J. Biol. Chem. 2002, 277, 48849–48857. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Tae, H.-S.; Huang, Y.-H.; Adams, D.J.; Craik, D.J.; Kaas, Q. Stoichiometry dependent inhibition of rat α3β4 nicotinic acetylcholine receptor by the ribbon isomer of α-conotoxin AuIB. Biochem. Pharmacol. 2018, 155, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Daly, N.L.; Callaghan, B.; Clark, R.J.; Nevin, S.T.; Adams, D.J.; Craik, D.J. Structure and activity of α-conotoxin PeIA at nicotinic acetylcholine receptor subtypes and GABAB receptor-coupled N-type calcium channels. J. Biol. Chem. 2011, 286, 10233–10237. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.M.; Plazas, P.V.; Watkins, M.; Gomez-Casati, M.E.; Olivera, B.M.; Elgoyhen, A.B. A novel α-conotoxin, PeIA, cloned from Conus pergrandis, discriminates between rat α9α10 and α7 nicotinic cholinergic receptors. J. Biol. Chem. 2005, 280, 30107–30112. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Tae, H.-S.; Xu, X.; Jiang, T.; Adams, D.J.; Yu, R. Dimerization of α-conotoxins as a strategy to enhance the inhibition of the human α7 and α9α10 nicotinic acetylcholine receptors. J. Med. Chem. 2020, 63, 2974–2985. [Google Scholar] [CrossRef] [PubMed]
- Bordas, A.; Cedillo, J.L.; Arnalich, F.; Esteban-Rodriguez, I.; Guerra-Pastrián, L.; De Castro, J.; Martín-Sánchez, C.; Atienza, G.; Fernández-Capitan, C.; Rios, J.J.; et al. Expression patterns for nicotinic acetylcholine receptor subunit genes in smoking-related lung cancers. Oncotarget 2017, 8, 67878–67890. [Google Scholar] [CrossRef]
- Yuan, D.-D.; Han, Y.-H.; Wang, C.-G.; Chi, C.-W. From the identification of gene organization of α conotoxins to the cloning of novel toxins. Toxicon 2007, 49, 1135–1149. [Google Scholar] [CrossRef]
- Carstens, B.B.; Berecki, G.; Daniel, J.T.; Lee, H.S.; Jackson, K.A.; Tae, H.S.; Sadeghi, M.; Castro, J.; O’Donnell, T.; Deiteren, A. Structure–activity studies of cysteine-rich α-conotoxins that inhibit high-voltage-activated calcium channels via GABAB receptor activation reveal a minimal functional motif. Angew. Chem. Int. Ed. 2016, 55, 4692–4696. [Google Scholar] [CrossRef]
- Sadeghi, M.; McArthur, J.R.; Finol-Urdaneta, R.K.; Adams, D.J. Analgesic conopeptides targeting G protein-coupled receptors reduce excitability of sensory neurons. Neuropharmacology 2017, 127, 116–123. [Google Scholar] [CrossRef]
- Davis, J.H.; Bradley, E.K.; Miljanich, G.P.; Nadasdi, L.; Ramachandran, J.; Basus, V.J. Solution structure of ω-conotoxin GVIA using 2-D NMR spectroscopy and relaxation matrix analysis. Biochemistry 1993, 32, 7396–7405. [Google Scholar] [CrossRef]
- Clark, R.J.; Akcan, M.; Kaas, Q.; Daly, N.L.; Craik, D.J. Cyclization of conotoxins to improve their biopharmaceutical properties. Toxicon 2012, 59, 446–455. [Google Scholar] [CrossRef] [PubMed]
- McDougal, O.M.; Granum, D.M.; Swartz, M.; Rohleder, C.; Maupin, C.M. pKa determination of histidine residues in α-conotoxin MII peptides by 1H NMR and constant pH molecular dynamics simulation. J. Phys. Chem. B 2013, 117, 2653–2661. [Google Scholar] [CrossRef] [PubMed]
- Grant, M.; Morelli, X.; Rigby, A. Conotoxins and structural biology: A prospective paradigm for drug discovery. Curr. Protein. Pept. Sci. 2004, 5, 235–248. [Google Scholar] [CrossRef]
- Bulaj, G.; Olivera, B.M. Folding of conotoxins: Formation of the native disulfide bridges during chemical synthesis and biosynthesis of Conus peptides. Antioxid. Redox Signal 2008, 10, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.-Y.; Wei, H.; Whyment, A.; Michael, N.; Robinson, A.; Adams, D.J.; Spanswick, D. Vc1.1, an α-conotoxin, reverses mechanical allodynia and inhibits ectopic discharge in neuropathic pain models. In Proceedings of the International Association for the Study of Pain (IASP), Yokohama, Japan, 26–30 September 2016; p. PW0211. [Google Scholar]
- Schmidt, B.; Hogg, P.J. Search for allosteric disulfide bonds in NMR structures. BMC Struct. Biol. 2007, 7, 49. [Google Scholar] [CrossRef]
- Chiu, J.; Hogg, P.J. Allosteric disulfides: Sophisticated molecular structures enabling flexible protein regulation. J. Biol. Chem. 2019, 294, 2949–2960. [Google Scholar] [CrossRef]
- Schmidt, B.; Ho, L.; Hogg, P.J. Allosteric disulfide bonds. Biochemistry 2006, 45, 7429–7433. [Google Scholar] [CrossRef]
- Gehrmann, J.; Alewood, P.F.; Craik, D.J. Structure determination of the three disulfide bond isomers of α-conotoxin GI: A model for the role of disulfide bonds in structural stability. J. Mol. Biol. 1998, 278, 401–415. [Google Scholar] [CrossRef]
- Fázio, M.A.; Oliveira, V.X., Jr.; Bulet, P.; Miranda, M.T.M.; Daffre, S.; Miranda, A. Structure–activity relationship studies of gomesin: Importance of the disulfide bridges for conformation, bioactivities, and serum stability. Biopolymers 2006, 84, 205–218. [Google Scholar] [CrossRef]
- Conibear, A.C.; Rosengren, K.J.; Daly, N.L.; Henriques, S.T.; Craik, D.J. The cyclic cystine ladder in θ-defensins is important for structure and stability, but not antibacterial activity. J. Biol. Chem. 2013, 288, 10830–10840. [Google Scholar] [CrossRef]
- Wouters, M.A.; Fan, S.W.; Haworth, N.L. Disulfides as redox switches: From molecular mechanisms to functional significance. Antioxid. Redox Signal 2010, 12, 53–91. [Google Scholar] [CrossRef] [PubMed]
- Hogg, P.J. Disulfide bonds as switches for protein function. Trends Biochem. Sci. 2003, 28, 210–214. [Google Scholar] [CrossRef]
- Wiedemann, C.; Kumar, A.; Lang, A.; Ohlenschläger, O. Cysteines and disulfide bonds as structure-forming units: Insights from different domains of life and the potential for characterization by NMR. Front. Chem. 2020, 8, 280. [Google Scholar] [CrossRef] [PubMed]
- Koide, T.; Itoh, H.; Otaka, A.; Yasui, H.; Kuroda, M.; Esaki, N.; Soda, K.; Fujii, N. Synthetic study on selenocystine-containing peptides. Chem. Pharm. Bull. 1993, 41, 502–506. [Google Scholar] [CrossRef]
- Besse, D.; Moroder, L. Synthesis of selenocysteine peptides and their oxidation to diselenide-bridged compounds. J. Pept. Sci. 1997, 3, 442–453. [Google Scholar] [CrossRef]
- Patora-Komisarska, K.; Jadwiga Podwysocka, D.; Seebach, D. Preparation of the β2-homoselenocysteine derivatives Fmoc-(s)-β2hSec(PMB)-OH and Boc-(s)-β2hSec(PMB)-OH for solution and solid-phase peptide synthesis. Helv. Chim. Acta 2011, 94, 1–17. [Google Scholar] [CrossRef]
- Armishaw, C.J.; Daly, N.L.; Nevin, S.T.; Adams, D.J.; Craik, D.J.; Alewood, P.F. α-Selenoconotoxins, a new class of potent α7 neuronal nicotinic receptor antagonists. J. Biol. Chem. 2006, 281, 14136–14143. [Google Scholar] [CrossRef]
- Johnson, D.S.; Martinez, J.; Elgoyhen, A.B.; Heinemann, S.F.; McIntosh, J.M. alpha-Conotoxin ImI exhibits subtype-specific nicotinic acetylcholine receptor blockade: Preferential inhibition of homomeric alpha 7 and alpha 9 receptors. Mol. Pharmacol. 1995, 48, 194. [Google Scholar]
- McIntosh, J.M.; Yoshikami, D.; Mahe, E.; Nielsen, D.B.; Rivier, J.E.; Gray, W.R.; Olivera, B.M. A nicotinic acetylcholine receptor ligand of unique specificity, alpha-conotoxin ImI. J. Biol. Chem. 1994, 269, 16733–16739. [Google Scholar]
- Armishaw, C.J.; Banerjee, J.; Ganno, M.L.; Reilley, K.J.; Eans, S.O.; Mizrachi, E.; Gyanda, R.; Hoot, M.R.; Houghten, R.A.; McLaughlin, J.P. Discovery of novel antinociceptive α-conotoxin analogues from the direct in vivo screening of a synthetic mixture-based combinatorial library. ACS Comb. Sci. 2013, 15, 153–161. [Google Scholar] [CrossRef]
- Muttenthaler, M.; Nevin, S.T.; Grishin, A.A.; Ngo, S.T.; Choy, P.T.; Daly, N.L.; Hu, S.-H.; Armishaw, C.J.; Wang, C.-I.A.; Lewis, R.J.; et al. Solving the α-conotoxin folding problem: Efficient selenium-directed on-resin generation of more potent and stable nicotinic acetylcholine receptor antagonists. J. Am. Chem. Soc. 2010, 132, 3514–3522. [Google Scholar] [CrossRef] [PubMed]
- Van Lierop, B.J.; Robinson, S.D.; Kompella, S.N.; Belgi, A.; McArthur, J.R.; Hung, A.; MacRaild, C.A.; Adams, D.J.; Norton, R.S.; Robinson, A.J. Dicarba α-conotoxin Vc1.1 analogues with differential selectivity for nicotinic acetylcholine and GABAB receptors. ACS Chem. Biol. 2013, 8, 1815–1821. [Google Scholar] [CrossRef] [PubMed]
- Azam, L.; Dowell, C.; Watkins, M.; Stitzel, J.A.; Olivera, B.M.; McIntosh, J.M. α-Conotoxin BuIA, a novel peptide from Conus bullatus, distinguishes among neuronal nicotinic acetylcholine receptors. J. Biol. Chem. 2005, 280, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Pegoraro, S.; Fiori, S.; Cramer, J.; Rudolph-Böhner, S.; Moroder, L. The disulfide-coupled folding pathway of apamin as derived from diselenide-quenched analogs and intermediates. Protein Sci. 1999, 8, 1605–1613. [Google Scholar] [CrossRef] [PubMed]
- Pegoraro, S.; Fiori, S.; Rudolph-Böhner, S.; Watanabe, T.X.; Moroder, L. Isomorphous replacement of cystine with selenocystine in endothelin: Oxidative refolding, biological and conformational properties of [Sec3,Sec11,Nle7]-endothelin-11. J. Mol. Biol. 1998, 284, 779–792. [Google Scholar] [CrossRef]
- De Araujo, A.D.; Callaghan, B.; Nevin, S.T.; Daly, N.L.; Craik, D.J.; Moretta, M.; Hopping, G.; Christie, M.J.; Adams, D.J.; Alewood, P.F. Total synthesis of the analgesic conotoxin MrVIB through selenocysteine-assisted folding. Angew. Chem. Int. Ed. 2011, 50, 6527–6529. [Google Scholar] [CrossRef]
- Walewska, A.; Zhang, M.-M.; Skalicky, J.J.; Yoshikami, D.; Olivera, B.M.; Bulaj, G. Integrated oxidative folding of cysteine/selenocysteine containing peptides: Improving chemical synthesis of conotoxins. Angew. Chem. Int. Ed. 2009, 48, 2221–2224. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Sanderson, S.J.; Mottram, J.C.; Coombs, G.H.; Meldal, M. Combinatorial library of peptidotriazoles: Identification of [1,2,3]-triazole inhibitors against a recombinant Leishmania mexicana cysteine protease. J. Comb. Chem. 2004, 6, 312–324. [Google Scholar] [CrossRef]
- Gori, A.; Gagni, P.; Rinaldi, S. Disulfide bond mimetics: Strategies and challenges. Chem. Eur. J. 2017, 23, 14987–14995. [Google Scholar] [CrossRef]
- Holland-Nell, K.; Meldal, M. Maintaining biological activity by using triazoles as disufide bond mimetics. Angew. Chem. Int. Ed. 2011, 50, 5204–5206. [Google Scholar] [CrossRef]
- Bonandi, E.; Christodoulou, M.S.; Fumagalli, G.; Perdicchia, D.; Rastelli, G.; Passarella, D. The 1, 2, 3-triazole ring as a bioisostere in medicinal chemistry. Drug Discov. Today 2017, 22, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, D.S.; Abell, A. 1,2,3-Triazoles in peptidomimetic chemistry. Eur. J. Org. Chem. 2011, 2011, 2399–2411. [Google Scholar] [CrossRef]
- Gori, A.; Wang, C.-I.A.; Harvey, P.J.; Rosengren, K.J.; Bhola, R.F.; Gelmi, M.L.; Longhi, R.; Christie, M.J.; Lewis, R.J.; Alewood, P.F.; et al. Stabilization of the cysteine-rich conotoxin MrIA by using a 1,2,3-triazole as a disulfide bond mimetic. Angew. Chem. Int. Ed. 2015, 54, 1361–1364. [Google Scholar] [CrossRef] [PubMed]
- Pacifico, S.; Kerckhoffs, A.; Fallow, A.J.; Foreman, R.E.; Guerrini, R.; McDonald, J.; Lambert, D.G.; Jamieson, A.G. Urotensin-II peptidomimetic incorporating a non-reducible 1,5-triazole disulfide bond reveals a pseudo-irreversible covalent binding mechanism to the urotensin G-protein coupled receptor. Org. Biomol. Chem. 2017, 15, 4704–4710. [Google Scholar] [CrossRef] [PubMed]
- Empting, M.; Avrutina, O.; Meusinger, R.; Fabritz, S.; Reinwarth, M.; Biesalski, M.; Voigt, S.; Buntkowsky, G.; Kolmar, H. “Triazole Bridge”: Disulfide-bond replacement by ruthenium-catalyzed formation of 1,5-disubstituted 1,2,3-triazoles. Angew. Chem. Int. Ed. 2011, 50, 5207–5211. [Google Scholar] [CrossRef] [PubMed]
- Knuhtsen, A.; Whitmore, C.; McWhinnie, F.S.; McDougall, L.; Whiting, R.; Smith, B.O.; Timperley, C.M.; Green, A.C.; Kinnear, K.I.; Jamieson, A.G. α-Conotoxin GI triazole-peptidomimetics: Potent and stable blockers of a human acetylcholine receptor. Chem. Sci. 2019, 10, 1671–1676. [Google Scholar] [CrossRef]
- Gray, W.R.; Luque, A.; Olivera, B.M.; Barrett, J.; Cruz, L.J. Peptide toxins from Conus geographus venom. J. Biol. Chem. 1981, 256, 4734–4740. [Google Scholar]
- Kobayashi, Y.; Ohkubo, T.; Kyogoku, Y.; Nishiuchi, Y.; Sakakibara, S.; Braun, W.; Go, N. Solution conformation of conotoxin GI determined by proton nuclear magnetic resonance spectroscopy and distance geometry calculations. Biochemistry 1989, 28, 4853–4860. [Google Scholar] [CrossRef]
- Bondebjerg, J.; Grunnet, M.; Jespersen, T.; Meldal, M. Solid-phase synthesis and biological activity of a thioether analogue of conotoxin GI. ChemBioChem 2003, 4, 186–194. [Google Scholar] [CrossRef]
- Dekan, Z.; Vetter, I.; Daly, N.L.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. α-Conotoxin ImI incorporating stable cystathionine bridges maintains full potency and identical three-dimensional structure. J. Am. Chem. Soc. 2011, 133, 15866–15869. [Google Scholar] [CrossRef]
- Almquist, R.G.; Kadambi, S.R.; Yasuda, D.M.; Weitl, F.L.; Polgar, W.E.; Toll, L.R. Paralytic activity of (des-Glu1) conotoxin GI analogs in the mouse diaphragm. Int. J. Pept. Protein Res. 1989, 34, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Hargittai, B.; Solé, N.A.; Groebe, D.R.; Abramson, S.N.; Barany, G. Chemical syntheses and biological activities of lactam analogues of α-conotoxin SI. J. Med. Chem. 2000, 43, 4787–4792. [Google Scholar] [CrossRef] [PubMed]
- MacRaild, C.A.; Illesinghe, J.; Van Lierop, B.J.; Townsend, A.L.; Chebib, M.; Livett, B.G.; Robinson, A.J.; Norton, R.S. Structure and activity of (2, 8)-dicarba-(3, 12)-cystino α-ImI, an α-conotoxin containing a nonreducible cystine analogue. J. Med. Chem. 2009, 52, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, S.; Belgi, A.; Bartels, P.; Van Lierop, B.J.; Robinson, S.D.; Kompella, S.N.; Hung, A.; Callaghan, B.P.; Adams, D.J.; Robinson, A.J.; et al. Dicarba analogues of α-conotoxin RgIA. Structure, stability, and activity at potential pain targets. J. Med. Chem. 2014, 57, 9933–9944. [Google Scholar] [CrossRef]
- Yu, L.; Lai, Y.; Wade, J.V.; Coutts, S.M. A simple and efficient method for the syntheses of thioether cyclic peptides. Tetrahedron Lett. 1998, 39, 6633–6636. [Google Scholar] [CrossRef]
- Adachi, Y.; Sakimura, K.; Shimizu, Y.; Nakayama, M.; Terao, Y.; Yano, T.; Asami, T. Potent and selective oxytocin receptor agonists without disulfide bridges. Biorg. Med. Chem. Lett. 2017, 27, 2331–2335. [Google Scholar] [CrossRef]
- Lebl, M.; Hruby, V.J. Synthesis of cyclic peptides by solid phase methodology. Tetrahedron Lett. 1984, 25, 2067–2068. [Google Scholar] [CrossRef]
- Mayer, J.P.; Heil, J.R.; Zhang, J.; Munson, M.C. An alternative solid-phase approach to C1-oxytocin. Tetrahedron Lett. 1995, 36, 7387–7390. [Google Scholar] [CrossRef]
- Muttenthaler, M.; Andersson, A.; De Araujo, A.D.; Dekan, Z.; Lewis, R.J.; Alewood, P.F. Modulating oxytocin activity and plasma stability by disulfide bond engineering. J. Med. Chem. 2010, 53, 8585–8596. [Google Scholar] [CrossRef]
- Procházka, Z.; Jošt, K. Synthesis of the amino-terminal decapeptide of human calcitonin, in which the disulfide bond is replaced by a thioether group. Collect. Czechoslov. Chem. Commun. 1980, 45, 1305–1314. [Google Scholar] [CrossRef]
- Knerr, P.J.; Tzekou, A.; Ricklin, D.; Qu, H.; Chen, H.; Van Der Donk, W.A.; Lambris, J.D. Synthesis and activity of thioether-containing analogues of the complement inhibitor compstatin. ACS Chem. Biol. 2011, 6, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Polinsky, A.; Cooney, M.G.; Toy-Palmer, A.; Osapay, G.; Goodman, M. Synthesis and conformational properties of the lanthionine-bridged opioid peptide [D-AlaL2, AlaL5] enkephalin as determined by NMR and computer simulations. J. Med. Chem. 1992, 35, 4185–4194. [Google Scholar] [CrossRef] [PubMed]
- Rew, Y.; Malkmus, S.; Svensson, C.; Yaksh, T.L.; Chung, N.N.; Schiller, P.W.; Cassel, J.A.; DeHaven, R.N.; Goodman, M. Synthesis and biological activities of cyclic lanthionine enkephalin analogues: δ-opioid receptor selective ligands. J. Med. Chem. 2002, 45, 3746–3754. [Google Scholar] [CrossRef] [PubMed]
- Daniel, J.T.; Clark, R.J. Molecular engineering of Conus peptides as therapeutic leads. In Peptides and Peptide-Based Biomaterials and Their Biomedical Applications; Springer: Cham, Germany, 2017; pp. 229–254. [Google Scholar]
- Houston, M.E., Jr.; Gannon, C.L.; Kay, C.M.; Hodges, R.S. Lactam bridge stabilization of α-helical peptides: Ring size, orientation and positional effects. J. Pept. Sci. 1995, 1, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.W. The synthesis and study of side-chain lactam-bridged peptides. Biopolymers 2002, 66, 49–75. [Google Scholar] [CrossRef] [PubMed]
- Khoo, K.K.; Wilson, M.J.; Smith, B.J.; Zhang, M.-M.; Gulyas, J.; Yoshikami, D.; Rivier, J.E.; Bulaj, G.; Norton, R.S. Lactam-stabilized helical analogues of the analgesic μ-conotoxin KIIIA. J. Med. Chem. 2011, 54, 7558–7566. [Google Scholar] [CrossRef] [PubMed]
- Grieco, P.; Carotenuto, A.; Patacchini, R.; Maggi, C.A.; Novellino, E.; Rovero, P. Design, synthesis, conformational analysis, and biological studies of urotensin-II lactam analogues. Biorg. Med. Chem. 2002, 10, 3731–3739. [Google Scholar] [CrossRef]
- Spinella, M.J.; Malik, A.B.; Everitt, J.; Andersen, T.T. Design and synthesis of a specific endothelin 1 antagonist: Effects on pulmonary vasoconstriction. Proc. Natl. Acad. Sci. USA 1991, 88, 7443–7446. [Google Scholar] [CrossRef]
- Thurieau, C.; Janiak, P.; Krantic, S.; Guyard, C.; Pillon, A.; Kucharczyk, N.; Vilaine, J.; Fauchere, J. A new somatostatin analog with optimized ring size inhibits neointima formation induced by balloon injury in rats without altering growth hormone release. Eur. J. Med. Chem. 1995, 30, 115–122. [Google Scholar] [CrossRef]
- Limal, D.; Briand, J.P.; Dalbon, P.; Jolivet, M. Solid-phase synthesis and on-resin cyclization of a disulfide bond peptide and lactam analogues corresponding to the major antigenic site of HIV gp41 protein. J. Pept. Res. 1998, 52, 121–129. [Google Scholar] [CrossRef]
- Gray, W.R.; Olivera, B.M.; Cruz, L.J. Peptide toxins from venomous Conus snails. Annu. Rev. Biochem. 1988, 57, 665–700. [Google Scholar] [CrossRef] [PubMed]
- Zafaralla, G.C.; Ramilo, C.; Gray, W.R.; Karlstrom, R.; Olivera, B.M.; Cruz, L.J. Phylogenetic specificity of cholinergic ligands: Alpha-conotoxin SI. Biochemistry 1988, 27, 7102–7105. [Google Scholar] [CrossRef] [PubMed]
- Benie, A.J.; Whitford, D.; Hargittai, B.; Barany, G.; Janes, R.W. Solution structure of α-conotoxin SI. FEBS Lett. 2000, 476, 287–295. [Google Scholar] [CrossRef]
- Barth, T.; Krejčí, I.; Kupková, B.; Jošt, K. Pharmacology of cyclic analogues of deamino-oxytocin not containing a disulphide bond (carba analogues). Eur. J. Pharamcol. 1973, 24, 183–188. [Google Scholar] [CrossRef]
- Keller, O.; Rudinger, J. Synthesis of [1, 6-α, α′-diaminosuberic acid]oxytocin (‘dicarba-oxytocin’). Helv. Chim. Acta 1974, 57, 1253–1259. [Google Scholar] [CrossRef]
- Gleeson, E.C.; Wang, Z.J.; Robinson, S.D.; Chhabra, S.; MacRaild, C.A.; Jackson, W.R.; Norton, R.S.; Robinson, A.J. Stereoselective synthesis and structural elucidation of dicarba peptides. Chem. Commun. 2016, 52, 4446–4449. [Google Scholar] [CrossRef]
- Miller, S.J.; Blackwell, H.E.; Grubbs, R.H. Application of ring-closing metathesis to the synthesis of rigidified amino acids and peptides. J. Am. Chem. Soc. 1996, 118, 9606–9614. [Google Scholar] [CrossRef]
- Pérez de Vega, M.J.; García-Aranda, M.I.; González-Muñiz, R. A role for ring-closing metathesis in medicinal chemistry: Mimicking secondary architectures in bioactive peptides. Med. Res. Rev. 2011, 31, 677–715. [Google Scholar] [CrossRef]
- Gleeson, E.C.; Jackson, W.R.; Robinson, A.J. Ring-closing metathesis in peptides. Tetrahedron Lett. 2016, 57, 4325–4333. [Google Scholar] [CrossRef]
- Quiram, P.A.; Sine, S.M. Structural elements in α-conotoxin ImI essential for binding to neuronal α7 receptors. J. Biol. Chem. 1998, 273, 11007–11011. [Google Scholar] [CrossRef]
- Ren, J.; Zhu, X.; Xu, P.; Li, R.; Fu, Y.; Dong, S.; Zhangsun, D.; Wu, Y.; Luo, S. D-amino acid substitution of α-conotoxin RgIA identifies its critical residues and improves the enzymatic stability. Mar. Drugs 2019, 17, 142. [Google Scholar] [CrossRef] [PubMed]
- Cai, F.; Xu, N.; Liu, Z.; Ding, R.; Yu, S.; Dong, M.; Wang, S.; Shen, J.; Tae, H.-S.; Adams, D.J. Targeting of N-type calcium channels via GABAB-receptor activation by α-conotoxin Vc1. 1 variants displaying improved analgesic activity. J. Med. Chem. 2018, 61, 10198–10205. [Google Scholar] [CrossRef] [PubMed]
- Loughnan, M.L.; Nicke, A.; Jones, A.; Adams, D.J.; Alewood, P.F.; Lewis, R.J. Chemical and functional identification and characterization of novel sulfated α-conotoxins from the cone snail conus a nemone. J. Med. Chem. 2004, 47, 1234–1241. [Google Scholar] [CrossRef] [PubMed]
- Nicke, A.; Loughnan, M.L.; Millard, E.L.; Alewood, P.F.; Adams, D.J.; Daly, N.L.; Craik, D.J.; Lewis, R.J. Isolation, structure, and activity of GID, a novel α4/7-conotoxin with an extended N-terminal sequence. J. Biol. Chem. 2003, 278, 3137–3144. [Google Scholar] [CrossRef]
- Sadeghi, M.; Carstens, B.B.; Callaghan, B.P.; Daniel, J.T.; Tae, H.-S.; O’Donnell, T.; Castro, J.; Brierley, S.M.; Adams, D.J.; Craik, D.J. Structure–activity studies reveal the molecular basis for GABAB-receptor mediated inhibition of high voltage-activated calcium channels by α-conotoxin Vc1. 1. ACS Chem. Biol. 2018, 13, 1577–1587. [Google Scholar] [CrossRef]
- Hone, A.J.; Ruiz, M.; Scadden, M.l.; Christensen, S.; Gajewiak, J.; Azam, L.; McIntosh, J.M. Positional scanning mutagenesis of α-conotoxin PeIA identifies critical residues that confer potency and selectivity for α6/α3β2β3 and α3β2 nicotinic acetylcholine receptors. J. Biol. Chem. 2013, 288, 25428–25439. [Google Scholar] [CrossRef]
- Grishin, A.A.; Cuny, H.; Hung, A.; Clark, R.J.; Brust, A.; Akondi, K.; Alewood, P.F.; Craik, D.J.; Adams, D.J. Identifying key amino acid residues that affect α-conotoxin AuIB inhibition of α3β4 nicotinic acetylcholine receptors. J. Biol. Chem. 2013, 288, 34428–34442. [Google Scholar] [CrossRef]
- McIntosh, J.M.; Azam, L.; Staheli, S.; Dowell, C.; Lindstrom, J.M.; Kuryatov, A.; Garrett, J.E.; Marks, M.J.; Whiteaker, P. Analogs of α-conotoxin MII are selective for α6-containing nicotinic acetylcholine receptors. Mol. Pharmacol. 2004, 65, 944–952. [Google Scholar] [CrossRef]
- Everhart, D.; Cartier, G.E.; Malhotra, A.; Gomes, A.V.; McIntosh, J.M.; Luetje, C.W. Determinants of potency on α-conotoxin MII, a peptide antagonist of neuronal nicotinic receptors. Biochemistry 2004, 43, 2732–2737. [Google Scholar] [CrossRef]
- Azam, L.; Maskos, U.; Changeux, J.-P.; Dowell, C.D.; Christensen, S.; Biasi, M.D.; McIntosh, J.M. α-Conotoxin BuIA[T5A;P6O]: A novel ligand that discriminates between α6β4 and α6β2 nicotinic acetylcholine receptors and blocks nicotine-stimulated norepinephrine release. FASEB J. 2010, 24, 5113–5123. [Google Scholar] [CrossRef]
- Halai, R.; Clark, R.J.; Nevin, S.T.; Jensen, J.E.; Adams, D.J.; Craik, D.J. Scanning mutagenesis of α-conotoxin Vc1. 1 reveals residues crucial for activity at the α9α10 nicotinic acetylcholine receptor. J. Biol. Chem. 2009, 284, 20275–20284. [Google Scholar] [CrossRef]
- Chu, X.; Tae, H.-S.; Xu, Q.; Jiang, T.; Adams, D.J.; Yu, R. α-Conotoxin Vc1.1 structure–activity relationship at the human α9α10 nicotinic acetylcholine receptor investigated by minimal side chain replacement. ACS Chem. Neurosci. 2019, 10, 4328–4336. [Google Scholar] [CrossRef] [PubMed]
- Armishaw, C.J.; Alewood, P.F. Conotoxins as research tools and drug leads. Curr. Protein Pept. Sci. 2005, 6, 221–240. [Google Scholar] [CrossRef] [PubMed]
- Gehrmann, J.; Daly, N.L.; Alewood, P.F.; Craik, D.J. Solution structure of α-conotoxin ImI by 1H nuclear magnetic resonance. J. Med. Chem. 1999, 42, 2364–2372. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, R.B.; DelaCruz, R.G.; Grose, J.H.; McIntosh, J.M.; Yoshikami, D.; Olivera, B.M. Critical residues influence the affinity and selectivity of α-conotoxin MI for nicotinic acetylcholine receptors. Biochemistry 1999, 38, 13310–13315. [Google Scholar] [CrossRef] [PubMed]
- Lamthanh, H.; Jegou-Matheron, C.; Servent, D.; Ménez, A.; Lancelin, J.-M. Minimal conformation of the α-conotoxin ImI for the α7 neuronal nicotinic acetylcholine receptor recognition: Correlated CD, NMR and binding studies. FEBS Lett. 1999, 454, 293–298. [Google Scholar] [CrossRef]
- Ellison, M.; McIntosh, J.M.; Olivera, B.M. α-Conotoxins ImI and ImII similar α7 nicotinic receptor antagonists act at different sites. J. Biol. Chem. 2003, 278, 757–764. [Google Scholar] [CrossRef]
- Kang, T.S.; Radić, Z.; Talley, T.T.; Jois, S.D.S.; Taylor, P.; Kini, R.M. Protein folding determinants: Structural features determining alternative disulfide pairing in α- and χ/λ-conotoxins. Biochemistry 2007, 46, 3338–3355. [Google Scholar] [CrossRef]
- Improta, R.; Benzi, C.; Barone, V. Understanding the role of stereoelectronic effects in determining collagen stability. 1. A quantum mechanical study of proline, hydroxyproline, and fluoroproline dipeptide analogues in aqueous solution. J. Am. Chem. Soc. 2001, 123, 12568–12577. [Google Scholar] [CrossRef]
- Panasik, N.; Eberhardt, E.S.; Edison, A.S.; Powell, D.R.; Raines, R.T. Inductive effects on the structure of proline residues. Int. J. Pept. Protein Res. 2018, 44, 262–269. [Google Scholar] [CrossRef]
- Mooney, S.D.; Kollman, P.A.; Klein, T.E. Conformational preferences of substituted prolines in the collagen triple helix. Biopolymers 2002, 64, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Inouye, K.; Kobayashi, Y.; Kyogoku, Y.; Kishida, Y.; Sakakibara, S.; Prockop, D.J. Synthesis and physical properties of (hydroxyproline-proline-glycine)10: Hydroxyproline in the X-position decreases the melting temperature of the collagen triple helix. Arch. Biochem. Biophys. 1982, 219, 198. [Google Scholar] [CrossRef]
- Nevin, S.T.; Clark, R.J.; Klimis, H.; Christie, M.J.; Craik, D.J.; Adams, D.J. Are α9α10 nicotinic acetylcholine receptors a pain target for α-conotoxins? Mol. Pharmacol. 2007, 72, 1406–1410. [Google Scholar] [CrossRef]
- Zhang, B.-B.; Zhao, C.; Wang, X.-S.; He, L.; Du, W.-H. Effects of 4-hydroxyproline stereochemistry on α-conotoxin solution conformation. Acta Phys-Chim. Sin. 2013, 29, 1080–1087. [Google Scholar] [CrossRef]
- Armishaw, C.; Jensen, A.A.; Balle, T.; Clark, R.J.; Harpsøe, K.; Skonberg, C.; Liljefors, T.; Strømgaard, K. Rational design of α-conotoxin analogues targeting α7 nicotinic acetylcholine receptors improved antagonistic activity by incorporation of proline derivatives. J. Biol. Chem. 2009, 284, 9498–9512. [Google Scholar] [CrossRef] [PubMed]
- Chierici, S.; Jourdan, M.; Figuet, M.; Dumy, P. A case study of 2,2-dimethylthiazolidine as locked cis proline amide bond: Synthesis, NMR and molecular modeling studies of a δ-conotoxin EVIA peptide analog. Org. Biomol. Chem. 2004, 2, 2437–2441. [Google Scholar] [CrossRef]
- Ellison, M.; Gao, F.; Wang, H.L.; Sine, S.M.; McIntosh, J.M.; Olivera, B.M. α-Conotoxins ImI and ImII target distinct regions of the human α7 nicotinic acetylcholine receptor and distinguish human nicotinic receptor subtypes. Biochemistry 2004, 43, 16019. [Google Scholar] [CrossRef]



| Conotoxin | Sequence | Analgesia | UniProt ID | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
 | |||||||||||||||||||||
| AnIB | G | G | C | C | S | H | P | A | C | A | A | N | N | Q | D | Y | C | * | n.r. | P0C1V7 | |
| AuIB | - | G | C | C | S | Y | P | P | C | F | A | T | N | P | D | - | C | * | ✓ | P56640 | |
| BuIA | - | G | C | C | S | T | P | P | C | A | V | L | Y | - | - | - | C | * | ✓ | P69657 | |
| EpI | - | G | C | C | S | D | P | R | C | N | M | N | N | P | D | Y | C | * | n.r. | P56638 | |
| GI | - | E | C | C | N | - | P | A | C | G | R | H | Y | S | - | - | C | * | ✓ | P01519 | |
| ImI | - | G | C | C | S | D | P | R | C | A | W | R | - | - | - | - | C | * | ✗ | P50983 | |
| Kn1.2 | P | G | C | C | N | N | P | A | C | V | K | H | R | - | - | - | C | G | n.r. | D4HRK7 | |
| MI | G | R | C | C | H | - | P | A | C | G | K | N | Y | S | - | - | C | * | ✓ | P01521 | |
| MII | - | G | C | C | S | N | P | V | C | H | L | E | H | S | N | L | C | * | ✓ | P56636 | |
| MrI.I | - | G | C | C | S | H | P | A | C | S | V | N | N | P | D | I | C | * | ✓ | Q6PTD1 | |
| PeIA | - | G | C | C | S | H | P | A | C | S | V | N | H | P | E | L | C | * | ✓ | Q1L777 | |
| [A10L]-PnIA | - | G | C | C | S | L | P | P | C | A | L | N | N | P | D | Y | C | * | n.r. | P50984 | |
| Pu1.2 | G | G | C | C | S | Y | P | P | C | I | A | N | N | P | L | - | C | * | ✓ | A1X8D8 | |
| Reg1d | - | G | C | C | S | D | P | R | C | K | H | E | - | - | - | - | C | * | n.r. | P85010 | |
| RgIA | - | G | C | C | S | D | P | R | C | R | Y | R | - | - | - | - | C | R | ✓ | P0C1D0 | |
| SI | - | I | C | C | N | - | P | A | C | G | P | K | Y | S | - | - | C | * | n.r. | P15471 | |
| Tx1.2 | P | Q | C | C | S | H | P | A | C | N | V | D | H | P | E | I | C | * | n.r. | P0DPL9 | |
| Vc1.1 | - | G | C | C | S | D | P | R | C | N | Y | D | H | P | E | I | C | * | ✓ | P69747 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kennedy, A.C.; Belgi, A.; Husselbee, B.W.; Spanswick, D.; Norton, R.S.; Robinson, A.J. α-Conotoxin Peptidomimetics: Probing the Minimal Binding Motif for Effective Analgesia. Toxins 2020, 12, 505. https://doi.org/10.3390/toxins12080505
Kennedy AC, Belgi A, Husselbee BW, Spanswick D, Norton RS, Robinson AJ. α-Conotoxin Peptidomimetics: Probing the Minimal Binding Motif for Effective Analgesia. Toxins. 2020; 12(8):505. https://doi.org/10.3390/toxins12080505
Chicago/Turabian StyleKennedy, Adam C., Alessia Belgi, Benjamin W. Husselbee, David Spanswick, Raymond S. Norton, and Andrea J. Robinson. 2020. "α-Conotoxin Peptidomimetics: Probing the Minimal Binding Motif for Effective Analgesia" Toxins 12, no. 8: 505. https://doi.org/10.3390/toxins12080505
APA StyleKennedy, A. C., Belgi, A., Husselbee, B. W., Spanswick, D., Norton, R. S., & Robinson, A. J. (2020). α-Conotoxin Peptidomimetics: Probing the Minimal Binding Motif for Effective Analgesia. Toxins, 12(8), 505. https://doi.org/10.3390/toxins12080505