Endothelial Damage, Inflammation and Immunity in Chronic Kidney Disease

 , , and
, , and 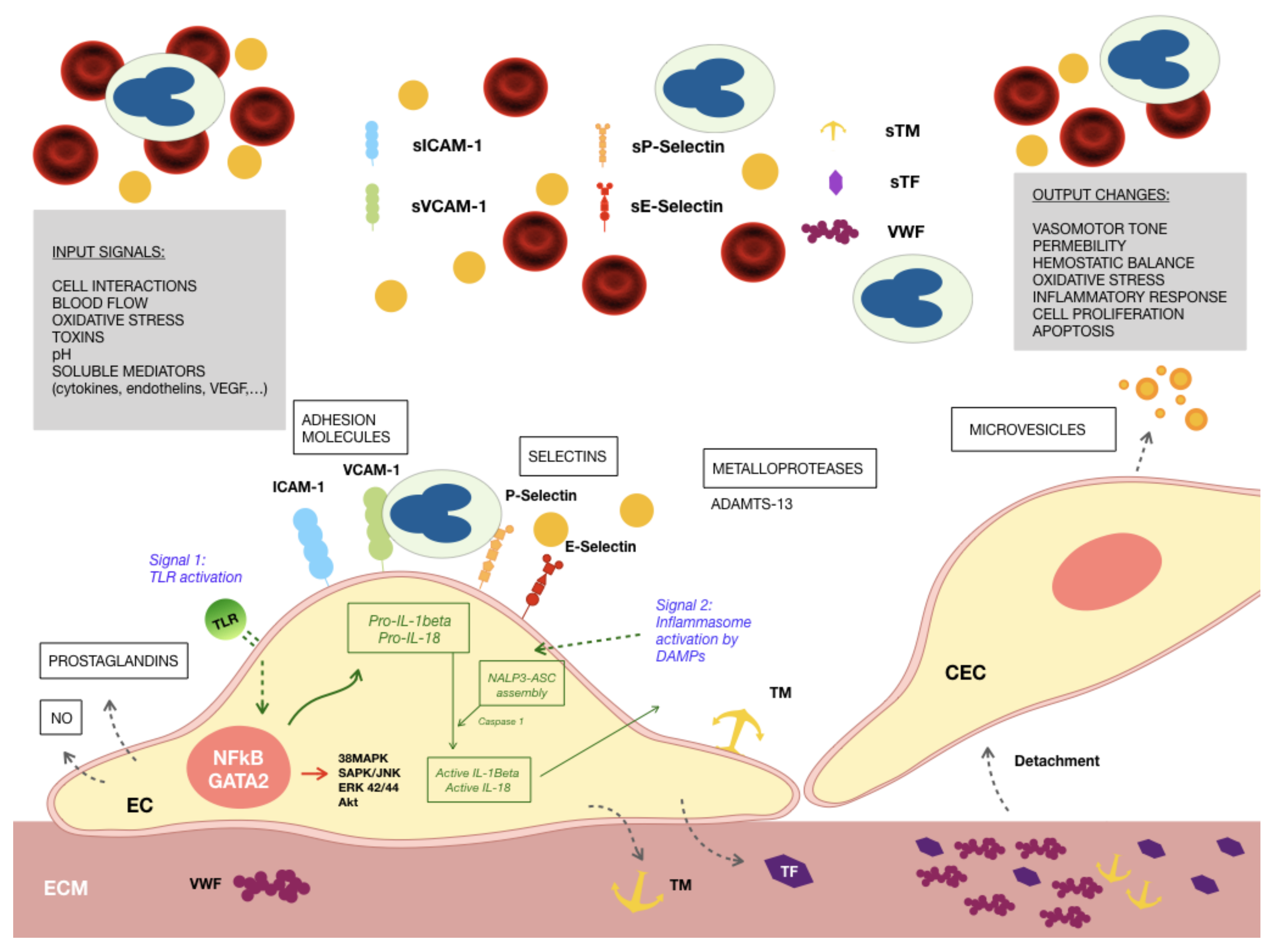{kind=link}
{kind=link}
Abstract
1. Introduction
2. Endothelial Activation and Damage in Uremia
3. The Endothelium, Inflammation and Immunity
4. Inflammasomes, TLRs, Endothelium and Chronic Kidney Disease
5. Gut Dysbiosis in CKD and Uremic Toxins: Role in Inflammation, Oxidative Stress and Endothelial Activation
6. Uremia, Platelet Dysfunction and Alterations in Immunity
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CKD | Chronic Kidney Disease |
| ESRD | End-stage renal disease |
| ED | Endothelial dysfunction |
| ECs | Endothelial cells |
| eGFR | Estimated glomerular filtration rate |
| PAMPs | Pathogen-associated molecular patterns |
| DAMPs | Damage-associated molecular patterns |
| HMGB1 | High-mobility group box 1 |
| ICAM-1 | Intercellular Adhesion Molecule 1 |
| VCAM-1 | Vascular Cell Adhesion Molecule 1 |
| MCP-1 | Monocyte chemoattractant protein-1 |
| VWF ATP | Von Willebrand Factor Adenosine 5’-triphosphate |
| LDL NET CASP1 | Low density lipoproteins Neutrophil extracellular traps Caspase-1 |
| IS | Indoxyl sulfate |
| PCS | p-Cresyl sulfate |
| IAA | Indole-3 acetic acid |
| TMAO | Trimethylamine N-oxide |
| ROS | Reactive oxygen species |
| AGEs | Advanced glycation end-products |
| AHR | Aryl hydrocarbon receptor |
| MAPK | Mitogen-activated protein kinase |
| ELAM-1 | Endothelial-leukocyte adhesion molecule |
| NFκB | Nuclear factor kappa B |
| TLR4 | Toll-like receptor 4 |
| NLRP3, NALP3 | NOD-like receptor prying domain-containing-3 |
| SIRT1 | Sirtuin 1 |
| KLF2 | Krüppel-like factor 2 |
| KLF4 | Krüppel-like factor 4 |
| eNOS | Endothelial nitric oxide synthase |
| VE-cadherin | Vascular endothelial cadherin |
| ZO1 | Zonula occludens |
| EPCs | Endothelial progenitor cells |
| MV | Microvesicles |
| EMV | Endothelial microvesicles |
| ECM | Extracellular matrix |
| TF | Tissue factor |
| IL | Interleukin |
| CECs | Circulating endothelial cells |
| PMNs | Polymorphonuclear leukocytes |
| TLRs | Toll-like receptors |
| RTT | Renal replacement therapy |
| PRRs | Pattern recognition receptors |
| ASC | Apoptosis-associated speck-like protein containing a CARD |
| TXNIP | Thioredoxin-interacting protein |
| GSDMD | Gasdermin D |
| VSMCs | Vascular smooth muscle cells |
| MMPs | Matrix metalloproteinases |
| TNF-α | Tumor necrosis factor alpha |
| LPSs | Lipopolysaccharides |
| HD | Hemodialysis |
| HDL | High-density lipoprotein |
| SCFA | Short-chain fatty acids |
| GI | Gastrointestinal tract |
| CV | Cardiovascular |
| PMPs | Platelet microparticles |
References
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.M.; Yang, C.W. Chronic kidney disease: Global dimension and perspectives. Lancet 2013, 382, 260–272. [Google Scholar] [CrossRef]
- Bikbov, B.; Purcell, C.A.; Levey, A.S.; Smith, M.; Abdoli, A.; Abebe, M.; Adebayo, O.M.; Afarideh, M.; Agarwal, S.K.; Agudelo-Botero, M.; et al. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the global burden of disease study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef]
- Levey, A.S.; Coresh, J. Chronic kidney disease. Lancet 2012, 379, 165–180. [Google Scholar] [CrossRef]
- Matsushita, K.; van der Velde, M.; Astor, B.C.; Woodward, M.; Levey, A.S.; de Jong, P.E.; Coresh, J.; Gansevoort, R.T.; El-Nahas, M.; Eckardt, K.U.; et al. Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: A collaborative meta-analysis. Lancet 2010, 375, 2073–2081. [Google Scholar] [CrossRef] [PubMed]
- Weiner, D.E.; Tighiouart, H.; Elsayed, E.F.; Griffith, J.L.; Salem, D.N.; Levey, A.S.; Sarnak, M.J. The Framingham predictive instrument in chronic kidney disease. J. Am. Coll. Cardiol. 2007, 50, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Stenvinkel, P.; Carrero, J.J.; Axelsson, J.; Lindholm, B.; Heimbürger, O.; Massy, Z. Emerging biomarkers for evaluating cardiovascular risk in the chronic kidney disease patient: How do new pieces fit into the uremic puzzle? Clin. J. Am. Soc. Nephrol. 2008, 3, 505–521. [Google Scholar] [CrossRef]
- Goligorsky, M.S. Pathogenesis of endothelial cell dysfunction in chronic kidney disease: A retrospective and what the future may hold. Kidney Res. Clin. Pr. 2015, 34, 76–82. [Google Scholar] [CrossRef]
- Cines, D.B.; Pollak, E.S.; Buck, C.A.; Loscalzo, J.; Zimmerman, G.A.; McEver, R.P.; Pober, J.S.; Wick, T.M.; Konkle, B.A.; Schwartz, B.S.; et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood 1998, 91, 3527–3561. [Google Scholar] [CrossRef]
- National Institutes of Health; National Institute of Diabetes and Digestive and Kidney; Diseases United States Renal Data System. 2018 USRDS Annual Data Report: Epidemiology of Kidney Disease in the United States; Bethesda: Rockville, MD, USA, 2018. [Google Scholar]
- Vaziri, N.D.; Pahl, M.V.; Crum, A.; Norris, K. Effect of uremia on structure and function of immune system. J. Ren. Nutr. 2012, 22, 149–156. [Google Scholar] [CrossRef]
- Kato, S.; Chmielewski, M.; Honda, H.; Pecoits-Filho, R.; Matsuo, S.; Yuzawa, Y.; Tranaeus, A.; Stenvinkel, P.; Lindholm, B. Aspects of immune dysfunction in end-stage renal disease. Clin. J. Am. Soc. Nephrol. 2008, 3, 1526–1533. [Google Scholar] [CrossRef]
- Gupta, J.; Mitra, N.; Kanetsky, P.A.; Devaney, J.; Wing, M.R.; Reilly, M.; Shah, V.O.; Balakrishnan, V.S.; Guzman, N.J.; Girndt, M.; et al. Association between albuminuria, kidney function, and inflammatory biomarker profile in CKD in CRIC. Clin. J. Am. Soc. Nephrol. 2012, 7, 1938–1946. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.I.; Stenvinkel, P.; Sonmez, A.; Saglam, M.; Yaman, H.; Kilic, S.; Eyileten, T.; Caglar, K.; Oguz, Y.; Vural, A.; et al. Vascular health, systemic inflammation and progressive reduction in kidney function; Clinical determinants and impact on cardiovascular outcomes. Nephrol. Dial. Transpl. 2011, 26, 3537–3543. [Google Scholar] [CrossRef] [PubMed]
- Recio-Mayoral, A.; Banerjee, D.; Streather, C.; Kaski, J.C. Endothelial dysfunction, inflammation and atherosclerosis in chronic kidney disease-a cross-sectional study of predialysis, dialysis and kidney-transplantation patients. Atherosclerosis 2011, 216, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Hamm, L.L.; Mohler, E.R.; Hudaihed, A.; Arora, R.; Chen, C.S.; Liu, Y.; Browne, G.; Mills, K.T.; Kleinpeter, M.A.; et al. Interrelationship of multiple endothelial dysfunction biomarkers with chronic kidney disease. PLoS ONE 2015, 10, e0132047. [Google Scholar] [CrossRef]
- Stinghen, A.E.M.; Gonçalves, S.M.; Martines, E.G.; Nakao, L.S.; Riella, M.C.; Aita, C.A.; Pecoits-Filho, R. Increased plasma and endothelial cell expression of chemokines and adhesion molecules in chronic kidney disease. Nephron Clin. Pr. 2009, 111, c117–c126. [Google Scholar] [CrossRef]
- Tumur, Z.; Shimizu, H.; Enomoto, A.; Miyazaki, H.; Niwa, T. Indoxyl sulfate upregulates expression of ICAM-1 and MCP-1 by oxidative stress-induced NF-κB activation. Am. J. Nephrol. 2010, 31, 435–441. [Google Scholar] [CrossRef]
- David, S.; Kümpers, P.; Lukasz, A.; Fliser, D.; Martens-Lobenhoffer, J.; Bode-Böger, S.M.; Kliem, V.; Haller, H.; Kielstein, J.T. Circulating angiopoietin-2 levels increase with progress of chronic kidney disease. Nephrol. Dial. Transpl. 2010, 25, 2571–2576. [Google Scholar] [CrossRef]
- Serradell, M.; Díaz-Ricart, M.; Cases, A.; Zurbano, M.J.; Aznar-Salatti, J.; López-Pedret, J.; Ordinas, A.; Escolar, G. Uremic medium disturbs the hemostatic balance of cultured human endothelial cells. Thromb. Haemost. 2001, 86, 1099–1105. [Google Scholar] [CrossRef]
- Addi, T.; Dou, L.; Burtey, S. Tryptophan-derived uremic toxins and thrombosis in chronic kidney disease. Toxins 2018, 10, 412. [Google Scholar] [CrossRef]
- Gondouin, B.; Cerini, C.; Dou, L.; Sallée, M.; Duval-Sabatier, A.; Pletinck, A.; Calaf, R.; Lacroix, R.; Jourde-Chiche, N.; Poitevin, S.; et al. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int. 2013, 84, 733–744. [Google Scholar] [CrossRef]
- Thambyrajah, J.; Landray, M.J.; McGlynn, F.J.; Jones, H.J.; Wheeler, D.C.; Townend, J.N. Abnormalities of endothelial function in patients with predialysis renal failure. Heart 2000, 83, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Van Der Vorm, L.N.; Visser, R.; Huskens, D.; Veninga, A.; Adams, D.L.; Remijn, J.A.; Hemker, H.C.; Rensma, P.L.; Van Horssen, R.; De Laat, B. Circulating active von Willebrand factor levels are increased in chronic kidney disease and end-stage renal disease. Clin. Kidney J. 2019, 13, 72–74. [Google Scholar] [CrossRef] [PubMed]
- Vila Cuenca, M.; Hordijk, P.L.; Vervloet, M.G. Most exposed: The endothelium in chronic kidney disease. Nephrol. Dial. Transpl. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zanoli, L.; Lentini, P.; Briet, M.; Castellino, P.; House, A.A.; London, G.M.; Malatino, L.; McCullough, P.A.; Mikhailidis, D.P.; Boutouyrie, P. Arterial stiffness in the heart disease of CKD. J. Am. Soc. Nephrol. 2019, 30, 918–928. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, J.B.; Zibara, K.; Issa, H.; Gross, P.; Langlet, G.; Six, I.; Choukroun, G.; Kamel, S.; Bennis, Y. Endothelial cells exposed to uremic toxins secrete interleukin-8 which promotes vascular calcifications. J. Hypertens. 2018, 36, e223–e224. [Google Scholar] [CrossRef]
- Burkhardt, D.; Bartosova, M.; Schaefer, B.; Grabe, N.; Lahrmann, B.; Nasser, H.; Freise, C.; Schneider, A.; Lingnau, A.; Degenhardt, P.; et al. Reduced microvascular density in omental biopsies of children with chronic kidney disease. PLoS ONE 2016, 11, e0166050. [Google Scholar] [CrossRef]
- Poulikakos, D.; Ross, L.; Recio-Mayoral, A.; Cole, D.; Andoh, J.; Chitalia, N.; Sharma, R.; Carlos Kaski, J.; Banerjee, D. Left ventricular hypertrophy and endothelial dysfunction in chronic kidney disease. Eur. Heart J. Cardiovasc. Imaging 2014, 15, 56–61. [Google Scholar] [CrossRef]
- Ioannou, K.; Stel, V.S.; Dounousi, E.; Jager, K.J.; Papagianni, A.; Pappas, K.; Siamopoulos, K.C.; Zoccali, C.; Tsakiris, D. Inflammation, endothelial dysfunction and increased left ventricular mass in chronic kidney disease (CKD) patients: A longitudinal study. PLoS ONE 2015, 10, e0138461. [Google Scholar] [CrossRef]
- Maaten, T.J.M.; Damman, K.; Verhaar, M.C.; Paulus, W.J.; Duncker, D.J.; Cheng, C.; van Heerebeek, L.; Hillege, H.L.; Lam, C.S.P.; Navis, G.; et al. Connecting heart failure with preserved ejection fraction and renal dysfunction: The role of endothelial dysfunction and inflammation. Eur. J. Heart Fail. 2016, 18, 588–598. [Google Scholar] [CrossRef]
- Suliman, M.E.; Qureshi, A.R.; Heimbürger, O.; Lindholm, B.; Stenvinkel, P. Soluble adhesion molecules in end-stage renal disease: A predictor of outcome. Nephrol. Dial. Transpl. 2006, 21, 1603–1610. [Google Scholar] [CrossRef]
- Stam, F.; Van Guldener, C.; Decker, A.; Dekker, J.M.; Heine, R.J.; Bouter, L.M.; Stehouwer, C.D.A. Endothelial dysfunction contributes to renal function-associated cardiovascular mortality in a population with mild renal insufficiency: The Hoorn study. J. Am. Soc. Nephrol. 2006, 17, 537–545. [Google Scholar] [CrossRef] [PubMed]
- London, G.M.; Pannier, B.; Agharazii, M.; Guerin, A.P.; Verbeke, F.H.M.; Marchais, S.J. Forearm reactive hyperemia and mortality in end-stage renal disease. Kidney Int. 2004, 65, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Ayer, A.; Mills, C.; Donovan, C.; Christenson, R.H.; Ganz, P.; Dubin, R.F. Associations of microvascular dysfunction with cardiovascular outcomes: The cardiac, endothelial function and arterial stiffness in ESRD (CERES) cohort. Hemodial. Int. 2019, 23, 58–68. [Google Scholar] [CrossRef] [PubMed]
- David, S.; John, S.G.; Jefferies, H.J.; Sigrist, M.K.; Kümpers, P.; Kielstein, J.T.; Haller, H.; McIntyre, C.W. Angiopoietin-2 levels predict mortality in CKD patients. Nephrol. Dial. Transpl. 2012, 27, 1867–1872. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Effects of shear stress on endothelial cells: Go with the flow. Acta Physiol. 2017, 219, 382–408. [Google Scholar] [CrossRef]
- Vallance, P.; Leone, A.; Calver, A.; Collier, J.; Moncada, S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet 1992, 339, 572–575. [Google Scholar] [CrossRef]
- Zoccali, C.; Bode-Böger, S.M.; Mallamaci, F.; Benedetto, F.A.; Tripepi, G.; Malatino, L.S.; Cataliotti, A.; Bellanuova, I.; Fermo, I.; Frölich, J.C.; et al. Plasma concentration of asymmetrical dimethylarginine and mortality in patients with end-stage renal disease: A prospective study. Lancet 2001, 358, 2113–2117. [Google Scholar] [CrossRef]
- Dou, L.; Jourde-Chiche, N.; Faure, V.; Cerini, C.; Berland, Y.; Dignat-George, F.; Brunet, P. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J. Thromb. Haemost. 2007, 5, 1302–1308. [Google Scholar] [CrossRef]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef]
- Matsumoto, T.; Kojima, M.; Takayanagi, K.; Taguchi, K.; Kobayashi, T. Role of S-Equol, indoxyl sulfate, and trimethylamine N-oxide on vascular function. Am. J. Hypertens. 2020. [Google Scholar] [CrossRef]
- Dou, L.; Sallée, M.; Cerini, C.; Poitevin, S.; Gondouin, B.; Jourde-Chiche, N.; Fallague, K.; Brunet, P.; Calaf, R.; Dussol, B.; et al. The cardiovascular effect of the uremic solute indole-3 acetic acid. J. Am. Soc. Nephrol. 2015, 26, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, K.; Myśliwiec, M.; Pawlak, D. Kynurenine pathway—A new link between endothelial dysfunction and carotid atherosclerosis in chronic kidney disease patients. Adv. Med. Sci. 2010, 55, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, H.; McKenzie, G.; Witting, P.K.; Stasch, J.P.; Hahn, M.; Changsirivathanathamrong, D.; Wu, B.J.; Ball, H.J.; Thomas, S.R.; et al. Kynurenine is an endothelium-derived relaxing factor produced during inflammation. Nat. Med. 2010, 16, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; De Smet, R.; Waterloos, M.A.; Van Landschoot, N.; Vogeleere, P.; Hoste, E.; Ringoir, S. Mechanisms of uremic inhibition of phagocyte reactive species production: Characterization of the role of p-cresol. Kidney Int. 1995, 47, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Meijers, B.K.I.; Van kerckhoven, S.; Verbeke, K.; Dehaen, W.; Vanrenterghem, Y.; Hoylaerts, M.F.; Evenepoel, P. The uremic retention solute p-cresyl sulfate and markers of endothelial damage. Am. J. Kidney Dis. 2009, 54, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–65. [Google Scholar] [CrossRef]
- Li, D.; Mehta, J. Oxidized LDL, a critical factor in atherogenesis. Cardiovasc. Res. 2005, 68, 353–354. [Google Scholar] [CrossRef]
- Apostolov, E.O.; Shah, S.V.; Ray, D.; Basnakian, A.G. Scavenger receptors of endothelial cells mediate the uptake and cellular proatherogenic effects of carbamylated LDL. Arter. Thromb. Vasc. Biol. 2009, 29, 1622–1630. [Google Scholar] [CrossRef]
- Tan, K.C.B.; Cheung, C.L.; Lee, A.C.H.; Lam, J.K.Y.; Wong, Y.; Shiu, S.W.M. Carbamylated lipoproteins and progression of diabetic kidney disease. Clin. J. Am. Soc. Nephrol. 2020, 15, 359–366. [Google Scholar] [CrossRef]
- D’Apolito, M.; Du, X.; Pisanelli, D.; Pettoello-Mantovani, M.; Campanozzi, A.; Giacco, F.; Maffione, A.B.; Colia, A.L.; Brownlee, M.; Giardino, I. Urea-induced ROS cause endothelial dysfunction in chronic renal failure. Atherosclerosis 2015, 239, 393. [Google Scholar] [CrossRef]
- Vera, M.; Torramade-Moix, S.; Martin-Rodriguez, S.; Cases, A.; Cruzado, J.M.; Rivera, J.; Escolar, G.; Palomo, M.; Diaz-Ricart, M. Antioxidant and anti-inflammatory strategies based on the potentiation of glutathione peroxidase activity prevent endothelial dysfunction in chronic kidney disease. Cell. Physiol. Biochem. 2018, 51, 1287–1300. [Google Scholar] [CrossRef]
- Faure, V.; Dou, L.; Sabatier, F.; Cerini, C.; Sampol, J.; Berland, Y.; Brunet, P.; Dignat-George, F. Elevation of circulating endothelial microparticles in patients with chronic renal failure. J. Thromb. Haemost. 2006, 4, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Saum, K.; Campos, B.; Celdran--Bonafonte, D.; Nayak, L.; Sangwung, P.; Thakar, C.; Roy--Chaudhury, P.; Owens, A.P. Uremic advanced glycation end products and protein-bound solutes induce endothelial dysfunction through suppression of krüppel-like factor 2. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Jourde-Chiche, N.; Dou, L.; Cerini, C.; Dignat-George, F.; Brunet, P. Vascular incompetence in dialysis patients-protein-bound uremic toxins and endothelial dysfunction. Semin. Dial. 2011, 24, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Jiang, X.; Pansuria, M.; Fang, P.; Mai, J.; Mallilankaraman, K.; Gandhirajan, R.K.; Eguchi, S.; Scalia, R.; Madesh, M.; et al. Hyperhomocysteinemia and hyperglycemia induce and potentiate endothelial dysfunction via μ-calpain activation. Diabetes 2015, 64, 947–959. [Google Scholar] [CrossRef]
- Di Marco, G.S.; König, M.; Stock, C.; Wiesinger, A.; Hillebrand, U.; Reiermann, S.; Reuter, S.; Amler, S.; Köhler, G.; Buck, F.; et al. High phosphate directly affects endothelial function by downregulating annexin II. Kidney Int. 2013, 83, 213–222. [Google Scholar] [CrossRef]
- Shuvy, M.; Abedat, S.; Eliaz, R.; Abu-Rmeileh, I.; Abu-Snieneh, A.; Ben-Dov, I.Z.; Meir, K.; Pereg, D.; Beeri, R.; Lotan, C. Hyperphosphatemia is required for initiation but not propagation of kidney failure-induced calcific aortic valve disease. Am. J. Physiol. Hear. Circ. Physiol. 2019, 317, H695–H704. [Google Scholar] [CrossRef]
- Louise, C.B.; Obrig, T.G. Shiga toxin-associated hemolytic uremic syndrome: Combined cytotoxic effects of shiga toxin and lipopolysaccharide (endotoxin) on human vascular endothelial cells in vitro. Infect. Immun. 1992, 60, 1536–1543. [Google Scholar] [CrossRef]
- Zoja, C.; Buelli, S.; Morigi, M. Shiga toxin-associated hemolytic uremic syndrome: Pathophysiology of endothelial dysfunction. Pediatr. Nephrol. 2010, 25, 2231–2240. [Google Scholar] [CrossRef]
- Van Poelgeest, E.P.; Dillingh, M.R.; de Kam, M.; Malone, K.E.; Kemper, M.; Stroes, E.S.G.; Burggraaf, J.; Moerland, M. Characterization of immune cell, endothelial, and renal responses upon experimental human endotoxemia. J. Pharm. Toxicol. Methods 2018, 89, 39–46. [Google Scholar] [CrossRef]
- Herbelin, A.; Nguyen, A.T.; Zingraff, J.; Ureña, P.; Descamps-Latscha, B. Influence of uremia and hemodialysis on circulating interleukin-1 and tumor necrosis factor α. Kidney Int. 1990, 37, 116–125. [Google Scholar] [CrossRef]
- Lin, Y.F.; Chang, D.M.; Shaio, M.F.; Lu, K.C.; Chyr, S.H.; Li, B.L.; Sheih, S. Der cytokine production during hemodialysis: Effects of dialytic membrane and complement activation. Am. J. Nephrol. 1996, 16, 293–299. [Google Scholar] [CrossRef]
- Turner, C.M.; Arulkumaran, N.; Singer, M.; Unwin, R.J.; Tam, F.W. Is the inflammasome a potential therapeutic target in renal disease? BMC Nephrol. 2014, 15, 21. [Google Scholar] [CrossRef] [PubMed]
- Martin-Rodriguez, S.; Caballo, C.; Gutierrez, G.; Vera, M.; Cruzado, J.M.; Cases, A.; Escolar, G.; Diaz-Ricart, M. TLR4 and NALP3 inflammasome in the development of endothelial dysfunction in uraemia. Eur. J. Clin. Investig. 2015, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Eloueyk, A.; Osta, B.; Alameldinne, R.; Awad, D. Uremic serum induces inflammation in cultured human endothelial cells and triggers vascular repair mechanisms. Inflammation 2019, 42, 2003–2010. [Google Scholar] [CrossRef]
- Serradell, M.; Díaz-Ricart, M.; Cases, A.; Petriz, J.; Ordinas, A.; Escolar, G. Uraemic medium accelerates proliferation but does not induce apoptosis of endothelial cells in culture. Nephrol. Dial. Transpl. 2003, 18, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Serradell, M.; Díaz-Ricart, M.; Cases, A.; Zurbano, M.J.; López-Pedret, J.; Arranz, O.; Ordinas, A.; Escolar, G. Uremic medium causes expression, redistribution and shedding of adhesion molecules in cultured endothelial cells. Haematologica 2002, 87, 1053–1061. [Google Scholar]
- Caballo, C.; Palomo, M.; Cases, A.; Galán, A.M.; Molina, P.; Vera, M.; Bosch, X.; Escolar, G.; Diaz-Ricart, M. NFκB in the development of endothelial activation and damage in uremia: An in vitro approach. PLoS ONE 2012, 7, e43374. [Google Scholar] [CrossRef]
- Shang, F.; Wang, S.C.; Hsu, C.Y.; Miao, Y.; Martin, M.; Yin, Y.; Wu, C.C.; Wang, Y.T.; Wu, G.; Chien, S.; et al. MicroRNA-92a mediates endothelial dysfunction in CKD. J. Am. Soc. Nephrol. 2017, 28, 3251–3261. [Google Scholar] [CrossRef] [PubMed]
- Carbó, C.; Arderiu, G.; Escolar, G.; Fusté, B.; Cases, A.; Carrascal, M.; Abián, J.; Díaz-Ricart, M. Differential expression of proteins from cultured endothelial cells exposed to uremic versus normal serum. Am. J. Kidney Dis. 2008, 51, 603–612. [Google Scholar] [CrossRef]
- Cardinal, H.; Raymond, M.A.; Hébert, M.J.; Madore, F. Uraemic plasma decreases the expression of ABCA1, ABCG1 and cell-cycle genes in human coronary arterial endothelial cells. Nephrol. Dial. Transpl. 2007, 22, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, G.S.; Reuter, S.; Hillebrand, U.; Amler, S.; König, M.; Larger, E.; Oberleithner, H.; Brand, E.; Pavenstädt, H.; Brand, M. The soluble VEGF receptor sFlt1 contributes to endothelial dysfunction in CKD. J. Am. Soc. Nephrol. 2009, 20, 2235–2245. [Google Scholar] [CrossRef] [PubMed]
- Buendía, P.; Carracedo, J.; Soriano, S.; Madueño, J.A.; Ortiz, A.; Martín-Malo, A.; Aljama, P.; Ramírez, R. Klotho prevents NFκB translocation and protects endothelial cell from senescence induced by uremia. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 70, 1198–1209. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Xie, R.; Yu, C.; Ma, R.; Dong, W.; Meng, H.; Zhang, Y.; Si, Y.; Zhang, Z.; Novakovic, V.; et al. Thrombotic role of blood and endothelial cells in uremia through phosphatidylserine exposure and microparticle release. PLoS ONE 2015, 10, e0142835. [Google Scholar] [CrossRef] [PubMed]
- Cuenca, M.V.; Van Bezu, J.; Beelen, R.H.J.; Vervloet, M.G.; Hordijk, P.L. Stabilization of cell-cell junctions by active vitamin D ameliorates uraemia-induced loss of human endothelial barrier function. Nephrol. Dial. Transpl. 2019, 34, 252–264. [Google Scholar] [CrossRef]
- Maciel, R.A.P.; Cunha, R.S.; Busato, V.; Franco, C.R.C.; Gregório, P.C.; Dolenga, C.J.R.; Nakao, L.S.; Massy, Z.A.; Boullier, A.; Pecoits-Filho, R.; et al. Uremia impacts ve-cadherin and zo-1 expression in human endothelial cell-to-cell junctions. Toxins 2018, 10, 404. [Google Scholar] [CrossRef]
- Addi, T.; Poitevin, S.; McKay, N.; El Mecherfi, K.E.; Kheroua, O.; Jourde-Chiche, N.; de Macedo, A.; Gondouin, B.; Cerini, C.; Brunet, P.; et al. Mechanisms of tissue factor induction by the uremic toxin indole-3 acetic acid through aryl hydrocarbon receptor/nuclear factor-kappa B signaling pathway in human endothelial cells. Arch. Toxicol. 2019, 93, 121–136. [Google Scholar] [CrossRef]
- Han, Z.; Miwa, Y.; Obikane, H.; Mitsumata, M.; Takahashi-Yanaga, F.; Morimoto, S.; Sasaguri, T. Aryl hydrocarbon receptor mediates laminar fluid shear stress-induced CYP1A1 activation and cell cycle arrest in vascular endothelial cells. Cardiovasc. Res. 2007, 77, 809–818. [Google Scholar] [CrossRef]
- Lano, G.; Laforêt, M.; Von Kotze, C.; Perrin, J.; Addi, T.; Brunet, P.; Poitevin, S.; Burtey, S.; Dou, L. Aryl hydrocarbon receptor activation and tissue factor induction by fluid shear stress and indoxyl sulfate in endothelial cells. Int. J. Mol. Sci. 2020, 21, 2392. [Google Scholar] [CrossRef]
- Koç, M.; Bihorac, A.; Segal, M.S. Circulating endothelial cells as potential markers of the state of the endothelium in hemodialysis patients. Am. J. Kidney Dis. 2003, 42, 704–712. [Google Scholar] [CrossRef]
- Koc, M.; Richards, H.B.; Bihorac, A.; Ross, E.A.; Schold, J.D.; Segal, M.S. Circulating endothelial cells are associated with future vascular events in hemodialysis patients. Kidney Int. 2005, 67, 1078–1083. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Kim, K.L.; Huh, W.; Kim, B.; Byun, J.; Suh, W.; Sung, J.; Jeon, E.S.; Oh, H.Y.; Kim, D.K. Decreased number and impaired angiogenic function of endothelial progenitor cells in patients with chronic renal failure. Arter. Thromb. Vasc. Biol. 2004, 24, 1246–1252. [Google Scholar] [CrossRef] [PubMed]
- Soriano, S.; Carmona, A.; Triviño, F.; Rodriguez, M.; Alvarez-Benito, M.; Martín-Malo, A.; Alvarez-Lara, M.A.; Ramírez, R.; Aljama, P.; Carracedo, J. Endothelial damage and vascular calcification in patients with chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2014, 307, F1302–F1311. [Google Scholar] [CrossRef] [PubMed]
- Krenning, G.; Dankers, P.Y.W.; Drouven, J.W.; Waanders, F.; Franssen, C.F.M.; Van Luyn, M.J.A.; Harmsen, M.C.; Popa, E.R. Endothelial progenitor cell dysfunction in patients with progressive chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2009, 296, F1314–F1322. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.L.; Leu, J.G.; Liu, W.C.; Zheng, C.M.; Lin, Y.F.; Shyu, J.F.; Wu, C.C.; Lu, K.C. Endothelial progenitor cells predict long-term mortality in hemodialysis patients. Int. J. Med. Sci. 2016, 13, 240–247. [Google Scholar] [CrossRef]
- Lee, H.J.; Kim, W.; Kim, W.S.; Woo, J.S.; Kim, Y.G.; Moon, J.Y.; Lee, S.H.; Ihm, C.G.; Lee, T.W.; Jeong, K.H. Circulating endothelial progenitor cell levels predict cardiovascular events in end-stage renal disease patients on maintenance hemodialysis. Nephron 2015, 130, 151–158. [Google Scholar] [CrossRef]
- Lorenzen, J.; David, S.; Bahlmann, F.H.; Groot, K.; Bahlmann, E.; Kielstein, J.T.; Haller, H.; Fliser, D. Endothelial progenitor cells and cardiovascular events in patients with chronic kidney disease-A prospective follow-up study. PLoS ONE 2010, 5, e11477. [Google Scholar] [CrossRef]
- Ridger, V.C.; Boulanger, C.M.; Angelillo-Scherrer, A.; Badimon, L.; Blanc-Brude, O.; Bochaton-Piallat, M.L.; Boilard, E.; Buzas, E.I.; Caporali, A.; Dignat-George, F.; et al. Microvesicles in vascular homeostasis and diseases position paper of the european society of cardiology (ESC) working group on atherosclerosis and vascular biology. Thromb. Haemost. 2017, 117, 1296–1316. [Google Scholar] [CrossRef]
- Trappenburg, M.C.; Van Schilfgaarde, M.; Frerichs, F.C.P.; Spronk, H.M.H.; Ten Cate, H.; De Fijter, C.W.H.; Terpstra, W.E.; Leyte, A. Chronic renal failure is accompanied by endothelial activation and a large increase in microparticle numbers with reduced procoagulant capacity. Nephrol. Dial. Transpl. 2012, 27, 1446–1453. [Google Scholar] [CrossRef]
- Burton, J.O.; Hamali, H.A.; Singh, R.; Abbasian, N.; Parsons, R.; Patel, A.K.; Goodall, A.H.; Brunskill, N.J. Elevated levels of procoagulant plasma microvesicles in dialysis patients. PLoS ONE 2013, 8, e72663. [Google Scholar] [CrossRef]
- Amabile, N.; Guérin, A.P.; Tedgui, A.; Boulanger, C.M.; London, G.M. Predictive value of circulating endothelial microparticles for cardiovascular mortality in end-stage renal failure: A pilot study. Nephrol. Dial. Transpl. 2012, 27, 1873–1880. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.J.; Cai, G.Y.; Chen, X.M. Cellular senescence, senescence-associated secretory phenotype, and chronic kidney disease. Oncotarget 2017, 8, 64520–64533. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Dejana, E.; Fiocchi, C. Immune regulation by microvascular endothelial cells: Directing innate and adaptive immunity, coagulation, and inflammation. J. Immunol. 2007, 178, 6017–6022. [Google Scholar] [CrossRef] [PubMed]
- Marelli-Berg, F.M.; Jarmin, S.J. Antigen presentation by the endothelium: A green light for antigen-specific T cell trafficking? Immunol. Lett. 2004, 93, 109–113. [Google Scholar] [CrossRef]
- Granger, D.N.; Kubes, P. The microcirculation and inflammation: Modulation of leukocyte-endothelial cell adhesion. J. Leukoc. Biol. 1994, 55, 662–675. [Google Scholar] [CrossRef]
- Massberg, S.; Enders, G.; Leiderer, R.; Eisenmeng’Er, S.; Vestweber, D.; Krombach, F.; Messmer, K. Platelet-endothelial cell interactions during ischemia/reperfusion: The role of P-selectin. Blood 1998, 92, 507–515. [Google Scholar] [CrossRef]
- Weyrich, A.S.; Zimmerman, G.A. Platelets: Signaling cells in the immune continuum. Trends Immunol. 2004, 25, 489–495. [Google Scholar] [CrossRef]
- Kurts, C.; Panzer, U.; Anders, H.J.; Rees, A.J. The immune system and kidney disease: Basic concepts and clinical implications. Nat. Rev. Immunol. 2013, 13, 738–753. [Google Scholar] [CrossRef]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef]
- Schroder, K.; Zhou, R.; Tschopp, J. The NLRP3 inflammasome: A sensor for metabolic danger? Science 2010, 327, 296–300. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Kasza, A. IL-1 and EGF regulate expression of genes important in inflammation and cancer. Cytokine 2013, 62, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Komada, T.; Muruve, D.A. The role of inflammasomes in kidney disease. Nat. Rev. Nephrol. 2019, 15, 501–520. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K + efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef]
- Gong, T.; Yang, Y.; Jin, T.; Jiang, W.; Zhou, R. Orchestration of NLRP3 inflammasome activation by ion fluxes. Trends Immunol. 2018, 39, 393–406. [Google Scholar] [CrossRef]
- Pétrilli, V.; Papin, S.; Dostert, C.; Mayor, A.; Martinon, F.; Tschopp, J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007, 14, 1583–1589. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–226. [Google Scholar] [CrossRef]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.-L.L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 2015, 22, 1111–1129. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.J.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef]
- Kinnunen, K.; Piippo, N.; Loukovaara, S.; Hytti, M.; Kaarniranta, K.; Kauppinen, A. Lysosomal destabilization activates the NLRP3 inflammasome in human umbilical vein endothelial cells (HUVECs). J. Cell Commun. Signal. 2017, 11, 275–279. [Google Scholar] [CrossRef]
- Yu, G.; Bai, Z.; Chen, Z.; Chen, H.; Wang, G.; Wang, G.; Liu, Z. The NLRP3 inflammasome is a potential target of ozone therapy aiming to ease chronic renal inflammation in chronic kidney disease. Int. Immunopharmacol. 2017, 43, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.; Zhang, X.Y.; Edfeldt, K.; Lundberg, A.M.; Levin, M.C.; Borén, J.; Li, W.; Yuan, X.M.; Folkersen, L.; Eriksson, P.; et al. Innate immune receptor NOD2 promotes vascular inflammation and formation of lipid-rich necrotic cores in hypercholesterolemic mice. Eur. J. Immunol. 2014, 44, 3081–3092. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.; Yang, X.; Yan, Z.; Zhao, M.; Yue, X.; Cheng, X.; Zheng, Z.; Guan, K.; Dou, J.; Xu, T.; et al. Nalp3 inflammasome is activated and required for vascular smooth muscle cell calcification. Int. J. Cardiol. 2013, 168, 2242–2247. [Google Scholar] [CrossRef] [PubMed]
- Spillmann, F.; De Geest, B.; Muthuramu, I.; Amin, R.; Miteva, K.; Pieske, B.; Tschöpe, C.; Van Linthout, S. Apolipoprotein A-I gene transfer exerts immunomodulatory effects and reduces vascular inflammation and fibrosis in ob/ob mice. J. Inflamm. 2016, 13, 25. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Xing, S.; Gong, Z.; Xing, Q. NLRP3 inflammasomes show high expression in Aorta of patients with atherosclerosis. Hear. Lung Circ. 2013, 22, 746–750. [Google Scholar] [CrossRef] [PubMed]
- Loppnow, H.; Libby, P. Proliferating or interleukin 1-activated human vascular smooth muscle cells secrete copious interleukin 6. J. Clin. Investig. 1990, 85, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Lüscher, T.F. Anti-inflammatory therapies for cardiovascular disease. Eur. Heart J. 2014, 35, 1782–1791. [Google Scholar] [CrossRef]
- Kirii, H.; Niwa, T.; Yamada, Y.; Wada, H.; Saito, K.; Iwakura, Y.; Asano, M.; Moriwaki, H.; Seishima, M. Lack of interleukin-1ß decreases the severity of atherosclerosis in apoE-deficient mice. Arter. Thromb. Vasc. Biol. 2003, 23, 656–660. [Google Scholar] [CrossRef]
- Bevilacqua, M.P.; Pober, J.S.; Wheeler, M.E.; Cotran, R.S.; Gimbrone, M.A. Interleukin 1 acts on cultured human vascular endothelium to increase the adhesion of polymorphonuclear leukocytes, monocytes, and related leukocyte cell lines. J. Clin. Investig. 1985, 76, 2003–2011. [Google Scholar] [CrossRef]
- Barnes, M.J.; Farndale, R.W. Collagens and atherosclerosis. Exp. Gerontol. 1999, 34, 513–525. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Vascular smooth muscle cell in atherosclerosis. Acta Physiol. 2015, 214, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Warner, S.J.C.; Friedman, G.B. Interleukin 1: A mitogen for human vascular smooth muscle cells that induces the release of growth-inhibitory prostanoids. J. Clin. Investig. 1988, 81, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Y.; Xia, M.; Gulbins, E.; Boini, K.M.; Li, P.L. Activation of Nlrp3 inflammasomes enhances macrophage lipid-deposition and migration: Implication of a novel role of inflammasome in atherogenesis. PLoS ONE 2014, 9, e87552. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Kong, L.; Kang, J.; Vaughn, D.M.; Bush, G.D.; Walding, A.L.; Grigorian, A.A.; Robinson, J.S.; Nakayama, D.K. Interleukin-lβ induces migration of rat arterial smooth muscle cells through a mechanism involving increased matrix metalloproteinase-2 activity. J. Surg. Res. 2011, 169, 328–336. [Google Scholar] [CrossRef]
- Libby, P. Interleukin-1 beta as a target for atherosclerosis therapy: Biological basis of CANTOS and beyond. J. Am. Coll. Cardiol. 2017, 70, 2278–2289. [Google Scholar] [CrossRef]
- Miteva, K.; Madonna, R.; De Caterina, R.; Van Linthout, S. Innate and adaptive immunity in atherosclerosis. Vasc. Pharm. 2018, 107, 67–77. [Google Scholar] [CrossRef]
- Hutton, H.L.; Ooi, J.D.; Holdsworth, S.R.; Kitching, A.R. The NLRP3 inflammasome in kidney disease and autoimmunity. Nephrology 2016, 21, 736–744. [Google Scholar] [CrossRef]
- Vilaysane, A.; Chun, J.; Seamone, M.E.; Wang, W.; Chin, R.; Hirota, S.; Li, Y.; Clark, S.A.; Tschopp, J.; Trpkov, K.; et al. The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J. Am. Soc. Nephrol. 2010, 21, 1732–1744. [Google Scholar] [CrossRef]
- Anders, H.J.; Muruve, D.A. The inflammasomes in kidney disease. J. Am. Soc. Nephrol. 2011, 22, 1007–1018. [Google Scholar] [CrossRef]
- Li, L.; Tang, W.; Yi, F. Role of inflammasome in chronic kidney disease. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2019; Volume 1165, pp. 407–421. [Google Scholar]
- Gauer, S.; Sichler, O.; Obermüller, N.; Holzmann, Y.; Kiss, E.; Sobkowiak, E.; Pfeilschifter, J.; Geiger, H.; Mühl, H.; Hauser, I.A. IL-18 is expressed in the intercalated cell of human kidney. Kidney Int. 2007, 72, 1081–1087. [Google Scholar] [CrossRef]
- Shahzad, K.; Bock, F.; Dong, W.; Wang, H.; Kopf, S.; Kohli, S.; Al-Dabet, M.M.; Ranjan, S.; Wolter, J.; Wacker, C.; et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015, 87, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Guo, H.; Xu, C.; Wang, B.; Zhang, M.; Ding, F. Mitochondrial reactive oxygen species-mediated NLRP3 inflammasome activation contributes to aldosterone-induced renal tubular cells injury. Oncotarget 2016, 7, 17479–17491. [Google Scholar] [CrossRef]
- Kahlenberg, J.M.; Carmona-Rivera, C.; Smith, C.K.; Kaplan, M.J. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J. Immunol. 2013, 190, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Granata, S.; Masola, V.; Zoratti, E.; Scupoli, M.T.; Baruzzi, A.; Messa, M.; Sallustio, F.; Gesualdo, L.; Lupo, A.; Zaza, G. NLRP3 inflammasome activation in dialyzed chronic kidney disease patients. PLoS ONE 2015, 10, e0122272. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16, 343–353. [Google Scholar] [CrossRef]
- Girndt, M.; Sester, M.; Sester, U.; Kaul, H.; Köhler, H. Defective expression of B7-2 (CD86) on monocytes of dialysis patients correlates to the uremia-associated immune defect. Kidney Int. 2001, 59, 1382–1389. [Google Scholar] [CrossRef]
- Ando, M.; Shibuya, A.; Tsuchiya, K.; Akiba, T.; Nitta, K. Reduced expression of Toll-like receptor 4 contributes to impaired cytokine response of monocytes in uremic patients. Kidney Int. 2006, 70, 358–362. [Google Scholar] [CrossRef]
- Kuroki, Y.; Tsuchida, K.; Go, I.; Aoyama, M.; Naganuma, T.; Takemoto, Y.; Nakatani, T. A study of innate immunity in patients with end-stage renal disease: Special reference to toll-like receptor-2 and-4 expression in peripheral blood monocytes of hemodialysis patients. Int. J. Mol. Med. 2007, 19, 783–790. [Google Scholar] [CrossRef]
- Grabulosa, C.C.; Manfredi, S.R.; Canziani, M.E.; Quinto, B.M.R.; Barbosa, R.B.; Rebello, J.F.; Batista, M.C.; Cendoroglo, M.; Dalboni, M.A. Chronic kidney disease induces inflammation by increasing Toll-like receptor-4, cytokine and cathelicidin expression in neutrophils and monocytes. Exp. Cell Res. 2018, 365, 157–162. [Google Scholar] [CrossRef]
- Raby, A.C.; González-Mateo, G.T.; Williams, A.; Topley, N.; Fraser, D.; López-Cabrera, M.; Labéta, M.O. Targeting toll-like receptors with soluble toll-like receptor 2 prevents peritoneal dialysis solution-induced fibrosis. Kidney Int. 2018, 94, 346–362. [Google Scholar] [CrossRef] [PubMed]
- Verzola, D.; Bonanni, A.; Sofia, A.; Montecucco, F.; D’Amato, E.; Cademartori, V.; Parodi, E.L.; Viazzi, F.; Venturelli, C.; Brunori, G.; et al. Toll-like receptor 4 signalling mediates inflammation in skeletal muscle of patients with chronic kidney disease. J. Cachexia. Sarcopenia Muscle 2017, 8, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.B.; Zhang, Y.; Boini, K.M.; Koka, S. High mobility group box 1 mediates TMAO-induced endothelial dysfunction. Int. J. Mol. Sci. 2019, 20, 3570. [Google Scholar] [CrossRef]
- Chen, Y.; Pitzer, A.L.; Li, X.; Li, P.-L.L.; Wang, L.; Zhang, Y. Instigation of endothelial Nlrp3 inflammasome by adipokine visfatin promotes inter-endothelial junction disruption: Role of HMGB1. J. Cell. Mol. Med. 2015, 19, 2715–2727. [Google Scholar] [CrossRef] [PubMed]
- Speer, T.; Rohrer, L.; Blyszczuk, P.; Shroff, R.; Kuschnerus, K.; Kränkel, N.; Kania, G.; Zewinger, S.; Akhmedov, A.; Shi, Y.; et al. Abnormal high-density lipoprotein induces endothelial dysfunction via activation of toll-like receptor-2. Immunity 2013, 38, 754–768. [Google Scholar] [CrossRef] [PubMed]
- Iwami, K.; Matsuguchi, T.; Masuda, A.; Kikuchi, T.; Musikacharoen, T.; Yoshikai, Y. Cutting edge: Naturally occurring soluble form of mouse toll-like receptor 4 inhibits lipopolysaccharide signaling. J. Immunol. 2000, 165, 6682–6686. [Google Scholar] [CrossRef] [PubMed]
- Esposito, P.; La Porta, E.; Grignano, M.A.; Verzola, D.; Milanesi, S.; Ansaldo, F.; Gregorini, M.; Libetta, C.; Garibotto, G.; Rampino, T. Soluble toll-like receptor 4: A new player in subclinical inflammation and malnutrition in hemodialysis patients. J. Ren. Nutr. 2018, 28, 259–264. [Google Scholar] [CrossRef]
- Lynch, S.V.; Pedersen, O. The human intestinal microbiome in health and disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef]
- Cigarran, S.; González, E.; Cases, A. Microbiota intestinal en la enfermedad renal crónica. Nefrologia 2017, 37, 9–19. [Google Scholar] [CrossRef]
- Anders, H.J.; Andersen, K.; Stecher, B. The intestinal microbiota, a leaky gut, and abnormal immunity in kidney disease. Kidney Int. 2013, 83, 1010–1016. [Google Scholar] [CrossRef]
- Magnusson, M.; Magnusson, K.E.; Sundqvist, T.; Denneberg, T. Impaired intestinal barrier function measured by differently sized polyethylene glycols in patients with chronic renal failure. Gut 1991, 32, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Yuan, J.; Norris, K. Role of urea in intestinal barrier dysfunction and disruption of epithelial tight junction in chronic kidney disease. Am. J. Nephrol. 2013, 37, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Zhao, Y.Y.; Pahl, M.V. Altered intestinal microbial flora and impaired epithelial barrier structure and function in CKD: The nature, mechanisms, consequences and potential treatment. Nephrol. Dial. Transpl. 2016, 31, 737–746. [Google Scholar] [CrossRef] [PubMed]
- De Almeida-Duarte, J.B.; De Aguilar-Nascimento, J.E.; Nascimento, M.; Nochi, R.J. Bacterial translocation in experimental uremia. Urol. Res. 2004, 32, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Jiang, H.; Shi, K.; Ren, Y.; Zhang, P.; Cheng, S. Gut bacterial translocation is associated with microinflammation in end-stage renal disease patients. Nephrology 2012, 17, 733–738. [Google Scholar] [CrossRef]
- Shi, K.; Wang, F.; Jiang, H.; Liu, H.; Wei, M.; Wang, Z.; Xie, L. Gut bacterial translocation may aggravate microinflammation in hemodialysis patients. Dig. Dis. Sci. 2014, 59, 2109–2117. [Google Scholar] [CrossRef]
- McIntyre, C.W.; Harrison, L.E.A.; Eldehni, M.T.; Jefferies, H.J.; Szeto, C.C.; John, S.G.; Sigrist, M.K.; Burton, J.O.; Hothi, D.; Korsheed, S.; et al. Circulating endotoxemia: A novel factor in systemic inflammation and cardiovascular disease in chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 133–141. [Google Scholar] [CrossRef]
- Olivier, V.; Dunyach-Remy, C.; Corbeau, P.; Cristol, J.-P.; Sutra, T.; Burtey, S.; Lavigne, J.-P.; Moranne, O. Factors of microinflammation in non-diabetic chronic kidney disease: A pilot study. BMC Nephrol. 2020, 21, 141. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Wong, J.; Pahl, M.; Piceno, Y.M.; Yuan, J.; Desantis, T.Z.; Ni, Z.; Nguyen, T.H.; Andersen, G.L. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013, 83, 308–315. [Google Scholar] [CrossRef]
- Wong, J.; Piceno, Y.M.; DeSantis, T.Z.; Pahl, M.; Andersen, G.L.; Vaziri, N.D. Expansion of urease-and uricase-containing, indole-and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am. J. Nephrol. 2014, 39, 230–237. [Google Scholar] [CrossRef]
- Jiang, S.; Xie, S.; Lv, D.; Zhang, Y.; Deng, J.; Zeng, L.; Chen, Y. A reduction in the butyrate producing species Roseburia spp. and Faecalibacterium prausnitzii is associated with chronic kidney disease progression. Antonie Van Leeuwenhoek Int. J. Gen. Mol. Microbiol. 2016, 109, 1389–1396. [Google Scholar] [CrossRef] [PubMed]
- Meijers, B.; Evenepoel, P.; Anders, H.J. Intestinal microbiome and fitness in kidney disease. Nat. Rev. Nephrol. 2019, 15, 531–545. [Google Scholar] [CrossRef] [PubMed]
- Lano, G.; Burtey, S.; Sallée, M. Indoxyl sulfate, a uremic endotheliotoxin. Toxins 2020, 12, 229. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Yoo, T.H.; Hwang, Y.; Lee, G.H.; Kim, B.; Jang, J.; Yu, H.T.; Kim, M.C.; Cho, J.Y.; Lee, C.J.; et al. Indoxyl sulfate (IS)-mediated immune dysfunction provokes endothelial damage in patients with end-stage renal disease (ESRD). Sci. Rep. 2017, 7, 3057. [Google Scholar] [CrossRef]
- Chen, S.C.; Huang, S.Y.; Wu, C.C.; Hsu, C.F. P-cresylsulfate, the protein-bound uremic toxin, increased endothelial permeability partly mediated by src-induced phosphorylation of VE-cadherin. Toxins 2020, 12, 62. [Google Scholar] [CrossRef]
- Koka, S.; Xia, M.; Chen, Y.; Li, P.-L.; Boini, K.M. Trimethylamine-N-oxide, an intestinal microbial metabolite instigates NLRP3 inflammasome activation and endothelial dysfunction. FASEB J. 2016, 30, 1204.10. [Google Scholar] [CrossRef]
- Yang, S.; Li, X.; Yang, F.; Zhao, R.; Pan, X.; Liang, J.; Tian, L.; Li, X.; Liu, L.; Xing, Y.; et al. Gut microbiota-dependent marker TMAO in promoting cardiovascular disease: Inflammation mechanism, clinical prognostic, and potential as a therapeutic target. Front. Pharm. 2019, 10. [Google Scholar] [CrossRef]
- Escolar, G.; Díaz-Ricart, M.; Cases, A. Uremic platelet dysfunction: Past and present. Curr. Hematol. Rep. 2005, 4, 359–367. [Google Scholar]
- Landray, M.J.; Wheeler, D.C.; Lip, G.Y.H.; Newman, D.J.; Blann, A.D.; McGlynn, F.J.; Ball, S.; Townend, J.N.; Baigent, C. Inflammation, endothelial dysfunction, and platelet activation in patients with chronic kidney disease: The chronic renal impairment in Birmingham (CRIB) study. Am. J. Kidney Dis. 2004, 43, 244–253. [Google Scholar] [CrossRef]
- Abbasian, N.; Herbert, K.E.; Pawluczyk, I.; Burton, J.O.; Bevington, A. Vesicles bearing gifts: The functional importance of micro-RNA transfer in extracellular vesicles in chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2018, 315, F1430–F1443. [Google Scholar] [CrossRef]
- Yang, K.; Du, C.; Wang, X.; Li, F.; Xu, Y.; Wang, S.; Chen, S.; Chen, F.; Shen, M.; Chen, M.; et al. Indoxyl sulfate induces platelet hyperactivity and contributes to chronic kidney disease-associated thrombosis in mice. Blood 2017, 129, 2667–2679. [Google Scholar] [CrossRef] [PubMed]
- Hasan, R.A.; Koh, A.Y.; Zia, A. The gut microbiome and thromboembolism. Thromb. Res. 2020, 189, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Jerez-Dolz, D.; Torramade-Moix, S.; Palomo, M.; Moreno-Castaño, A.; Lopez-Vilchez, I.; Hernandez, R.; Badimon, J.J.; Zafar, M.U.; Diaz-Ricart, M.; Escolar, G. Internalization of microparticles by platelets is partially mediated by toll-like receptor 4 and enhances platelet thrombogenicity. Atherosclerosis 2020, 294, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, L.M.; Freedman, J.E. The role of inflammation in regulating platelet production and function: Toll-like receptors in platelets and megakaryocytes. Thromb. Res. 2010, 125, 205–209. [Google Scholar] [CrossRef]
- Cognasse, F.; Laradi, S.; Berthelot, P.; Bourlet, T.; Marotte, H.; Mismetti, P.; Garraud, O.; Hamzeh-Cognasse, H. Platelet inflammatory response to stress. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Escolar, G.; Diaz-Ricart, M.; Cases, A.; Castillo, R.; Ordinas, A.; White, J.G. Abnormal cytoskeletal assembly in platelets from uremic patients. Am. J. Pathol. 1993, 143, 823–831. [Google Scholar]
- Díaz-Ricart, M.; Estebanell, E.; Cases, A.; Calls, J.; López-Pedret, J.; Carretero, M.; Castillo, R.; Ordinas, A.; Escolar, G. Abnormal platelet cytoskeletal assembly in hemodialyzed patients results in deficient tyrosine phosphorylation signaling. Kidney Int. 2000, 57, 1905–1914. [Google Scholar] [CrossRef][Green Version]
- Van der Poll, T.; Parker, R.I. Platelet activation and endothelial cell dysfunction. Crit. Care Clin. 2020, 36, 233–253. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diaz-Ricart, M.; Torramade-Moix, S.; Pascual, G.; Palomo, M.; Moreno-Castaño, A.B.; Martinez-Sanchez, J.; Vera, M.; Cases, A.; Escolar, G. Endothelial Damage, Inflammation and Immunity in Chronic Kidney Disease. Toxins 2020, 12, 361. https://doi.org/10.3390/toxins12060361
Diaz-Ricart M, Torramade-Moix S, Pascual G, Palomo M, Moreno-Castaño AB, Martinez-Sanchez J, Vera M, Cases A, Escolar G. Endothelial Damage, Inflammation and Immunity in Chronic Kidney Disease. Toxins. 2020; 12(6):361. https://doi.org/10.3390/toxins12060361
Chicago/Turabian StyleDiaz-Ricart, Maribel, Sergi Torramade-Moix, Georgina Pascual, Marta Palomo, Ana Belen Moreno-Castaño, Julia Martinez-Sanchez, Manel Vera, Aleix Cases, and Gines Escolar. 2020. "Endothelial Damage, Inflammation and Immunity in Chronic Kidney Disease" Toxins 12, no. 6: 361. https://doi.org/10.3390/toxins12060361
APA StyleDiaz-Ricart, M., Torramade-Moix, S., Pascual, G., Palomo, M., Moreno-Castaño, A. B., Martinez-Sanchez, J., Vera, M., Cases, A., & Escolar, G. (2020). Endothelial Damage, Inflammation and Immunity in Chronic Kidney Disease. Toxins, 12(6), 361. https://doi.org/10.3390/toxins12060361