Development of a Sensitive and Reliable UHPLC-MS/MS Method for the Determination of Multiple Urinary Biomarkers of Mycotoxin Exposure


Abstract
1. Introduction
2. Results and Discussion
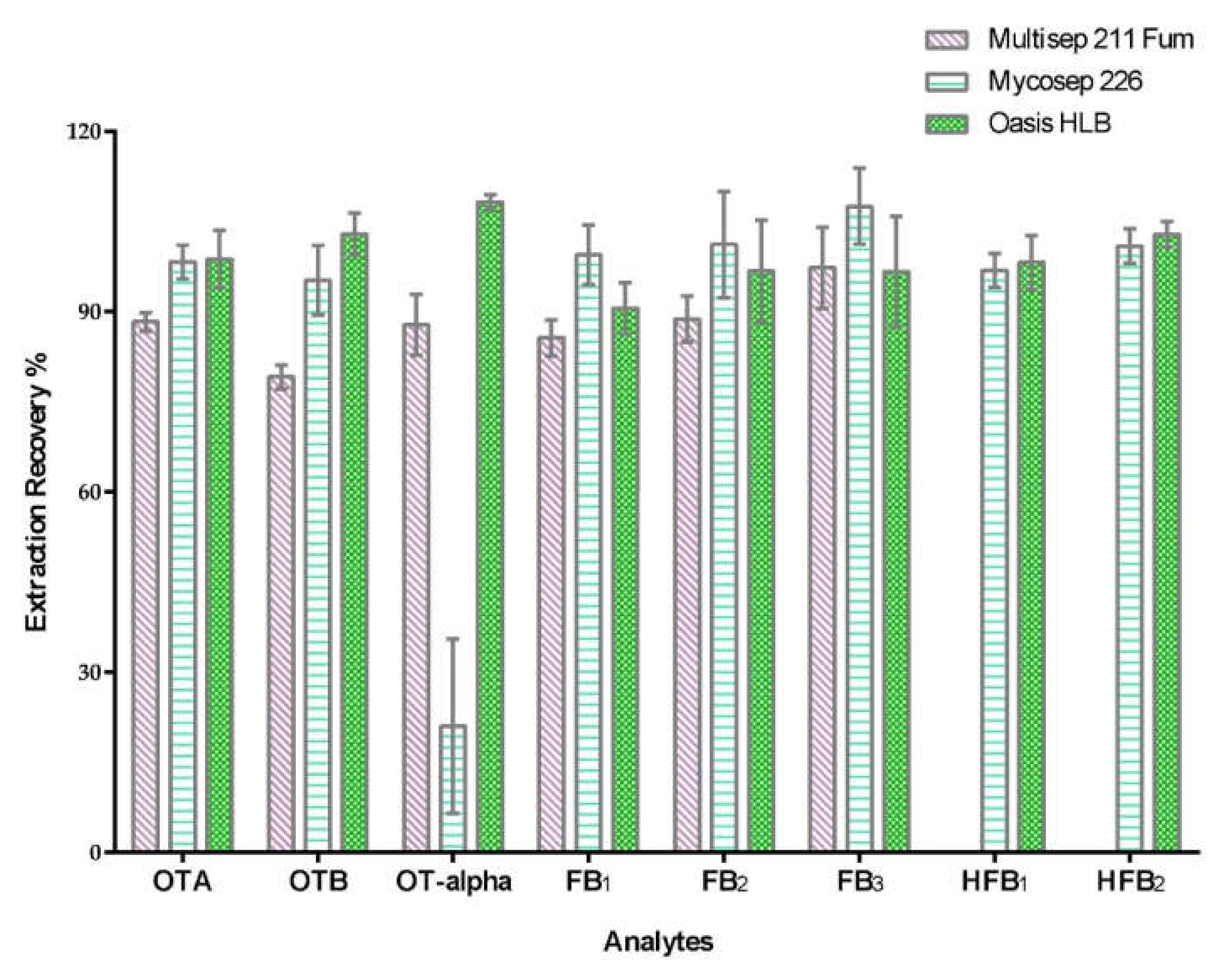
2.1. Sample Clean-Up
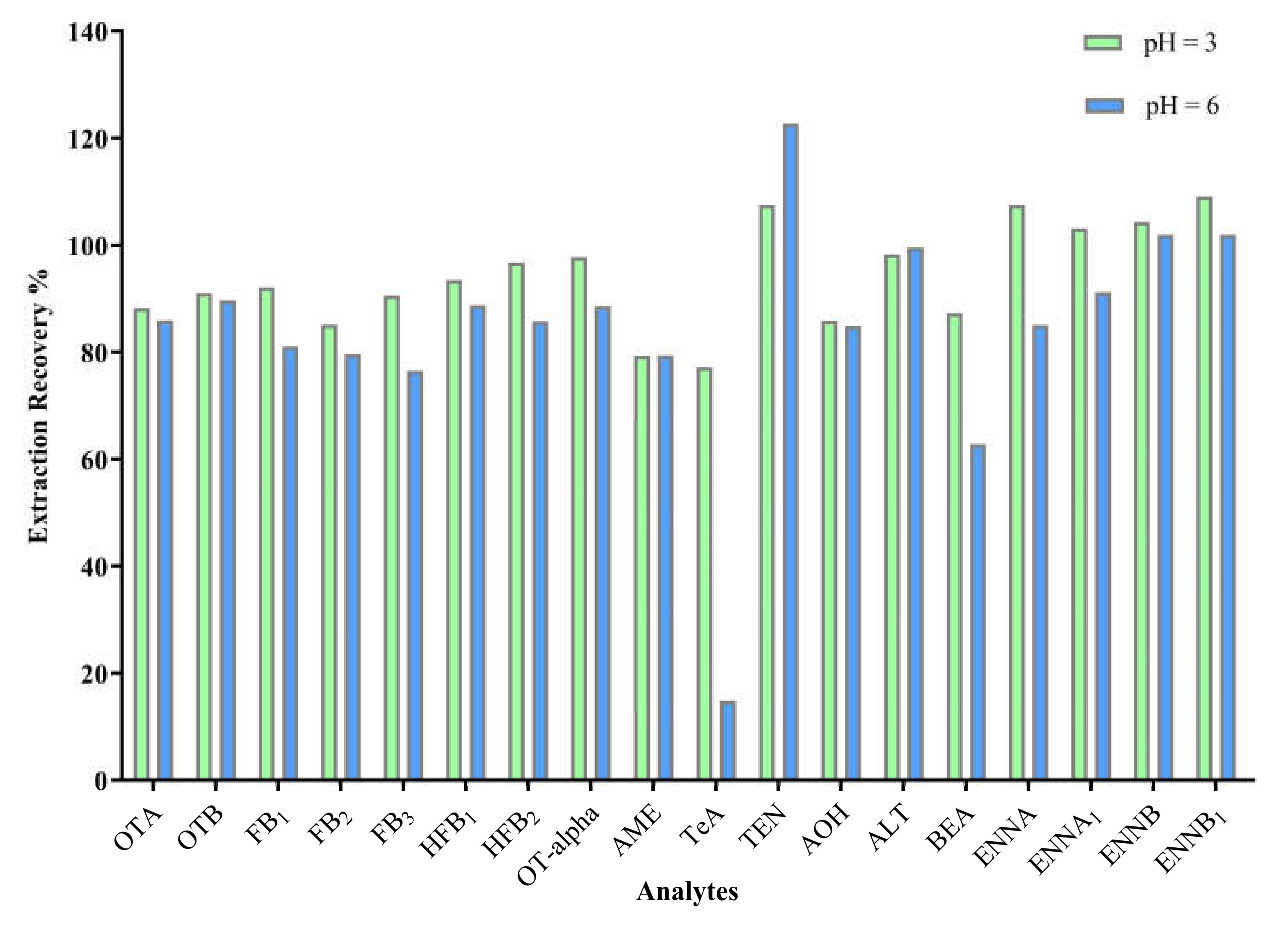
2.2. Enzyme Hydrolysis
2.3. Optimization of MS-MS Parameters
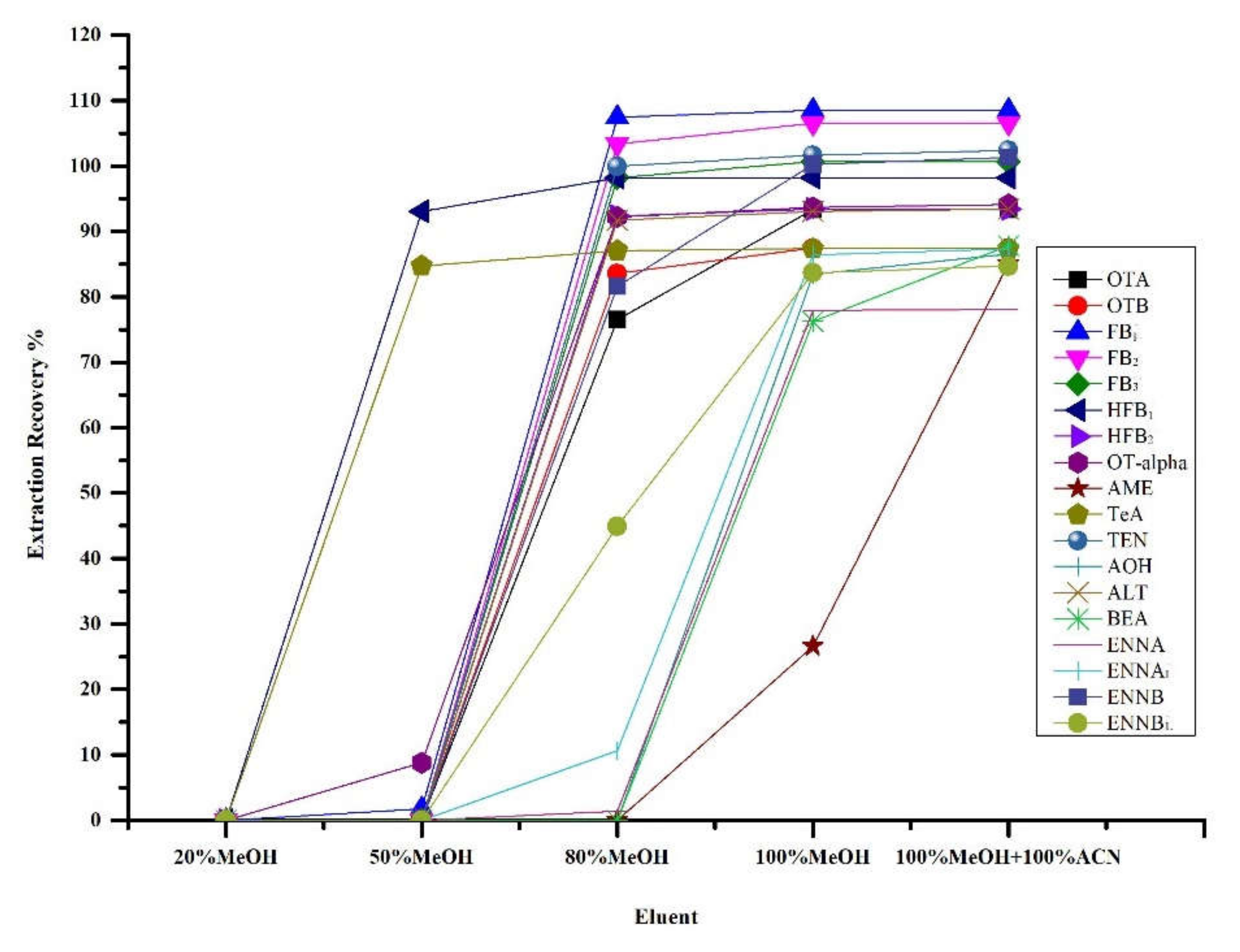
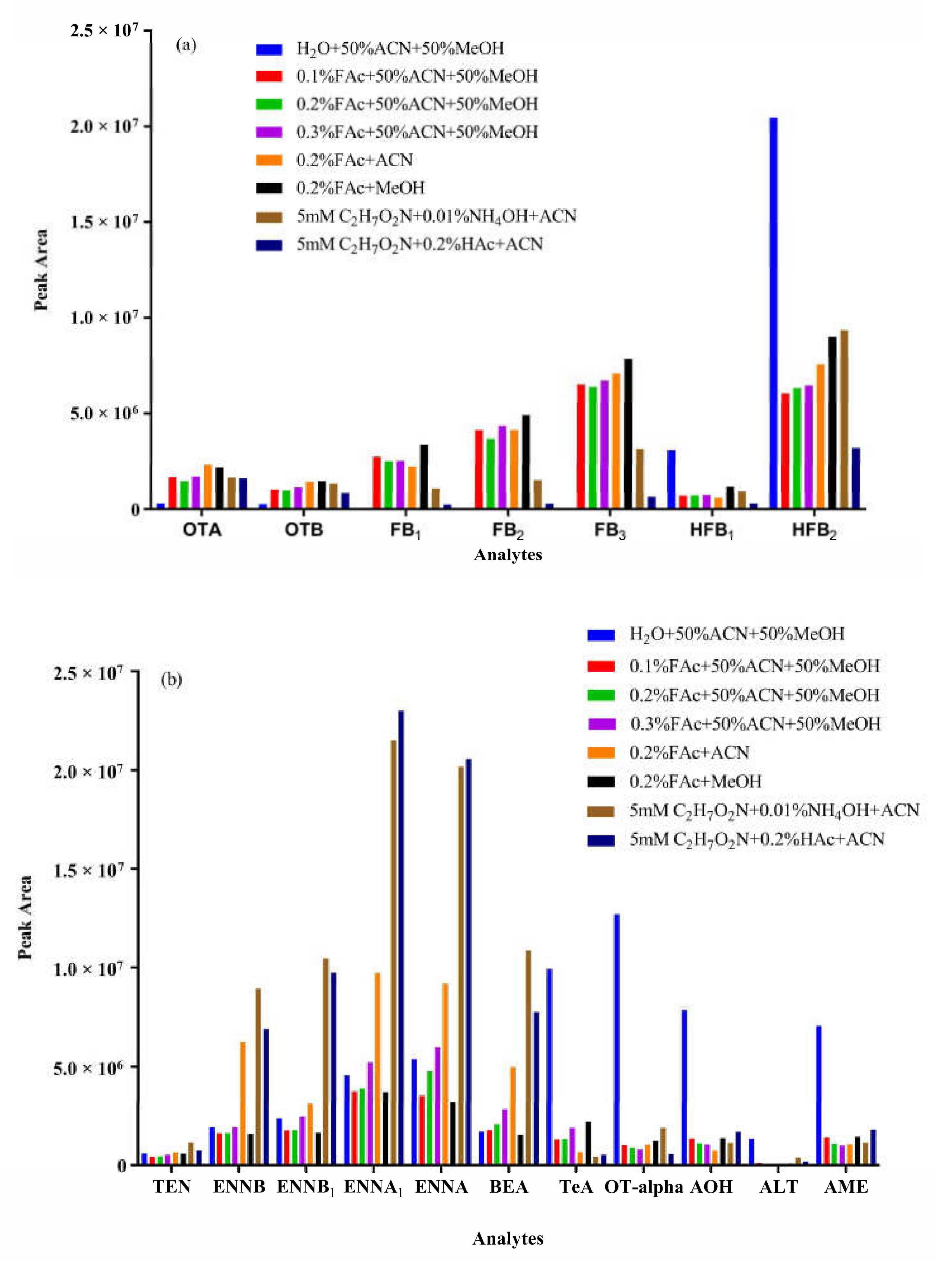
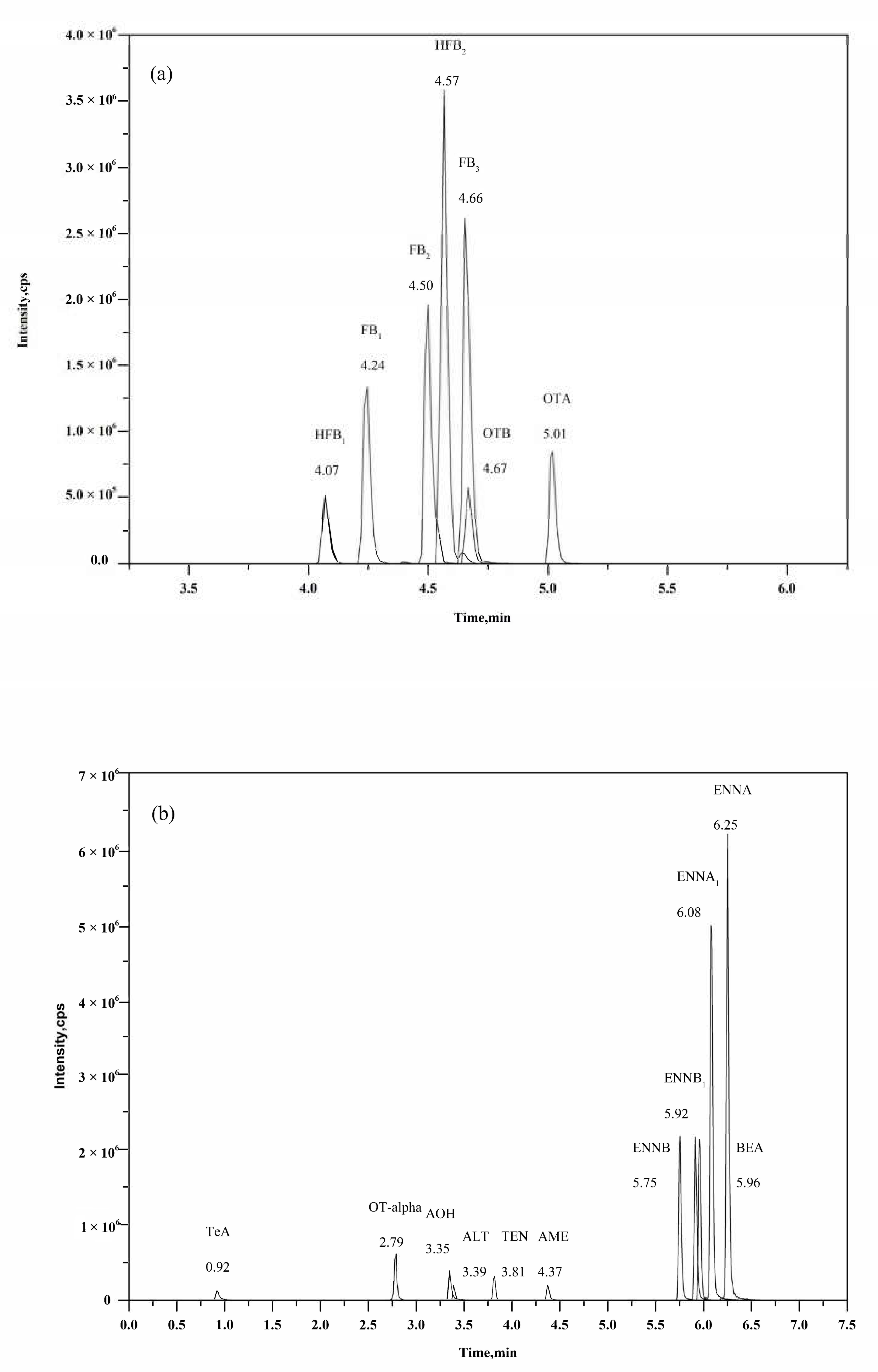
2.4. Optimization of Chromatographic Separation
2.5. Validation Experiments
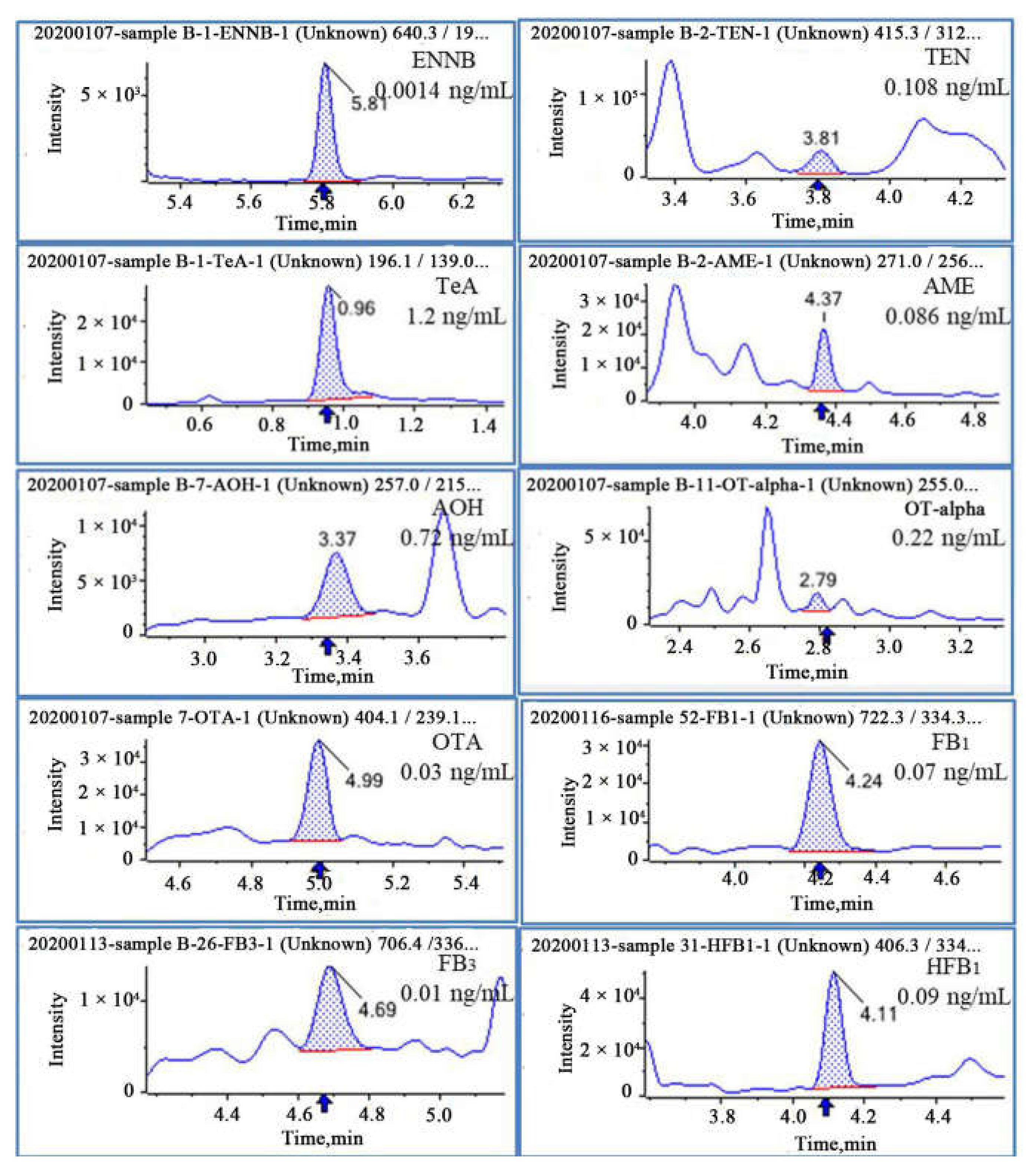
2.6. Application of the Method
3. Conclusions
4. Materials and Methods
4.1. Reagents and Chemicals
4.2. Samples
4.3. Preparation of Standard Soultions and Quality Control Samples
4.4. Sample Preparation
4.5. LC-MS/MS Conditions
4.5.1. Chromatographic Conditions
4.5.2. Mass Spectrometry Condition
4.6. Method Validation
Author Contributions
Funding
Conflicts of Interest
References
- Freire, L.; Sant’Ana, A.S. Modified mycotoxins: An updated review on their formation, detection, occurrence, and toxic effects. Food Chem. Toxicol. 2018, 111, 189–205. [Google Scholar] [PubMed]
- Da Rocha, M.E.B.; Freire, F.D.C.O.; Maia, F.E.F.; Guedes, M.I.F.; Rondina, D. Mycotoxins and their effects on human and animal health. Food Control. 2014, 36, 159–165. [Google Scholar] [CrossRef]
- Sarkanj, B.; Ezekiel, C.N.; Turner, P.C.; Abia, W.A.; Rychlik, M.; Krska, R.; Sulyok, M.; Warth, B. Ultra- sensitive, stable isotope assisted quantification of multiple urinary mycotoxin exposure biomarkers. Anal. Chim. Acta 2018, 1019, 84–92. [Google Scholar]
- Warth, B.; Sulyok, M.; Fruhmann, P.; Mikula, H.; Berthiller, F.; Schuhmacher, R.; Hametner, C.; Abia, W.A.; Adam, G.; Frohlich, J.; et al. Development and validation of a rapid multi-biomarker liquid chromatography/tandem mass spectrometry method to assess human exposure to mycotoxins. Rapid Commun. Mass. Spectrom. 2012, 26, 1533–1540. [Google Scholar]
- Cao, X.; Li, X.; Li, J.; Niu, Y.; Shi, L.; Fang, Z.; Zhang, T.; Ding, H. Quantitative determination of carcinogenic mycotoxins in human and animal biological matrices and animal-derived foods using multi-mycotoxin and analyte-specific high performance liquid chromatography-tandem mass spectrometric methods. J. Chromatogr. B 2018, 1073, 191–200. [Google Scholar]
- Marín, S.; Cano-Sancho, G.; Sanchis, V.; Ramos, A.J. The role of mycotoxins in the human exposome: Application of mycotoxin biomarkers in exposome-health studies. Food Chem. Toxicol. 2018, 121, 504–518. [Google Scholar] [CrossRef]
- Serrano, A.; Capriotti, A.; Cavaliere, C.; Piovesana, S.; Samperi, R.; Ventura, S.; Laganà, A. Development of a Rapid LC-MS/MS Method for the Determination of Emerging Fusarium mycotoxins Enniatins and Beauvericin in Human Biological Fluids. Toxins 2015, 7, 3554–3571. [Google Scholar] [CrossRef]
- Fan, K.; Xu, J.; Jiang, K.; Liu, X.; Meng, J.; Di Mavungu, J.D.; Guo, W.; Zhang, Z.; Jing, J.; Li, H.; et al. Determination of multiple mycotoxins in paired plasma and urine samples to assess human exposure in Nanjing, China. Environ. Pollut. 2019, 248, 865–873. [Google Scholar] [CrossRef]
- FAO. Worldwide Regulations for Mycotoxins in Food and Feed in 2003; FAO Food and Nutrition Paper No. 81; Food and Agriculture Organization of the United Nations: Rome, Italy, 2004. [Google Scholar]
- Commission of the European Communities. Commission Regulation (EC) No 1881/2006 of 19 December 2006 Setting Maximum Levels for Certain Contaminants in Foodstuffs; Commission of the European Communities: Brussel, Belgium, 2006; Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2006:364:0005:0024:EN:PDF (accessed on 16 March 2020).
- EFSA. Dietary exposure assessment to Alternaria toxins in the European population. EFSA J. 2016, 14, 04654. [Google Scholar]
- EFSA Panel on Contaminants in the Food Chain (CONTAM). Scientific opinion on the risks to human and animal health related to the presence of beauvericin and enniatins in food and feed. EFSA J. 2014, 12, 3802. [Google Scholar] [CrossRef]
- Warth, B.; Sulyok, M.; Krska, R. LC-MS/MS-based multibiomarker approaches for the assessment of human exposure to mycotoxins. Anal. Bioanal. Chem. 2013, 405, 5687–5695. [Google Scholar] [CrossRef]
- Ediage, E.N.; Di Mavungu, J.D.; Song, S.; Wu, A.; Van Peteghem, C.; De Saeger, S. A direct assessment of mycotoxin biomarkers in human urine samples by liquid chromatography tandem mass spectrometry. Anal. Chim. Acta 2012, 741, 58–69. [Google Scholar] [CrossRef]
- Martins, C.; Vidal, A.; De Boevre, M.; De Saeger, S.; Nunes, C.; Torres, D.; Goios, A.; Lopes, C.; Assuncao, R.; Alvito, P. Exposure assessment of Portuguese population to multiple mycotoxins: The human biomonitoring approach. Int. J. Hyg. Environ. Health 2019, 222, 913–925. [Google Scholar] [CrossRef]
- Vidal, A.; Cano-Sancho, G.; Marín, S.; Ramos, A.J.; Sanchis, V. Multidetection of urinary ochratoxin A, deoxynivalenol and its metabolites: Pilot time-course study and risk assessment in Catalonia, Spain. World Mycotoxin J. 2016, 9, 597–612. [Google Scholar] [CrossRef]
- Lauwers, M.; De Baere, S.; Letor, B.; Rychlik, M.; Croubels, S.; Devreese, M. Multi LC-MS/MS and LC-HRMS Methods for Determination of 24 Mycotoxins including Major Phase I and II Biomarker Metabolites in Biological Matrices from Pigs and Broiler Chickens. Toxins 2019, 11, 171. [Google Scholar] [CrossRef]
- Heyndrickx, E.; Sioen, I.; Huybrechts, B.; Callebaut, A.; De Henauw, S.; De Saeger, S. Human biomonitoring of multiple mycotoxins in the Belgian population: Results of the BIOMYCO study. Environ. Int. 2015, 84, 82–89. [Google Scholar] [CrossRef]
- Franco, L.T.; Petta, T.; Rottinghaus, G.E.; Bordin, K.; Gomes, G.A.; Alvito, P.; Assuncao, R.; Oliveira, C.A.F. Assessment of mycotoxin exposure and risk characterization using occurrence data in foods and urinary biomarkers in Brazil. Food Chem. Toxicol. 2019, 128, 21–34. [Google Scholar] [CrossRef]
- Klapec, T.; Sarkanj, B.; Banjari, I.; Strelec, I. Urinary ochratoxin A and ochratoxin alpha in pregnant women. Food Chem. Toxicol. 2012, 50, 4487–4492. [Google Scholar] [CrossRef]
- Munoz, K.; Blaszkewicz, M.; Degen, G.H. Simultaneous analysis of ochratoxin A and its major metabolite ochratoxin alpha in plasma and urine for an advanced biomonitoring of the mycotoxin. J. Chromatogr. B 2010, 878, 2623–2629. [Google Scholar] [CrossRef]
- De Girolamo, A.; Lattanzio, V.M.; Schena, R.; Visconti, A.; Pascale, M. Use of liquid chromatography -high-resolution mass spectrometry for isolation and characterization of hydrolyzed fumonisins and relevant analysis in maize-based products. J. Mass Spectrom. 2014, 49, 297–305. [Google Scholar] [CrossRef]
- Cawood, M.; Gelderblom, W.C.; Alberts, J.F.; Snyman, S.D. Mycotoxin Biomarkers of Exposure: A Comprehensive Review. Comprehensive Reviews in Food Science and Food Safety. Food Chem. Toxicol. 1994, 32, 627–632. [Google Scholar] [CrossRef]
- Gambacorta, S.; Solfrizzo, H.; Visconti, A.; Powers, S.; Cossalter, A.M.; Pinton, P.; Oswald, I.P. Validation study on urinary biomarkers of exposure for aflatoxin B1, ochratoxin A, fumonisin B1, deoxynivalenol and zearalenone in piglets. World Mycotoxin J. 2013, 6, 299–308. [Google Scholar] [CrossRef]
- Souto, P.; Jager, A.V.; Tonin, F.G.; Petta, T.; Di Gregorio, M.C.; Cossalter, A.M.; Pinton, P.; Oswald, I.P.; Rottinghaus, G.E.; Oliveira, C.A.F.; et al. Determination of fumonisin B1 levels in body fluids and hair from piglets fed fumonisin B1-contaminated diets. Food Chem. Toxicol. 2017, 108, 1–9. [Google Scholar] [CrossRef]
- Vidal, A.; Mengelers, M.; Yang, S.; De Saeger, S.; De Boevre, M. Mycotoxin Biomarkers of Exposure: A Comprehensive Review. Compr. Rev. Food Sci. Food Saf. 2018, 17, 1127–1155. [Google Scholar] [CrossRef]
- Shephard, G.; Thiel, P.G.; Sydenham, E.W.; Vleggaar, R.; Alberts, J.F. Determination of the mycotoxin fumonisin B1 and identification of its partially hydrolysed metabolites in the faeces of non-human primates. Food Chem. Toxicol. 1994, 32, 23–29. [Google Scholar] [CrossRef]
- Gordon, S.; Shephard, P.G.T.; Sydenham, E.W.; Savard, M.E. Fate of a single dose of [14C]-labelled fumonisin B1 in vervet monkeys. Nat. Toxins 1995, 3, 145–150. [Google Scholar]
- Berthiller, F.; Crews, C.; Dall’Asta, C.; Saeger, S.D.; Haesaert, G.; Karlovsky, P.; Oswald, I.P.; Seefelder, W.; Speijers, G.; Stroka, J.; et al. Masked mycotoxins: A review. Mol. Nutr. Food Res. 2013, 57, 165–186. [Google Scholar] [CrossRef]
- Righetti, L.; Fenclova, M.; Dellafiora, L.; Hajslova, J.; Stranska-Zachariasova, M.; Dall’Asta, C. High resolution-ion mobility mass spectrometry as an additional powerful tool for structural characterization of mycotoxin metabolites. Food Chem. 2018, 245, 768–774. [Google Scholar] [CrossRef]
- Duarte, S.C.; Lino, C.M.; Pena, A. Ochratoxin A in food and urine: A nationwide Portuguese two-year study. World Mycotoxin J. 2015, 8, 121–132. [Google Scholar] [CrossRef]
- Ali, N.; Munoz, K.; Degen, G.H. Ochratoxin A and its metabolites in urines of German adults-An assessment of variables in biomarker analysis. Toxicol. Lett. 2017, 275, 19–26. [Google Scholar] [CrossRef]
- Rodriguez-Carrasco, Y.; Molto, J.C.; Manes, J.; Berrada, H. Development of a GC-MS/MS strategy to determine 15 mycotoxins and metabolites in human urine. Talanta 2014, 128, 125–131. [Google Scholar] [CrossRef]
- Bordin, K.; Rottinghaus, G.E.; Landers, B.R.; Ledoux, D.R.; Kobashigawa, E.; Corassin, C.H.; Oliveira, C.A.F. Evaluation of fumonisin exposure by determination of fumonisin B1 in human hair and in Brazilian corn products. Food Control. 2015, 53, 67–71. [Google Scholar] [CrossRef]
- Slobodchikova, I.; Vuckovic, D. Liquid chromatography—High resolution mass spectrometry method for monitoring of 17 mycotoxins in human plasma for exposure studies. J. Chromatogr. A 2018, 1548, 51–63. [Google Scholar] [CrossRef]
- De Santis, B.; Raggi, M.E.; Moretti, G.; Facchiano, F.; Mezzelani, A.; Villa, L.; Bonfanti, A.; Campioni, A.; Rossi, S.; Camposeo, S.; et al. Study on the Association among Mycotoxins and other Variables in Children with Autism. Toxins 2017, 9, 203. [Google Scholar] [CrossRef]
- Ezekiel, C.N.; Warth, B.; Ogara, I.M.; Abia, W.A.; Ezekiel, V.C.; Atehnkeng, J.; Sulyok, M.; Turner, P.C.; Tayo, G.O.; Krska, R.; et al. Mycotoxin exposure in rural residents in northern Nigeria: A pilot study using multi-urinary biomarkers. Environ. Int. 2014, 66, 138–145. [Google Scholar] [CrossRef]
- Rodriguez-Carrasco, Y.; Izzo, L.; Gaspari, A.; Graziani, G.; Manes, J.; Ritieni, A. Urinary levels of enniatin B and its phase I metabolites: First human pilot biomonitoring study. Food Chem. Toxicol. 2018, 118, 454–459. [Google Scholar] [CrossRef]
- Solfrizzo, M.; Gambacorta, L.; Lattanzio, V.M.T.; Powers, S.; Visconti, A. Simultaneous LC–MS/MS determination of aflatoxin M1, ochratoxin A, deoxynivalenol, de-epoxydeoxynivalenol, α and β- zearalenols and fumonisin B1 in urine as a multi-biomarker method to assess exposure to mycotoxins. Anal. Bioanal. Chem. 2011, 401, 2831–2841. [Google Scholar] [CrossRef]
- Gazzotti, T.; Zironi, E.; Lugoboni, B.; Barbarossa, A.; Piva, A.; Pagliuca, G. Analysis of fumonisins B1, B2 and their hydrolysed metabolites in pig liver by LC–MS/MS. Food Chem. 2011, 125, 1379–1384. [Google Scholar] [CrossRef]
- Huybrechts, B.; Martins, J.C.; Debongnie, P.; Uhlig, S.; Callebaut, A. Fast and sensitive LC-MS/MS method measuring human mycotoxin exposure using biomarkers in urine. Arch. Toxicol. 2015, 89, 1993–2005. [Google Scholar] [CrossRef]
- Duarte, S.; Bento, J.; Pena, A.; Lino, C.M.; Delerue-Matos, C.; Oliva-Teles, T.; Morais, S.; Correia, M.; Oliveira, M.B.; Alves, M.R.; et al. Monitoring of ochratoxin A exposure of the Portuguese population through a nationwide urine survey--Winter 2007. Sci. Total Environ. 2010, 408, 1195–1198. [Google Scholar] [CrossRef]
- Gerding, J.; Ali, N.; Schwartzbord, J.; Cramer, B.; Brown, D.L.; Degen, G.H.; Humpf, H.U. A comparative study of the human urinary mycotoxin excretion patterns in Bangladesh, Germany, and Haiti using a rapid and sensitive LC-MS/MS approach. Mycotoxin Res. 2015, 31, 127–136. [Google Scholar] [CrossRef]
- Asam, S.; Habler, K.; Rychlik, M. Determination of tenuazonic acid in human urine by means of a stable isotope dilution assay. Anal. Bioanal. Chem. 2013, 405, 4149–4158. [Google Scholar] [CrossRef]
- Sun, D.; Qiu, N.; Zhou, S.; Lyu, B.; Zhang, S.; Li, J.; Zhao, Y.; Wu, Y. Development of Sensitive and Reliable UPLC-MS/MS Methods for Food Analysis of Emerging Mycotoxins in China Total Diet Study. Toxins 2019, 11, 166. [Google Scholar] [CrossRef]
- Commission Decision 2002/657/EC Implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and the Interpretation of Results. Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2002:221:0008:0036:EN:PDF (accessed on 17 March 2019).
- European Medicines Agency. Guideline on Bioanalytical Method Validation. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500109686.pdf (accessed on 12 January 2016).
- FDA. FDA Guidance for Industry: Bioanalytical Method Validation; Food and Drug Administration: Silver Spring, MD, USA, 2001. Available online: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070107.pdf (accessed on 16 March 2019).
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Precursor Ion | Q1(m/z) | Q3(m/z) | CE (eV) 1 | DP (V) 2 |
|---|---|---|---|---|---|
| OTA | [M + H]+ | 404.1 | 239.1 a 358.1 | 34 21 | 50 50 |
| OTB | [M + H]+ | 371.1 | 205.9 a 188.1 | 31 35 | 40 40 |
| OT-alpha | [M − H]− | 255.0 | 211.0 a 167.1 | −21 −32 | −30 −30 |
| FB1 | [M + H]+ | 722.3 | 334.3 a 352.3 | 55 51 | 50 50 |
| FB2 | [M + H]+ | 706.4 | 336.1 a 354.2 | 50 46 | 50 50 |
| FB3 | [M + H]+ | 706.4 | 336.3 a 318.1 | 50 51 | 50 50 |
| HFB1 | [M + H]+ | 406.3 | 334.1 a 352.1 | 35 31 | 40 40 |
| HFB2 | [M + H]+ | 390.3 | 336.2 a 238.1 | 33 37 | 35 35 |
| AOH | [M − H]− | 257.0 | 215.0 a 147.0 | −34 −43 | −130 −130 |
| ALT | [M + H]+ | 291.0 | 214.0 a 229.2 | −30 −21 | −100 −100 |
| TEN | [M + H]+ | 415.3 | 312.1 a 256.0 | 29 40 | 110 110 |
| TeA | [M − H]− | 196.1 | 139.0 a 112.0 | −25 −35 | −50 −50 |
| AME | [M − H]− | 271.0 | 256.0 a 228.0 | −30 −36 | −110 −110 |
| BEA | [M + H]+ | 784.5 | 244.2 a 134.3 | 41 92 | 240 240 |
| ENNA | [M + H]+ | 682.4 | 210.2 a 228.2 | 34 38 | 220 220 |
| ENNA1 | [M + H]+ | 668.3 | 210.0 a 228.1 | 32 33 | 200 200 |
| ENNB | [M + H]+ | 640.3 | 196.4 a 241.2 | 34 33 | 180 180 |
| ENNB1 | [M + H]+ | 654.3 | 196.0 a 210.2 | 33 35 | 180 180 |
| 13C20-OTA | [M + H]+ | 424.2 | 250.1 a 337.1 | 35 20 | 50 50 |
| 13C34-FB1 | [M + H]+ | 756.7 | 374.4 a 356.3 | 53 55 | 50 50 |
| 13C34-FB2 | [M + H]+ | 740.7 | 358.3 a 376.2 | 50 47 | 50 50 |
| 13C34-FB3 | [M + H]+ | 740.7 | 358.2 a 376.1 | 50 47 | 50 50 |
| TeA-d13 | [M − H]− | 210.3 | 143.0 a 115.0 | −27 −34 | −50 −50 |
| TEN-d3 | [M + H]+ | 418.2 | 314.9 a 305.4 | 30 19 | 140 140 |
| AME-d3 | [M − H]− | 274.0 | 256.0 a 228.1 | −32 −39 | −110 −110 |
| AOH-d2 | [M + H]+ | 258.9 | 215.1 a 148.0 | −31 −43 | −130 −130 |
| Ion Source Parameters | Settings |
|---|---|
| Curtain gas CUR (psi) | 20 |
| Collision Gas CAD | 10 |
| Ion Spray Voltage (IS) (V) | 5500/−4500 |
| Temperature (TEM) (°C) | 600 |
| Ion Source Gas 1 (GS1) (psi) | 65 |
| Ion Source Gas 2 (GS2) (psi) | 55 |
| Analyte | RE (%) | SSE (%) | RA (%) | Linear Range 1 (ng/mL) | R2 | LOD 2 (ng/mL) | LOQ 3 (ng/mL) |
|---|---|---|---|---|---|---|---|
| OTA | 107% | 49.1% | 52.3% | 0.05–50 | 0.999 | 0.01 | 0.02 |
| OTB | 91.6% | 64.3% | 58.9% | 0.25–250 | 0.998 | 0.01 | 0.04 |
| FB1 | 93.7% | 77.3% | 72.4% | 0.25–250 | 0.998 | 0.02 | 0.05 |
| FB2 | 93.7% | 69.3% | 64.9% | 0.25–250 | 0.999 | 0.02 | 0.05 |
| FB3 | 93.5% | 95.7% | 89.5% | 0.25–250 | 0.998 | 0.01 | 0.03 |
| HFB1 | 104% | 64.2% | 66.6% | 0.25–125 | 0.998 | 0.01 | 0.02 |
| HFB2 | 111% | 56.9% | 63.0% | 0.25–250 | 0.997 | 0.02 | 0.04 |
| TeA | 87.3% | 41.8% | 36.4% | 0.5–500 | 0.999 | 0.2 | 0.5 |
| OT-alpha | 99.0% | 12.2% | 12.1% | 0.25–125 | 0.998 | 0.02 | 0.05 |
| AOH | 75.2% | 16.4% | 12.4% | 0.25–250 | 0.998 | 0.01 | 0.04 |
| ALT | 85.2% | 93.4% | 79.6% | 0.5–500 | 0.998 | 0.02 | 0.07 |
| AME | 74.6% | 120% | 89.3% | 0.01–50 | 0.999 | 0.001 | 0.003 |
| TEN | 88.4% | 49.7% | 43.9% | 0.05–50 | 0.998 | 0.002 | 0.01 |
| ENNB | 90.0% | 101% | 90.8% | 0.001–25 | 0.998 | 0.0001 | 0.0002 |
| ENNB1 | 84.1% | 104% | 87.1% | 0.001–25 | 0.997 | 0.0001 | 0.0002 |
| ENNA | 71.6% | 101% | 72.5% | 0.005–50 | 0.997 | 0.0002 | 0.0008 |
| ENNA1 | 77.5% | 101% | 78.5% | 0.005–50 | 0.998 | 0.0001 | 0.0005 |
| BEA | 85.5% | 89.7% | 76.6% | 0.005–50 | 0.998 | 0.0002 | 0.0006 |
| Analyte | Spiked Level in Urine (ng/mL) | Measured Value in Urine (ng/mL) | Method Recovery (%) | RSD (%) | |
|---|---|---|---|---|---|
| Intra-day (n = 6) | Inter-day (n = 18) | ||||
| OTA | 0.1 | 0.10 | 100 | 2.8 | 4.8 |
| 1 | 1.11 | 111 | 3.3 | 3.5 | |
| 4 | 4.15 | 104 | 2.4 | 3.8 | |
| OTB | 0.5 | 0.41 | 82.1 | 1.8 | 10.8 |
| 5 | 4.18 | 83.5 | 2.8 | 4.6 | |
| 20 | 17.4 | 86.9 | 0.9 | 5.8 | |
| FB1 | 0.5 | 0.47 | 94.6 | 3.1 | 3.8 |
| 5 | 4.95 | 99.0 | 3.6 | 4.9 | |
| 20 | 19.5 | 97.4 | 2.5 | 3.4 | |
| FB2 | 0.5 | 0.49 | 98.0 | 3.7 | 4.8 |
| 5 | 5.28 | 106 | 3.8 | 10.0 | |
| 20 | 18.3 | 91.7 | 3.5 | 6.5 | |
| FB3 | 0.5 | 0.47 | 93.3 | 2.7 | 4.3 |
| 5 | 5.20 | 104 | 4.6 | 6.3 | |
| 20 | 19.4 | 97.1 | 2.5 | 6.0 | |
| HFB1 | 0.25 | 0.21 | 83.5 | 0.9 | 3.6 |
| 2.5 | 2.25 | 89.8 | 0.5 | 5.1 | |
| 10 | 8.83 | 88.3 | 3.5 | 5.6 | |
| HFB2 | 0.5 | 0.37 | 74.0 | 3.1 | 9.5 |
| 5 | 4.22 | 84.5 | 1.1 | 3.5 | |
| 20 | 16.4 | 82.0 | 0.6 | 6.6 | |
| TeA | 1 | 0.93 | 93.0 | 6.9 | 9.2 |
| 10 | 9.84 | 98.4 | 6.2 | 8.3 | |
| 40 | 38.3 | 95.8 | 1.3 | 4.4 | |
| OT-alpha | 0.25 | 0.21 | 84.4 | 1.2 | 8.7 |
| 2.5 | 2.00 | 79.9 | 4.1 | 6.5 | |
| 10 | 0.88 | 88.1 | 1.9 | 8.7 | |
| AOH | 0.5 | 0.538 | 108 | 1.4 | 7.5 |
| 5 | 5.33 | 107 | 2.2 | 4.4 | |
| 20 | 21.3 | 107 | 2.2 | 2.7 | |
| ALT | 1 | 1.15 | 115 | 8.7 | 13.4 |
| 10 | 13.3 | 133 | 6.3 | 11.1 | |
| 40 | 51.8 | 130 | 3.2 | 7.3 | |
| AME | 0.1 | 0.08 | 80.9 | 2.7 | 5.2 |
| 1 | 0.92 | 92.2 | 0.7 | 2.6 | |
| 4 | 3.81 | 95.2 | 1.7 | 2.4 | |
| TEN | 0.1 | 0.11 | 110 | 4.8 | 6.0 |
| 1 | 1.08 | 108 | 2.7 | 2.8 | |
| 4 | 4.12 | 103 | 1.2 | 3.2 | |
| ENNB | 0.05 | 0.05 | 94.6 | 1.7 | 4.9 |
| 0.5 | 0.49 | 98.5 | 2.8 | 4.9 | |
| 2 | 2.02 | 101 | 2.7 | 4.3 | |
| ENNB1 | 0.05 | 0.05 | 95.0 | 6.9 | 7.2 |
| 0.5 | 0.49 | 97.4 | 1.1 | 5.9 | |
| 2 | 1.92 | 96.0 | 3.2 | 6.2 | |
| ENNA | 0.1 | 0.09 | 84.3 | 1.7 | 8.2 |
| 1 | 0.90 | 89.9 | 3.6 | 7.0 | |
| 4 | 3.79 | 94.7 | 3.2 | 6.9 | |
| ENNA1 | 0.1 | 0.09 | 90.0 | 2.9 | 9.9 |
| 1 | 1.00 | 99.4 | 1.4 | 7.5 | |
| 4 | 3.90 | 97.5 | 3.8 | 7.5 | |
| BEA | 0.1 | 0.08 | 80.8 | 4.1 | 5.8 |
| 1 | 0.77 | 76.4 | 1.8 | 3.9 | |
| 4 | 3.20 | 80.1 | 2.7 | 4.3 | |
| Analyte | Sample Preparation | Analysis Method | LOQ (μg/L) | Reference |
|---|---|---|---|---|
| OTA, OT-alpha, FB1, FB2, FB3 HFB1, AOH, AME | QuEChERS | UPLC-MS/MS | 0.02–1.0 | [15] |
| OTA, OT-alpha, FB1, ENNB | Dilute and shoot | LC-MS/MS | 0.0005–0.1 | [43] |
| OTA, FB1, AOH | SPE | UHPLC-MS/MS | 0.001–0.03 | [3] |
| OTA, FB1 | LLE+ QuEChERS | UPLC–MS/MS | 0.16–1.1 | [36] |
| OTA, OT-alpha, FB1 | Direct method | UHPLC-MS/MS | 0.1–0.5 | [8] |
| OTA, OT-alpha | LLE | HPLC-FD | 0.02 | [32] |
| OTA, OT-alpha, FB1, FB2, FB3 | Direct method/ICA | LC–MS/MS | 0.003–0.2 | [41] |
| OTA, FB1, FB2 | Dilute and shoot | LC–MS/MS | 0.017–0.17 | [4] |
| OTA, FB1, FB2 | SPE + SPE | LC–MS/MS | 0.007–0.017 | [19] |
| ENNB | salting-out liquid–liquid extraction | UHPLC-Q-Orbitrap HRMS | 0.001 | [38] |
| ENNS, BEA | SPE | LC-MS/MS | 0.005–0.02 | [7] |
| OTA, OT-alpha, FB1, HFB1 | LLE + SPE | LC-MS/MS | 0.06–1.02 | [14] |
| Mycotoxin Biomarkers | Total Concentrations | |||
|---|---|---|---|---|
| Positive (%) | Mean ± SD (ng/mL) | Range (ng/mL) | Median (ng/mL) | |
| OTA | 24 (40%) | 0.02 ± 0.02 | <LOD~0.14 | 0.005 |
| OT-alpha | 20 (33.3%) | 0.22 ± 0.42 | <LOD~2.38 | 0.001 |
| FB1 | 2 (3.3%) | 0.01 ± 0.01 | <LOD~0.07 | 0.01 |
| FB3 | 1 (1.7%) | 0.01 | <LOD~0.01 | 0.005 |
| HFB1 | 1 (1.7%) | 0.01 ± 0.01 | <LOD~0.09 | 0.005 |
| TeA | 52 (86.7%) | 4.4 ± 8.7 | <LOD~54.1 | 1.4 |
| AOH | 17 (28.3%) | 0.34 ± 1.09 | <LOD~7.68 | 0.01 |
| AME | 50 (83.3%) | 0.059 ± 0.049 | <LOD~0.167 | 0.048 |
| TEN | 23 (38.3%) | 0.024 ± 0.042 | <LOD~0.193 | 0.001 |
| ENNB | 24 (40.0%) | 0.0002 ± 0.0002 | <LOD~0.0014 | 0.0001 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Zhao, X.; Wu, L.; Zhou, S.; Gong, Z.; Zhao, Y.; Wu, Y. Development of a Sensitive and Reliable UHPLC-MS/MS Method for the Determination of Multiple Urinary Biomarkers of Mycotoxin Exposure. Toxins 2020, 12, 193. https://doi.org/10.3390/toxins12030193
Liu Z, Zhao X, Wu L, Zhou S, Gong Z, Zhao Y, Wu Y. Development of a Sensitive and Reliable UHPLC-MS/MS Method for the Determination of Multiple Urinary Biomarkers of Mycotoxin Exposure. Toxins. 2020; 12(3):193. https://doi.org/10.3390/toxins12030193
Chicago/Turabian StyleLiu, Zhezhe, Xiaoxue Zhao, Libiao Wu, Shuang Zhou, Zhiyong Gong, Yunfeng Zhao, and Yongning Wu. 2020. "Development of a Sensitive and Reliable UHPLC-MS/MS Method for the Determination of Multiple Urinary Biomarkers of Mycotoxin Exposure" Toxins 12, no. 3: 193. https://doi.org/10.3390/toxins12030193
APA StyleLiu, Z., Zhao, X., Wu, L., Zhou, S., Gong, Z., Zhao, Y., & Wu, Y. (2020). Development of a Sensitive and Reliable UHPLC-MS/MS Method for the Determination of Multiple Urinary Biomarkers of Mycotoxin Exposure. Toxins, 12(3), 193. https://doi.org/10.3390/toxins12030193