Parathyroid Hormone: A Uremic Toxin


Abstract
1. Introduction
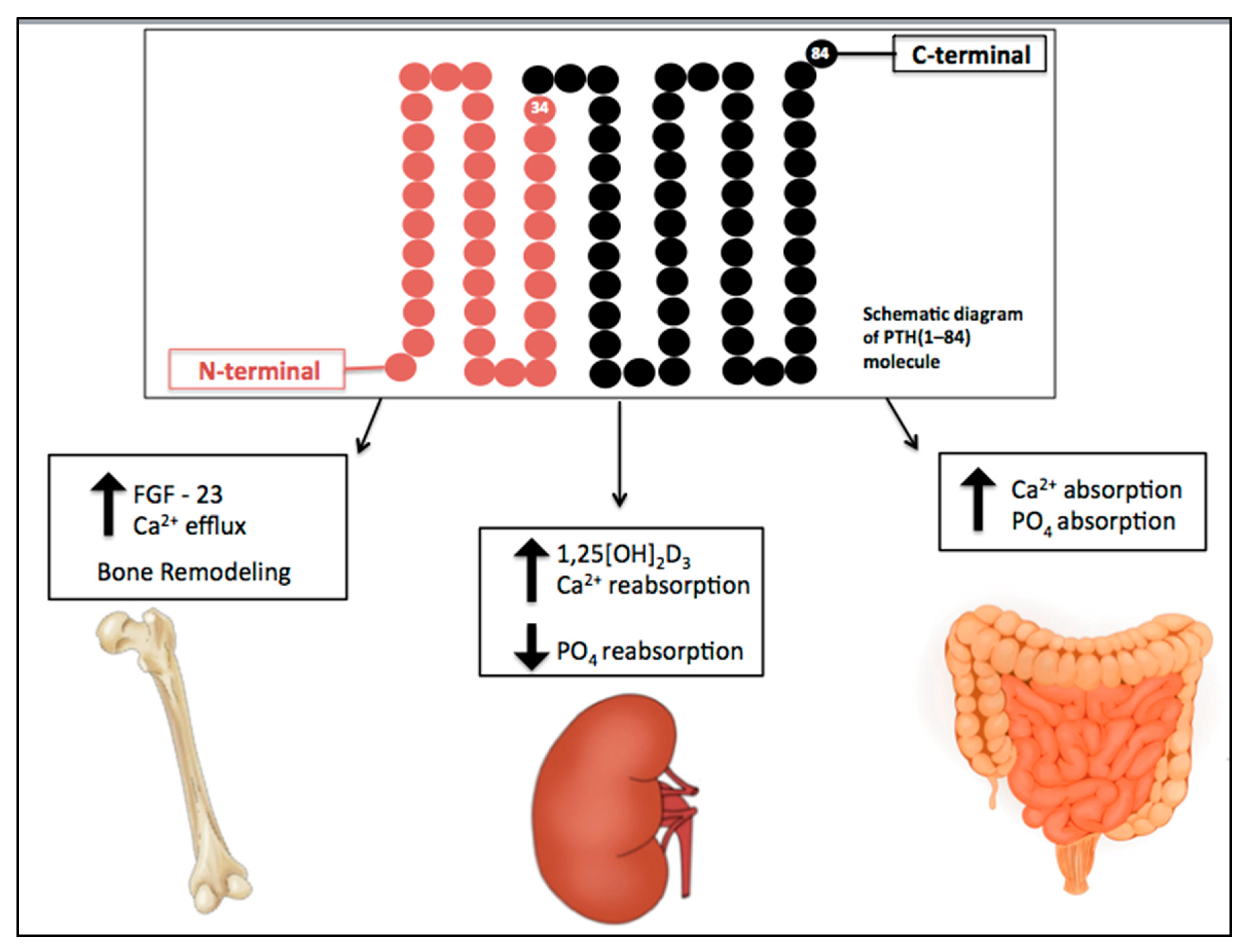
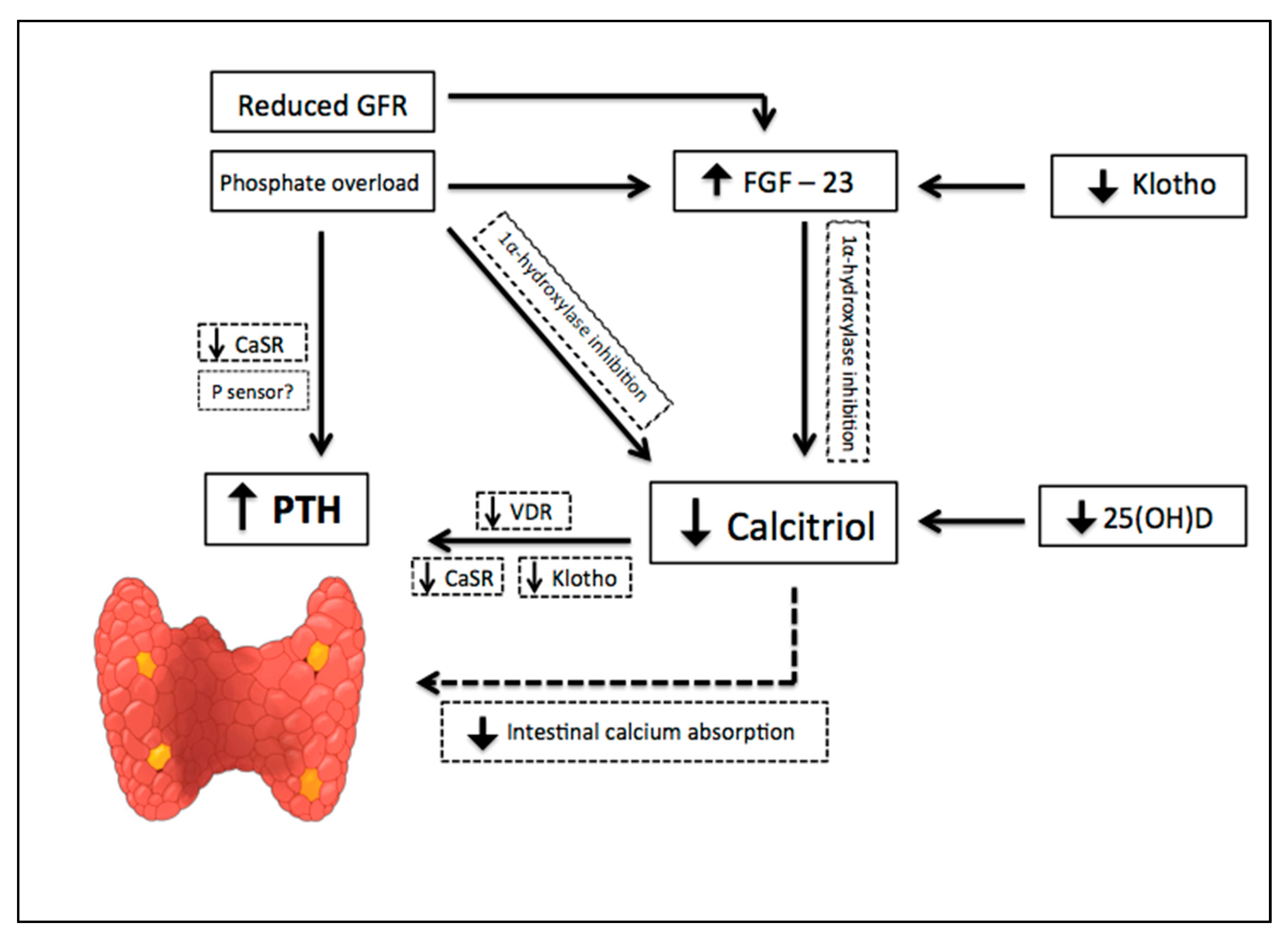
2. CKD-Associated Secondary Hyperparathyroidism
3. Effects of sHPT on BONE
4. Effects of sHPT on Cardiovascular System
5. Effects of sHPT on CKD Progression
6. Effects of sHPT on CKD-Related Caquexia and Energy Expenditure
7. Effect of sHPT on Glucose Metabolism
8. Effect of sHPT on Central Nervous System
9. Effect of sHPT on Hematopoietic and Immunological System
10. Conclusions
Funding
Conflicts of Interest
References
- Goodman, W.G.; Salusky, I.B.; Juppner, H. New lessons from old assays: Parathyroid hormone (PTH), its receptors, and the potential biological relevance of PTH fragments. Nephrol. Dial. Transplant. 2002, 17, 1731–1736. [Google Scholar] [CrossRef] [PubMed]
- Conigrave, A.D. The calcium-sensing receptor and the parathyroid: Past, present, future. Front. Physiol. 2016, 7, 563. [Google Scholar] [CrossRef] [PubMed]
- Blaine, J.; Chonchol, M.; Levi, M. Renal control of calcium, phosphate, and magnesium homeostasis. Clin. J. Am. Soc. Nephrol. CJASN 2015, 10, 1257–1272. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; Kneissel, M. WNT signaling in bone homeostasis and disease: From human mutations to treatments. Nat. Med. 2013, 19, 179–192. [Google Scholar] [CrossRef]
- Baron, R.; Rawadi, G. Targeting the Wnt/beta-catenin pathway to regulate bone formation in the adult skeleton. Endocrinology 2007, 148, 2635–2643. [Google Scholar] [CrossRef]
- Jilka, R.L. Molecular and cellular mechanisms of the anabolic effect of intermittent PTH. Bone 2007, 40, 1434–1446. [Google Scholar] [CrossRef]
- Hofbauer, L.C.; Khosla, S.; Dunstan, C.R.; Lacey, D.L.; Boyle, W.J.; Riggs, B.L. The roles of osteoprotegerin and osteoprotegerin ligand in the paracrine regulation of bone resorption. J. Bone Miner. Res. 2000, 15, 2–12. [Google Scholar] [CrossRef]
- National Kidney, F. K/DOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am. J. Kidney Dis. 2003, 42, S1–S201. [Google Scholar]
- Murer, H.; Hernando, N.; Forster, I.; Biber, J. Regulation of Na/Pi transporter in the proximal tubule. Annu. Rev. Physiol. 2003, 65, 531–542. [Google Scholar] [CrossRef]
- Silver, J.; Naveh-Many, T. FGF-23 and secondary hyperparathyroidism in chronic kidney disease. Nat. Rev. Nephrol. 2013, 9, 641–649. [Google Scholar] [CrossRef]
- Hu, M.C.; Shi, M.; Zhang, J.; Quinones, H.; Griffith, C.; Kuro-o, M.; Moe, O.W. Klotho deficiency causes vascular calcification in chronic kidney disease. J. Am. Soc. Nephrol. JASN 2011, 22, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; David, V.; Quarles, L.D. Regulation and function of the FGF23/klotho endocrine pathways. Physiol. Rev. 2012, 92, 131–155. [Google Scholar] [CrossRef] [PubMed]
- Olauson, H.; Lindberg, K.; Amin, R.; Sato, T.; Jia, T.; Goetz, R.; Mohammadi, M.; Andersson, G.; Lanske, B.; Larsson, T.E. Parathyroid-specific deletion of Klotho unravels a novel calcineurin-dependent FGF23 signaling pathway that regulates PTH secretion. PLoS Genet. 2013, 9, e1003975. [Google Scholar] [CrossRef]
- Tentori, F.; Blayney, M.J.; Albert, J.M.; Gillespie, B.W.; Kerr, P.G.; Bommer, J.; Young, E.W.; Akizawa, T.; Akiba, T.; Pisoni, R.L.; et al. Mortality risk for dialysis patients with different levels of serum calcium, phosphorus, and PTH: The Dialysis Outcomes and Practice Patterns Study (DOPPS). Am. J. Kidney Dis. 2008, 52, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Naveh-Many, T.; Rahamimov, R.; Livni, N.; Silver, J. Parathyroid cell proliferation in normal and chronic renal failure rats. The effects of calcium, phosphate, and vitamin D. J. Clin. Investig. 1995, 96, 1786–1793. [Google Scholar] [CrossRef] [PubMed]
- Denda, M.; Finch, J.; Slatopolsky, E. Phosphorus accelerates the development of parathyroid hyperplasia and secondary hyperparathyroidism in rats with renal failure. Am. J. Kidney Dis. 1996, 28, 596–602. [Google Scholar] [CrossRef]
- Centeno, P.P.; Herberger, A.; Mun, H.C.; Tu, C.; Nemeth, E.F.; Chang, W.; Conigrave, A.D.; Ward, D.T. Phosphate acts directly on the calcium-sensing receptor to stimulate parathyroid hormone secretion. Nat. Commun. 2019, 10, 4693. [Google Scholar] [CrossRef]
- Goto, S.; Fujii, H.; Hamada, Y.; Yoshiya, K.; Fukagawa, M. Association between indoxyl sulfate and skeletal resistance in hemodialysis patients. Ther. Apher. Dial. 2010, 14, 417–423. [Google Scholar] [CrossRef]
- Nickolas, T.L.; Cremers, S.; Zhang, A.; Thomas, V.; Stein, E.; Cohen, A.; Chauncey, R.; Nikkel, L.; Yin, M.T.; Liu, X.S.; et al. Discriminants of prevalent fractures in chronic kidney disease. J. Am. Soc. Nephrol. JASN 2011, 22, 1560–1572. [Google Scholar] [CrossRef]
- Araujo, M.J.; Karohl, C.; Elias, R.M.; Barreto, F.C.; Barreto, D.V.; Canziani, M.E.; Carvalho, A.B.; Jorgetti, V.; Moyses, R.M. The pitfall of treating low bone turnover: Effects on cortical porosity. Bone 2016, 91, 75–80. [Google Scholar] [CrossRef]
- Moe, S.M.; Chen, N.X.; Newman, C.L.; Gattone, V.H., 2nd; Organ, J.M.; Chen, X.; Allen, M.R. A comparison of calcium to zoledronic acid for improvement of cortical bone in an animal model of CKD. J. Bone Miner. Res. 2014, 29, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; De Luca, V.; Seeman, E. Parathyroid hormone deficiency and excess: Similar effects on trabecular bone but differing effects on cortical bone. J. Clin. Endocrinol. Metab. 1999, 84, 718–722. [Google Scholar] [CrossRef] [PubMed]
- Nickolas, T.L.; Stein, E.M.; Dworakowski, E.; Nishiyama, K.K.; Komandah-Kosseh, M.; Zhang, C.A.; McMahon, D.J.; Liu, X.S.; Boutroy, S.; Cremers, S.; et al. Rapid cortical bone loss in patients with chronic kidney disease. J. Bone Miner. Res. 2013, 28, 1811–1820. [Google Scholar] [CrossRef] [PubMed]
- Parfitt, A.M. Hormonal influences on bone remodeling and bone loss: Application to the management of primary hyperparathyroidism. Ann. Intern. Med. 1996, 125, 413–415. [Google Scholar] [CrossRef]
- Lanske, B.; Amling, M.; Neff, L.; Guiducci, J.; Baron, R.; Kronenberg, H.M. Ablation of the PTHrP gene or the PTH/PTHrP receptor gene leads to distinct abnormalities in bone development. J. Clin. Investig. 1999, 104, 399–407. [Google Scholar] [CrossRef]
- Jansz, T.T.; Goto, N.A.; van Ballegooijen, A.J.; Willems, H.C.; Verhaar, M.C.; van Jaarsveld, B.C. The prevalence and incidence of vertebral fractures in end-stage renal disease and the role of parathyroid hormone. Osteoporos. Int. 2019, 1–10. [Google Scholar] [CrossRef]
- Coco, M.; Rush, H. Increased incidence of hip fractures in dialysis patients with low serum parathyroid hormone. Am. J. Kidney Dis. 2000, 36, 1115–1121. [Google Scholar] [CrossRef]
- Iimori, S.; Mori, Y.; Akita, W.; Kuyama, T.; Takada, S.; Asai, T.; Kuwahara, M.; Sasaki, S.; Tsukamoto, Y. Diagnostic usefulness of bone mineral density and biochemical markers of bone turnover in predicting fracture in CKD stage 5D patients--a single-center cohort study. Nephrol. Dial. Transplant. 2012, 27, 345–351. [Google Scholar] [CrossRef]
- Danese, M.D.; Kim, J.; Doan, Q.V.; Dylan, M.; Griffiths, R.; Chertow, G.M. PTH and the risks for hip, vertebral, and pelvic fractures among patients on dialysis. Am. J. Kidney Dis. 2006, 47, 149–156. [Google Scholar] [CrossRef]
- Fukagawa, M.; Kazama, J.J.; Shigematsu, T. Skeletal resistance to pth as a basic abnormality underlying uremic bone diseases. Am. J. Kidney Dis. 2001, 38, S152–S155. [Google Scholar] [CrossRef]
- Iwasaki-Ishizuka, Y.; Yamato, H.; Nii-Kono, T.; Kurokawa, K.; Fukagawa, M. Downregulation of parathyroid hormone receptor gene expression and osteoblastic dysfunction associated with skeletal resistance to parathyroid hormone in a rat model of renal failure with low turnover bone. Nephrol. Dial. Transplant. 2005, 20, 1904–1911. [Google Scholar] [CrossRef] [PubMed]
- Graciolli, F.G.; Neves, K.R.; Barreto, F.; Barreto, D.V.; Dos Reis, L.M.; Canziani, M.E.; Sabbagh, Y.; Carvalho, A.B.; Jorgetti, V.; Elias, R.M.; et al. The complexity of chronic kidney disease-mineral and bone disorder across stages of chronic kidney disease. Kidney Int. 2017, 91, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Boltenstal, H.; Qureshi, A.R.; Behets, G.J.; Lindholm, B.; Stenvinkel, P.; D’Haese, P.C.; Haarhaus, M. Association of serum sclerostin with bone sclerostin in chronic kidney disease is lost in glucocorticoid treated patients. Calcif. Tissue Int. 2019, 104, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Picton, M.L.; Moore, P.R.; Mawer, E.B.; Houghton, D.; Freemont, A.J.; Hutchison, A.J.; Gokal, R.; Hoyland, J.A. Down-regulation of human osteoblast PTH/PTHrP receptor mRNA in end-stage renal failure. Kidney Int. 2000, 58, 1440–1449. [Google Scholar] [CrossRef] [PubMed]
- Manolagas, S.C. Birth and death of bone cells: Basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr. Rev. 2000, 21, 115–137. [Google Scholar] [CrossRef] [PubMed]
- Bonewald, L.F.; Dallas, S.L. Role of active and latent transforming growth factor beta in bone formation. J. Cell. Biochem. 1994, 55, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Noda, M.; Camilliere, J.J. In vivo stimulation of bone formation by transforming growth factor-beta. Endocrinology 1989, 124, 2991–2994. [Google Scholar] [CrossRef]
- Filvaroff, E.; Erlebacher, A.; Ye, J.; Gitelman, S.E.; Lotz, J.; Heillman, M.; Derynck, R. Inhibition of TGF-beta receptor signaling in osteoblasts leads to decreased bone remodeling and increased trabecular bone mass. Development 1999, 126, 4267–4279. [Google Scholar]
- Qiu, T.; Wu, X.; Zhang, F.; Clemens, T.L.; Wan, M.; Cao, X. TGF-beta type II receptor phosphorylates PTH receptor to integrate bone remodelling signalling. Nat. Cell Biol. 2010, 12, 224–234. [Google Scholar] [CrossRef]
- Mace, M.L.; Gravesen, E.; Nordholm, A.; Hofman-Bang, J.; Secher, T.; Olgaard, K.; Lewin, E. Kidney fibroblast growth factor 23 does not contribute to elevation of its circulating levels in uremia. Kidney Int. 2017, 92, 165–178. [Google Scholar] [CrossRef]
- Jiang, X.; Kanai, H.; Shigehara, T.; Maezawa, A.; Yano, S.; Naruse, T. Metabolism of transforming growth factor-beta in patients receiving hemodialysis especially those with renal osteodystrophy. Ren. Fail. 1998, 20, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Santos, F.R.; Moyses, R.M.; Montenegro, F.L.; Jorgetti, V.; Noronha, I.L. IL-1beta, TNF-alpha, TGF-beta, and bFGF expression in bone biopsies before and after parathyroidectomy. Kidney Int. 2003, 63, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Ritz, E.; Stefanski, A.; Rambausek, M. The role of the parathyroid glands in the uremic syndrome. Am. J. Kidney Dis. 1995, 26, 808–813. [Google Scholar] [CrossRef]
- Lishmanov, A.; Dorairajan, S.; Pak, Y.; Chaudhary, K.; Chockalingam, A. Elevated serum parathyroid hormone is a cardiovascular risk factor in moderate chronic kidney disease. Int. Urol. Nephrol. 2012, 44, 541–547. [Google Scholar] [CrossRef]
- Jorde, R.; Sundsfjord, J.; Haug, E.; Bonaa, K.H. Relation between low calcium intake, parathyroid hormone, and blood pressure. Hypertension 2000, 35, 1154–1159. [Google Scholar] [CrossRef]
- Jorde, R.; Svartberg, J.; Sundsfjord, J. Serum parathyroid hormone as a predictor of increase in systolic blood pressure in men. J Hypertens 2005, 23, 1639–1644. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, D.Z. Circulating parathyroid hormone and risk of hypertension: A meta-analysis. Clin. Chim. Acta 2018, 482, 40–45. [Google Scholar] [CrossRef]
- Leiba, A.; Cohen, M.S.; Dinour, D.; Holtzman, E.J. Severe and long-lasting hypotension occuring immediately after parathyroidectomy in hypertensive hemodialysis patients: A case series. J. Hum. Hypertens. 2013, 27, 399–401. [Google Scholar] [CrossRef]
- Heyliger, A.; Tangpricha, V.; Weber, C.; Sharma, J. Parathyroidectomy decreases systolic and diastolic blood pressure in hypertensive patients with primary hyperparathyroidism. Surgery 2009, 146, 1042–1047. [Google Scholar] [CrossRef]
- Custodio, M.R.; Koike, M.K.; Neves, K.R.; dos Reis, L.M.; Graciolli, F.G.; Neves, C.L.; Batista, D.G.; Magalhaes, A.O.; Hawlitschek, P.; Oliveira, I.B.; et al. Parathyroid hormone and phosphorus overload in uremia: Impact on cardiovascular system. Nephrol. Dial. Transplant. 2012, 27, 1437–1445. [Google Scholar] [CrossRef]
- Saleh, F.N.; Schirmer, H.; Sundsfjord, J.; Jorde, R. Parathyroid hormone and left ventricular hypertrophy. Eur. Heart J. 2003, 24, 2054–2060. [Google Scholar] [CrossRef]
- Schlüter, K.-D.; Piper, H.M. Cardiovascular actions of parathyroid hormone and parathyroid hormone-related peptide. Cardiovasc. Res. 1998, 37, 34–41. [Google Scholar] [CrossRef]
- Amann, K.; Ritz, E.; Wiest, G.; Klaus, G.; Mall, G. A role of parathyroid hormone for the activation of cardiac fibroblasts in uremia. J. Am. Soc. Nephrol. JASN 1994, 4, 1814–1819. [Google Scholar] [PubMed]
- Huntgeburth, M.; Tiemann, K.; Shahverdyan, R.; Schluter, K.D.; Schreckenberg, R.; Gross, M.L.; Modersheim, S.; Caglayan, E.; Muller-Ehmsen, J.; Ghanem, A.; et al. Transforming growth factor beta(1) oppositely regulates the hypertrophic and contractile response to beta-adrenergic stimulation in the heart. PLoS ONE 2011, 6, e26628. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, O.M.; Januzzi, J.L.; Isakova, T.; Laliberte, K.; Smith, K.; Collerone, G.; Sarwar, A.; Hoffmann, U.; Coglianese, E.; Christenson, R.; et al. Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation 2009, 119, 2545–2552. [Google Scholar] [CrossRef]
- Faul, C.; Amaral, A.P.; Oskouei, B.; Hu, M.C.; Sloan, A.; Isakova, T.; Gutierrez, O.M.; Aguillon-Prada, R.; Lincoln, J.; Hare, J.M.; et al. FGF23 induces left ventricular hypertrophy. J. Clin. Investig. 2011, 121, 4393–4408. [Google Scholar] [CrossRef]
- Rashid, G.; Plotkin, E.; Klein, O.; Green, J.; Bernheim, J.; Benchetrit, S. Parathyroid hormone decreases endothelial osteoprotegerin secretion: Role of protein kinase A and C. Am. J. Physiol. Ren. Physiol. 2009, 296, F60–F66. [Google Scholar] [CrossRef]
- Shao, J.S.; Cheng, S.L.; Charlton-Kachigian, N.; Loewy, A.P.; Towler, D.A. Teriparatide (human parathyroid hormone (1–34)) inhibits osteogenic vascular calcification in diabetic low density lipoprotein receptor-deficient mice. J. Biol. Chem. 2003, 278, 50195–50202. [Google Scholar] [CrossRef]
- Linefsky, J.P.; O’Brien, K.D.; Katz, R.; de Boer, I.H.; Barasch, E.; Jenny, N.S.; Siscovick, D.S.; Kestenbaum, B. Association of serum phosphate levels with aortic valve sclerosis and annular calcification: The cardiovascular health study. J. Am. Coll. Cardiol. 2011, 58, 291–297. [Google Scholar] [CrossRef]
- Neves, K.R.; Graciolli, F.G.; dos Reis, L.M.; Graciolli, R.G.; Neves, C.L.; Magalhaes, A.O.; Custodio, M.R.; Batista, D.G.; Jorgetti, V.; Moyses, R.M. Vascular calcification: Contribution of parathyroid hormone in renal failure. Kidney Int. 2007, 71, 1262–1270. [Google Scholar] [CrossRef]
- McCarthy, J.T.; El-Azhary, R.A.; Patzelt, M.T.; Weaver, A.L.; Albright, R.C.; Bridges, A.D.; Claus, P.L.; Davis, M.D.; Dillon, J.J.; El-Zoghby, Z.M.; et al. Survival, risk factors, and effect of treatment in 101 patients with calciphylaxis. Mayo Clin. Proc. 2016, 91, 1384–1394. [Google Scholar] [CrossRef] [PubMed]
- Nigwekar, S.U.; Zhao, S.; Wenger, J.; Hymes, J.L.; Maddux, F.W.; Thadhani, R.I.; Chan, K.E. A nationally representative study of calcific uremic arteriolopathy risk factors. J. Am. Soc. Nephrol. JASN 2016, 27, 3421–3429. [Google Scholar] [CrossRef] [PubMed]
- Hofbauer, L.C.; Brueck, C.C.; Shanahan, C.M.; Schoppet, M.; Dobnig, H. Vascular calcification and osteoporosis--from clinical observation towards molecular understanding. Osteoporos. Int. 2007, 18, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Da, J.; Xie, X.; Wolf, M.; Disthabanchong, S.; Wang, J.; Zha, Y.; Lv, J.; Zhang, L.; Wang, H. Serum phosphorus and progression of CKD and mortality: A meta-analysis of cohort studies. Am. J. Kidney Dis. 2015, 66, 258–265. [Google Scholar] [CrossRef]
- Neves, K.R.; Graciolli, F.G.; dos Reis, L.M.; Pasqualucci, C.A.; Moyses, R.M.; Jorgetti, V. Adverse effects of hyperphosphatemia on myocardial hypertrophy, renal function, and bone in rats with renal failure. Kidney Int. 2004, 66, 2237–2244. [Google Scholar] [CrossRef]
- Lee, Y.J.; Okuda, Y.; Sy, J.; Obi, Y.; Kang, D.H.; Nguyen, S.; Hsiung, J.T.; Park, C.; Rhee, C.M.; Kovesdy, C.P.; et al. Association of mineral bone disorder with decline in residual kidney function in incident hemodialysis patients. J. Bone Miner. Res. 2019, 35, 317–325. [Google Scholar] [CrossRef]
- Schumock, G.T.; Andress, D.L.; Marx, S.E.; Sterz, R.; Joyce, A.T.; Kalantar-Zadeh, K. Association of secondary hyperparathyroidism with CKD progression, health care costs and survival in diabetic predialysis CKD patients. Nephron. Clin. Pract. 2009, 113, c54–c61. [Google Scholar] [CrossRef]
- Adey, D.; Kumar, R.; McCarthy, J.T.; Nair, K.S. Reduced synthesis of muscle proteins in chronic renal failure. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E219–E225. [Google Scholar] [CrossRef]
- Workeneh, B.T.; Mitch, W.E. Review of muscle wasting associated with chronic kidney disease. Am. J. Clin. Nutr. 2010, 91, 1128S–1132S. [Google Scholar] [CrossRef]
- Molina, P.; Carrero, J.J.; Bover, J.; Chauveau, P.; Mazzaferro, S.; Torres, P.U.; European Renal, N.; Chronic Kidney, D.-M.; Bone Disorder Working Groups of the European Renal Association-European Dialysis Transplant Association. Vitamin D, a modulator of musculoskeletal health in chronic kidney disease. J. Cachexia Sarcopenia Muscle 2017, 8, 686–701. [Google Scholar] [CrossRef]
- Baczynski, R.; Massry, S.G.; Magott, M.; el-Belbessi, S.; Kohan, R.; Brautbar, N. Effect of parathyroid hormone on energy metabolism of skeletal muscle. Kidney Int. 1985, 28, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Fernandez, P.; Sanchez Agudo, L.; Calatrava, J.M.; Martinez, M.E.; Escuin Sancho, F.; Selgas, R.; Sanchez Sicilia, L. Parathormone as a uremic toxin. Possible effect on respiratory muscle function in uremia. Med. Clin. 1984, 82, 395–397. [Google Scholar]
- Khajehdehi, P.; Ali, M.; Al-Gebory, F.; Henry, G.; Bastani, B. The effects of parathyroidectomy on nutritional and biochemical status of hemodialysis patients with severe secondary hyperparathyroidism. J. Ren. Nutr. 1999, 9, 186–191. [Google Scholar] [CrossRef]
- Cuppari, L.; de Carvalho, A.B.; Avesani, C.M.; Kamimura, M.A.; Dos Santos Lobao, R.R.; Draibe, S.A. Increased resting energy expenditure in hemodialysis patients with severe hyperparathyroidism. J. Am. Soc. Nephrol. JASN 2004, 15, 2933–2939. [Google Scholar] [CrossRef] [PubMed]
- Kir, S.; Komaba, H.; Garcia, A.P.; Economopoulos, K.P.; Liu, W.; Lanske, B.; Hodin, R.A.; Spiegelman, B.M. PTH/PTHrP receptor mediates cachexia in models of kidney failure and cancer. Cell Metab. 2016, 23, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Fadda, G.Z.; Akmal, M.; Premdas, F.H.; Lipson, L.G.; Massry, S.G. Insulin release from pancreatic islets: Effects of CRF and excess PTH. Kidney Int. 1988, 33, 1066–1072. [Google Scholar] [CrossRef]
- Amend, W.J., Jr.; Steinberg, S.M.; Lowrie, E.G.; Lazarus, J.M.; Soeldner, J.S.; Hampers, C.L.; Merrill, J.P. The influence of serum calcium and parathyroid hormone upon glucose metabolism in uremia. J. Lab. Clin. Med. 1975, 86, 435–444. [Google Scholar]
- Ahamed, N.A.; Abdul-Aziz, M.Y.; El-Bauomy, A.; Salem, T.S. Parathyroid hormone: eFfects on glucose homeostasis and insulin sensitivity in chronic renal failure patients on regular hemodialysis. J. Taibah Univ. Med. Sci. 2008, 3, 44–54. [Google Scholar] [CrossRef][Green Version]
- Mak, R.H. Intravenous 1,25 dihydroxycholecalciferol corrects glucose intolerance in hemodialysis patients. Kidney Int. 1992, 41, 1049–1054. [Google Scholar] [CrossRef]
- Mak, R.H.; Turner, C.; Haycock, G.B.; Chantler, C. Secondary hyperparathyroidism and glucose intolerance in children with uremia. Kidney Int. Suppl. 1983, 16, S128–S133. [Google Scholar]
- Wolf, G. Energy regulation by the skeleton. Nutr. Rev. 2008, 66, 229–233. [Google Scholar] [CrossRef]
- Fulzele, K.; Riddle, R.C.; DiGirolamo, D.J.; Cao, X.; Wan, C.; Chen, D.; Faugere, M.C.; Aja, S.; Hussain, M.A.; Bruning, J.C.; et al. Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell 2010, 142, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Goldenstein, P.T.; Graciolli, F.G.; Antunes, G.L.; Dominguez, W.V.; Dos Reis, L.M.; Moe, S.; Elias, R.M.; Jorgetti, V.; Moyses, R.M.A. A prospective study of the influence of the skeleton on calcium mass transfer during hemodialysis. PLoS ONE 2018, 13, e0198946. [Google Scholar] [CrossRef]
- Fraser, C.L.; Sarnacki, P.; Arieff, A.I. Calcium transport abnormality in uremic rat brain synaptosomes. J. Clin. Investig. 1985, 76, 1789–1795. [Google Scholar] [CrossRef] [PubMed]
- Guisado, R.; Arieff, A.I.; Massry, S.G.; Lazarowitz, V.; Kerian, A. Changes in the electroencephalogram in acute uremia. Effects of parathyroid hormone and brain electrolytes. J. Clin. Investig. 1975, 55, 738–745. [Google Scholar] [CrossRef] [PubMed]
- Lourida, I.; Thompson-Coon, J.; Dickens, C.M.; Soni, M.; Kuzma, E.; Kos, K.; Llewellyn, D.J. Parathyroid hormone, cognitive function and dementia: A systematic review. PLoS ONE 2015, 10, e0127574. [Google Scholar] [CrossRef] [PubMed]
- Meytes, D.; Bogin, E.; Ma, A.; Dukes, P.P.; Massry, S.G. Effect of parathyroid hormone on erythropoiesis. J. Clin. Investig. 1981, 67, 1263–1269. [Google Scholar] [CrossRef]
- Levi, J.; Malachi, T.; Djaldetti, M.; Bogin, E. Biochemical changes associated with the osmotic fragility of young and mature erythrocytes caused by parathyroid hormone in relation to the uremic syndrome. Clin. Biochem. 1987, 20, 121–125. [Google Scholar] [CrossRef]
- Brickman, A.S.; Sherrard, D.J.; Jowsey, J.; Singer, F.R.; Baylink, D.J.; Maloney, N.; Massry, S.G.; Norman, A.W.; Coburn, J.W. 1,25-dihydroxycholecalciferol. Effect on skeletal lesions and plasma parathyroid hormone levels in uremic osteodystrophy. Arch. Intern. Med. 1974, 134, 883–888. [Google Scholar] [CrossRef]
- Klinger, M.; Alexiewicz, J.M.; Linker-Israeli, M.; Pitts, T.O.; Gaciong, Z.; Fadda, G.Z.; Massry, S.G. Effect of parathyroid hormone on human T cell activation. Kidney Int. 1990, 37, 1543–1551. [Google Scholar] [CrossRef]
- Yasunaga, C.; Nakamoto, M.; Matsuo, K.; Nishihara, G.; Yoshida, T.; Goya, T. Effects of a parathyroidectomy on the immune system and nutritional condition in chronic dialysis patients with secondary hyperparathyroidism. Am. J. Surg. 1999, 178, 332–336. [Google Scholar] [CrossRef]
- Hory, B.G.; Roussanne, M.C.; Rostand, S.; Bourdeau, A.; Drueke, T.B.; Gogusev, J. Absence of response to human parathyroid hormone in athymic mice grafted with human parathyroid adenoma, hyperplasia or parathyroid cells maintained in culture. J. Endocrinol. Investig. 2000, 23, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Pettway, G.J.; Schneider, A.; Koh, A.J.; Widjaja, E.; Morris, M.D.; Meganck, J.A.; Goldstein, S.A.; McCauley, L.K. Anabolic actions of PTH (1-34): Use of a novel tissue engineering model to investigate temporal effects on bone. Bone 2005, 36, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Chou, F.F.; Chen, J.B.; Lee, C.H.; Chen, S.H.; Sheen-Chen, S.M. Parathyroidectomy can improve bone mineral density in patients with symptomatic secondary hyperparathyroidism. Arch. Surg. 2001, 136, 1064–1068. [Google Scholar] [CrossRef]
- Chou, F.F.; Chen, J.B.; Hsieh, K.C.; Liou, C.W. Cognitive changes after parathyroidectomy in patients with secondary hyperparathyroidism. Surgery 2008, 143, 526–532. [Google Scholar] [CrossRef]
- Mandolfo, S.; Malberti, F.; Farina, M.; Villa, G.; Scanziani, R.; Surian, M.; Imbasciati, E. Parathyroidectomy and response to erythropoietin therapy in anaemic patients with chronic renal failure. Nephrol. Dial. Transplant. 1998, 13, 2708–2709. [Google Scholar] [CrossRef]
- Goldenstein, P.T.; Elias, R.M.; Pires de Freitas do Carmo, L.; Coelho, F.O.; Magalhaes, L.P.; Antunes, G.L.; Custodio, M.R.; Montenegro, F.L.; Titan, S.M.; Jorgetti, V.; et al. Parathyroidectomy improves survival in patients with severe hyperparathyroidism: A comparative study. PLoS ONE 2013, 8, e68870. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| PTH-Related Manifestations | Parathyroidectomy Effects | Ref. |
|---|---|---|
| Hypertension Myocardial hypertrophy | Blood pressure reduction Beneficial effect on cardiovascular mortality | [44,46] |
| Abnormal bone density (mainly in cortical compartment) | Improvement of bone mineral density at lumbar spine and femoral neck | [19,94] |
| Increase of levels of cytosolic calcium in brain synaptic terminals | Improvement of cognitive function Prevention of electroencephalogram abnormalities in uremic animals | [85,95] |
| Hematopoietic dysfunction Accelerate erythrocyte sedimentation rate Increase of osmotic fragility of erythrocytes | Improvement of anemia Regression of medullar fibrosis | [87,96] |
| Reduction of T lymphocyte proliferation and cytokine production Impairment of immunoglobulins production | Improvement on serum immunoglobulins and complement titles | [90,91] |
| Increase in all-cause mortality risk | Improve of survival in patients with severe secondary Hyperparathyroidism Improvement of quality of life | [14,97] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duque, E.J.; Elias, R.M.; Moysés, R.M.A. Parathyroid Hormone: A Uremic Toxin. Toxins 2020, 12, 189. https://doi.org/10.3390/toxins12030189
Duque EJ, Elias RM, Moysés RMA. Parathyroid Hormone: A Uremic Toxin. Toxins. 2020; 12(3):189. https://doi.org/10.3390/toxins12030189
Chicago/Turabian StyleDuque, Eduardo J., Rosilene M. Elias, and Rosa M. A. Moysés. 2020. "Parathyroid Hormone: A Uremic Toxin" Toxins 12, no. 3: 189. https://doi.org/10.3390/toxins12030189
APA StyleDuque, E. J., Elias, R. M., & Moysés, R. M. A. (2020). Parathyroid Hormone: A Uremic Toxin. Toxins, 12(3), 189. https://doi.org/10.3390/toxins12030189