Abstract
Perforation of cellular membranes by pore-forming proteins can affect cell physiology, tissue integrity, or immune response. Since many pore-forming proteins are toxins or highly potent virulence factors, they represent an attractive target for the development of molecules that neutralize their actions with high efficacy. There has been an assortment of inhibitors developed to specifically obstruct the activity of pore-forming proteins, in addition to vaccination and antibiotics that serve as a plausible treatment for the majority of diseases caused by bacterial infections. Here we review a wide range of potential inhibitors that can specifically and effectively block the activity of pore-forming proteins, from small molecules to more specific macromolecular systems, such as synthetic nanoparticles, antibodies, antibody mimetics, polyvalent inhibitors, and dominant negative mutants. We discuss their mechanism of inhibition, as well as advantages and disadvantages.
Keywords:
pore-forming proteins; pore-forming toxins; anthrax toxin; lipid membranes; pore formation; inhibitor Key Contribution:
The manuscript reviews current possibilities for inhibition of pore-forming proteins with an emphasis on alternative approaches.
1. Introduction to Toxic Pore-Forming Proteins
1.1. Different Modes of Creating a Pore in Cellular Membranes
Plasma as well as organelle membranes are vital for cells. They protect cells from the environment, including invading organisms, enable exchange of substances either between cells and their surroundings or between different cellular compartments, cell adhesion, transport, metabolism, and flow of information via cell signaling. Thus, interfering with the integrity of membranes can disturb cellular processes and can, in extreme cases, be detrimental. During evolution, organisms from all kingdoms of life have evolved mechanisms to form pores in membranes, in order to attack other organisms or defend against them, to digest their prey, or as a part of the immune system to remove unwanted cells. Excellent reviews are available describing diverse modes of transmembrane pore formation by proteins [1,2,3,4,5,6,7,8].
Pore-forming proteins (PFPs) are generally secreted by cells as soluble monomers that assemble into structured oligomeric complexes at the target membrane surface. Upon binding to the lipid membrane, monomers oligomerize on its surface to form structured assemblies called prepores and undergo conformational changes in order to expose hydrophobic surfaces, leading to spontaneous insertion into the lipid bilayer, pore formation, and membrane permeabilization. Pores made by PFPs are largely diverse in inner diameter, ranging from 0.7 nm as in the case of colicins [9] to the largest known pores of cholesterol-dependent cytolysins (CDCs) with diameters of 25–40 nm [8]. Depending on the size of the pore, different substances pass through, such as ions (e.g., Ca2+, K+), small molecules (e.g., adenosine triphosphate (ATP)), or large molecules (e.g., proteins) [10]. Structurally, PFPs are divided into two major classes based on secondary structure elements that frame the transmembrane channel of their pores either with α-helices (i.e., α-PFPs) [11] or β-barrels (β-PFPs) [1,2,12] (Figure 1). Three dimensional structures of soluble monomeric PFPs, prepores and pores from different families have been known to date [13,14,15,16,17,18,19,20,21,22] and several excellent reviews describe their features [10,23,24]. These structural models provide a valuable insight into the mechanism of action by PFPs and crucially contribute to a rational design of their potential inhibitors. The shapes of pores are quite diverse. Some PFPs form matrix-type toroidal pores, where the transmembrane protein units are interspersed by lipids [3], such as actinoporins [25,26], colicins [27], and proteins from the Bcl-2 family of apoptotic proteins [28]. In contrast to toroidal formations, pore walls can also be completely built of proteins, forming either compact α-barrels as in the case of α-PFP cytolysin A from Escherichia coli (Figure 1), or β-barrels, formed by β-PFPs such as α- and γ-toxin from Staphylococcus aureus, membrane attack complex/perforin (MACPF)/cholesterol-dependent cytolysins (CDCs) protein superfamily, or aerolysin-like proteins [2]. β-barrels are also formed by bacterial secretion systems of type III and IV [1,29] and binary AB type toxins, where the B component is pore-forming and allows translocation of A subunits that possess catalytic activity (e.g., diphtheria; anthrax; α-, ε-, ι-, and C2 toxins) [14,30,31,32] (Figure 1). In the case of β-barrel proteins such as MACPF/CDCs, arc-type toroidal pores partly lined with lipids as well as multimeric pores can also be built in addition to fully proteinaceous ring-shaped pores [3]. The majority of work regarding PFP inhibitors has been done on the B component (protective antigen or PA) of the anthrax toxin from Bacillus anthracis, hence a short description of this toxin follows. For an in-depth understanding of individual PFPs and their characteristics, an interested reader can choose from recent reviews about that topic [1,2,3,7,8,33,34,35,36,37,38].
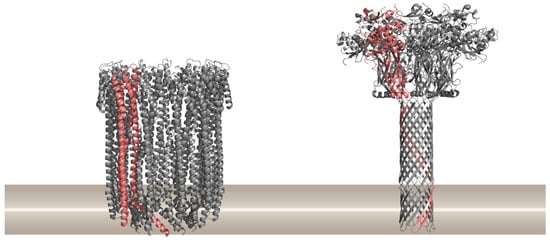
Figure 1.
Two major classes of pore-forming proteins (PFPs) based on the structural element present in the final pore, α-helical PFPs exemplified by the cytolysin A from Escherichia coli (PDB ID 2WCD) on the left, and β-barrel PFPs exemplified by the anthrax toxin protective antigen pore from Bacillus anthracis (PDB ID 3J9C) on the right. Ribbon representations of proteins are drawn by using PyMOL [39]. A single protomer in the pore is shown in pink. The approximate position of the lipid membrane is shown in brown.
Anthrax is a deadly disease and is considered a biological threat due to the antecedent weaponization of this agent. The component B of an anthrax toxin is responsible for the cell surface binding, whereas the component A is enzymatically active [40]. The component B is known as protective antigen (PA), while there are two distinct A components, a lethal factor (LF) and an edema factor (EF). Association between PA an LF forms the lethal toxin (LT), and interaction of PA with EF the edema toxin (ET) [41]. The pore is formed by a precursor PA83 binding to cell surface receptors [42,43], followed by proteolytic cleavage of PA83 by the protease furin, resulting in PA63, which oligomerizes and forms a homo-heptameric [15,16] and/or homo-octameric [44] PA prepore, which undergoes conformational changes to insert in the membrane and form a functional pore. The pore allows binding and transportation of LF or EF to the cytosol [45]. Vaccines against anthrax are available [46], but despite its poor prognosis, a widespread public immunization is unlikely due to its low incidence [47]. Consequently, searching for new strategies to protect against this disease is therefore warranted [48].
1.2. Effects of PFPs on Target Cells and Their Biological Roles
The best characterized and the largest group of PFPs are bacterial PFPs [10], many of which are the key virulence factors of deadly diseases and are also referred to as pore-forming toxins (PFTs). They can act on host cell physiology, tissue integrity, and immune response and cause inflammation that may interfere with antimicrobial treatment [49,50]. PFPs produced by a particular bacterium can form pores in the membrane of other bacteria, plants, animals, or humans, thereby causing disruption of membrane integrity and ion imbalance [29]. To kill other bacteria, some bacteria produce proteins such as colicins [51,52]. To attack eukaryotic cells, some bacteria express CDCs, hemolysins, and aerolysin-like proteins to promote colonization, spread, and survival within the hostile environment of a host organism [29]. In addition to bacteria, PFPs with (potential) toxic function are excreted also by eukaryotic organisms such as fungi, parasites, cnidarians, arachnids, earthworms, or plants, for the purposes of feeding or to defend against their predators. PFPs that are used in defense are also produced by vertebrates, for instance the complement membrane attack complex (MAC) to kill bacteria [17,53], or perforin to kill malignant or virus-infected cells [54], as well as proteins of the Bcl-2 family that cause apoptosis (e.g., Bak and Bax proteins) [55,56].
In this review, we describe various ways of preventing pore formation, especially of toxic PFPs. For majority of toxic PFPs, there are no effective antidotes or antitoxins developed and approved for human use [57]. These different ways and means of inhibition of PFPs can on one side help in studies of the pore-forming mechanism at the molecular level, as well as in the design of novel agents and innovative strategies for therapeutic, diagnostic, labeling, or biosensing purposes.
2. Modes of Preventing Pore Formation
Although structural features and properties of pores formed by PFPs are substantially diverse, their activity can be targeted in a similar manner, as the molecular mechanism of action basically follows a common pathway. Generalized steps in the molecular mechanism of pore formation of PFPs together with steps allowing potential inhibition are illustrated in Figure 2.
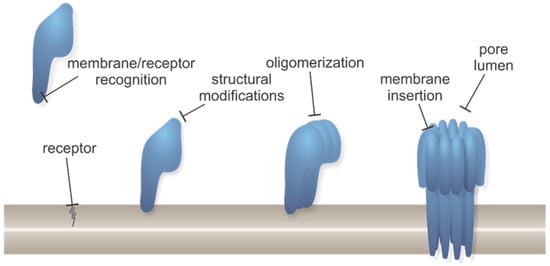
Figure 2.
Generalized pore formation process by different types of PFPs with marked positions for possible inhibitors interfering. Protein monomers are shown in blue, lipid membrane is shown in brown, receptor for PFP binding (which can be either a specific lipid as shown here, or protein, etc.) is shown in gray.
A range of various molecules has been developed that neutralizes the activity of toxins, the majority of them aiming for therapeutic potential [58,59]. Possible ways to inhibit the virulent effects of PFPs are by interfering either with their expression (inhibition of transcription regulators [60], protein synthesis, quorum sensing [61]), or interaction with a cognate receptor [62,63,64,65], structural modifications of membrane-bound precursors, oligomerization, membrane insertion, or pore lumen and, consequently, with the transport of molecules or ions through formed pores [66] (Figure 2). Additionally, PFP function can be also targeted indirectly by counteracting the effects of PFPs [58], such as by membrane repair [67,68,69], enhancement of blebbing and microvesicle shedding [70], and by specifically boosting or pre-activating host defense that neutralizes PFPs [38].
Antibiotics in conjunction with vaccination are set as the first line of treatment for some diseases caused by toxic PFPs (anthrax, pneumonia, etc.). Vaccination has some drawbacks, such as inconsistent efficacy, economical impracticality, or unavailability. Furthermore, the antibiotic treatment can provoke several side-effects [71], it must be given early when symptoms are nonspecific, the interval between initial exposure and the onset of treatment can be lengthy [72]. Moreover, there is a growing number of multidrug resistant bacteria secreting highly potent exotoxins with no antitoxins currently available on the market [73,74]. Therefore, development of new inhibitor scaffolds, virulence-targeted antimicrobial prophylactics, and therapeutics with a narrower spectrum, and combination therapies are needed to find more efficient treatments of increasingly resistant bacteria [75,76,77]. Here we review possible approaches for direct inhibition of pore formation by intervening with various steps of pore formation process, as outlined in Figure 2. Those strategies are worth pursuing as they offer several advantages compared with targeting the bacteria themselves: (i) Organisms are less likely to develop resistance to such scaffolds and normal microbiota remain undisturbed [73,78], (ii) mechanism of action of PFPs is well defined and is composed of distinct steps, which allow specific and targeted activity of inhibitors and consequently reduced probability of side-effects, (iii) broad application against many bacterial infections, and (iv) acting directly on a particular virulence factor to prevent as well as cure the disease [38,57,66,79,80,81]. However, it has to be noted that since alternative approaches for inhibition of PFPs have not been as widely used as antibiotics and vaccines, side-effects have not been recognized to a similar extent yet.
Below we describe possible approaches, from small organic molecules to relatively large organic particles, peptides, or proteins and discuss their relevance. However, the standard approach used in drug design, of targeting proteins with small molecules, is not commonly employed in the case of PFPs. PFPs have a specific mode of action that involves large protein surfaces and only rarely provides cavities or binding sites that could be used for small-molecule drug development. For a successful blockage, an important role is played by the size, conformation, symmetry, and structural plasticity of the inhibitor. Therefore, the diversity of potential inhibitors encompasses small organic molecules to relatively large organic particles, peptides, or proteins. Lately, there has been an ascent of alternative binding scaffolds with similar binding characteristics as antibodies, yet overcoming some of their weaknesses, such as high cost, challenging production protocols, and low production yield [82]. Furthermore, there are some less conventional ways for the neutralization of toxin activity, for instance the utilization of dominant negative mutants [83] and receptor decoys [84,85,86]. The representatives of all discussed inhibition strategies are presented in Figure 3.
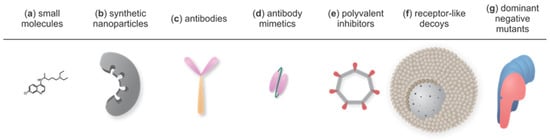
Figure 3.
Overview of various strategies to inhibit PFPs: (a) Small molecules. Chloroquine as a representative molecule. (b) Synthetic nanoparticles. A mold with pockets for PFP binding. (c) Antibodies (Abs), a fragment crystallizable (Fc) region shown in orange and fragment antigen-binding (Fab) regions in pink. (d) Antibody mimetics. Smaller protein molecules derived from antibodies that overcome some of their weaknesses. scFv (single chain fragment variable), a fusion protein of interconnected variable regions of Fab as a representative molecule. (e) Polyvalent inhibitors. Cyclic scaffold (gray) with PFP-binding moieties (red) that positionally match with monomeric protein units in a pore. (f) Receptor-like decoys. Polymeric core enclosed by lipid bilayer containing PFP receptors. (g) Dominant negative mutants. Mutant protein monomer (pink) forms oligomers with the wild-type protein (blue), but such complexes fail to form pores. For clarity, individual schemes are not drawn in proportion.
2.1. Small Molecules
The discovery and development of small-molecule antitoxins represent a high-priority task in modern drug design and medicinal chemistry [57,73,87], mostly because of their small size, excellent tissue penetration, long room-temperature shelf life, ease of analogue design and preparation of high-purity molecules in large quantities. Although this is an attractive research avenue, the number of studies of small-molecule PFPs inhibitors is very limited. Assorted trials are described in the review by Nestorovich and Bezrukov, 2012 [57].
Inhibition of a completely formed pore of oligomerized PFP units by small molecules is usually non-specific, meaning that the small molecule sterically hinders ion fluxes through the pore either by electrostatic or hydrophobic interactions in the lumen of the channel without binding to a specific binding site on the protein [88]. Such inhibitors can probably block only PFPs with a small pore diameter and not larger ones, e.g., CDCs. The most recognized small molecules acting that way are various chloride channel blockers. For example, they inhibit VacA, a vacuolating pore-forming binary AB type cytotoxin produced by the human pathogen Helicobacter pylori, as well as a variety of other non-homologous anion-selective channels [88]. Other binary toxins such as C2II component of C2 toxin, Ib of ι-toxin, and PA63 of anthrax toxin, can be efficiently blocked in vitro and in vivo by a drug chloroquine (Figure 3a) or its analogues with the same backbone architecture and the side-chain diversity, containing at least one positively charged quaternary ammonium group [89,90,91,92,93]. Chloroquine and other quinoline derivatives have been used in the treatment of malaria. Besides PFPs, they can also block endogenous chloride channels [94], as well as nicotinic acetylcholine receptors (nAChRs) [95] and have several recognized side-effects, such as sensorineural hearing loss, tinnitus, and vertigo [96].
Inhibition of a toxic PFP activity can also be achieved by targeting receptors and specific sites or “hot spots” involved in protein–protein or protein–lipid interactions. Small-molecule inhibitors can also occupy receptors and therefore attenuate the possibility of PFP binding. PFP receptors can be involved in diseases (e.g., cancer, Alzheimer’s disease, cystic fibrosis, or auto-immune diseases) [65,97,98,99,100] and small-molecule therapeutics, used to cure these diseases, can help develop blockers for PFP receptors [101]. In another case, inhibitors of ATP-gated purinergic receptors (P2XR) were found to inhibit S. aureus α-toxin membrane binding and oligomerization [102]. Examples of small-molecule inhibitors that bind PFP monomers and attenuate the binding to receptors are calixarenes (p-sulfonato-calix[n]arenes), inhibiting leukotoxins [49]. Moreover, small molecules can be also targeted to hot spots on the PFP surface responsible for oligomerization. Several natural compounds have been found that prevent different steps in pore-forming process of S. aureus α-toxin [103,104,105], Streptococcus pneumoniae pneumolysin [106,107], Streptococcus pyogenes streptolysin O [108], and Listeria monocytogenes listeriolysin O [109]. Recently, necrosulfonamide, a small-molecule inhibitor of pyroptotic PFP gasdermin D was identified, which disables protein dimers to oligomerize and form pores [110]. Cisplatin, which is one of the most effective chemotherapeutic anticancer agents, can inhibit proper heptamer assembly of anthrax toxin PA component in a noncovalent reversible manner, preventing toxicity of both factors, LF and EF [66,111]. Additionally, hexa-D-arginine can be used as a blocker of PA proteolytic cleavage and oligomerization [112]. Another strategy for inhibition is to impair membrane insertion once the prepore is already formed. For example, this can be achieved by amiodarone and bepridil, which have been used to treat cardiac arrhythmia or angina. These drugs interfere with the insertion of the PA heptamer into the endosomal membrane via neutralization of the endosomal pH, thereby blocking toxin entry into the cytosol. Those drugs, however, can have severe side-effects in high doses. For efficient PFP inhibition and reduced risk of side-effects Sanchez et al., 2007, propose a combination of different drugs at lower concentrations [113]. Toxic PFP action can also be diminished by inhibition of a non-pore-forming component of the toxin, such as numerous examples of small-molecule inhibitors of anthrax toxin component LF [18,81,114,115,116,117,118].
Perforin is one of the most important proteins in the immune system of vertebrates. It belongs to the MACPF/CDC superfamily and is able to form pores in target cells. During the process of elimination of cancer or virus-infected cells, perforin is released into the immunological synapse by cytotoxic T lymphocytes and natural killer cells. It forms transmembrane β-barrel pores on target cells and enables the passage of apoptotic proteins, which leads to cell death [19]. Specific inhibitors of perforin were identified by using high-throughput screens, particularly dihydrofuro [3,4-c]pyridinones [119], 1-amino-2,4-dicyanopyrido[1,2-a]benzimidazoles [120], and aryl-substituted isobenzofuran-1(3H)-ones [121,122]. Some of the substances were shown to have a half maximal inhibitory concentration (IC50) in the micromolar range for lysis of Jurkat cells and showed considerable inhibitory potency in the killings of target cells in cell-based cytotoxic assays. However, there has been a limited success in further development of potent inhibitors due to toxicity of compounds to the cells, poor solubility, or loss of activity in the presence of serum.
2.2. Synthetic Nanoparticles
Synthetic polymer nanoparticles (NPs) or “plastic antidotes” (Figure 3b) are synthetic scaffolds with affinity for target biomacromolecules and can be thus used as a tool for the inhibition of PFPs. They are synthesized by precipitation polymerization of different acrylamide monomers in the presence of a PFP [123]. Based on their architecture, they can interact with target molecules through multiple sites, however, they retain small size and ability to diffuse to many locations throughout the body. In comparison to larger bulk materials, nano-sized materials have larger surface areas and thus possess a substantial adsorbing capacity [124,125]. Hoshino et al. demonstrated that imprinted nanoparticles, which are custom-made plastic antidotes comprised of polymeric matrix including functional binders for melittin, aligned in a sense that they form a mold for a specific target, is a very efficient way to inhibit melittin [123]. Such NPs can be prepared by screening a library of NPs composed of various ratios of monomers containing functional groups complementary to the target peptide to select one with the highest intrinsic affinity [76,125]. Compared with biologic materials such as antibodies, synthetic materials offer an advantage because of their robustness and inexpensive production [126]. Limitations regarding nanoparticle utilization in therapeutic or imaging purposes are their potential toxicity [127], adsorption of serum proteins to NPs [128] that can alter or suppress their function, which leads to opsonization, followed by clearance from the bloodstream [129]. Many strategies were developed to prolong their blood circulation time and enhance tissue-specific uptake, mostly focusing on composition, size, surface charge, PEGylation and targeting functionality of NPs [130,131,132], but those can only be used to a limited extent in order to retain their binding characteristics [125].
2.3. Neutralizing Antibodies
Polyclonal antibodies (pAbs) have been utilized before for anti-PFP action, for example in passive protection of guinea pigs against anthrax infection with guinea pig pAbs [133]. In contrast to pAbs, monoclonal antibodies (mAbs) provide a continuous supply of homogeneous, well-characterized antibodies and represent an exquisite tool for specific binding. The traditional way of utilizing mAbs against toxins is by direct inhibition of their function [66], with several possible ways of preventing pore formation: Blockage of the toxin binding to its receptor, interference with oligomer assembly, or, in cases of PFPs with additional catalytic domains, binding to catalytic subunits of the toxin [133].
PFP-neutralizing antibodies were developed for pathogenic PFPs to complement the antibiotic therapy of various diseases, caused by those toxins. The first mAbs with PFP-neutralizing activity were developed in 1960s and inhibited pore formation of a CDC member streptolysin O [134,135,136,137]. In the 1990s, several mAbs were developed that neutralized the pore-forming activity of listeriolysin O and other CDCs, either by prevention of membrane binding, or preventing subsequent stages in the pore-forming process [137,138]. Jacobs et al. found a mAb that successfully bound to an undecapeptide, a peptide sequence conserved in all CDCs mediating the attachment of CDC monomers to the membrane, thereby neutralizing pore-forming activity of all tested toxins from the CDC family [139]. mAbs represent an excellent research tool. For example, it was possible to assess various conformational states of human perforin by using several mAbs [140].
Comprehensive reviews of anthrax-neutralizing mAbs have been published [45,141]. Anthrax toxin inhibitory mAbs can inhibit either receptor recognition by PA83 [142,143,144,145], proteolytic cleavage of PA83 [146], oligomerization step of the PA63 [147,148], or LF interaction with oligomerized PA63 [142,143,144,145,146,147,148]. A cooperative effect between two mAbs, one directed against LF and another against PA [146], or mAbs that exhibit synergistic protection when combined with established antibiotics [149,150], may prevent antibody-dependent enhancement of pathogenicity.
In addition to the anthrax toxin, many studies have been done on finding neutralizing mAbs against other pathogenic PFPs, such as toxin B from Clostridium difficile [151], S. aureus α-toxin [152,153], Clostridium perfringens ε-toxin [154,155], and others. Interestingly, some broad-spectrum mAbs were found to inhibit disparate PFPs, for example α-toxin together with four different types of leukotoxins [156]. In the case of human perforin, it was shown that commercially available monoclonal antibody Pf-80 (Mabtech) could inhibit its permeabilizing activity without affecting its binding to membranes [140,157].
Drawbacks of mAbs encompass their difficult production, accessibility of suitable hybridoma cell line, instability, and low yield of some hybridomas, side-effects, low tissue penetration, and high production cost [158,159,160]. However, those limitations are very well handled nowadays with utilization of different expression systems, and alternative selection techniques, such as phage display, and production of smaller antibody mimetics with retained or even improved antibody characteristics [158,161,162].
2.4. Antibody-Derived Scaffolds and Antibody Mimetics
Due to their high specificity and binding affinity, antibodies are still the most abundantly used proteins in various diagnostic assays and other molecular recognition purposes [163]. Bivalency, completely human origin, and long plasma half-life are undoubtedly the very desired advantages of IgG molecules, but on the other hand they are also relatively large and unstable, composed of multiple domains, and need disulfide formation and glycosylation for their activity, which makes their production laborious and costly [164]. These drawbacks, together with intellectual property rights that are bound to the majority of antibodies present on the market, motivate the development of alternative scaffolds which present an increasing role in biotechnology and medicine [164,165,166].
Most frequently used antibody fragments and non-immunoglobulin binders are antigen-binding fragments (Fabs and F(ab’)2s), single chain variable fragments (scFvs) (Figure 3d), variable fragments of heavy chain antibodies (VHHs, also known as nanobodies), variable domains of sharks’ immunoglobulin new antigen receptors (VNAR), and adnectins (10th fibronectin type III domain derivatives, called also monobodies). They are easy to produce as recombinant proteins by bacterial cells in a fully functional form due to their small size, absence of disulfides, and no need for post-translational modifications [167]. Their advantages are also high solubility, excellent thermal stability, and allowance of complex sequence variation [168]. They are selected mostly by directed evolution of naïve libraries of a chosen scaffold, where the consensus areas important for correct folding are preserved, and the predicted epitope is subjected to diversification. This is performed by panning against the desired target with one of the display techniques, most commonly phage, cell-surface, mRNA, or ribosome display [169].
There are several examples of antibody fragment Fabs inhibiting different toxic PFPs. For example, Wild et al., 2003, selected a Fab that bound to a conformational epitope formed by PA63, inhibited LF interaction with PA63, and neutralized toxin substoichiometrically [170]. Moreover, a human/murine chimeric Fab was developed against LF [171] as well as against PA83 [172] of B. anthracis, all showing therapeutic potential for treatment of anthrax. Among others, scFvs directed against LF [173] and Cry1Ab toxin from Bacillus thuringiensis [174] were also developed. There are reports of isolated VHHs and IgNARs against cholera toxin [175,176], C. difficile toxins [177,178], and others. Efficient neutralization of toxic PFPs can be achieved by using fusion proteins or bispecific binders that bind two subunits of PFP simultaneously and in such a manner inhibit the formation of pores [142]. Yang et al., 2015, produced a bispecific neutralizing construct, consisting of a mAb against epidermal growth factor receptor (EGFR) and a member of CDC, perfringolysin O (PFO), reversibly inhibited by an adnectin. The mAb enabled targeted delivery, whereas the adnectin ensured inactivity of PFO in the extracellular environment. After endocytosis, the adnectin dissociated from PFO, which lead to pore formation on membranes of endocytic compartments and the release of co-targeted protein with therapeutic effect [179]. Protein fusions can also aim for improved stability, such as scFv fused to the human antibody light chain constant κ domain (fusion called scAb), that bound to the PA83 of the anthrax toxin and as such acted as a competitor of the cellular receptor for PA83 binding [133]. It protected against anthrax toxin challenge in vitro and in vivo and was stable at elevated temperatures and highly resistant to deactivation in serum.
2.5. Polyvalent Inhibitors
Polyvalency refers to simultaneous binding of multiple ligands provided by one entity (inhibitor) to complementary receptors on the other (PFPs) [180]. Such polyvalent interactions are much stronger than the corresponding monovalent interactions and the principle has been successfully applied to block PFP activity with synthetic polymeric molecules, so called polyvalent inhibitors (PVIs).
Adequate matching when designing PVIs for pore formations can be achieved with a symmetric rigid cyclic scaffold (Figure 3e), one of the most studied being cyclodextrins [181]. Based on the number of sugar units in a molecule, cyclodextrins are divided into three groups, namely α, β, and γ. β-cyclodextrins are seven-fold symmetrical cyclic molecules with a hydrophobic cavity that mimic the symmetry of heptameric pores. They partially block pores of staphylococcal toxin α-toxin [20]. Their blocking efficiency can be further enhanced by addition of positively charged groups [182,183] or methylation and combination with cholesterol [184]. PA63 and LF of anthrax toxin were inhibited by β- as well as eight-fold γ-cyclodextrin derivatives [21,185,186,187,188], whereas six-fold α-cyclodextrins were ineffective. Additionally, β-cyclodextrins effectively inhibited C2 toxin of Clostridium botulinum, ι-toxin of C. perfringens and CDT binary toxin of C. difficile [189,190,191]. Mourez et al., 2001, identified a 12-amino acid residue peptide that weakly bound to the heptameric PA63 but not to monomeric PA. The peptide was synthesized chemically and shown to inhibit the interaction between PA63 and its ligands, EF and LF, albeit weakly. To generate a more potent form, they produced a PVI consisting of multiple copies of the synthetic peptide covalently linked to a flexible polyacrylamide backbone. The resulting construct was 7000-fold more potent than the monomeric form, owing to its ability to form multiple links to the oligomeric target [192]. The potency of inhibition can be further increased by a combination of the approaches mentioned above. For example, seven copies of an inhibitory peptide against heptameric PA63 were attached to a β-cyclodextrin scaffold, thereby producing a heptavalent inhibitor. This resulted in more than a 105-fold increase in inhibition in comparison to the monomeric peptide [22].
Furthermore, prepores can be neutralized by peptide-functionalized liposomes. In the case of anthrax (PA63 heptamer) inhibitors, the liposomes measured approximately 50 nm in diameter and contained specific prepore-binding peptides fused to phospholipid headgroups [193]. The inhibitory effect was achieved at very low concentrations, they were active in vivo [191] and their further development has been made easier due to several FDA-approved liposome formulations as adjuvants [194] and drug deliverers [195,196]. Other forms of PVIs are dendrimers with functionalized ligands [197,198], rigid scaffolds fused with carbohydrate (instead of peptide) ligands [199], and receptor-directed PVIs [200] that diminish the consequences of toxic PFPs’ action by blocking host proteins. PVIs of PFPs have been recently reviewed by Yamini and Nestorovich, 2016 [201].
2.6. Receptor-Like Decoys for Pore-Forming Toxins
Another approach with a clinical potential for inhibition of various different types of PFPs was proposed by Bradley et al., 2001 [42], and further developed by several research groups [84,85,202,203,204,205]. The idea is to capture proteins that physiologically bind to receptors or cellular membranes with a surrogate system that mimics the natural one and consequently make the PFP unavailable for binding to its biological receptor in vivo [84,206]. This approach offers significant advantages compared to conventional strategies relying primarily on structure-specific epitope binding (e.g., antibodies) where we are usually faced with a high cost of antibodies and dosage requirements, the highly malleable nature of some of the PFPs, which implies they can easily mutate to become resistant to antibody-binding [73,207], and a serious challenge to devise an effective detoxification platform against bacterial infections of a very diverse range of toxic PFPs [208,209]. The superiority of receptor-like decoys over antibody-based antitoxins lies in their ability to accurately mimic the natural receptor while being less sensitive to natural or artificial changes in the PFPs’ primary structure [210,211].
There are numerous examples of surrogate receptors or membrane mimics that act as decoys to preoccupy and neutralize toxin actions. Soluble forms of extracellular domains of both anthrax receptors (TEM8 and CMG2) function as potent antitoxins that can protect cultured cells from intoxication, presumably by acting as receptor decoys to prevent PA83 from binding to cell-surface receptors [42,43,86,207,210]. There are various improved versions of receptor-like decoys. Wycoff et al., 2011, for example, fused the extracellular domain of CMG2 with a human Fc (fragment crystallizable) of IgG. The inclusion of the Fc domain allows efficient purification with protein A, causes dimerization (which increases size), and induces recycling and retention by interaction with the neonatal Fc receptor. The fusion protein efficiently neutralized PA83 as well as mutant forms of PA83 that were not successfully recognized by anti-PA monoclonal antibodies in vitro and in vivo [211].
An ingenious rationale that serum lipoproteins and membrane lipid extracts dispersed in water bind to and inhibit lethal and cytolytic activity of PFPs [212,213] was further developed by Hu et al., who named their receptor-like decoys “nanosponges” (Figure 3f). Consisting of erythrocyte membrane-coated nanoparticle system, they nonspecifically absorbed a broad spectrum of PFPs, thereby targeting the universal membrane-binding mechanism and offering an all-purpose PFP decoy strategy to absorb various types of PFPs regardless of their molecular structures [84,85]. Nanosponges have been so far effective in vivo at neutralizing α-toxin (also MRSA strain infections), streptolysin-O, and melittin [84,205,209], however a similar approach would probably be effective for all PFPs. Instead of an erythrocyte membrane, Henry et al. utilized artificial liposomes, containing higher than in vivo concentrations of cholesterol and sphingomyelin, resulting in inhibition of several staphylococcal and streptococcal toxins [206]. A similar approach can be practiced to capture PFPs that bind to specific receptors rather than to the membrane itself. Polyzos et al., 2007, for example, entrapped gangliosides GM1 into surfactant mesophase and the construct functioned as a polyvalent inhibitor of cholera toxin [214].
2.7. Dominant Negative Mutants
Dominant negative (DN) mutants (Figure 3g) are defective proteins that retain interaction capabilities but are inactive and possess the ability to inhibit the phenotype of the wild-type protein when mixed together [87]. For example, the DN mutant of the monomeric unit of VacA that is involved in oligomerization in the pore-forming process was able to block the pore-forming activity. The mutant PFP lacked the amino-terminal hydrophobic segment and did not exhibit any detectable defects in secretion, binding to membranes, oligomerization, or uptake by cells, but it failed to induce the vacuolization of the toxin, and was consequently non-cytotoxic. When combined with the wild-type toxin, the dysfunctional mixed oligomers comprised of both mutant and wild-type VacA monomeric components were formed and the cytotoxic activity of the latter was inhibited [215].
Inhibition of the anthrax toxin PA component pore formation by DN mutants was reported independently by two groups. Sellman, Mourez, and Collier, 2001, identified a PA with double mutation with properties of a DN mutant. It co-assembled with the wild-type PA and generated defective prepores impaired in pore formation and in translocating EF and LF across the endosomal membrane. Despite these malfunctions, the proteolytic activation of PA and self-assembly of the toxin remained unaffected [216]. Singh et al., 2001, found another mutated PA with similar effects. They showed that a mixture of DN mutant PA and wild-type PA completely inhibited toxin activity in vitro and in vivo [217]. Later, a whole map of possible sites where a single amino acid replacement on PA63 can give a DN phenotype was proposed [218]. They found that DN mutations are only feasible in pore-forming domain II of PA and specifically affect pore formation and translocation. Furthermore, none of the DN mutations tested significantly impaired the immunogenic properties of PA. Hence, DN forms of PA may have the potential to serve both as direct inhibitors of toxin action and as inducers of protective antibodies. These properties may make them useful for post-exposure therapy and prophylaxis against anthrax [41,48]. As antibiotic treatment cannot provide full protection against relapse or subsequent exposure to anthrax, some claim conjunctive antibiotic treatment and vaccination with DN inhibitors would be an ideal option [219]. There are numerous other examples of DN mutants inhibiting pore formation by anthrax [220,221] and other pore-forming toxins, including C. perfringens ε-toxin [83], B. thuringiensis Cry1Ab [222], and E. coli cytolysin A [223]. These examples imply that toxins acting through oligomeric complexes are amenable to dominant-negative inhibition, a paradigm that could be broadly applied.
Notably, many of toxin inhibitors devised so far are either derivatives of a toxin component or receptor. In addition to DN mutants and receptor decoys, toxin derivatives can act as competitive inhibitors for receptor binding. For example, PA with a mutated furin site competes with the wild-type protein for receptors, and the mutation in the N-terminal domain of LF competitively inhibits binding of EF and LF to PA63 [41]. However, DN forms of toxins are far more potent toxin inhibitors than those that merely compete for receptor binding, and for this reason the competitive inhibition of receptor binding is not widely used for toxin neutralization.
3. Conclusions
The biological activity of toxic PFPs, which represent the largest group of bacterial cytotoxic proteins, can be blocked by various strategies and inhibitor designs. Here we describe general approaches by which the pore-formation step can be inhibited together with a number of studies in which these approaches have been employed. All presented strategies possess the ability to potently inhibit PFPs. The suitability of the strategy hence depends mostly on the aim of application.
Specific inhibitors of pore-formation have been primarily developed to affect the course of disease and its symptoms. As such, drugs targeting toxic PFPs could help limit the extent of infection, aid in preventing systemic spreading when a localized infection is present, and prevent toxic PFP-mediated tissue destruction (e.g., in S. pneumoniae or S. aureus pneumonia or clostridial myonecrosis). Such drugs could also be used to prevent problematic nosocomial infections (e.g., preventive administration during surgery or the use of catheters) [38] or as an adjunct to antibiotic therapy—for co-administration with existing antibiotics for delaying the infection and therefore providing time for antibacterial agents or the immune system to clear an infection [86,170,192,200,207]. Probably, a cocktail of peptides or protein fragments that interfere with several of the protein–protein interactions required for toxin action would be the most efficacious therapy [224].
Besides utilization of inhibitors that prevent specific steps in pore formation of PFPs in therapeutic applications, these inhibitors are also a valuable tool in studies of the mechanism of action of PFPs [137,140,217], protein expression in vitro and in vivo [157,225,226], determination of structure–function relationship [81,218,227,228], detection of cytotoxic cells that express PFPs [225], testing PFPs’ activity with functional assays [157], probing the distribution, orientation, and mobility of membrane receptors [229], etc. PFPs also play an important role in a wide range of recent advances in nanotechnology [230,231]. By gaining new findings about their pore-forming mechanisms, we could use them more effectively in developing technologies, such as molecular sensing and detection [232,233,234,235], DNA sequencing [236,237,238], monitoring of chemical and biochemical reactions, development of biocompatible nanotransistors [239], and novel drug delivery systems and targeted killing of cells [179,230,240], as well as develop new applications. As the technology proceeds, the potential space for applications of PFP inhibitors will also grow.
Funding
This research was funded by the Slovenian Research Agency, grant number P1-0391 and J4-8225.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bischofberger, M.; Iacovache, I.; van der Goot, F.G. Pathogenic pore-forming proteins: Function and host response. Cell Host Microbe 2012, 12, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Anderluh, G.; Lakey, J.H. Disparate proteins use similar architectures to damage membranes. Trends Biochem. Sci. 2008, 33, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.J.C.; Dalla Serra, M.; Froelich, C.J.; Wallace, M.I.; Anderluh, G. Membrane pore formation at protein-lipid interfaces. Trends Biochem. Sci. 2014, 39, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Cajnko, M.M.; Mikelj, M.; Turk, T.; Podobnik, M.; Anderluh, G. Membrane Interactions and Cellular Effects of MACPF/CDC Proteins. In MACPF/CDC Proteins—Agents of Defence, Attack and Invasion; Anderluh, G., Gilbert, R., Eds.; Springer: Dordrecht, The Netherlands, 2014; pp. 119–144. [Google Scholar]
- Lakey, J.H.; Anderluh, G. Membrane-Disrupting Proteins. In Biogenesis of Fatty Acids, Lipids and Membranes; Springer International Publishing: Dordrecht, The Netherlands, 2019; pp. 729–739. [Google Scholar]
- Ros, U.; García-Sáez, A.J. More Than a Pore: The Interplay of Pore-Forming Proteins and Lipid Membranes. J. Membr. Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Dunstone, M.A.; Tweten, R.K. Packing a punch: The mechanism of pore formation by cholesterol dependent cytolysins and membrane attack complex/perforin-like proteins. Curr. Opin. Struct. Biol. 2012, 22, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Tweten, R.K.; Hotze, E.M.; Wade, K.R. The unique molecular choreography of giant pore formation by the cholesterol-dependent cytolysins of Gram-positive bacteria. Annu. Rev. Microbiol. 2015, 69, 323–340. [Google Scholar] [CrossRef]
- Krasilnikov, O.V.; Da Cruz, J.B.; Yuldasheva, L.N.; Varanda, W.A.; Nogueira, R.A. A novel approach to study the geometry of the water lumen of ion channels: Colicin Ia channels in planar lipid bilayers. J. Membr. Biol. 1998, 161, 83–92. [Google Scholar] [CrossRef]
- Dal Peraro, M.; van der Goot, F.G. Pore-forming toxins: ancient, but never really out of fashion. Nat. Rev. Microbiol. 2016, 14, 77–92. [Google Scholar] [CrossRef]
- Cosentino, K.; Ros, U.; García-Sáez, A.J. Assembling the puzzle: Oligomerization of α-pore forming proteins in membranes. Biochim. Biophys. Acta Biomembr. 2016, 1858, 457–466. [Google Scholar] [CrossRef]
- Gouaux, E. Channel-forming toxins: Tales of transformation. Curr. Opin. Struct. Biol. 1997, 7, 566–573. [Google Scholar] [CrossRef]
- Podobnik, M.; Savory, P.; Rojko, N.; Kisovec, M.; Wood, N.; Hambley, R.; Pugh, J.; Wallace, E.J.; McNeill, L.; Bruce, M.; et al. Crystal structure of an invertebrate cytolysin pore reveals unique properties and mechanism of assembly. Nat. Commun. 2016, 7, 11598. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, J.; Nagahama, M.; Hisatsune, J.; Katunuma, N.; Tsuge, H. Clostridium perfringens ι-toxin, ADP-ribosyltransferase: Structure and mechanism of action. Adv. Enzym. Regul. 2003, 43, 361–377. [Google Scholar] [CrossRef]
- Petosa, C.; Collier, R.J.; Klimpel, K.R.; Leppla, S.H.; Liddington, R.C. Crystal structure of the anthrax toxin protective antigen. Nature 1997, 385, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Pentelute, B.L.; Collier, R.J.; Hong Zhou, Z. Atomic structure of anthrax protective antigen pore elucidates toxin translocation. Nature 2015, 521, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Serna, M.; Giles, J.L.; Morgan, B.P.; Bubeck, D. Structural basis of complement membrane attack complex formation. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Turk, B.E.; Wong, T.Y.; Schwarzenbacher, R.; Jarrell, E.T.; Leppla, S.H.; Collier, R.J.; Liddington, R.C.; Cantley, L.C. The structural basis for substrate and inhibitor selectivity of the anthrax lethal factor. Nat. Struct. Mol. Biol. 2004, 11, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Voskoboinik, I.; Dunstone, M.A.; Baran, K.; Whisstock, J.C.; Trapani, J.A. Perforin: Structure, function, and role in human immunopathology. Immunol. Rev. 2010, 235, 35–54. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Hobaugh, M.R.; Shustak, C.; Cheley, S.; Bayley, H.; Gouaux, J.E. Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science 1996, 274, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Karginov, V.A.; Nestorovich, E.M.; Moayeri, M.; Leppla, S.H.; Bezrukov, S.M. Blocking anthrax lethal toxin at the protective antigen channel by using structure-inspired drug design. Proc. Natl. Acad. Sci. USA 2005, 102, 15075–15080. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.; Kate, S.; Poon, V.; Mondal, D.; Boggara, M.B.; Saraph, A.; Martin, J.T.; McAlpine, R.; Day, R.; Garcia, A.E.; et al. Structure-based design of a heptavalent anthrax toxin inhibitor. Biomacromolecules 2011, 12, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.W.; Feil, S.C. Pore-forming protein toxins: From structure to function. Prog. Biophys. Mol. Biol. 2005, 88, 91–142. [Google Scholar] [CrossRef] [PubMed]
- Christie, M.P.; Johnstone, B.A.; Tweten, R.K.; Parker, M.W.; Morton, C.J. Cholesterol-dependent cytolysins: From water-soluble state to membrane pore. Biophys. Rev. 2018, 10, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Rojko, N.; Dalla Serra, M.; Maček, P.; Anderluh, G. Pore formation by actinoporins, cytolysins from sea anemones. Biochim. Biophys. Acta Biomembr. 2016, 1858, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Anderluh, G.; Dalla Serra, M.; Viero, G.; Guella, G.; Maček, P.; Menestrina, G. Pore formation by equinatoxin II, a eukaryotic protein toxin, occurs by induction of nonlamellar lipid structures. J. Biol. Chem. 2003, 278, 45216–45223. [Google Scholar] [CrossRef] [PubMed]
- Sobko, A.A.; Kotova, E.A.; Antonenko, Y.N.; Zakharov, S.D.; Cramer, W.A. Effect of lipids with different spontaneous curvature on the channel activity of colicin E1: Evidence in favor of a toroidal pore. FEBS Lett. 2004, 576, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.F.; Martinou, J.C.; Montessuit, S.; Epand, R.M.; Yip, C.M. Direct evidence for membrane pore formation by the apoptotic protein Bax. Biochem. Biophys. Res. Commun. 2002, 298, 744–749. [Google Scholar] [CrossRef]
- Henkel, J.S.; Baldwin, M.R.; Barbieri, J.T. Toxins from bacteria. In Molecular, Clinical and Environmental Toxicology. Experientia Supplementum; Luch, A., Ed.; Birkhäuser: Basel, Switzerland, 2010; Volume 100. [Google Scholar]
- Bhakdi, S.; Tranum-Jensen, J. Alpha-Toxin of Staphylococcus aureus. Microbiol. Rev. 1991, 55, 733–751. [Google Scholar]
- Popoff, M.R. Epsilon toxin: A fascinating pore-forming toxin. FEBS J. 2011, 278, 4602–4615. [Google Scholar] [CrossRef] [PubMed]
- Aktories, K.; Barth, H. The actin-ADP-ribosylating Clostridium botulinum C2 toxin. Anaerobe 2004, 10, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Anderluh, G.; Gilbert, R. MACPF/CDC Proteins—Agents of Defence, Attack and Invasion; Anderluh, G., Gilbert, R., Eds.; Springer: Berlin, Germany, 2014. [Google Scholar]
- van der Goot, G. Pore Forming Toxins; Springer: Berlin, Germany, 2001. [Google Scholar]
- Bubeck Wardenburg, J.; Whisstock, J.; Tweten, R.K. Pore-forming toxins. In Bacterial Toxins: Genetics, Cellular Biology and Practical Applications; Proft, T., Ed.; Caister Academic Press: Wymondham, UK, 2013. [Google Scholar]
- Dalla Serra, M.; Teyuca Martinez, M. Pore-forming Toxins. In Encyclopedia of Life Sciences; John Wiley&Sons, Ltd.: Hoboken, NJ, USA, 2011. [Google Scholar]
- Iacovache, I.; van der Goot, F.G.; Pernot, L. Pore formation: An ancient yet complex form of attack. Biochim. Biophys. Acta 2008, 1778, 1611–1623. [Google Scholar] [CrossRef]
- Los, F.C.O.; Randis, T.M.; Aroian, R.V.; Ratner, A.J. Role of pore-forming toxins in bacterial infectious diseases. Microbiol. Mol. Biol. Rev. 2013, 77, 173–207. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC. Available online: https://pymol.org/2/ (accessed on 9 September 2019).
- Barth, H.; Aktories, K.; Popoff, M.R.; Stiles, B.G. Binary bacterial toxins: Biochemistry, biology, and applications of common Clostridium and Bacillus proteins. Microbiol. Mol. Biol. Rev. 2004, 68, 373–402. [Google Scholar] [CrossRef] [PubMed]
- Collier, R.J.; Young, J.A.T. Anthrax toxin. Annu. Rev. Cell Dev. Biol. 2003, 19, 45–70. [Google Scholar] [CrossRef] [PubMed]
- Bradley, K.A.; Mogridge, J.; Mourez, M.; Collier, R.J.; Young, J.A. Identification of the cellular receptor for anthrax toxin. Nature 2001, 414, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Scobie, H.M.; Rainey, G.J.A.; Bradley, K.A.; Young, J.A.T. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 5170–5174. [Google Scholar] [CrossRef] [PubMed]
- Kintzer, A.F.; Thoren, K.L.; Sterling, H.J.; Dong, K.C.; Feld, G.K.; Tang, I.I.; Zhang, T.T.; Williams, E.R.; Berger, J.M.; Krantz, B.A. The Protective Antigen Component of Anthrax Toxin Forms Functional Octameric Complexes. J. Mol. Biol. 2009, 392, 614–629. [Google Scholar] [CrossRef]
- Froude, J.W.; Thullier, P.; Pelat, T. Antibodies Against Anthrax: Mechanisms of Action and Clinical Applications. Toxins 2011, 3, 1433–1452. [Google Scholar] [CrossRef]
- Little, S.F. Anthrax vaccines: A development update. BioDrugs 2005, 19, 233–245. [Google Scholar] [CrossRef]
- Ambrose, E.A. Botulinum Neurotoxin, Tetanus Toxin, and Anthrax Lethal Factor Countermeasures. In Topics in Medicinal Chemistry; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Bouzianas, D.G. Current and future medical approaches to combat the anthrax threat. J. Med. Chem. 2010, 53, 4305–4331. [Google Scholar] [CrossRef]
- Laventie, B.; Potrich, C.; Atmanène, C.; Saleh, M.; Joubert, O.; Viero, G.; Bachmeyer, C.; Antonini, V.; Mancini, I.; Cianferani-Sanglier, S.; et al. p-Sulfonato-calix[n]arenes inhibit staphylococcal bicomponent leukotoxins by supramolecular interactions. Biochem. J. 2013, 450, 559–571. [Google Scholar] [CrossRef]
- LaRosa, S.P.; Opal, S.M. Sepsis Strategies in Development. Clin. Chest Med. 2008, 29, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Lakey, J.H.; van der Goot, F.G.; Pattus, F. All in the family: The toxic activity of pore-forming colicins. Toxicology 1994, 87, 85–108. [Google Scholar] [CrossRef]
- Bullock, J.O.; Kolen, E.R.; Shear, J.L. Ion Selectivity of Colicin El: II. Permeability to Organic Cations. J. Membr. Biol. 1992, 128. [Google Scholar] [CrossRef] [PubMed]
- Podack, E.R. Molecular composition of the tubular structure of the membrane attack complex of complement. J. Biol. Chem. 1984, 259, 8641–8647. [Google Scholar] [PubMed]
- Thiery, J.; Keefe, D.; Boulant, S.; Boucrot, E.; Martinvalet, D.; Goping, I.S.; Bleackley, R.C.; Lieberman, J. Perforin pores in the endosomal membrane trigger release of endocytosed granzyme B to the cytosol of target cells. Nat. Immunol. 2011, 12, 770–777. [Google Scholar] [CrossRef]
- Gross, A.; Mcdonnell, J.M.; Korsmeyer, S.J. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999, 13, 1899–1911. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. Life-or-death decisions by the Bcl-2 protein family. Trends Biochem. Sci. 2001, 26, 61–66. [Google Scholar] [CrossRef]
- Nestorovich, E.M.; Bezrukov, S.M. Obstructing toxin pathways by targeted pore blockage. Chem. Rev. 2012, 112, 6388–6430. [Google Scholar] [CrossRef]
- Escajadillo, T.; Nizet, V. Pharmacological Targeting of Pore-Forming Toxins as Adjunctive Therapy for Invasive Bacterial Infection. Toxins 2018, 10. [Google Scholar] [CrossRef]
- Bezrukov, S.M.; Nestorovich, E.M. Inhibiting bacterial toxins by channel blockage. FEMS Pathog. Dis. 2016, 74. [Google Scholar] [CrossRef]
- Hung, D.T.; Shakhnovich, E.A.; Pierson, E.; Mekalanos, J.J. Small-molecule inhibitor of Vibrio cholerae virulence and intestinal colonization. Science 2005, 310, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Hentzer, M.; Wu, H.; Andersen, J.B.; Riedel, K.; Rasmussen, T.B.; Bagge, N.; Kumar, N.; Schembri, M.A.; Song, Z.; Kristoffersen, P.; et al. Attenuation of Pseudomonas aeruginosa virulence by quorum sensing inhibitors. EMBO J. 2003, 22, 3803–3815. [Google Scholar] [CrossRef] [PubMed]
- Statt, S.; Ruan, J.W.; Hung, L.Y.; Chang, C.Y.; Huang, C.T.; Lim, J.H.; Li, J.D.; Wu, R.; Kao, C.Y. Statin-conferred enhanced cellular resistance against bacterial pore-forming toxins in airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2015, 53, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Shewell, L.K.; Harvey, R.M.; Higgins, M.A.; Day, C.J.; Hartley-Tassell, L.E.; Chen, A.Y.; Gillen, C.M.; James, D.B.A.; Alonzo, F.; Torres, V.J.; et al. The cholesterol-dependent cytolysins pneumolysin and streptolysin O require binding to red blood cell glycans for hemolytic activity. Proc. Natl. Acad. Sci. USA 2014. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, A.; Hundhausen, C.; Lambert, M.; Broadway, N.; Andrews, R.; Bickett, D.; Leesnitzer, M.; Becherer, J. Metalloproteinase Inhibitors for the Disintegrin-Like Metalloproteinases ADAM10 and ADAM17 that Differentially Block Constitutive and Phorbol Ester-Inducible Shedding of Cell Surface Molecules. Comb. Chem. High Throughput Screen. 2005, 8, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Alonzo, F.; Kozhaya, L.; Rawlings, S.A.; Reyes-Robles, T.; Dumont, A.L.; Myszka, D.G.; Landau, N.R.; Unutmaz, D.; Torres, V.J. CCR5 is a receptor for Staphylococcus aureus leukotoxin ED. Nature 2013, 493, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Clatworthy, A.E.; Pierson, E.; Hung, D.T. Targeting virulence: A new paradigm for antimicrobial therapy. Nat. Chem. Biol. 2007, 3, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Gurcel, L.; Abrami, L.; Girardin, S.; Tschopp, J.; van der Goot, F.G. Caspase-1 Activation of Lipid Metabolic Pathways in Response to Bacterial Pore-Forming Toxins Promotes Cell Survival. Cell 2006, 126, 1135–1145. [Google Scholar] [CrossRef]
- Yarovinsky, T.O.; Monick, M.M.; Husmann, M.; Hunninghake, G.W. Interferons increase cell resistance to staphylococcal alpha-toxin. Infect. Immun. 2008, 76, 571–577. [Google Scholar] [CrossRef]
- McNeil, P.L.; Kirchhausen, T. An emergency response team for membrane repair. Nat. Rev. Mol. Cell Biol. 2005, 6, 499–505. [Google Scholar] [CrossRef]
- Romero, M.; Keyel, M.; Shi, G.; Bhattacharjee, P.; Roth, R.; Heuser, J.E.; Keyel, P.A. Intrinsic repair protects cells from pore-forming toxins by microvesicle shedding. Cell Death Differ. 2017, 24, 798–808. [Google Scholar] [CrossRef]
- Cunha, B.A. Antibiotic Side Effects. Med. Clin. N. Am. 2001, 85, 149–185. [Google Scholar] [CrossRef]
- Bromberg-White, J.L.; Duesbery, N.S. Biological and Biochemical Characterization of Anthrax Lethal Factor, a Proteolytic Inhibitor of MEK Signaling Pathways. Methods Enzymol. 2008, 438, 355–365. [Google Scholar] [CrossRef]
- Ivarsson, M.E.; Leroux, J.C.; Castagner, B. Targeting bacterial toxins. Angew. Chem. Int. Ed. 2012, 51, 4024–4045. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.D.; Wright, G.D. Antibacterial drug discovery in the resistance era. Nature 2016, 529, 336–343. [Google Scholar] [CrossRef]
- Ventola, C. The antibiotic resistance crisis: Part 1: Causes and threats. Pharmacol. Ther. 2015, 40, 277–283. [Google Scholar]
- Yoshimatsu, K.; Koide, H.; Hoshino, Y.; Shea, K.J. Preparation of abiotic polymer nanoparticles for sequestration and neutralization of a target peptide toxin. Nat. Protoc. 2015, 10, 595–604. [Google Scholar] [CrossRef]
- Fischbach, M.A.; Walsh, C.T. Antibiotics for emerging pathogens. Science 2009, 325, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Rasko, D.A.; Sperandio, V. Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug Discov. 2010, 9, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Burnett, J.C.; Ruthel, G.; Stegmann, C.M.; Panchal, R.G.; Nguyen, T.L.; Hermone, A.R.; Stafford, R.G.; Lane, D.J.; Kenny, T.A.; McGrath, C.F.; et al. Inhibition of metalloprotease botulinum serotype A from a pseudo-peptide binding mode to a small molecule that is active in primary neurons. J. Biol. Chem. 2007, 282, 5004–5014. [Google Scholar] [CrossRef]
- Cegelski, L.; Marshall, G.R.; Eldridge, G.R.; Hultgren, S.J. The biology and future prospects of antivirulence therapies. Nat. Rev. Microbiol. 2008, 6, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Shoop, W.L.; Xiong, Y.; Wiltsie, J.; Woods, A.; Guo, J.; Pivnichny, J.V.; Felcetto, T.; Michael, B.F.; Bansal, A.; Cummings, R.T.; et al. Anthrax lethal factor inhibition. Proc. Natl. Acad. Sci. USA 2005, 102, 7958–7963. [Google Scholar] [CrossRef] [PubMed]
- Škrlec, K.; Štrukelj, B.; Berlec, A. Non-immunoglobulin scaffolds: A focus on their targets. Trends Biotechnol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Pelish, T.M.; McClain, M.S. Dominant-negative inhibitors of the Clostridium perfringens ε-toxin. J. Biol. Chem. 2009, 284, 29446–29453. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.J.; Fang, R.H.; Copp, J.; Luk, B.T.; Zhang, L. A biomimetic nanosponge that absorbs pore-forming toxins. Nat. Nanotechnol. 2013, 8, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.J.; Zhang, L.; Aryal, S.; Cheung, C.; Fang, R.H. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc. Natl. Acad. Sci. USA 2011, 108, 10980–10985. [Google Scholar] [CrossRef] [PubMed]
- Scobie, H.M.; Thomas, D.; Marlett, J.M.; Destito, G.; Wigelsworth, D.J.; Collier, R.J.; Young, J.A.T.; Manchester, M. A soluble receptor decoy protects rats against anthrax lethal toxin challenge. J. Infect. Dis. 2005, 192, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Rainey, G.J.A.; Young, J.A.T. Antitoxins: Novel strategies to target agents of bioterrorism. Nat. Rev. Microbiol. 2004, 2, 721–726. [Google Scholar] [CrossRef]
- Tombola, F.; Oregna, F.; Brutsche, S.; Szabò, I.; Del Giudice, G.; Rappuoli, R.; Montecucco, C.; Papini, E.; Zoratti, M. Inhibition of the vacuolating and anion channel activities of the VacA toxin of Helicobacter pylori. FEBS Lett. 1999, 460, 221–225. [Google Scholar] [CrossRef]
- Bachmeyer, C.; Benz, R.; Barth, H.; Aktories, K.; Gilbert, M.; Popoff, M.R. Interaction of Clostridium botulinum C2 toxin with lipid bilayer membranes and Vero cells: Inhibition of channel function by chloroquine and related compounds in vitro and intoxification in vivo. FASEB J. 2001, 15, 1658–1660. [Google Scholar] [CrossRef]
- Bachmeyer, C.; Orlik, F.; Barth, H.; Aktories, K.; Benz, R. Mechanism of C2-toxin inhibition by fluphenazine and related compounds: Investigation of their binding kinetics to the C2II-channel using the current noise analysis. J. Mol. Biol. 2003, 333, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Kronhardt, A.; Beitzinger, C.; Barth, H.; Benz, R. Chloroquine Analog Interaction with C2- and Iota-Toxin in Vitro and in Living Cells. Toxins 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Blaustein, R.O.; Lea, E.J.; Finkelstein, A. Voltage-dependent block of anthrax toxin channels in planar phospholipid bilayer membranes by symmetric tetraalkylammonium ions. Single-channel analysis. J. Gen. Physiol. 1990, 96, 921–942. [Google Scholar] [CrossRef] [PubMed]
- Blaustein, R.O.; Koehler, T.M.; Collier, R.J.; Finkelstein, A. Anthrax toxin: Channel-forming activity of protective antigen in planar phospholipid bilayers. Proc. Natl. Acad. Sci. USA 1989, 86, 2209–2213. [Google Scholar] [CrossRef] [PubMed]
- Voets, T.; Droogmans, G.; Nilius, B. Potent block of volume-activated chloride currents in endothelial cells by the uncharged form of quinine and quinidine. Br. J. Pharmacol. 1996, 118, 1869–1871. [Google Scholar] [CrossRef] [PubMed]
- Ballestero, J.A. Effects of Quinine, Quinidine and Chloroquine on α9α10 Nicotinic Cholinergic Receptors. Mol. Pharmacol. 2005, 68, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.T.; Rhee, C.K.; Lee, C.S.; Park, Y.S.; Choi, D.C. Ototoxicity of salicylate, nonsteroidal antiinflammatory drugs, and quinine. Otolaryngol. Clin. North Am. 1993, 26, 791–810. [Google Scholar] [PubMed]
- Manzine, P.R.; Ettcheto, M.; Cano, A.; Busquets, O.; Marcello, E.; Pelucchi, S.; Di Luca, M.; Endres, K.; Olloquequi, J.; Camins, A.; et al. ADAM10 in Alzheimer’s disease: Pharmacological modulation by natural compounds and its role as a peripheral marker. Biomed. Pharmacother. 2019, 113, 108661. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.; Debnath, B.; Neamati, N. Role of the CXCL8-CXCR1/2 axis in cancer and inflammatory diseases. Theranostics 2017, 7, 1543–1588. [Google Scholar] [CrossRef]
- Cheng, Y.; Ma, X.; Wei, Y.; Wei, X.-W. Potential roles and targeted therapy of the CXCLs/CXCR2 axis in cancer and inflammatory diseases. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 289–312. [Google Scholar] [CrossRef]
- Wilke, G.A.; Wardenburg, J.B. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus α-hemolysin-mediated cellular injury. Proc. Natl. Acad. Sci. USA 2010, 107, 13473–13478. [Google Scholar] [CrossRef] [PubMed]
- Seilie, E.S.; Bubeck Wardenburg, J. Staphylococcus aureus pore-forming toxins: The interface of pathogen and host complexity. Semin. Cell Dev. Biol. 2017, 72, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Schwiering, M.; Husmann, M.; Hellmann, N. P2X-receptor antagonists inhibit the interaction of S. aureus hemolysin A with membranes. Toxins 2017, 9. [Google Scholar] [CrossRef]
- Qiu, J.; Wang, D.; Zhang, Y.; Dong, J.; Wang, J.; Niu, X. Molecular modeling reveals the novel inhibition mechanism and binding mode of three natural compounds to Staphylococcal α-hemolysin. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.S.; Lee, J.H.; Cho, M.H.; Lee, J. Red wines and flavonoids diminish Staphylococcus aureus virulence with anti-biofilm and anti-hemolytic activities. Biofouling 2014, 31, 1–11. [Google Scholar] [CrossRef]
- Qiu, J.; Niu, X.; Dong, J.; Wang, D.; Wang, J.; Li, H.; Luo, M.; Li, S.; Feng, H.; Deng, X. Baicalin protects mice from Staphylococcus aureus pneumonia via inhibition of the cytolytic activity of α-hemolysin. J. Infect. Dis. 2012, 206, 292–301. [Google Scholar] [CrossRef]
- Zhao, X.; Li, H.; Wang, J.; Guo, Y.; Liu, B.; Deng, X.; Niu, X. Verbascoside Alleviates Pneumococcal Pneumonia by Reducing Pneumolysin Oligomers. Mol. Pharmacol. 2016, 89, 376–387. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, B.; Liu, S.; Wang, L.; Wang, J. Anticytotoxin Effects of Amentoflavone to Pneumolysin. Biol. Pharm. Bull. 2017, 40, 61–67. [Google Scholar] [CrossRef]
- Arzanlou, M.; Bohlooli, S. Inhibition of streptolysin O by allicin—An active component of garlic. J. Med. Microbiol. 2010, 59, 1044–1049. [Google Scholar] [CrossRef]
- Wang, J.; Qiu, J.; Tan, W.; Zhang, Y.; Wang, H.; Zhou, X.; Liu, S.; Feng, H.; Li, W.; Niu, X.; et al. Fisetin inhibits Listeria monocytogenes virulence by interfering with the oligomerization of Listeriolysin O. J. Infect. Dis. 2015, 211, 1376–1387. [Google Scholar] [CrossRef]
- Rathkey, J.K.; Zhao, J.; Liu, Z.; Chen, Y.; Yang, J.; Kondolf, H.C.; Benson, B.L.; Chirieleison, S.M.; Huang, A.Y.; Dubyak, G.R.; et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Moayeri, M.; Wiggins, J.F.; Lindeman, R.E.; Leppla, S.H. Cisplatin inhibition of anthrax lethal toxin. Antimicrob. Agents Chemother. 2006, 50, 2658–2665. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sarac, M.S.; Peinado, J.R.; Leppla, S.H.; Lindberg, I. Protection against Anthrax Toxemia by Hexa-D-Arginine In Vitro and In Vivo. Infect. Immun. 2004, 72, 602–605. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.M.; Thomas, D.; Gillespie, E.J.; Damoiseaux, R.; Rogers, J.; Saxe, J.P.; Huang, J.; Manchester, M.; Bradley, K.A. Amiodarone and bepridil inhibit anthrax toxin entry into host cells. Antimicrob. Agents Chemother. 2007, 51, 2403–2411. [Google Scholar] [CrossRef] [PubMed]
- Dell’Aica, I.; Donà, M.; Tonello, F.; Piris, A.; Mock, M.; Montecucco, C.; Garbisa, S. Potent inhibitors of anthrax lethal factor from green tea. EMBO Rep. 2004, 5, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Numa, M.M.D.; Lee, L.V.; Hsu, C.C.; Bower, K.E.; Wong, C.H. Identification of novel anthrax lethal factor inhibitors generated by combinatorial Pictet-Spengler reaction followed by screening in situ. ChemBioChem 2005, 6, 1002–1006. [Google Scholar] [CrossRef] [PubMed]
- Panchal, R.G.; Hermone, A.R.; Nguyen, T.L.; Wong, T.Y.; Schwarzenbacher, R.; Schmidt, J.; Lane, D.; McGrath, C.; Turk, B.E.; Burnett, J.; et al. Identification of small molecule inhibitors of anthrax lethal factor. Nat. Struct. Mol. Biol. 2004, 11, 67–72. [Google Scholar] [CrossRef]
- Tonello, F.; Seveso, M.; Marin, O.; Mock, M.; Montecucco, C. Screening inhibitors of anthrax lethal factor. Nature 2002, 418, 386. [Google Scholar] [CrossRef]
- Xiong, Y.; Wiltsie, J.; Woods, A.; Guo, J.; Pivnichny, J.V.; Tang, W.; Bansal, A.; Cummings, R.T.; Cunningham, B.R.; Friedlander, A.M.; et al. The discovery of a potent and selective lethal factor inhibitor for adjunct therapy of anthrax infection. Bioorg. Med. Chem. Lett. 2006, 16, 964–968. [Google Scholar] [CrossRef]
- Lena, G.; Trapani, J.A.; Sutton, V.R.; Ciccone, A.; Browne, K.A.; Smyth, M.J.; Denny, W.A.; Spicer, J.A. Dihydrofuro[3,4-c]pyridinones as inhibitors of the cytolytic effects of the pore-forming glycoprotein perforin. J. Med. Chem. 2008, 51, 7614–7624. [Google Scholar] [CrossRef]
- Lyons, D.M.; Huttunen, K.M.; Browne, K.A.; Ciccone, A.; Trapani, J.A.; Denny, W.A.; Spicer, J.A. Inhibition of the cellular function of perforin by 1-amino-2,4-dicyanopyrido[1,2-a]benzimidazoles. Bioorg. Med. Chem. 2011, 19, 4091–4100. [Google Scholar] [CrossRef] [PubMed]
- Spicer, J.A.; Huttunen, K.M.; Miller, C.K.; Denny, W.A.; Ciccone, A.; Browne, K.A.; Trapani, J.A. Inhibition of the pore-forming protein perforin by a series of aryl-substituted isobenzofuran-1(3H)-ones. Bioorg. Med. Chem. 2012, 20, 1319–1336. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.K.; Huttunen, K.M.; Denny, W.A.; Jaiswal, J.K.; Ciccone, A.; Browne, K.A.; Trapani, J.A.; Spicer, J.A. Diarylthiophenes as inhibitors of the pore-forming protein perforin. Bioorg. Med. Chem. Lett. 2016, 26, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, Y.; Kodama, T.; Okahata, Y.; Shea, K.J. Peptide imprinted polymer nanoparticles: A plastic antibody. J. Am. Chem. Soc. 2008, 130, 15242–15243. [Google Scholar] [CrossRef] [PubMed]
- Mahon, C.S.; Fulton, D.A. Mimicking nature with synthetic macromolecules capable of recognition. Nat. Chem. 2014, 6, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, Y.; Koide, H.; Furuya, K.; Haberaecker, W.W.; Lee, S.; Kodama, T.; Kanazawa, H.; Oku, N.; Shea, K.J. The rational design of a synthetic polymer nanoparticle that neutralizes a toxic peptide in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Yoshimatsu, K.; Yamazaki, T.; Hoshino, Y.; Rose, P.E.; Epstein, L.F.; Miranda, L.P.; Tagari, P.; Beierle, J.M.; Yonamine, Y.; Shea, K.J. Epitope discovery for a synthetic polymer nanoparticle: A new strategy for developing a peptide tag. J. Am. Chem. Soc. 2014, 136, 1194–1197. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolskaia, M.A.; Germolec, D.R.; Weaver, J.L. Evaluation of nanoparticle immunotoxicity. Nat. Nanotechnol. 2009, 4, 411–414. [Google Scholar] [CrossRef]
- Cedervall, T.; Lynch, I.; Lindman, S.; Berggard, T.; Thulin, E.; Nilsson, H.; Dawson, K.A.; Linse, S. Understanding the nanoparticle–protein corona using methods to quantify exchange rates and affinities of proteins for nanoparticles. Proc. Natl. Acad. Sci. USA 2007, 104, 2050–2055. [Google Scholar] [CrossRef]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors Affecting the Clearance and Biodistribution of Polymeric Nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef]
- Petros, R.A.; Desimone, J.M. Strategies in the design of nanoparticles for therapeutic applications. Nat. Rev. Drug Discov. 2010, 9, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Jokerst, J.V.; Lobovkina, T.; Zare, R.N.; Gambhir, S.S. Nanoparticle PEGylation for imaging and therapy. Nanomedicine 2011, 6, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Maynard, J.A.; Maassen, C.B.M.; Leppla, S.H.; Brasky, K.; Patterson, J.L.; Iverson, B.L.; Georgiou, G. Protection against anthrax toxin by recombinant antibody fragments correlates with antigen affinity. Nat. Biotechnol. 2002, 20, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Zettervall, O.; Sjöquist, J.; Waldenström, J.; Winblad, S. Serological activity in myeloma type globulins. Clin. Exp. Immunol. 1966, 1, 213–222. [Google Scholar] [PubMed]
- Seligmann, M.; Danon, F.; Basch, A.; Bernard, J. IgG myeloma cryoglobulin with antistreptolysin activity. Nature 1968, 220, 711–712. [Google Scholar] [CrossRef]
- Michaelsen, T.E.; Forre, O.; Hoyby, A.; Lea, T. Streptolysin O neutralizing capacity and idiotypic properties of fragments, subunits and reassociated H and L chains from three human IgG monoclonal proteins. Mol. Immunol. 1980, 17, 1143–1153. [Google Scholar] [CrossRef]
- Darji, A.; Niebuhr, K.; Hense, M.; Wehland, J.; Chakraborty, T. Neutralizing monoclonal antibodies against listeriolysin: Mapping of epitopes involved in pore formation. Infect. Immun. 1996, 64, 2356–2358. [Google Scholar]
- Nato, F.; Reich, K.; Lhopital, S.; Rouyre, S.; Geoffroy, C.; Mazie, J.C.; Cossart, P. Production and characterization of neutralizing and nonneutralizing monoclonal antibodies against listeriolysin O. Infect. Immun. 1991, 59, 4641–4646. [Google Scholar]
- Jacobs, T.; Cima-Cabal, M.D.; Darji, A.; Méndez, F.J.; Vázquez, F.; Jacobs, A.A.; Shimada, Y.; Ohno-Iwashita, Y.; Weiss, S.; de los Toyos, J.R. The conserved undecapeptide shared by thiol-activated cytolysins is involved in membrane binding. FEBS Lett. 1999, 459, 463–466. [Google Scholar] [CrossRef]
- Praper, T.; Beseničar Podlesnik, M.; Istinič, H.; Podlesek, Z.; Metkar, S.S.; Froelich, C.J.; Anderluh, G. Human perforin permeabilizing activity, but not binding to lipid membranes, is affected by pH. Mol. Immunol. 2010, 47, 2492–2504. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Moayeri, M.; Purcell, R. Monoclonal antibody therapies against anthrax. Toxins 2011, 3, 1004–1019. [Google Scholar] [CrossRef] [PubMed]
- Little, S.F.; Leppla, S.H.; Cora, E. Production and Characterization of Monoclonal Antibodies to the Protective Antigen Component of Bacillus anthracis Toxin. Infect. Immun. 1988, 56, 1807–1813. [Google Scholar] [PubMed]
- Little, S.F.; Novak, J.M.; Lowe, J.R.; Leppla, S.H.; Singh, Y.; Klimpel, K.R.; Lidgerding, B.C.; Friedlanderl, A.M. Characterization of lethal factor binding and cell receptor binding domains of protective antigen of Bacillus anthracis using monoclonal antibodies. Microbiology 1996, 142, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Migone, T.; Subramanian, G.M.; Zhong, J.; Healey, L.M.; Corey, A.; Devalaraja, M.; Lo, L.; Ullrich, S.; Zimmerman, J.; Chen, A.; et al. Raxibacumab for the treatment of inhalational anthrax. N. Engl. J. Med. 2009, 361, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, N.; Clagett, M.; Li, J.; Jones, S.; Pincus, S.; D’Alia, G.; Nardone, L.; Babin, M.; Spitalny, G.; Casey, L. A high-affinity monoclonal antibody to anthrax protective antigen passively protects rabbits before and after aerosolized Bacillus anthracis spore challenge. Infect. Immun. 2005, 73, 795–802. [Google Scholar] [CrossRef]
- Brossier, F.; Le, M.; Landier, A.; Lafaye, P. Functional Analysis of Bacillus anthracis Protective Antigen by Using Neutralizing Monoclonal Antibodies. Infect. Immun. 2004, 72, 6313–6317. [Google Scholar] [CrossRef]
- Wang, F.; Ruther, P.; Jiang, I.; Sawada-Hirai, R.; Sun, S.M.; Nedellec, R.; Morrow, P.R.; Kang, A.S. Human monoclonal antibodies that neutralize anthrax toxin by inhibiting heptamer assembly. Hum. Antib. 2004, 13, 105–110. [Google Scholar] [CrossRef]
- Vitale, L.; Blanset, D.; Lowy, I.; O’Neill, T.; Goldstein, J.; Little, S.F.; Andrews, G.P.; Dorough, G.; Taylor, R.K.; Keler, T. Prophylaxis and therapy of inhalational anthrax by a novel monoclonal antibody to protective antigen that mimics vaccine-induced immunity. Infect. Immun. 2006, 74, 5840–5847. [Google Scholar] [CrossRef]
- Peterson, J.W.; Comer, J.E.; Noffsinger, D.M.; Wenglikowski, A.; Walberg, K.G.; Chatuev, B.M.; Chopra, A.K.; Stanberry, L.R.; Kang, A.S.; Scholz, W.W.; et al. Human monoclonal anti-protective antigen antibody completely protects rabbits and is synergistic with ciprofloxacin in protecting mice and guinea pigs against inhalation anthrax. Infect. Immun. 2006, 74, 1016–1024. [Google Scholar] [CrossRef]
- Karginov, V.A.; Robinson, T.M.; Riemenschneider, J.; Golding, B.; Kennedy, M.; Shiloach, J.; Alibek, K. Treatment of anthrax infection with combination of ciprofloxacin and antibodies to protective antigen of Bacillus anthracis. FEMS Immunol. Med. Microbiol. 2004, 40, 71–74. [Google Scholar] [CrossRef]
- Orth, P.; Xiao, L.; Hernandez, L.D.; Reichert, P.; Sheth, P.R.; Beaumont, M.; Yang, X.; Murgolo, N.; Ermakov, G.; Dinunzio, E.; et al. Mechanism of action and epitopes of Clostridium difficile toxin B-neutralizing antibody bezlotoxumab revealed by X-ray crystallography. J. Biol. Chem. 2014, 289, 18008–18021. [Google Scholar] [CrossRef] [PubMed]
- Harshman, S.; Alouf, J.E.; Siffert, O.; Baleux, F. Reaction of staphylococcal alpha-toxin with peptide-induced antibodies. Infect. Immun. 1989, 57, 3856–3862. [Google Scholar] [PubMed]
- Ragle, B.E.; Wardenburg, J.B. Anti-alpha-hemolysin monoclonal antibodies mediate protection against Staphylococcus aureus pneumonia. Infect. Immun. 2009, 77, 2712–2718. [Google Scholar] [CrossRef] [PubMed]
- McClain, M.S.; Cover, T.L. Functional analysis of neutralizing antibodies against Clostridium perfringens epsilon-toxin. Infect. Immun. 2007, 75, 1785–1793. [Google Scholar] [CrossRef] [PubMed]
- Percival, D.A.; Shuttleworth, A.D.; Williamson, E.D.; Kelly, D.C. Anti-idiotypic antibody-induced protection against Clostridium perfringens type D. Infect. Immun. 1990, 58, 2487–2492. [Google Scholar] [PubMed]
- Rouha, H.; Badarau, A.; Visram, Z.C.; Battles, M.B.; Prinz, B.; Magyarics, Z.; Nagy, G.; Mirkina, I.; Stulik, L.; Zerbs, M.; et al. Five birds, one stone: Neutralization of α-hemolysin and 4 bi-component leukocidins of Staphylococcus aureus with a single human monoclonal antibody. mAbs 2015, 7, 243–254. [Google Scholar] [CrossRef]
- Schlesinger, B.C.; Cheng, L. Characterization of a novel monoclonal antibody against human perforin using transfected cell lines. Immunology 1994, 81, 291–295. [Google Scholar]
- Liu, J.K.H. The history of monoclonal antibody development—Progress, remaining challenges and future innovations. Ann. Med. Surg. 2014, 3, 113–116. [Google Scholar] [CrossRef]
- Chames, P.; Van Regenmortel, M.; Weiss, E.; Baty, D. Therapeutic antibodies: Successes, limitations and hopes for the future. Br. J. Pharmacol. 2009, 157, 220–233. [Google Scholar] [CrossRef]
- Beckman, R.A.; Weiner, L.M.; Davis, H.M. Antibody constructs in cancer therapy: Protein engineering strategies to improve exposure in solid tumors. Cancer 2007, 109, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Clackson, T.; Hoogenboomt, H.R.; Griffithst, A.D.; Winter, G. Making antibody fragments using phage display libraries. Nature 1991, 352, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Frenzel, A.; Schirrmann, T.; Hust, M. Phage display-derived human antibodies in clinical development and therapy. mAbs 2016, 8, 1177–1194. [Google Scholar] [CrossRef] [PubMed]
- Borrebaeck, C.A.K. Antibodies in diagnostics - from immunoassays to protein chips. Immunol. Today 2000, 21, 379–382. [Google Scholar] [CrossRef]
- Binz, H.K.; Amstutz, P.; Plückthun, A. Engineering novel binding proteins from nonimmunoglobulin domains. Nat. Biotechnol. 2005, 23, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Skerra, A. Alternative non-antibody scaffolds for molecular recognition. Curr. Opin. Biotechnol. 2007, 18, 295–304. [Google Scholar] [CrossRef]
- Frejd, F.Y.; Kim, K.T. Affibody molecules as engineered protein drugs. Exp. Mol. Med. 2017, 49, e306. [Google Scholar] [CrossRef]
- Gebauer, M.; Skerra, A. Engineered protein scaffolds as next-generation antibody therapeutics. Curr. Opin. Chem. Biol. 2009, 13, 245–255. [Google Scholar] [CrossRef]
- Liu, J.L.; Anderson, G.P.; Goldman, E.R. Isolation of anti-toxin single domain antibodies from a semi-synthetic spiny dogfish shark display library. BMC Biotechnol. 2007, 7. [Google Scholar] [CrossRef]
- He, M.; Taussig, M.J. Emerging Technologies for Antibody Selection. In Handbook of Therapeutic Antibodies; Dübel, S., Reichert, J.M., Eds.; Wiley-VCH Verlag & Co. KGaA: Weinheim, Germany, 2014; pp. 393–405. [Google Scholar]
- Wild, M.A.; Xin, H.; Maruyama, T.; Nolan, M.J.; Calveley, P.M.; Malone, J.D.; Wallace, M.R.; Bowdish, K.S. Human antibodies from immunized donors are protective against anthrax toxin in vivo. Nat. Biotechnol. 2003, 21, 1305–1306. [Google Scholar] [CrossRef]
- Ding, G.; Chen, X.; Zhu, J.; Duesbery, N.S.; Cheng, X.; Cao, B. A human/murine chimeric fab antibody neutralizes anthrax lethal toxin in vitro. Clin. Dev. Immunol. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.; Zheng, F.; Xiong, S.; Hu, D.; Lv, H.; Tang, Q.; Yang, J.; Feng, Z.; Wang, C.; Zhu, J. Preparation and evaluation of human-murine chimeric antibody against protective antigen of Bacillus anthracis. Int. J. Mol. Sci. 2014, 15, 18496–18507. [Google Scholar] [CrossRef] [PubMed]
- Pelat, T.; Hust, M.; Laffly, E.; Condemine, F.; Bottex, C.; Vidal, D.; Lefranc, M.P.; Dübel, S.; Thullier, P. High-affinity, human antibody-like antibody fragment (single-chain variable fragment) neutralizing the lethal factor (LF) of Bacillus anthracis by inhibiting protective antigen-LF complex formation. Antimicrob. Agents Chemother. 2007, 51, 2758–2764. [Google Scholar] [CrossRef] [PubMed]
- Gómez, I.; Miranda-Ríos, J.; Arenas, I.; Grande, R.; Becerril, B.; Bravo, A. Identification of scFv Molecules that Recognize Loop 3 of Domain II and Domain III of Cry1Ab Toxin from Bacillus thuringiensis. In Proceedings of the 6th Pacific Rim Conference on the Biotechnology of Bacillus thuringiensis and its Environmental Impact, Victoria, BC, Canada, 30 October–3 November 2005; pp. 12–14. [Google Scholar]
- Goldman, E.R.; Andersen, G.P.; Liu, J.L.; Delehanty, J.B.; Sherwood, L.J.; Osborn, L.E.; Cummins, L.B.; Hayhurst, A. Facile generation of heat-stable antiviral and antitoxin single domain antibodies from a semisynthetic llama library. Anal. Chem. 2006, 78, 8245–8255. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.L.; Anderson, G.P.; Delehanty, J.B.; Baumann, R.; Hayhurst, A.; Goldman, E.R. Selection of cholera toxin specific IgNAR single-domain antibodies from a naive shark library. Mol. Immunol. 2007, 44, 1775–1783. [Google Scholar] [CrossRef] [PubMed]
- Hussack, G.; Arbabi-Ghahroudi, M.; Van Faassen, H.; Songer, J.G.; Ng, K.K.S.; MacKenzie, R.; Tanha, J. Neutralization of Clostridium difficile toxin A with single-domain antibodies targeting the cell receptor binding domain. J. Biol. Chem. 2011, 286, 8961–8976. [Google Scholar] [CrossRef] [PubMed]
- Unger, M.; Eichhoff, A.M.; Schumacher, L.; Strysio, M.; Menzel, S.; Schwan, C.; Alzogaray, V.; Zylberman, V.; Seman, M.; Brandner, J.; et al. Selection of Nanobodies that Block the Enzymatic and Cytotoxic Activities of the Binary Clostridium Difficile Toxin CDT. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- Yang, N.J.; Liu, D.V.; Sklaviadis, D.; Gui, D.Y.; Vander Heiden, M.G.; Wittrup, K.D. Antibody-Mediated Neutralization of Perfringolysin O for Intracellular Protein Delivery. Mol. Pharm. 2015, 12, 1992–2000. [Google Scholar] [CrossRef]
- Mammen, M.; Choi, S.; Whitesides, G.M. Polyvalent Interactions in Biological Systems: Implications for Design and Use of Multivalent Ligands and Inhibitors. Angew. Chem. Int. Ed. 1998, 37, 2754–2794. [Google Scholar] [CrossRef]
- Crini, G. Review: A history of cyclodextrins. Chem. Rev. 2014, 114, 10940–10975. [Google Scholar] [CrossRef]
- Karginov, V.A.; Nestorovich, E.M.; Schmidtmann, F.; Robinson, T.M.; Yohannes, A.; Fahmi, N.E.; Bezrukov, S.M.; Hecht, S.M. Inhibition of S. aureus alpha-hemolysin and B. anthracis lethal toxin by beta-cyclodextrin derivatives. Bioorg. Med. Chem. 2007, 15, 5424–5431. [Google Scholar] [CrossRef] [PubMed]
- Ragle, B.E.; Karginov, V.A.; Wardenburg, J.B. Prevention and treatment of Staphylococcus aureus pneumonia with a β-cyclodextrin derivative. Antimicrob. Agents Chemother. 2010, 54, 298–304. [Google Scholar] [CrossRef] [PubMed]
- McCormick, C.C.; Caballero, A.R.; Balzli, C.L.; Tang, A.; O’Callaghan, R.J. Chemical inhibition of alpha-toxin, a key corneal virulence factor of Staphylococcus aureus. Invest. Ophthalmol. Vis. Sci. 2009, 50, 2848–2854. [Google Scholar] [CrossRef] [PubMed]
- Backer, M.V.; Patel, V.; Jehning, B.T.; Claffey, K.P.; Karginov, V.A.; Backer, J.M. Inhibition of anthrax protective antigen outside and inside the cell. Antimicrob. Agents Chemother. 2007, 51, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Karginov, V.A.; Nestorovich, E.M.; Yohannes, A.; Robinson, T.M.; Fahmi, N.E.; Schmidtmann, F.; Hecht, S.M.; Bezrukov, S.M. Search for cyclodextrin-based inhibitors of anthrax toxins: Synthesis, structural features, and relative activities. Antimicrob. Agents Chemother. 2006, 50, 3740–3753. [Google Scholar] [CrossRef] [PubMed]
- Karginov, V.A.; Yohannes, A.; Robinson, T.M.; Fahmi, N.E.; Alibek, K.; Hecht, S.M. β-Cyclodextrin derivatives that inhibit anthrax lethal toxin. Bioorg. Med. Chem. 2006, 14, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Moayeri, M.; Robinson, T.M.; Leppla, S.H.; Karginov, V.A. In vivo efficacy of beta-cyclodextrin derivatives against anthrax lethal toxin. Antimicrob. Agents Chemother. 2008, 52, 2239–2241. [Google Scholar] [CrossRef]
- Bezrukov, S.M.; Liu, X.; Karginov, V.A.; Wein, A.N.; Leppla, S.H.; Popoff, M.R.; Barth, H.; Nestorovich, E.M. Interactions of high-affinity cationic blockers with the translocation pores of B. anthracis, C. botulinum, and C. perfringens binary toxins. Biophys. J. 2012, 103, 1208–1217. [Google Scholar] [CrossRef]
- Roeder, M.; Nestorovich, E.M.; Karginov, V.A.; Schwan, C.; Aktories, K.; Barth, H. Tailored Cyclodextrin Pore Blocker Protects Mammalian Cells from Clostridium difficile Binary Toxin CDT. Toxins 2014, 6, 2097–2114. [Google Scholar] [CrossRef]
- Nestorovich, E.M.; Karginov, V.A.; Popoff, M.R.; Bezrukov, S.M.; Barth, H. Tailored ß-cyclodextrin blocks the translocation pores of binary exotoxins from C. botulinum and C. perfringens and protects cells from intoxication. PLoS ONE 2011, 6, e23927. [Google Scholar] [CrossRef]
- Mourez, M.; Kane, R.S.; Mogridge, J.; Metallo, S.; Deschatelets, P.; Sellman, B.R.; Whitesides, G.M.; Collier, R.J. Designing a polyvalent inhibitor of anthrax toxin. Nat. Biotechnol. 2001, 19, 958–961. [Google Scholar] [CrossRef] [PubMed]
- Rai, P.; Padala, C.; Poon, V.; Saraph, A.; Basha, S.; Kate, S.; Tao, K.; Mogridge, J.; Kane, R.S. Statistical pattern matching facilitates the design of polyvalent inhibitors of anthrax and cholera toxins. Nat. Biotechnol. 2006, 24, 582–586. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.; Smith Korsholm, K.; Andersen, P.; Agger, E.M. Cationic liposomes as vaccine adjuvants. Expert Rev. Vaccines 2011, 10, 513–521. [Google Scholar] [CrossRef] [PubMed]
- González-Rodríguez, M.L.; Rabasco, A.M. Charged liposomes as carriers to enhance the permeation through the skin. Expert Opin. Drug Deliv. 2011, 8, 857–871. [Google Scholar] [CrossRef]
- Henriksen-Lacey, M.; Smith Korsholm, K.; Andersen, P.; Perrie, Y.; Christensen, D. Liposomal vaccine delivery systems. Expert Opin. Drug Deliv. 2011, 8, 505–519. [Google Scholar] [CrossRef]
- Förstner, P.; Bayer, F.; Kalu, N.; Felsen, S.; Förtsch, C.; Aloufi, A.; Ng, D.Y.W.; Weil, T.; Nestorovich, E.M.; Barth, H. Cationic PAMAM dendrimers as pore-blocking binary toxin inhibitors. Biomacromolecules 2014, 15, 2461–2474. [Google Scholar] [CrossRef]
- Yamini, G.; Kalu, N.; Nestorovich, E.M. Impact of dendrimer terminal group chemistry on blockage of the anthrax toxin channel: A single molecule study. Toxins 2016, 8. [Google Scholar] [CrossRef]
- Kitov, P.I.; Sadowska, J.M.; Mulvey, G.; Armstrong, G.D.; Ling, H.; Pannu, N.S.; Read, R.J.; Bundle, D.R. Shiga-like toxins are neutralized by tailored multivalent carbohydrate ligands. Nature 2000, 403, 669–672. [Google Scholar] [CrossRef]
- Basha, S.; Rai, P.; Poon, V.; Saraph, A.; Gujraty, K.; Go, M.Y.; Sadacharan, S.; Frost, M.; Mogridge, J.; Kane, R.S. Polyvalent inhibitors of anthrax toxin that target host receptors. Proc. Natl. Acad. Sci. USA 2006, 103, 13509–13513. [Google Scholar] [CrossRef]
- Yamini, G.; Nestorovich, E.M. Multivalent Inhibitors of Channel-Forming Bacterial Toxins. In Current topics in microbiology and immunology; Barth, H., Ed.; Springer International Publishing: Basel, Switzerland, 2016. [Google Scholar]
- Rummel, A.; Karnath, T.; Henke, T.; Bigalke, H.; Binz, T. Synaptotagmins I and II act as nerve cell receptors for botulinum neurotoxin G. J. Biol. Chem. 2004, 279, 30865–30870. [Google Scholar] [CrossRef]
- Dong, M.; Richards, D.A.; Goodnough, M.C.; Tepp, W.H.; Johnson, E.A.; Chapman, E.R. Synaptotagmins I and II mediate entry of botulinum neurotoxin B into cells. J. Cell Biol. 2003, 162, 1293–1303. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.M.; Wang, J.-L.; Kang, L.; Gao, S.; Liu, Y.; Hu, T.M. Construction and analysis of high-complexity ribosome display random peptide libraries. PLoS ONE 2008, 3, e2092. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gao, W.; Chen, Y.; Escajadillo, T.; Ungerleider, J.; Fang, R.H.; Christman, K.; Nizet, V.; Zhang, L. Self-Assembled Colloidal Gel Using Cell Membrane-Coated Nanosponges as Building Blocks. ACS Nano 2017, 11, 11923–11930. [Google Scholar] [CrossRef] [PubMed]
- Henry, B.D.; Neill, D.R.; Becker, K.A.; Gore, S.; Bricio-Moreno, L.; Ziobro, R.; Edwards, M.J.; Mühlemann, K.; Steinmann, J.; Kleuser, B.; et al. Engineered liposomes sequester bacterial exotoxins and protect from severe invasive infections in mice. Nat. Biotechnol. 2015, 33, 81–88. [Google Scholar] [CrossRef]
- Sharma, S.; Thomas, D.; Marlett, J.; Manchester, M.; Young, J.A.T. Efficient neutralization of antibody-resistant forms of anthrax toxin by a soluble receptor decoy inhibitor. Antimicrob. Agents Chemother. 2009, 53, 1210–1212. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.H.; Luk, B.T.; Hu, C.J.; Zhang, L. Engineered nanoparticles mimicking cell membranes for toxin neutralization. Adv. Drug Deliv. Rev. 2015, 90, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Gao, W.; Thamphiwatana, S.; Luk, B.T.; Angsantikul, P.; Zhang, Q.; Hu, C.J.; Fang, R.H.; Copp, J.A.; Pornpattananangkul, D.; et al. Hydrogel Retaining Toxin-Absorbing Nanosponges for Local Treatment of Methicillin-Resistant Staphylococcus aureus Infection. Adv. Mater. 2015, 27, 3437–3443. [Google Scholar] [CrossRef]
- Cai, C.; Che, J.; Xu, L.; Guo, Q.; Kong, Y.; Fu, L.; Xu, J.; Cheng, Y.; Chen, W. Tumor endothelium marker-8 based decoys exhibit superiority over capillary morphogenesis protein-2 based decoys as anthrax toxin inhibitors. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Wycoff, K.L.; Belle, A.; Deppe, D.; Schaefer, L.; MacLean, J.M.; Haase, S.; Trilling, A.K.; Liu, S.; Leppla, S.H.; Geren, I.N.; et al. Recombinant anthrax toxin receptor-Fc fusion proteins produced in plants protect rabbits against inhalational anthrax. Antimicrob. Agents Chemother. 2011, 55, 132–139. [Google Scholar] [CrossRef]
- Narat, M.; Maček, P.; Kotnik, V.; Sedmak, B. The humoral and cellular immune response to a lipid attenuated pore-forming toxin from the sea anemone Actinia equina L. Toxicon 1994, 32, 65–71. [Google Scholar] [CrossRef]
- Turk, T.; Maček, P.; Gubenšek, F. Chemical modification of equinatoxin II, a lethal and cytolytic toxin from the sea anemone Actinia equina L. Toxicon 1989, 27, 375–384. [Google Scholar] [CrossRef]
- Polyzos, A.; Alderton, M.R.; Dawson, R.M.; Hartley, P.G. Biofunctionalized surfactant mesophases as polyvalent inhibitors of cholera toxin. Bioconjug. Chem. 2007, 18, 1442–1449. [Google Scholar] [CrossRef] [PubMed]
- Vinion-Dubiel, A.D.; McClain, M.S.; Czajkowsky, D.M.; Iwamoto, H.; Ye, D.; Cao, P.; Schraw, W.; Szabo, G.; Blanke, S.R.; Shao, Z.; et al. A dominant negative mutant of Helicobacter pylori vacuolating toxin (VacA) inhibits VacA-induced cell vacuolation. J. Biol. Chem. 1999, 274, 37736–37742. [Google Scholar] [CrossRef] [PubMed]
- Sellman, B.R.; Mourez, M.; Collier, R.J. Dominant-negative mutants of a toxin subunit: An approach to therapy of anthrax. Science 2001, 292, 695–697. [Google Scholar] [CrossRef] [PubMed]
- Singh, Y.; Khanna, H.; Chopra, A.P.; Mehra, V. A Dominant Negative Mutant of Bacillus anthracis Protective Antigen Inhibits Anthrax Toxin Action in Vivo. J. Biol. Chem. 2001, 276, 22090–22094. [Google Scholar] [CrossRef] [PubMed]
- Mourez, M.; Yan, M.; Lacy, D.B.; Dillon, L.; Bentsen, L.; Marpoe, A.; Maurin, C.; Hotze, E.; Wigelsworth, D.; Pimental, R.; et al. Mapping dominant-negative mutations of anthrax protective antigen by scanning mutagenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 13803–13808. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Roehrl, M.H. Anthrax vaccine design: Strategies to achieve comprehensive protection against spore, bacillus, and toxin. Med. Immunol. 2005, 4. [Google Scholar] [CrossRef]
- Cao, S.; Guo, A.; Liu, Z.; Tan, Y.; Wu, G.; Zhang, C.; Zhao, Y.; Chen, H. Investigation of new dominant-negative inhibitors of anthrax protective antigen mutants for use in therapy and vaccination. Infect. Immun. 2009, 77, 4679–4687. [Google Scholar] [CrossRef]
- Yan, M.; Collier, R.J. Characterization of Dominant-Negative Forms of Anthrax Protective Antigen. Mol. Med. 2003, 9, 46–51. [Google Scholar] [CrossRef]
- Rodríguez-Almazán, C.; Zavala, L.E.; Muñoz-Garay, C.; Jiménez-Juárez, N.; Pacheco, S.; Masson, L.; Soberón, M.; Bravo, A. Dominant negative mutants of Bacillus thuringiensis Cry1Ab toxin function as anti-toxins: Demonstration of the role of oligomerization in toxicity. PLoS ONE 2009, 4. [Google Scholar] [CrossRef]
- Wai, S.N.; Westermark, M.; Oscarsson, J.; Jass, J.; Maier, E.; Benz, R.; Uhlin, B.E. Characterization of dominantly negative mutant ClyA cytotoxin proteins in Escherichia coli. J. Bacteriol. 2003, 185, 5491–5499. [Google Scholar] [CrossRef] [PubMed]
- Leppla, S.H. A dominant-negative therapy for anthrax. Nat. Med. 2001, 7, 659–660. [Google Scholar] [CrossRef] [PubMed]
- Hameed, A.; Olsen, K.J.; Cheng, L.; Fox, W.M.; Hruban, R.H.; Podackt, E.R. Immunohistochemical Identification of Cytotoxic Lymphocytes Using Human Perforin Monoclonal Antibody. Am. J. Pathol. 1992, 140, 1025–1030. [Google Scholar] [PubMed]
- Portman, J.L.; Huang, Q.; Reniere, M.L.; Iavarone, A.T.; Portnoy, D.A. Activity of the pore-forming virulence factor Listeriolysin O is reversibly inhibited by naturally occurring S-glutathionylation. Infect. Immun. 2017, 85. [Google Scholar] [CrossRef] [PubMed]
- Geisberg, M.; Trapani, J.A.; Dupont, B. Monoclonal antibodies detecting discrete epitopes of human perforin. Tissue Antigens 1990, 35, 229–233. [Google Scholar] [CrossRef] [PubMed]
- McComb, R.C.; Martchenko, M. Neutralizing antibody and functional mapping of Bacillus anthracis protective antigen—The first step toward a rationally designed anthrax vaccine. Vaccine 2016, 34, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Hugo, F.; Reichwein, J.; Arvand, M.; Krämer, S.; Bhakdi, S. Use of a monoclonal antibody to determine the mode of transmembrane pore formation by streptolysin O. Infect. Immun. 1986, 54, 641–645. [Google Scholar] [PubMed]
- Majd, S.; Yusko, E.C.; Billeh, Y.N.; Macrae, M.X.; Yang, J.; Mayer, M. Applications of biological pores in nanomedicine, sensing, and nanoelectronics. Curr. Opin. Biotechnol. 2010, 21, 439–476. [Google Scholar] [CrossRef] [PubMed]
- Misawa, N.; Osaki, T.; Takeuchi, S. Membrane protein-based biosensors. J. R. Soc. Interface 2018, 15, 20170952. [Google Scholar] [CrossRef]
- Robertson, J.W.F.; Rodrigues, C.G.; Stanford, V.M.; Rubinson, K.A.; Krasilnikov, O.V.; Kasianowicz, J.J. Single-molecule mass spectrometry in solution using a solitary nanopore. Proc. Natl. Acad. Sci. USA 2007, 104, 8207–8211. [Google Scholar] [CrossRef]
- Rauf, S.; Zhang, L.; Ali, A.; Liu, Y.; Li, J. Label-Free Nanopore Biosensor for Rapid and Highly Sensitive Cocaine Detection in Complex Biological Fluids. ACS Sensors 2017, 2, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Montana, V.; Grubišić, V.; Stout, R.F.; Parpura, V.; Gu, L.Q. Nanopore sensing of botulinum toxin type B by discriminating an enzymatically cleaved peptide from a synaptic protein synaptobrevin 2 derivative. ACS Appl. Mater. Interfaces 2015, 7, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Acharya, S.; Edwards, S.; Schmidt, J. Research highlights: Nanopore protein detection and analysis. Lab. Chip. 2015, 15, 3424–3427. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.; Wu, H.; Jayasinghe, L.; Patel, A.; Reid, S.; Bayley, H. Continuous base identification for single-molecule nanopore DNA sequencing. Nat. Nanotechnol. 2009, 4, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Ayub, M.; Bayley, H. Engineered transmembrane pores. Curr. Opin. Chem. Biol. 2016, 34, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Bayley, H. Nanopore sequencing: From imagination to reality. Clin. Chem. 2015, 61, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Misra, N.; Martinez, J.A.; Huang, S.J.; Wang, Y.; Stroeve, P.; Grigoropoulos, C.P. Bioelectronic silicon nanowire devices using functional membrane proteins. Proc. Natl. Acad. Sci. USA 2009, 106, 13780–13784. [Google Scholar] [CrossRef] [PubMed]
- Provoda, C.J.; Stier, E.M.; Lee, K.D. Tumor cell killing enabled by listeriolysin O-liposome-mediated delivery of the protein toxin gelonin. J. Biol. Chem. 2003, 278, 35102–35108. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).