Naja atra Cardiotoxin 3 Elicits Autophagy and Apoptosis in U937 Human Leukemia Cells through the Ca2+/PP2A/AMPK Axis


Abstract
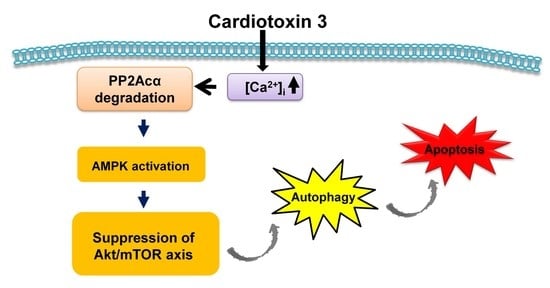
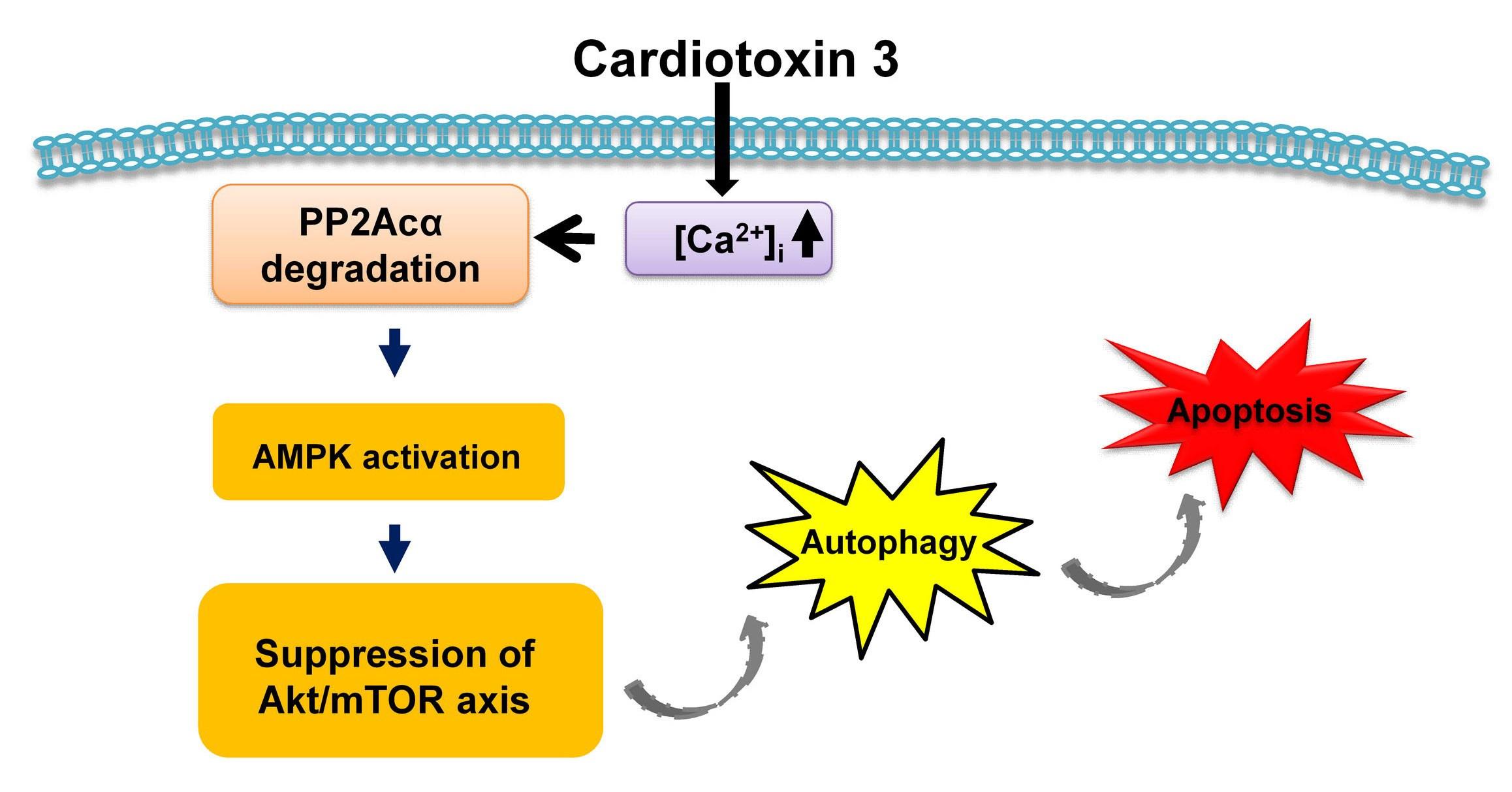{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
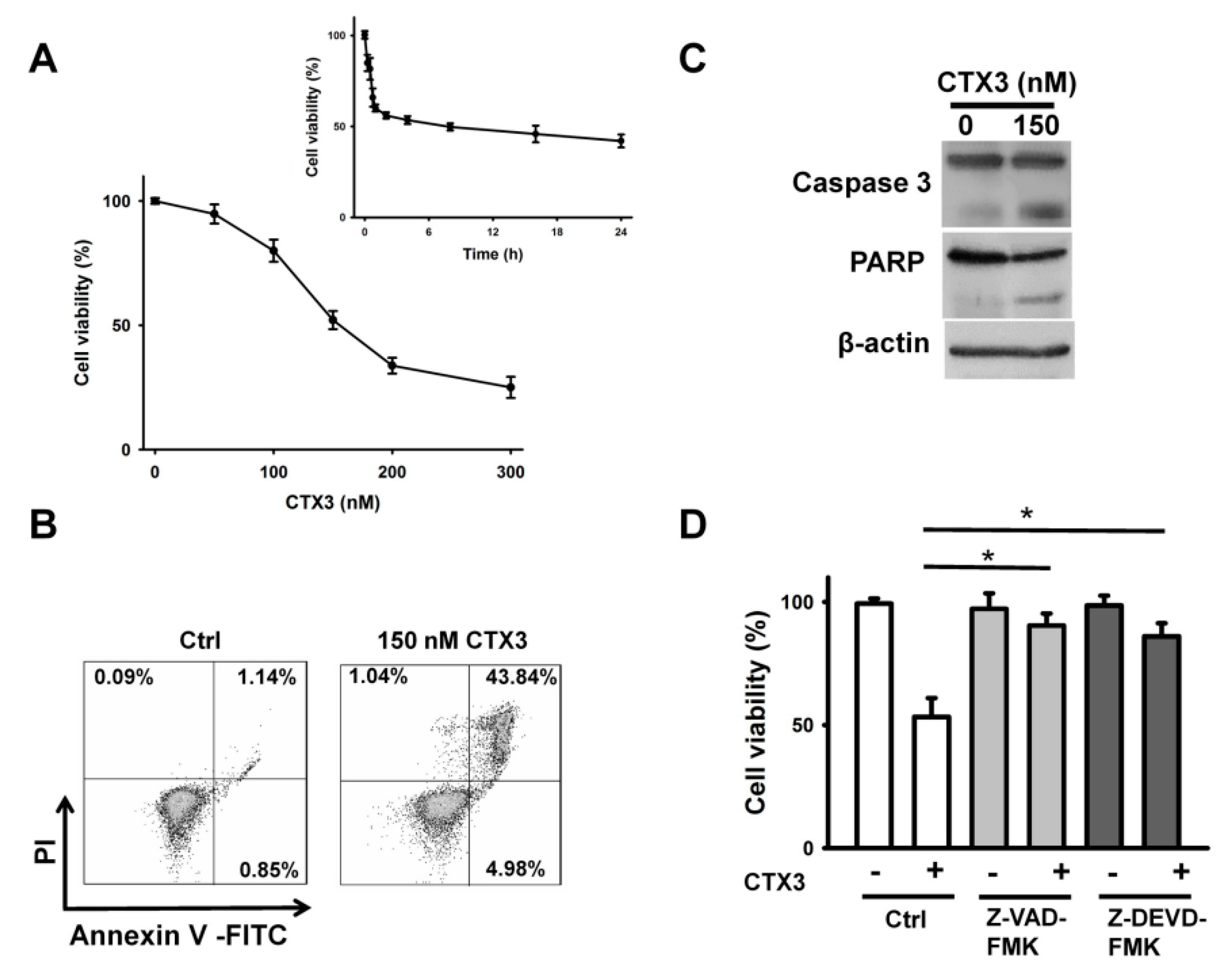
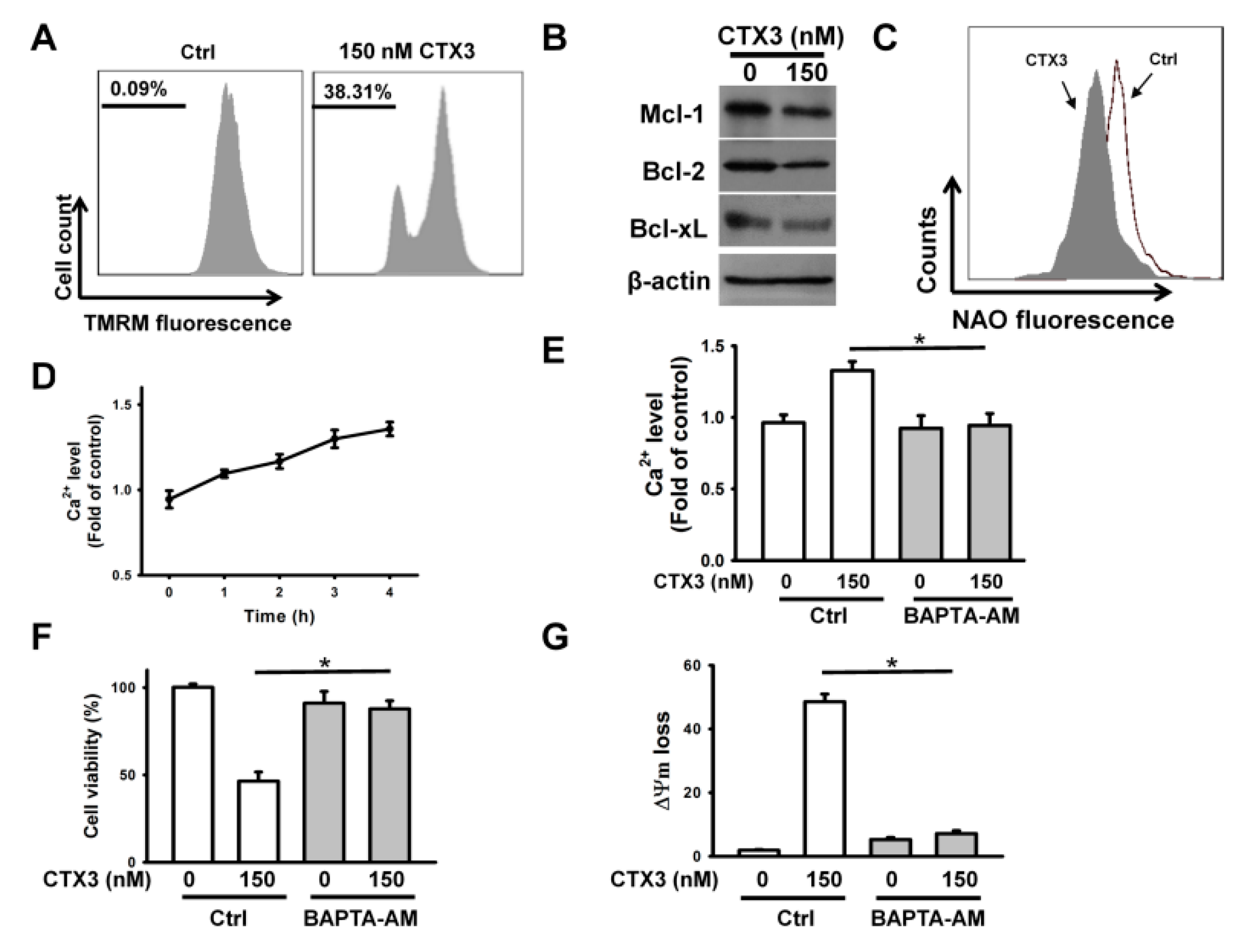
2. Results
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Chemicals and Reagents
5.2. Cell Culture
5.3. Measurement of Mitochondrial Depolarization
5.4. Measurement of Intracellular Ca2+ Concentration ([Ca2+]i)
5.5. Measurement of Mitochondrial Mass
5.6. Quantitative RT-PCR
5.7. Transfection of DNA
5.8. Membrane Permeability Assay
5.9. Leakage of Calcein-Loaded Cells
5.10. Other Tests
5.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dufton, M.J.; Hider, R.C. The structure and pharmacology of Elapid cytotoxins. In Snake Toxins; Harvey, A.L., Ed.; Pergamon Press: New York, NY, USA, 1999; pp. 259–302. [Google Scholar]
- Konshina, A.G.; Dubovskii, P.V.; Efremov, R.G. Structure and dynamics of cardiotoxins. Curr. Protein Pept. Sci. 2012, 13, 570–584. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, S.; Cheng, X.W.; Inoue, A.; Hu, L.; Piao, L.; Yu, C.; Goto, H.; Xu, W.; Zhao, G.; Lei, Y.; et al. Cathepsin K activity controls cardiotoxin-induced skeletal muscle repair in mice. J. Cachexia Sarcopenia Muscle 2018, 9, 160–175. [Google Scholar] [CrossRef] [PubMed]
- Feofanov, A.V.; Sharonov, G.V.; Astapova, M.V.; Rodionov, D.I.; Utkin, Y.N.; Arseniev, A.S. Cancer cell injury by cytotoxins from cobra venom is mediated through lysosomal damage. Biochem. J. 2005, 390, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Chien, C.M.; Yang, S.H.; Yang, C.C.; Chang, L.S.; Lin, S.R. Cardiotoxin III induces c-jun N-terminal kinase-dependent apoptosis in HL-60 human leukaemia cells. Cell Biochem. Funct. 2008, 26, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Vyas, V.K.; Brahmbhatt, K.; Bhatt, H.; Parmar, U. Therapeutic potential of snake venom in cancer therapy: Current perspectives. Asian Pac. J. Trop. Biomed. 2013, 3, 156–162. [Google Scholar] [CrossRef]
- Wu, M.; Ming, W.; Tang, Y.; Zhou, S.; Kong, T.; Dong, W. The anticancer effect of cytotoxin 1 from Naja atra Cantor venom is mediated by a lysosomal cell death pathway involving lysosomal membrane permeabilization and cathepsin B release. Am. J. Chin. Med. 2013, 41, 643–663. [Google Scholar] [CrossRef]
- Chaisakul, J.; Hodgson, W.C.; Kuruppu, S.; Prasongsook, N. Effects of animal venoms and toxins on hallmarks of cancer. J. Cancer 2016, 7, 1571–1578. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.G.; Tjong, S.C.; Wu, P.L.; Kuo, J.H.; Wu, K. Role of heparan sulfates and glycosphingolipids in the pore formation of basic polypeptides of cobra cardiotoxin. Adv. Exp. Med. Biol. 2010, 677, 143–149. [Google Scholar] [PubMed]
- Wang, C.H.; Wu, W.G. Amphiphilic β-sheet cobra cardiotoxin targets mitochondria and disrupts its network. FEBS Lett. 2005, 579, 3169–3174. [Google Scholar] [CrossRef]
- Zhang, B.; Li, F.; Chen, Z.; Shrivastava, I.H.; Gasanoff, E.S.; Dagda, R.K. Naja mossambica mossambica cobra cardiotoxin targets mitochondria to disrupt mitochondrial membrane structure and function. Toxins 2019, 11, 152. [Google Scholar] [CrossRef]
- Langone, F.; Cannata, S.; Fuoco, C.; Lettieri Barbato, D.; Testa, S.; Nardozza, A.P.; Ciriolo, M.R.; Castagnoli, L.; Gargioli, C.; Cesareni, G. Metformin protects skeletal muscle from cardiotoxin induced degeneration. PLoS ONE 2014, 9, e114018. [Google Scholar] [CrossRef] [PubMed]
- Marin, T.L.; Gongol, B.; Zhang, F.; Martin, M.; Johnson, D.A.; Xiao, H.; Wang, Y.; Subramaniam, S.; Chien, S.; Shyy, J.Y. AMPK promotes mitochondrial biogenesis and function by phosphorylating the epigenetic factors DNMT1, RBBP7, and HAT1. Sci. Signal 2017, 10, eaaf7478. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell. Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef]
- Eichner, L.J.; Brun, S.N.; Herzig, S.; Young, N.P.; Curtis, S.D.; Shackelford, D.B.; Shokhirev, M.N.; Leblanc, M.; Vera, L.I.; Hutchins, A.; et al. Genetic analysis reveals AMPK is required to support tumor growth in murine Kras-dependent lung cancer models. Cell Metab. 2019, 29, 285–302. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Mills, G.B. AMPK: A contextual oncogene or tumor suppressor? Cancer Res. 2013, 73, 2929–2935. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef]
- Chiou, S.H.; Raynor, R.L.; Zheng, B.; Chambars, T.C.; Kuo, J.F. Cobra venom cardiotoxin (cytotoxin) isoforms and neurotoxin: Comparative potency of protein kinase C inhibition and cancer cell cytotoxicity and model of enzyme inhibition. Biochemistry 1993, 32, 2062–2067. [Google Scholar] [CrossRef]
- Morciano, G.; Giorgi, C.; Balestra, D.; Marchi, S.; Perrone, D.; Pinotti, M.; Pinton, P. Mcl-1 involvement in mitochondrial dynamics is associated with apoptotic cell death. Mol. Biol. Cell 2016, 27, 20–34. [Google Scholar] [CrossRef]
- Park, S.; Scheffler, T.L.; Rossie, S.S.; Gerrard, D.E. AMPK activity is regulated by calcium-mediated protein phosphatase 2A activity. Cell Calcium 2013, 53, 217–223. [Google Scholar] [CrossRef]
- Joseph, B.K.; Liu, H.Y.; Francisco, J.; Pandya, D.; Donigan, M.; Gallo-Ebert, C.; Giordano, C.; Bata, A.; Nickels, J.T., Jr. Inhibition of AMP kinase by the protein phosphatase 2A heterotrimer, PP2APpp2r2d. J. Biol. Chem. 2015, 290, 10588–10598. [Google Scholar] [CrossRef]
- Guo, S.; Chen, C.; Ji, F.; Mao, L.; Xie, Y. PP2A catalytic subunit silence by microRNA-429 activates AMPK and protects osteoblastic cells from dexamethasone. Biochem. Biophys. Res. Commun. 2017, 487, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Kao, P.H.; Lin, S.R.; Chang, L.S. Differential binding to phospholipid bilayers modulates membrane-damaging activity of Naja naja atra cardiotoxins. Toxicon 2009, 54, 321–328. [Google Scholar] [CrossRef]
- Chen, K.C.; Kao, P.H.; Lin, S.R.; Chang, L.S. The mechanism of cytotoxicity by Naja naja atra cardiotoxin 3 is physically distant from its membrane-damaging effect. Toxicon 2007, 50, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Su, S.H.; Su, S.J.; Lin, S.R.; Chang, K.L. Cardiotoxin-III selectively enhances activation-induced apoptosis of human CD8+ T lymphocytes. Toxicol. Appl. Pharmacol. 2003, 193, 97–105. [Google Scholar] [CrossRef]
- Wang, C.H.; Monette, R.; Lee, S.C.; Morley, P.; Wu, W.G. Cobra cardiotoxin-induced cell death in fetal rat cardiomyocytes and cortical neurons: Different pathway but similar cell surface target. Toxicon 2005, 46, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.H.; Chang, L.S. Reactive oxygen species and p38 mitogen-activated protein kinase induce apoptotic death of U937 cells in response to Naja nigricollis toxin-γ. J. Cell. Mol. Med. 2009, 13, 1695–1705. [Google Scholar] [CrossRef]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L., Jr.; Losón, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef]
- Sekar, P.; Huang, D.Y.; Hsieh, S.L.; Chang, S.F.; Lin, W.W. AMPK-dependent and independent actions of P2X7 in regulation of mitochondrial and lysosomal functions in microglia. Cell Commun. Signal. 2018, 16, 83. [Google Scholar] [CrossRef]
- Trockenbacher, A.; Suckow, V.; Foerster, J.; Winter, J.; Krauss, S.; Ropers, H.H.; Schneider, R.; Schweiger, S. MID1, mutated in Opitz syndrome, encodes an ubiquitin ligase that targets phosphatase 2A for degradation. Nat. Genet. 2001, 29, 287–294. [Google Scholar] [CrossRef]
- Kong, M.; Ditsworth, D.; Lindsten, T.; Thompson, C.B. α4 is an essential regulator of PP2A phosphatase activity. Mol. Cell 2009, 36, 51–60. [Google Scholar] [CrossRef]
- Jiang, L.; Stanevich, V.; Satyshur, K.A.; Kong, M.; Watkins, G.R.; Wadzinski, B.E.; Sengupta, R.; Xing, Y. Structural basis of protein phosphatase 2A stable latency. Nat. Commun. 2013, 4, 1699. [Google Scholar] [CrossRef]
- Shi, Y.J.; Huang, C.H.; Lee, Y.C.; Wang, L.J.; Chiou, J.T.; Chang, L.S. Naja atra cardiotoxins enhance the protease activity of chymotrypsin. Int. J. Biol. Macromol. 2019, 136, 512–520. [Google Scholar] [CrossRef]
- Liu, W.H.; Chou, W.M.; Chang, L.S. p38 MAPK/PP2Acα/TTP pathway on the connection of TNF-α and caspases activation on hydroquinone-induced apoptosis. Carcinogenesis 2013, 34, 818–827. [Google Scholar] [CrossRef]
- Lee, Y.C.; Chen, Y.J.; Huang, C.H.; Chang, L.S. Amsacrine-induced apoptosis of human leukemia U937 cells is mediated by the inhibition of AKT- and ERK-induced stabilization of MCL1. Apoptosis 2017, 22, 406–420. [Google Scholar] [CrossRef]
- Lee, Y.C.; Wang, L.J.; Huang, C.H.; Shi, Y.J.; Chang, L.S. ABT-263-induced MCL1 upregulation depends on autophagy-mediated 4EBP1 downregulation in human leukemia cells. Cancer Lett. 2018, 432, 191–204. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiou, J.-T.; Shi, Y.-J.; Wang, L.-J.; Huang, C.-H.; Lee, Y.-C.; Chang, L.-S. Naja atra Cardiotoxin 3 Elicits Autophagy and Apoptosis in U937 Human Leukemia Cells through the Ca2+/PP2A/AMPK Axis. Toxins 2019, 11, 527. https://doi.org/10.3390/toxins11090527
Chiou J-T, Shi Y-J, Wang L-J, Huang C-H, Lee Y-C, Chang L-S. Naja atra Cardiotoxin 3 Elicits Autophagy and Apoptosis in U937 Human Leukemia Cells through the Ca2+/PP2A/AMPK Axis. Toxins. 2019; 11(9):527. https://doi.org/10.3390/toxins11090527
Chicago/Turabian StyleChiou, Jing-Ting, Yi-Jun Shi, Liang-Jun Wang, Chia-Hui Huang, Yuan-Chin Lee, and Long-Sen Chang. 2019. "Naja atra Cardiotoxin 3 Elicits Autophagy and Apoptosis in U937 Human Leukemia Cells through the Ca2+/PP2A/AMPK Axis" Toxins 11, no. 9: 527. https://doi.org/10.3390/toxins11090527
APA StyleChiou, J.-T., Shi, Y.-J., Wang, L.-J., Huang, C.-H., Lee, Y.-C., & Chang, L.-S. (2019). Naja atra Cardiotoxin 3 Elicits Autophagy and Apoptosis in U937 Human Leukemia Cells through the Ca2+/PP2A/AMPK Axis. Toxins, 11(9), 527. https://doi.org/10.3390/toxins11090527