Molecular Determinants of Brevetoxin Binding to Voltage-Gated Sodium Channels


Abstract
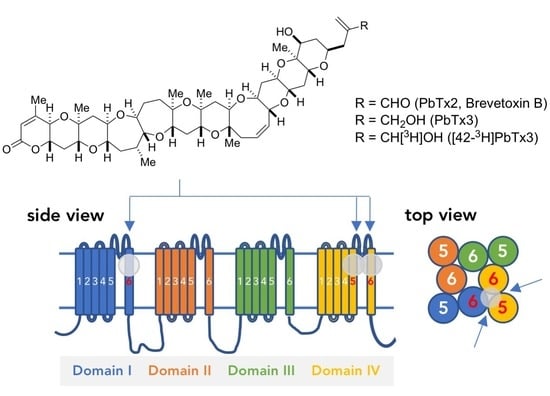
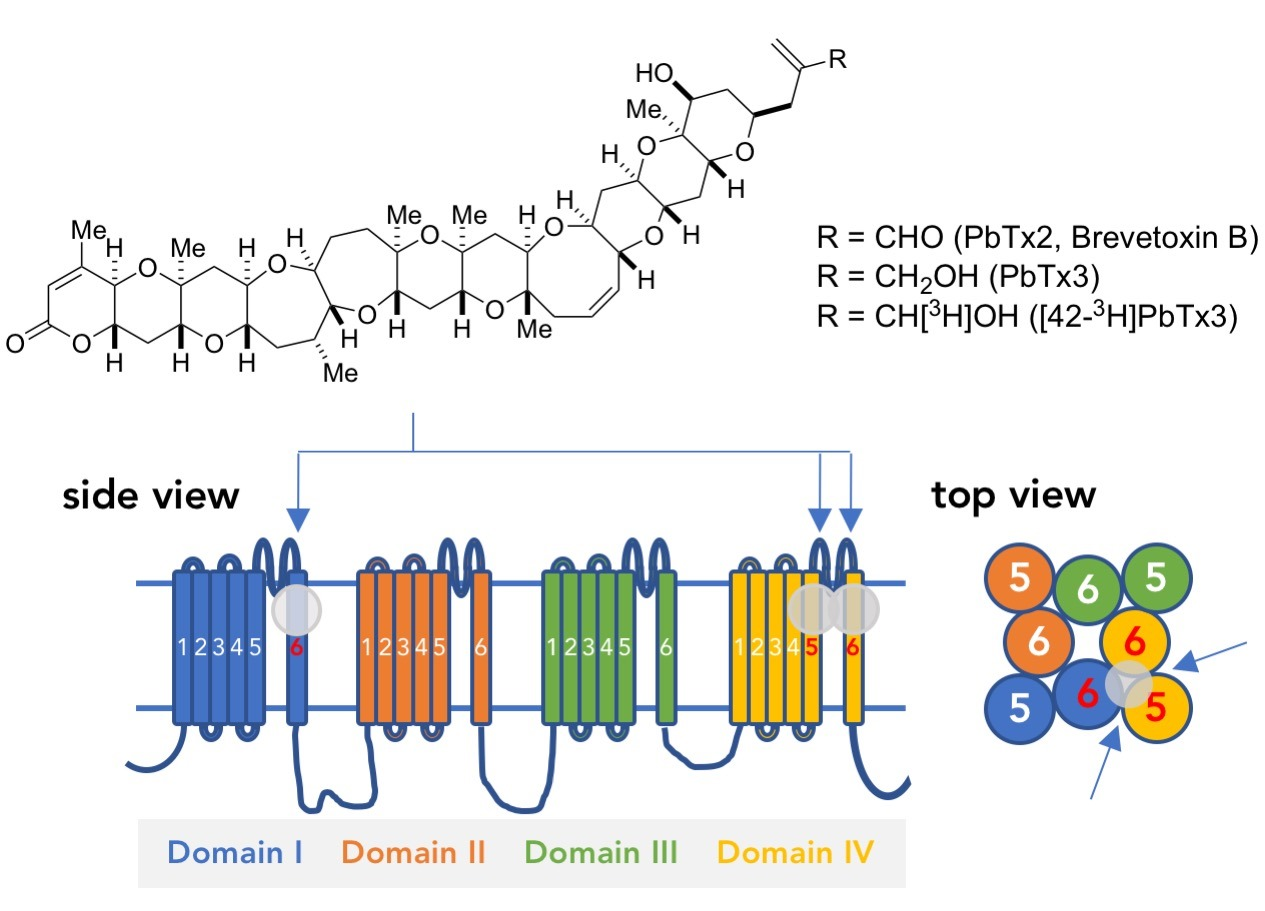
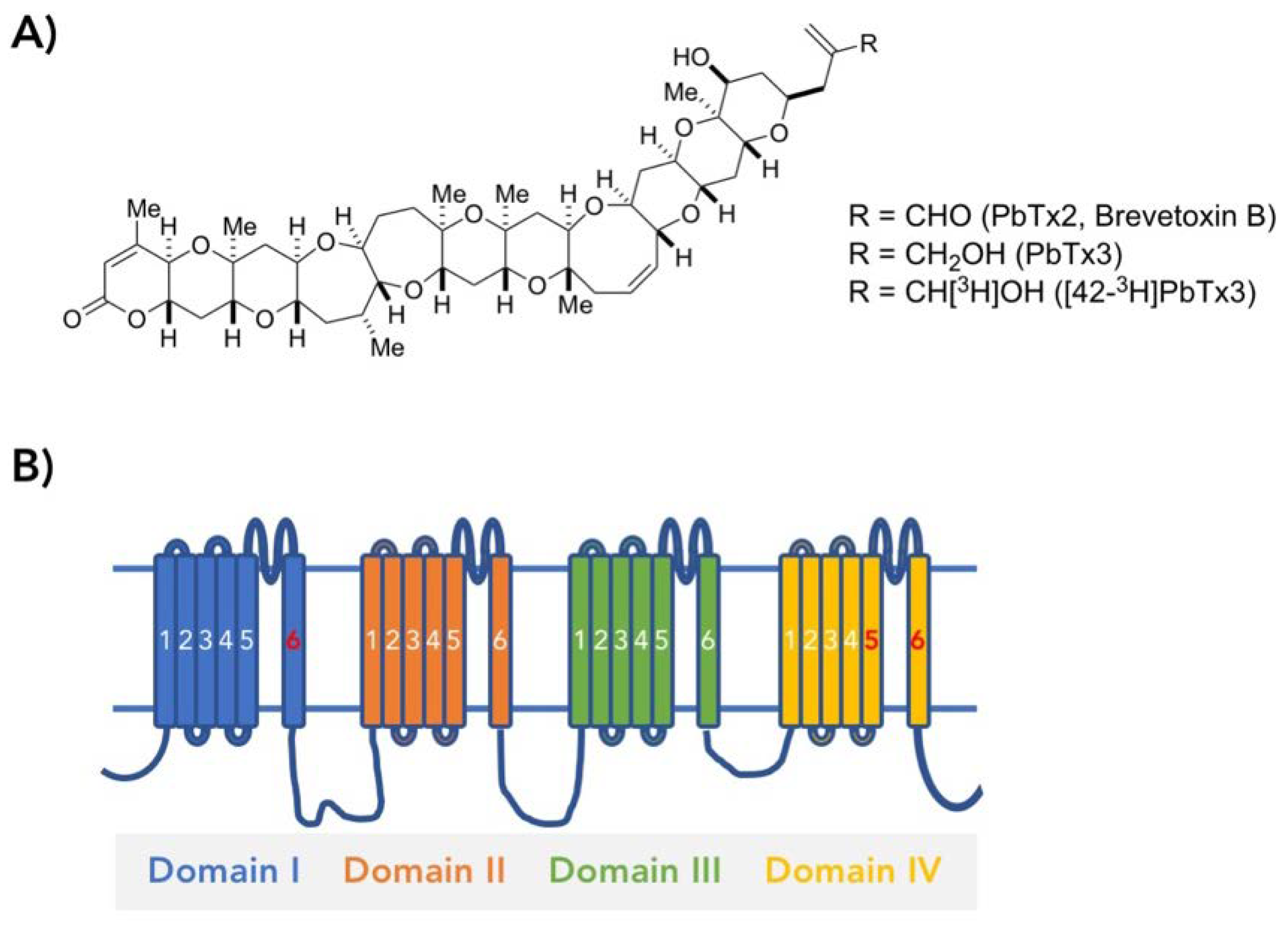
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
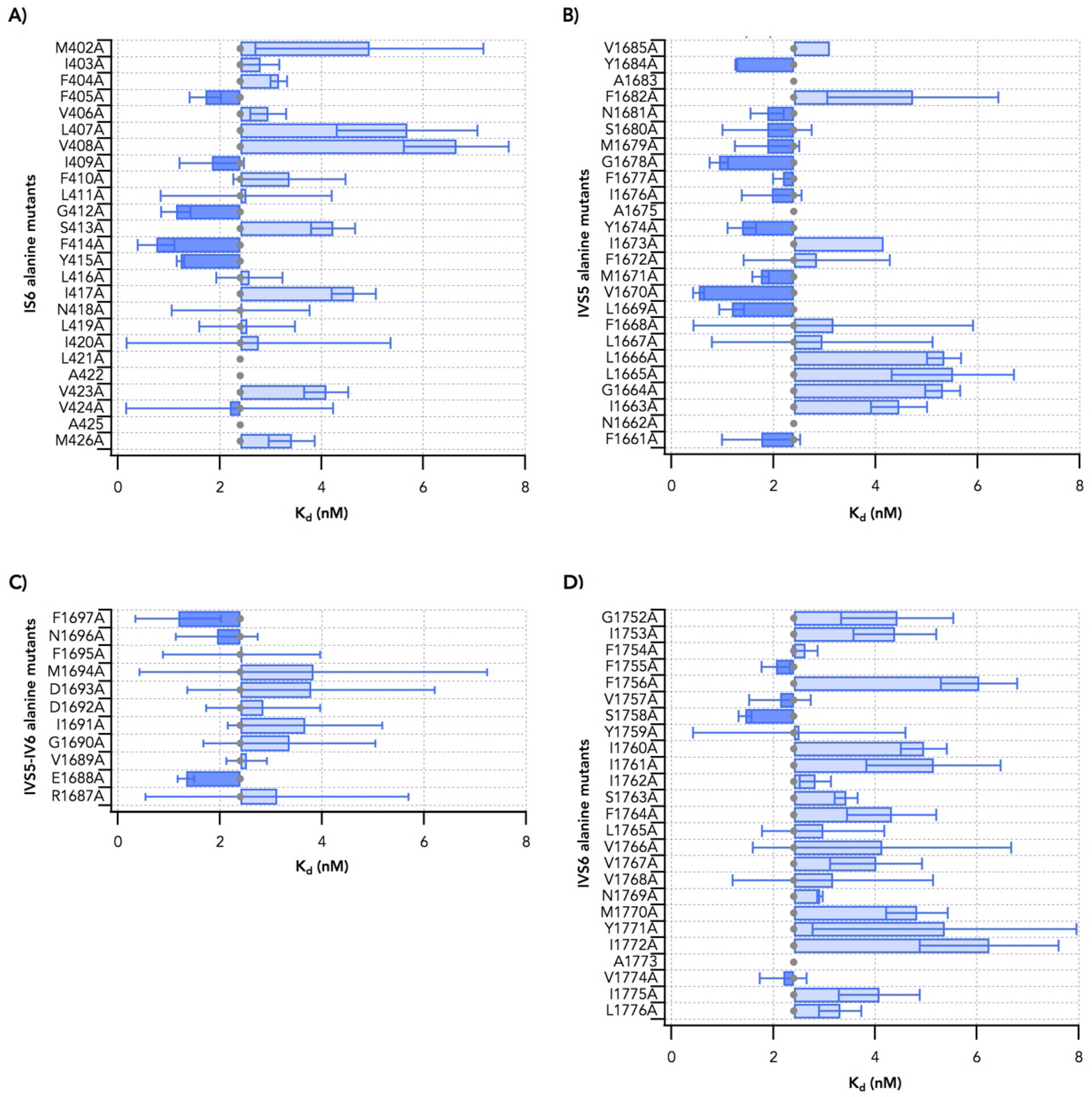
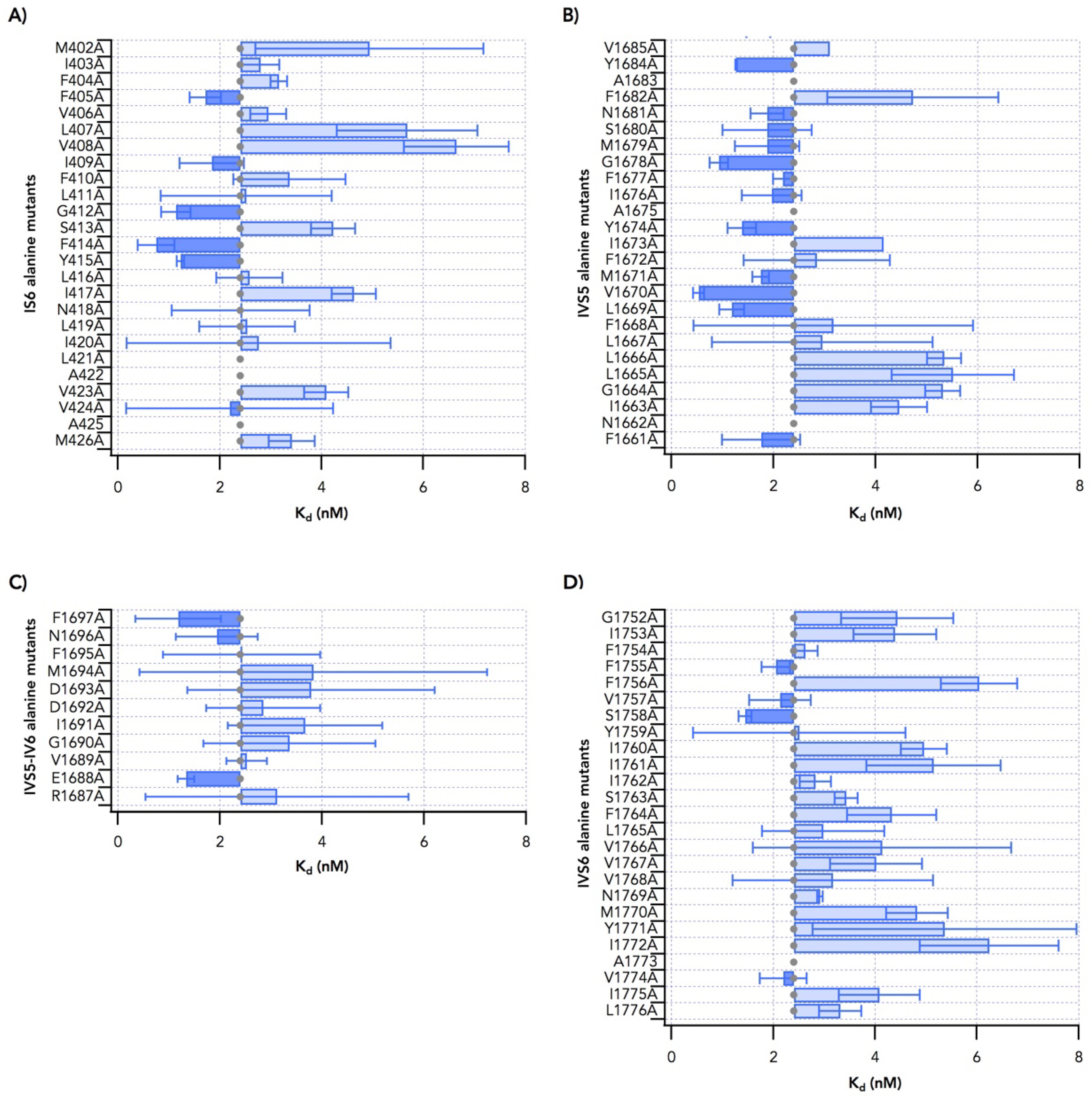
2.1. Alanine Scanning of Transmembrane Segments
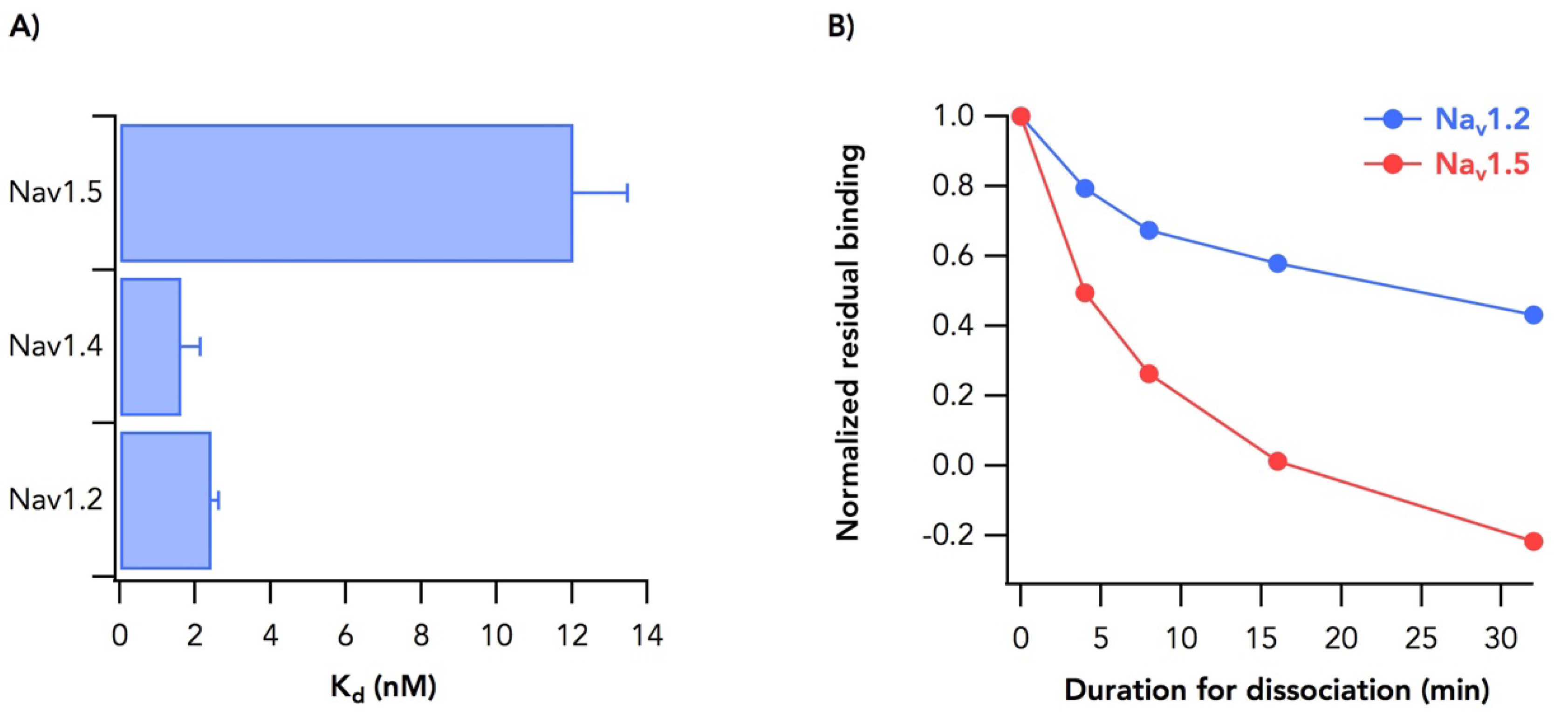
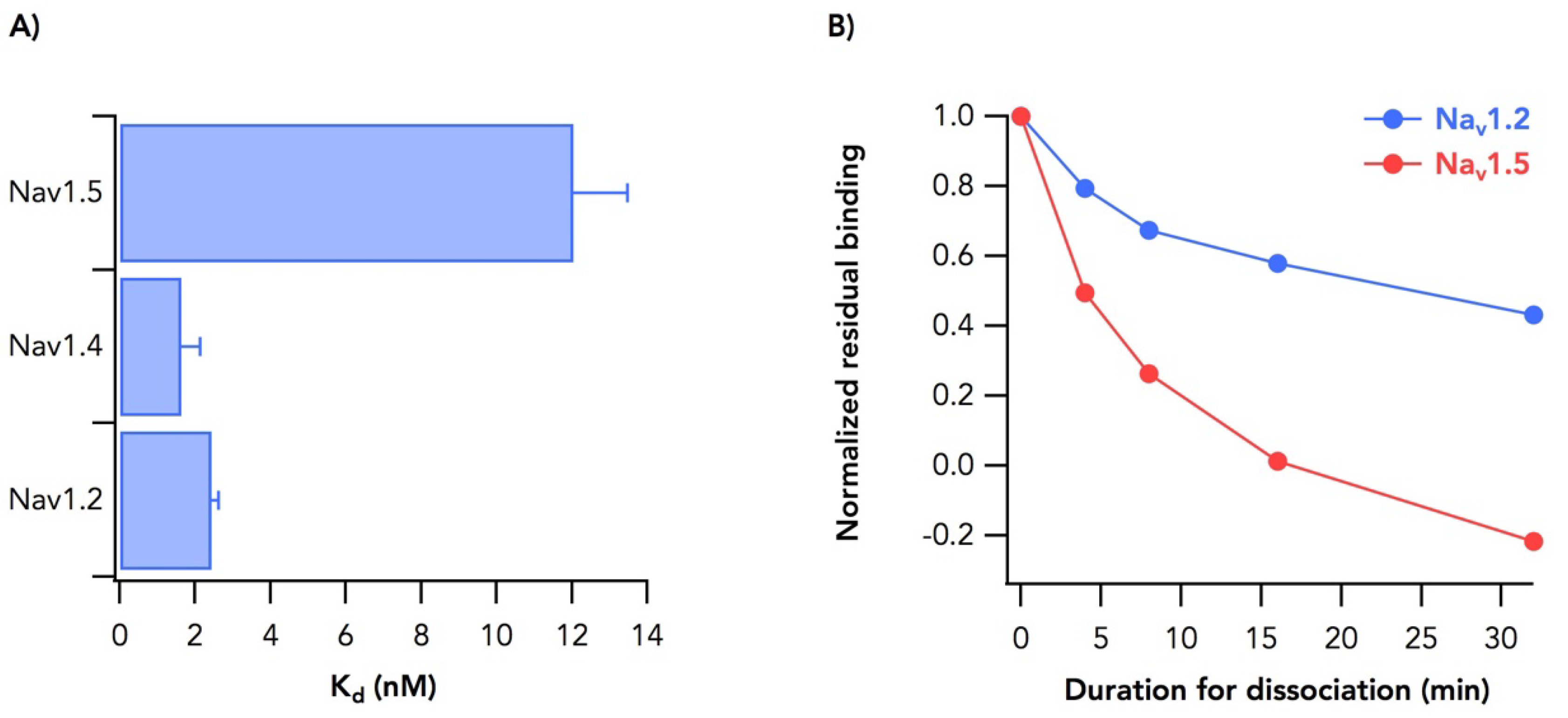
2.2. Sodium Channel Isoform-Dependent Differences in Toxin Binding
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Molecular Biology
4.3. Transfection of Nav1.2 into tsA-201 Cells
4.4. Preparation of Membrane Fractions
4.5. In Vitro Binding
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lin, Y.Y.; Risk, M.; Ray, S.M.; Van Engen, D.; Clardy, J.; Golik, J.; James, J.C.; Nakanishi, K. Isolation and structure of brevetoxin B from the red tide dinoflagellate Ptychodiscus brevis (Gymnodinium breve). J. Am. Chem. Soc. 1981, 103, 6773–6775. [Google Scholar] [CrossRef]
- Shimizu, Y.; Chou, H.N.; Bando, H.; Vanduyne, G.; Clardy, J.C. Structure of brevetoxin A (Gb-1 Toxin), the most potent toxin in the Florida red tide organism Gymnodinium breve (Ptychodiscus brevis). J. Am. Chem. Soc. 1986, 108, 514–515. [Google Scholar] [CrossRef] [PubMed]
- Poli, M.A.; Mende, T.J.; Baden, D.G. Brevetoxins, unique activators of voltage-sensitive sodium channels, bind to specific sites in rat brain synaptosomes. Mol. Pharmacol. 1986, 30, 129–135. [Google Scholar] [PubMed]
- Morohashi, A.; Satake, M.; Murata, K.; Naoki, H.; Kaspar, H.F.; Yasumoto, T. Brevetoxin B3, a new brevetoxin analog isolated from the greenshell mussel Perna canaliculus involved in neurotoxic shellfish poisoning in New Zealand. Tetrahedron Lett. 1995, 36, 8995–8998. [Google Scholar] [CrossRef]
- Morohashi, A.; Satake, M.; Naoki, H.; Kaspar, H.F.; Oshima, Y.; Yasumoto, T. Brevetoxin B4 isolated from greenshell mussels Perna canaliculus, the major toxin involved in neurotoxic shellfish poisoning in New Zealand. Nat. Toxins 1999, 7, 45–48. [Google Scholar] [CrossRef]
- Murata, K.; Satake, M.; Naoki, H.; Kaspar, H.F.; Yasumoto, T. Isolation and structure of a new brevetoxin analog, brevetoxin B2, from greenshell mussels from New Zealand. Tetrahedron 1998, 54, 735–742. [Google Scholar] [CrossRef]
- Murata, M.; Naoki, H.; Iwashita, T.; Matsunaga, S.; Sasaki, M.; Yokoyama, A.; Yasumoto, T. Structure of Maitotoxin. J. Am. Chem. Soc. 1993, 115, 2060–2062. [Google Scholar] [CrossRef]
- Murata, M.; Legrand, A.M.; Ishibashi, Y.; Fukui, M.; Yasumoto, T. Structures and configurations of ciguatoxin from the moray eel Gymnothorax javanicus and its likely precursor from the dinoflagellate Gambierdiscus toxicus. J. Am. Chem. Soc. 1990, 112, 4380–4386. [Google Scholar] [CrossRef]
- Igarashi, T.; Satake, M.; Yasumoto, T. Structures and partial stereochemical assignments for prymnesin-1 and prymnesin-2: Potent hemolytic and ichthyotoxic glycosides isolated from the red tide alga Prymnesium parvum. J. Am. Chem. Soc. 1999, 121, 8499–8511. [Google Scholar] [CrossRef]
- Satake, M.; Murata, M.; Yasumoto, T. Gambierol—A new toxic polyether compound isolated from the marine dinoflagellate Gambierdiscus toxicus. J. Am. Chem. Soc. 1993, 115, 361–362. [Google Scholar] [CrossRef]
- Nagai, H.; Torigoe, K.; Satake, M.; Murata, M.; Yasumoto, T.; Hirota, H. Gambieric acids—Unprecedented potent antifungal substances isolated from cultures of a marine dinoflagellate Gambierdiscus toxicus. J. Am. Chem. Soc. 1992, 114, 1102–1103. [Google Scholar] [CrossRef]
- Murata, M.; Kumagai, M.; Lee, J.S.; Yasumoto, T. Isolation and structure of yessotoxin, a novel polyether compound implicated in diarrhetic shellfish poisoning. Tetrahedron Lett. 1987, 28, 5869–5872. [Google Scholar] [CrossRef]
- Bourdelais, A.J.; Jacocks, H.M.; Wright, J.L.C.; Bigwarfe, P.M.; Baden, D.G. A new polyether ladder compound produced by the dinoflagellate Karenia brevis. J. Nat. Prod. 2005, 68, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Satake, M.; Bourdelais, A.J.; Van Wagoner, R.M.; Baden, D.G.; Wright, J.L.C. Brevisamide: An unprecedented monocyclic ether alkaloid from the dinoflagellate Karenia brevis that provides a potential model for ladder-frame initiation. Org. Lett. 2008, 10, 3465–3468. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Risk, M. Toxin T46 from Ptychodiscus brevis (formerly Gymnodinium breve) enhances activation of voltage-sensitive sodium channels by veratridine. Mol. Pharmacol. 1981, 19, 345–348. [Google Scholar] [PubMed]
- Ghiaroni, V.; Sasaki, M.; Fuwa, H.; Rossini, G.P.; Scalera, G.; Yasumoto, T.; Pietra, P.; Bigiani, A. Inhibition of voltage-gated potassium currents by gambierol in mouse taste cells. Toxicol. Sci. 2005, 85, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Alfonso, A.; de la Rosa, L.; Vieytes, M.R.; Yasumoto, T.; Botana, L.M. Yessotoxin, a novel phycotoxin, activates phosphodiesterase activity—Effect of yessotoxin on cAMP levels in human lymphocytes. Biochem. Pharmacol. 2003, 65, 193–208. [Google Scholar] [CrossRef]
- Catterall, W.A.; Goldin, A.L.; Waxman, S.G. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 2005, 57, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, R.E.; Adler, M. The actions of a red tide toxin from Ptychodiscus brevis on single sodium channels in mammalian neuroblastoma cells. FEBS Lett. 1989, 247, 448–452. [Google Scholar] [CrossRef]
- Jeglitsch, G.; Rein, K.; Baden, D.G.; Adams, D.J. Brevetoxin-3 (PbTx-3) and its derivatives modulate single tetrodotoxin-sensitive sodium channels in rat sensory neurons. J. Pharmacol. Exp. Ther. 1998, 284, 516–525. [Google Scholar]
- Schreibmayer, W.; Jeglitsch, G. The sodium channel activator brevetoxin-3 uncovers a multiplicity of different open states of the cardiac sodium channel. Biochim. Biophys. Acta 1992, 1104, 233–242. [Google Scholar] [CrossRef]
- Trainer, V.L.; Moreau, E.; Guedin, D.; Baden, D.G.; Catterall, W.A. Neurotoxin binding and allosteric modulation at receptor site 2 and site 5 on purified and reconstituted rat brain sodium channels. J. Biol. Chem. 1993, 268, 17114–17119. [Google Scholar] [PubMed]
- Al-Kouder, G.; Nawaz, A.; Gerami, C.; Matta, H.; Jacobsz, A.W.; Al-Salem, A.H. Volvulus of the sigmoid colon in a child. Saudi Med. J. 2002, 23, 594–596. [Google Scholar] [PubMed]
- Lombet, A.; Bidard, J.N.; Lazdunski, M. Ciguatoxin and brevetoxins share a common receptor site on the neuronal voltage-dependent Na+ channel. FEBS Lett. 1987, 219, 355–359. [Google Scholar] [CrossRef]
- Dechraoui, M.Y.; Naar, J.; Pauillac, S.; Legrand, A.M. Ciguatoxins and brevetoxins, neurotoxic polyether compounds active on sodium channels. Toxicon 1999, 37, 125–143. [Google Scholar] [CrossRef]
- Hirama, M.; Oishi, T.; Uehara, H.; Inoue, M.; Maruyama, M.; Guri, H.; Satake, M. Total synthesis of ciguatoxin CTX3C. Science 2001, 294, 1904–1907. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, K.; Inoue, M.; Miyahara, H.; Miyazaki, K.; Hirama, M. A quantitative and comparative study of the effects of a synthetic ciguatoxin CTX3C on the kinetic properties of voltage-dependent sodium channels. Br. J. Pharmacol. 2004, 142, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, K.; Inoue, M.; Miyazaki, K.; Hirama, M.; Kondo, C.; Kinoshita, E.; Miyoshi, H.; Seyama, I. Synthetic ciguatoxins selectively activate Nav1.8-derived chimeric sodium channels expressed in HEK293 Cells. J. Biol. Chem. 2009, 284, 7597–7605. [Google Scholar] [CrossRef]
- Trainer, V.L.; Baden, D.G.; Catterall, W.A. Identification of peptide components of the brevetoxin receptor site of rat brain sodium channels. J. Biol. Chem. 1994, 269, 19904–19909. [Google Scholar]
- Trainer, V.L.; Brown, G.B.; Catterall, W.A. Site of covalent labeling by a photoreactive batrachotoxin derivative near transmembrane segment IS6 of the sodium channel alpha subunit. J. Biol. Chem. 1996, 271, 11261–11267. [Google Scholar] [CrossRef]
- Tejedor, F.J.; Catterall, W.A. Site of covalent attachment of α scorpion toxin derivatives in domain I of the sodium channel α subunit. Proc. Natl. Acad. Sci. USA 1988, 85, 8742–8746. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.C.; Qu, Y.S.; Tanada, T.N.; Scheuer, T.; Catterall, W.A. Molecular determinants of high affinity binding of α-scorpion toxin and sea anemone toxin in the S3-S4 extracellular loop in domain IV of the Na+ channel alpha subunit. J. Biol. Chem. 1996, 271, 15950–15962. [Google Scholar] [CrossRef] [PubMed]
- Doyle, D.A.; Cabral, J.M.; Pfuetzner, R.A.; Kuo, A.L.; Gulbis, J.M.; Cohen, S.L.; Chait, B.T.; MacKinnon, R. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science 1998, 280, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Cestele, S.; Qu, Y.S.; Rogers, J.C.; Rochat, H.; Scheuer, T.; Catterall, W.A. Voltage sensor-trapping: Enhanced activation of sodium channels by beta-scorpion toxin bound to the S3-S4 loop in domain II. Neuron 1998, 21, 919–931. [Google Scholar] [PubMed]
- Dechraoui, M.Y.B.; Ramsdell, J.S. Type B brevetoxins show tissue selectivity for voltage-gated sodium channels: Comparison of brain, skeletal muscle and cardiac sodium channels. Toxicon 2003, 41, 919–927. [Google Scholar] [CrossRef]
- Dechraoui, M.Y.B.; Wacksman, J.J.; Ramsdell, J.S. Species selective resistance of cardiac muscle voltage gated sodium channels: Characterization of brevetoxin and ciguatoxin binding sites in rats and fish. Toxicon 2006, 48, 702–712. [Google Scholar] [CrossRef]
- Torikai, K.; Oishi, T.; Ujihara, S.; Matsumori, N.; Konoki, K.; Murata, M.; Aimoto, S. Design and synthesis of ladder-shaped tetracyclic, heptacyclic, and decacyclic ethers and evaluation of the interaction with transmembrane proteins. J. Am. Chem. Soc. 2008, 130, 10217–10226. [Google Scholar] [CrossRef]
- Oishi, T.; Hasegawa, F.; Torikai, K.; Konoki, K.; Matsumori, N.; Murata, M. Convergent synthesis and biological activity of the WXYZA’B’C’ ring system of maitotoxin. Org. Lett. 2008, 10, 3599–3602. [Google Scholar] [CrossRef]
- Shen, H.Z.; Liu, D.L.; Wu, K.; Lei, J.L.; Yan, N. Structures of human Nav1.7 channel in complex with auxiliary subunits and animal toxins. Science 2019, 363, 1303–1308. [Google Scholar] [CrossRef]
- Clairfeuille, T.; Cloake, A.; Infield, D.T.; Llongueras, J.P.; Arthur, C.P.; Li, Z.R.; Jian, Y.W.; Martin-Eauclaire, M.F.; Bougis, P.E.; Ciferri, C.; et al. Structural basis of α-scorpion toxin action on Nav channels. Science 2019, 363, eaav8573. [Google Scholar] [CrossRef]
- Ujihara, S.; Oishi, T.; Torikai, K.; Konoki, K.; Matsumori, N.; Murata, M.; Oshima, Y.; Aimoto, S. Interaction of ladder-shaped polyethers with transmembrane α-helix of glycophorin A as evidenced by saturation transfer difference NMR and surface plasmon resonance. Bioorg. Med. Chem. Lett. 2008, 18, 6115–6118. [Google Scholar] [CrossRef] [PubMed]
- White, S.H.; Wimley, W.C. Membrane protein folding and stability: Physical principles. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 319–365. [Google Scholar] [CrossRef] [PubMed]
- Mcphee, J.C.; Ragsdale, D.S.; Scheuer, T.; Catterall, W.A. Critical role for transmembrane segment Ivs6 of the sodium channel α subunit in fast inactivation. J. Biol. Chem. 1995, 270, 12025–12034. [Google Scholar] [CrossRef] [PubMed]
- Yarov-Yarovoy, V.; McPhee, J.C.; Idsvoog, D.; Pate, C.; Scheuer, T.; Catterall, W.A. Role of amino acid residues in transmembrane segments IS6 and IIS6 of the Na+ channel alpha subunit in voltage-dependent gating and drug block. J. Biol. Chem. 2002, 277, 35393–35401. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Okayama, H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol. Cell. Biol. 1987, 7, 2745–2752. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Konoki, K.; Baden, D.G.; Scheuer, T.; Catterall, W.A. Molecular Determinants of Brevetoxin Binding to Voltage-Gated Sodium Channels. Toxins 2019, 11, 513. https://doi.org/10.3390/toxins11090513
Konoki K, Baden DG, Scheuer T, Catterall WA. Molecular Determinants of Brevetoxin Binding to Voltage-Gated Sodium Channels. Toxins. 2019; 11(9):513. https://doi.org/10.3390/toxins11090513
Chicago/Turabian StyleKonoki, Keiichi, Daniel G. Baden, Todd Scheuer, and William A. Catterall. 2019. "Molecular Determinants of Brevetoxin Binding to Voltage-Gated Sodium Channels" Toxins 11, no. 9: 513. https://doi.org/10.3390/toxins11090513
APA StyleKonoki, K., Baden, D. G., Scheuer, T., & Catterall, W. A. (2019). Molecular Determinants of Brevetoxin Binding to Voltage-Gated Sodium Channels. Toxins, 11(9), 513. https://doi.org/10.3390/toxins11090513