Membrane Permeabilization by Pore-Forming RTX Toxins: What Kind of Lesions Do These Toxins Form?

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
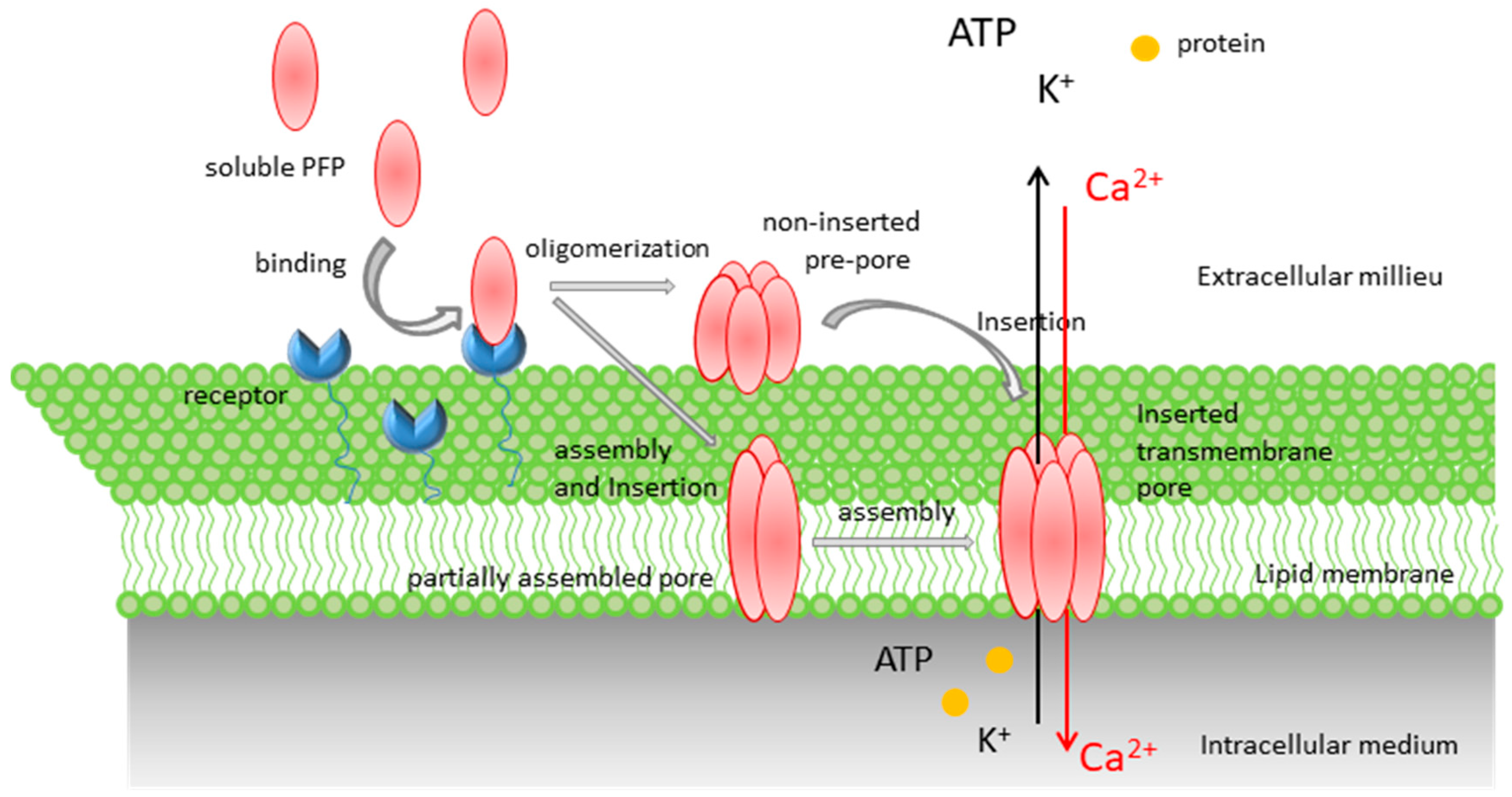
Pore-Forming Proteins
2. RTX Protein Family
2.1. Pore-Forming RTX Toxins: Still Little-Known Orphans
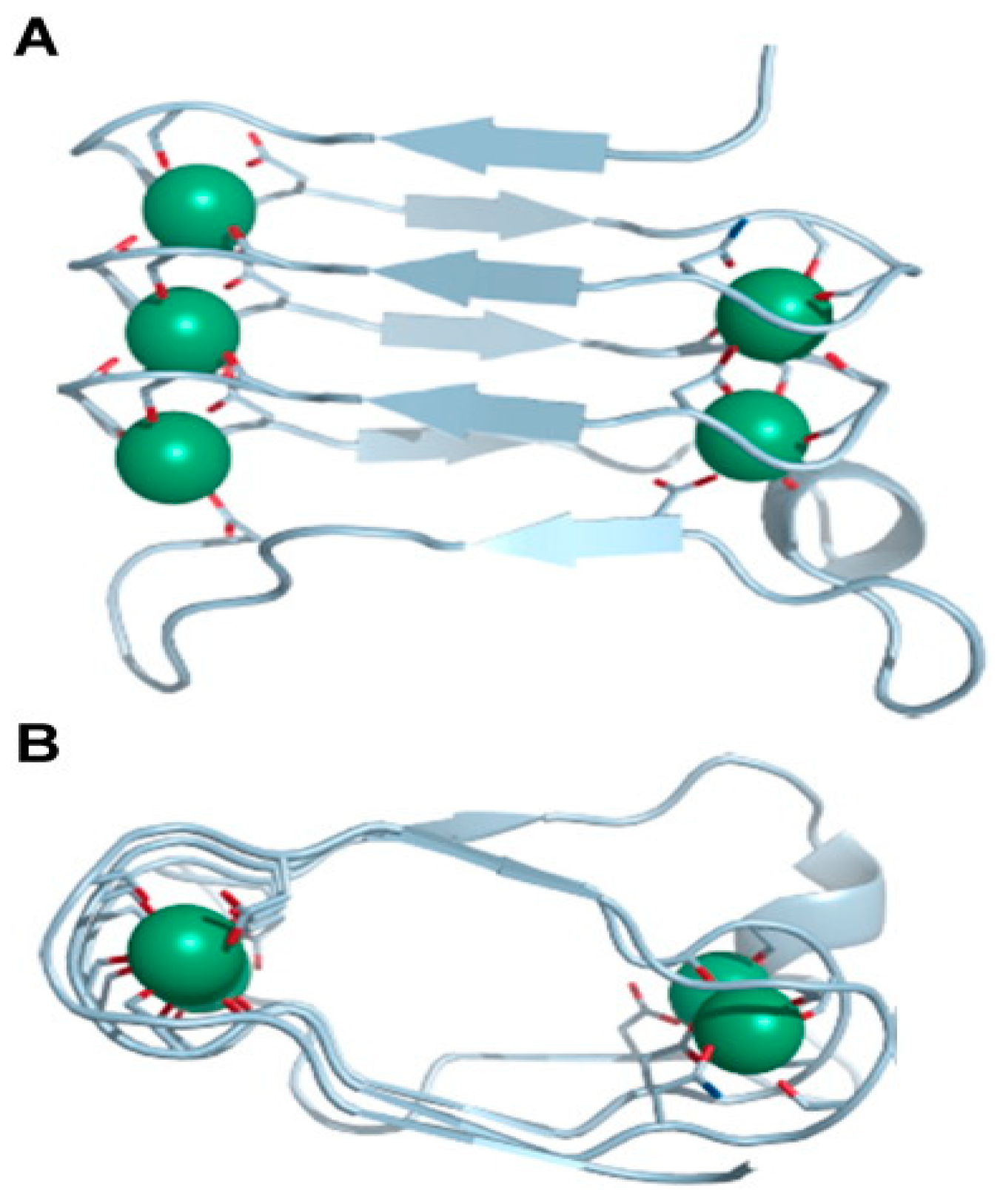
2.1.1. Pore Formation by RTX Toxins
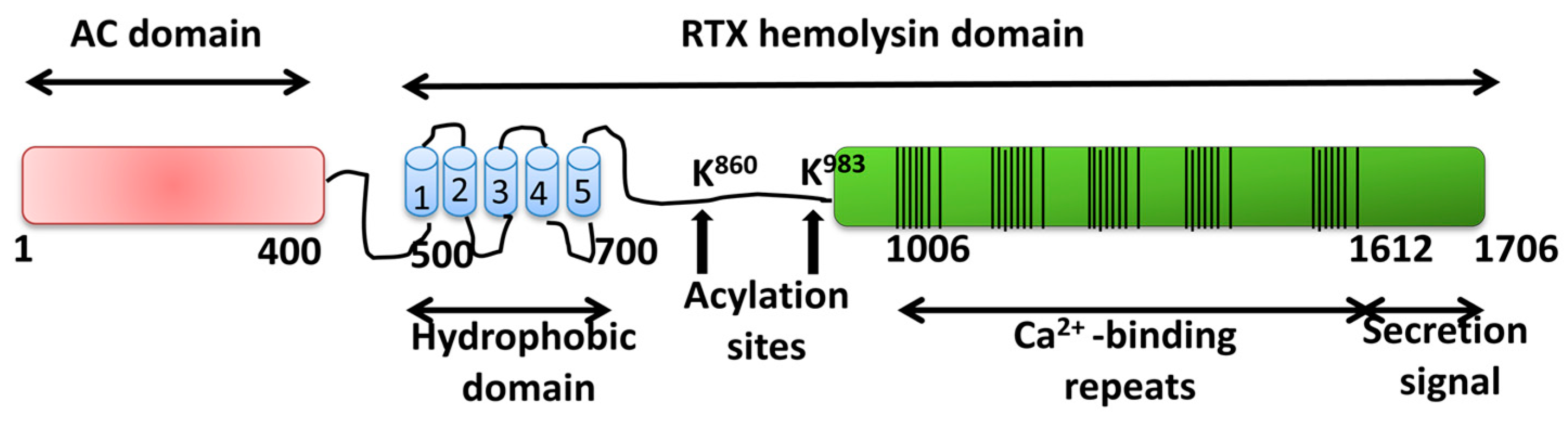
2.1.2. Adenylate Cyclase Toxin from Bordetella pertussis
Pore-Forming Activity of ACT
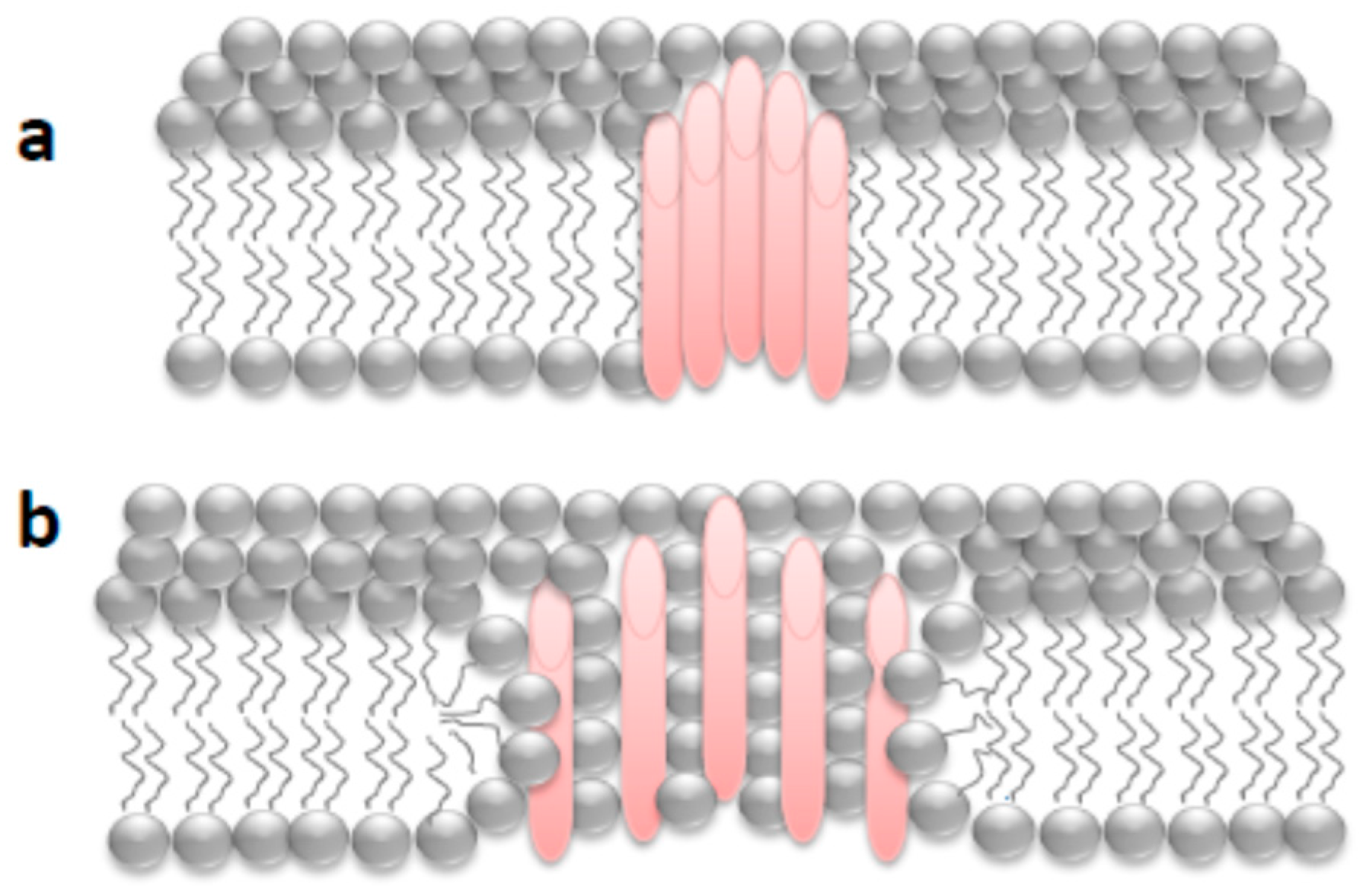
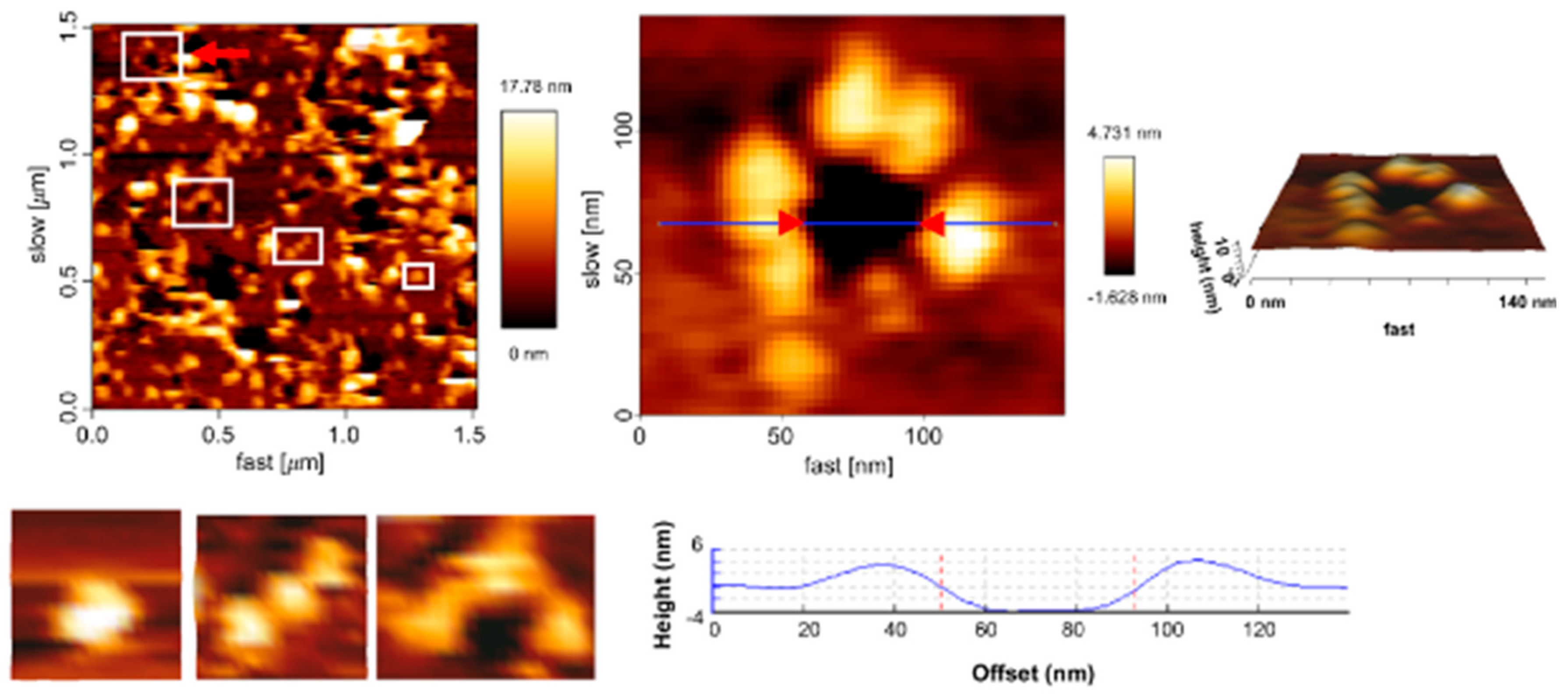
First Nanoscale Pictures of ACT Lytic Pores in Membranes
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Iacovache, I.; Bischofberger, M.; van der Goot, F.G. Structure and assembly of pore-forming proteins. Curr. Opin. Struct. Biol. 2010, 20, 241–246. [Google Scholar] [CrossRef]
- Bischofberger, M.; Iacovache, I.; Gisou Van Der Goot, F. Pathogenic pore-forming proteins: Function and host response. Cell Host Microbe 2012, 12, 266–275. [Google Scholar] [CrossRef]
- Peraro, M.D.; Van Der Goot, F.G. Pore-forming toxins: Ancient, but never really out of fashion. Nat. Rev. Microbiol. 2016, 14, 77–92. [Google Scholar] [CrossRef]
- Etxaniz, A.; González-Bullón, D.; Martín, C.; Ostolaza, H. Membrane repair mechanisms against permeabilization by pore-forming toxins. Toxins 2018, 10, 234. [Google Scholar] [CrossRef]
- Parker, M.W.; Feil, S.C. Pore-forming protein toxins: From structure to function. Prog. Biophys. Mol. Biol. 2005, 88, 91–142. [Google Scholar] [CrossRef]
- Iacovache, I.; van der Goot, F.G.; Pernot, L. Pore formation: An ancient yet complex form of attack. Biochim. Biophys. Acta Biomembr. 2008, 1778, 1611–1623. [Google Scholar] [CrossRef]
- Song, L.; Hobaugh, M.R.; Shustak, C.; Cheley, S.; Bayley, H.; Gouaux, J.E. Structure of staphylococcal a-hemolysin, a heptameric transmembrane pore. Science 1996, 274, 1859–1866. [Google Scholar] [CrossRef]
- Gurcel, L.; Iacovache, I.; van der Goot, F.G. Aerolysin and related Aeromonas toxins. In The Comprehensive Sourcebook of Bacterial Protein Toxins; Elsevier: Amsterdam, The Netherlands, 2006; pp. 608–622. [Google Scholar]
- Young, J.A.T.; Collier, R.J. Anthrax toxin: Receptor binding, internalization, pore formation, and translocation. Annu. Rev. Biochem. 2007, 76, 243–265. [Google Scholar] [CrossRef]
- Hotze, E.M.; Tweten, R.K. Membrane assembly of the cholesterol-dependent cytolysin pore complex. Biochim. Biophys. Acta Biomembr. 2012, 1818, 1028–1038. [Google Scholar] [CrossRef]
- Cosentino, K.; García-Sáez, A.J. Bax and Bak Pores: Are We Closing the Circle? Trends Cell Biol. 2017, 27, 266–275. [Google Scholar] [CrossRef]
- Cao, C.; Li, M.; Cirauqui, N.; Wang, Y.; Dal Peraro, M.; Tian, H.; Long, Y. Mapping the sensing spots of aerolysin for single oligonucleotides analysis. Nat. Commun. 2018, 9, 2823. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, N.; Bryant, S.L.; Thomas, C.; Richtsmeier, D.; Pu, X.; Tinker, J.; Fologea, D. Stochastic sensing of Angiotensin II with lysenin channels. Sci. Rep. 2017, 7, 2448. [Google Scholar] [CrossRef] [PubMed]
- Kasianowicz, J.J.; Brandin, E.; Branton, D.; Deamer, D.W. Characterization of individual polynucleotide molecules using a membrane channel. Proc. Natl. Acad. Sci. USA 1996, 93, 13770–13773. [Google Scholar] [CrossRef] [PubMed]
- Welch, R.A. Pore-forming cytolysins of Gram-negative bacteria. Mol. Microbiol. 1991, 5, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Baumann, U.; Wu, S.; Flaherty, K.M.; McKay, D.B. Three-dimensional structure of the alkaline protease of Pseudomonas aeruginosa: A two-domain protein with a calcium binding parallel beta roll motif. EMBO J. 1993, 12, 3357–3364. [Google Scholar] [CrossRef] [PubMed]
- Goebel, W.; Hedgpeth, J. Cloning and functional characterization of the plasmid-encoded hemolysin determinant of Escherichia coli. J. Bacteriol. 1982, 151, 1290–1298. [Google Scholar] [PubMed]
- Muller, D.; Hughes, C.; Goebel, W. Relationship between plasmid and chromosomal hemolysin determinants of Escherichia coli. J. Bacteriol. 1983, 153, 846–851. [Google Scholar] [PubMed]
- Felmlee, T.; Pellett, S.; Welch, R.A. Nucleotide sequence of an Escherichia coli chromosomal hemolysin. J. Bacteriol. 1985, 163, 94–105. [Google Scholar] [PubMed]
- Forestier, C.; Welch, R.A. Identification of RTX toxin target cell specificity domains by use of hybrid genes. Infect. Immun. 1991, 59, 4212–4220. [Google Scholar] [PubMed]
- Linhartová, I.; Bumba, L.; Mašn, J.; Basler, M.; Osicka, R.; Kamanová, J.; Procházková, K.; Adkins, I.; HejnováHolubová, J.; Sadílková, L.; et al. RTX proteins: A highly diverse family secreted bya common mechanism. FEMS Microbiol. Rev. 2010, 34, 1076–1112. [Google Scholar] [CrossRef]
- Thomas, S.; Holland, I.B.; Schmitt, L. The Type 1 secretion pathway—The hemolysin system and beyond. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 1629–1641. [Google Scholar] [CrossRef] [PubMed]
- Fullner, K.J.; Mekalanos, J.J. In vivo covalent cross-linking of cellular actin by the Vibrio cholerae RTX toxin. EMBO J. 2000, 19, 5315–5323. [Google Scholar] [CrossRef] [PubMed]
- Coote, J.G. Structural and functional relationships among the RTX toxin determinants of Gram-negative bacteria. FEMS Microbiol. Lett. 1992, 88, 137–161. [Google Scholar] [CrossRef] [PubMed]
- Gadeberg, O.V.; Orskov, I. In vitro cytotoxic effect of a-hemolytic Escherichia coli on human blood granulocytes. Infect. Immun. 1984, 45, 255–260. [Google Scholar] [PubMed]
- Keane, W.F.; Welch, R.; Gekker, G.; Peterson, P.K. Mechanism of Escherichia coli a-hemolysin-induced injury to isolated renal tubular cells. Am. J. Pathol. 1987, 126, 350–357. [Google Scholar] [PubMed]
- Mobley, H.L.; Green, D.M.; Trifillis, A.L.; Johnson, D.E.; Chippendale, G.R.; Lockatell, C.V.; Jones, B.D.; Warren, J.W. Pyelonephritogenic Escherichia coli and killing of cultured human renal proximal tubular epithelial cells: Role of hemolysin in some strains. Infect. Immun. 1990, 58, 1281–1289. [Google Scholar] [PubMed]
- Suttorp, N.; Floer, B.; Schnittler, H.; Seeger, W.; Bhakdi, S. Effects of Escherichia coli hemolysin on endothelial cell function. Infect. Immun. 1990, 58, 3796–3801. [Google Scholar] [PubMed]
- Grimminger, F.; Walmrath, D.; Birkemeyer, R.G.; Bhakdi, S.; Seeger, W. Leukotriene and hydroxyeicosatetraenoic acid generation elicited by low doses of Escherichia coli hemolysin in rabbit lungs. Infect. Immun. 1990, 58, 2659–2663. [Google Scholar]
- Bhakdi, S.; Muhly, M.; Korom, S.; Schmidt, G. Effects of Escherichia coli hemolysin on human monocytes. Cytocidal action and stimulation of interleukin 1 release. J. Clin. Investig. 1990, 85, 1746–1753. [Google Scholar] [CrossRef]
- Shewen, P.E.; Wilkie, B.N. Cytotoxin of Pasteurella haemolytica acting on bovine leukocytes. Infect. Immun. 1982, 35, 91–94. [Google Scholar]
- Taichman, N.S.; Shenker, B.J.; Tsai, C.; Glickman, L.T.; Baehni, P.C.; Stevens, R.; Hammond, B.F. Cytopathic effects of Actinobacillus actinomycetemcomitans on monkey blood leukocytes. J. Periodontal Res. 1984, 19, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Strathdee, C.A.; Lo, R.Y.C. Cloning, nucleotide sequence, and characterization of genes encoding the secretion function of the Pasteurella haemolytica leukotoxin determinant. J. Bacteriol. 1989, 171, 916–928. [Google Scholar] [CrossRef] [PubMed]
- Guermonprez, P.; Khelef, N.; Blouin, E.; Rieu, P.; Ricciardi-Castagnoli, P.; Guiso, N.; Ladant, D.; Leclerc, C. The adenylate cyclase toxin of Bordetella pertussis binds to target cells via the αMβ2 integrin (CD11b/CD18). J. Exp. Med. 2001, 193, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Lally, E.T.; Kieba, I.R.; Sato, A.; Green, C.L.; Rosenbloom, J.; Korostoff, J.; Wang, J.F.; Shenker, B.J.; Ortlepp, S.; Robinson, M.K.; et al. RTX toxins recognize a β2 integrin on the surface of human target cells. J. Biol. Chem. 1997, 272, 30463–30469. [Google Scholar] [CrossRef] [PubMed]
- Masin, J.; Osicka, R.; Bumba, L.; Sebo, P. Bordetella adenylate cyclase toxin: A unique combination of a pore-forming moiety with a cell-invading adenylate cyclase enzyme. Pathog. Dis. 2015, 73. [Google Scholar] [CrossRef] [PubMed]
- Bakás, L.; Ostolaza, H.; Vaz, W.L.C.; Goñi, F.M. Reversible adsorption and nonreversible insertion of Escherichia coli a-hemolysin into lipid bilayers. Biophys. J. 1996, 71, 1869–1876. [Google Scholar] [CrossRef]
- Ostolaza, H.; Bakás, L.; Goñi, F.M. Balance of electrostatic and hydrophobic interactions in the lysis of model membranes by E. coli a-haemolysin. J. Membr. Biol. 1997, 158, 137–145. [Google Scholar] [CrossRef]
- Iwase, M.; Lally, E.T.; Berthold, P.; Korchak, H.M.; Taichman, N.S. Effects of cations and osmotic protectants on cytolytic activity of Actinobacillus actinomycetemcomitans leukotoxin. Infect. Immun. 1990, 58, 1782–1788. [Google Scholar]
- Taichman, N.S.; Iwase, M.; Lally, E.T.; Shattil, S.J.; Cunningham, M.E.; Korchak, H.M. Early changes in cytosolic calcium and membrane potential induced by Actinobacillus actinomycetemcomitans leukotoxin in susceptible and resistant target cells. J. Immunol. 1991, 147, 3587–3594. [Google Scholar]
- Sánchez-Magraner, L.; Viguera, A.R.; García-Pacios, M.; Garcillán, M.P.; Arrondo, J.-R.; De La Cruz, F.; Goñi, F.M.; Ostolaza, H. The calcium-binding C-terminal domain of Escherichia coli a-hemolysin is a major determinant in the surface-active properties of the protein. J. Biol. Chem. 2007, 282, 11827–11835. [Google Scholar] [CrossRef]
- Basler, M.; Knapp, O.; Masin, J.; Fiser, R.; Maier, E.; Benz, R.; Sebo, P.; Osicka, R. Segments crucial for membrane translocation and pore-forming activity of Bordetella adenylate cyclase toxin. J. Biol. Chem. 2007, 282, 12419–12429. [Google Scholar] [CrossRef] [PubMed]
- Bellalou, J.; Sakamoto, H.; Ladant, D.; Geoffroy, C.; Ullmann, A. Deletions affecting hemolytic and toxin activities of Bordetella pertussis adenylate cyclase. Infect. Immun. 1990, 58, 3242–3247. [Google Scholar] [PubMed]
- Ludwig, A.; Schmid, A.; Benz, R.; Goebel, W. Mutations affecting pore formation by haemolysin from Escherichia coli. Mol. Gen. Genet. 1991, 226, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, A.; Vogel, M.; Goebel, W. Mutations affecting activity and transport of haemolysin in Escherichia coli. Mol. Gen. Genet. 1987, 206, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Schindel, C.; Zitzer, A.; Schulte, B.; Gerhards, A.; Stanley, P.; Hughes, C.; Koronakis, V.; Bhakdi, S.; Palmer, M. Interaction of Escherichia coli hemolysin with biological membranes: A study using cysteine scanning mutagenesis. Eur. J. Biochem. 2001, 268, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Hyland, C.; Vuillard, L.; Hughes, C.; Koronakis, V. Membrane interaction of Escherichia coli hemolysin: Flotation and insertion-dependent labeling by phospholipid vesicles. J. Bacteriol. 2001, 183, 5364–5370. [Google Scholar] [CrossRef] [PubMed]
- Benz, R.; Maier, E.; Bauer, S.; Ludwig, A. The deletion of several amino acid stretches of Escherichia coli alpha-hemolysin (HlyA) suggests that the channel-forming domain contains beta-strands. PLoS ONE 2014, 9, e112248. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.C.; Boesze-Battaglia, K.; Balashova, N.V.; Gómez, N.M.; Speicher, K.; Tang, H.; Duszyk, M.E.; Lally, E.T. Membrane localization of the Repeats-in-Toxin (RTX) Leukotoxin (LtxA) produced by Aggregatibacter actinomycetemcomitans. PLoS ONE 2018, 13, e0205871. [Google Scholar] [CrossRef] [PubMed]
- Bhakdi, S.; Mackman, N.; Nicaud, J.; Holland, I.B. Escherichia coli hemolysin may damage target cell membranes by generating transmembrane pores. Infect. Immun. 1986, 52, 63–69. [Google Scholar] [PubMed]
- Menestrina, G.; Mackman, N.; Holland, I.B.; Bhakdi, S. Escherichia coli haemolysin forms voltage-dependent ion channels in lipid membranes. Biochim. Biophys. Acta Biomembr. 1987, 905, 109–117. [Google Scholar] [CrossRef]
- Benz, R.; Schmid, A.; Wagner, W.; Goebel, W. Pore formation by the Escherichia coli hemolysin: Evidence for an association-dissociation equilibrium of the pore-forming aggregates. Infect. Immun. 1989, 57, 887–895. [Google Scholar] [PubMed]
- Menestrina, G.; Dalla Serra, M.; Pederzolli, C.; Bregante, M.; Gambale, F. Bacterial hemolysins and leukotoxins affect target cells by forming large exogenous pores into their plasma membrane. Escherichia coli hemolysin a as a case example. Biosci. Rep. 1995, 15, 543–551. [Google Scholar]
- Lalonde, G.; McDonald, T.V.; Gardner, P.; O’Hanley, P.D. Identification of a hemolysin from Actinobacillus pleuropneumoniae and characterization of its channel properties in planar phospholipid bilayers. J. Biol. Chem. 1989, 264, 13559–13564. [Google Scholar] [PubMed]
- Clinkenbeard, K.D.; Mosier, D.A.; Confer, A.W. Transmembrane pore size and role of cell swelling in cytotoxicity caused by Pasteurella haemolytica leukotoxin. Infect. Immun. 1989, 57, 420–425. [Google Scholar] [PubMed]
- Ehrmann, I.E.; Gray, M.C.; Gordon, V.M.; Gray, L.S.; Hewlett, E.L. Hemolytic activity of adenylate cyclase toxin from Bordetella pertussis. FEBS Lett. 1991, 278, 79–83. [Google Scholar] [PubMed]
- Szabo, G.; Gray, M.C.; Hewlett, E.L. Adenylate cyclase toxin from Bordetella pertussis produces ion conductance across artificial lipid bilayers in a calcium- and polarity-dependent manner. J. Biol. Chem. 1994, 269, 22496–22499. [Google Scholar] [PubMed]
- Benz, R.; Hardie, K.R.; Hughes, C. Pore formation in artificial membranes by the secreted hemolysins of Proteus vulgaris and Morganella morganii. Eur. J. Biochem. 1994, 220, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.; Maier, E.; Karch, H.; Benz, R. Pore-forming properties of the plasmid-encoded hemolysin of enterohemorrhagic Escherichia coli O157:H7. Eur. J. Biochem. 1996, 241, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Maier, E.; Reinhard, N.; Benz, R.; Frey, J. Channel-forming activity and channel size of the RTX toxins ApxI, ApxII, and ApxIII of Actinobacillus pleuropneumoniae. Infect. Immun. 1996, 64, 4415–4423. [Google Scholar] [PubMed]
- Lear, J.D.; Furblur, U.G.; Lally, E.T.; Tanaka, J.C. Actinobacillus actinomycetemcomitans leukotoxin forms large conductance, voltage-gate ion channels when incorporated into planar lipid bilayers. Biochim. Biophys. Acta Biomembr. 1995, 1238, 34–41. [Google Scholar] [CrossRef]
- Karakelian, D.; Lear, J.D.; Lally, E.T.; Tanaka, J.C. Characterization of Actinobacillus actinomycetemcomitans leukotoxin pore formation in HL60 cells. Biochim. Biophys. Acta Mol. Basis Dis. 1998, 1406, 175–187. [Google Scholar] [CrossRef]
- Bárcena-Uribarri, I.; Benz, R.; Winterhalter, M.; Zakharian, E.; Balashova, N. Pore forming activity of the potent RTX-toxin produced by pediatric pathogen Kingella kingae: Characterization and comparison to other RTX-family members. Biochim. Biophys. Acta Biomembr. 2015, 1848, 1536–1544. [Google Scholar] [CrossRef] [PubMed]
- Benz, R.; Maier, E.; Ladant, D.; Ullmann, A.; Sebo, P. Adenylate cyclase toxin (CyaA) of Bordetella pertussis. Evidence for the formation of small ion-permeable channels and comparison with HlyA of Escherichia coli. J. Biol. Chem. 1994, 269, 27231–27239. [Google Scholar] [PubMed]
- Bejerano, M.; Nisan, I.; Ludwig, A.; Goebel, W.; Hanski, E. Characterization of the C-terminal domain essential for toxic activity of adenylate cyclase toxin. Mol. Microbiol. 1999, 31, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, A.; Benz, R.; Goebel, W. Oligomerization of Escherichia coli haemolysin (HlyA) is involved in pore formation. Mol. Gen. Genet. 1993, 241, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, M.; Ullmann, A.; Šebo, P. Identification by in vitro complementation of regions required for cell-invasive activity of Bordetella pertussis adenylate cyclase toxin. Mol. Microbiol. 1995, 17, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Eberspacher, B.; Hugo, F.; Bhakdi, S. Quantitative study of the binding and hemolytic efficiency of Escherichia coli hemolysin. Infect. Immun. 1989, 57, 983–988. [Google Scholar]
- Vojtova-Vodolanova, J.; Basler, M.; Osicka, R.; Knapp, O.; Maier, E.; Cerny, J.; Benada, O.; Benz, R.; Sebo, P. Oligomerization is involved in pore formation by Bordetella adenylate cyclase toxin. FASEB J. 2009, 23, 2831–2843. [Google Scholar] [CrossRef] [PubMed]
- Bumba, L.; Masin, J.; Macek, P.; Wald, T.; Motlova, L.; Bibova, I.; Klimova, N.; Bednarova, L.; Veverka, V.; Kachala, M.; et al. Calcium-Driven Folding of RTX Domain ß-Rolls Ratchets Translocation of RTX Proteins through Type I Secretion Ducts. Mol. Cell 2016, 62, 47–62. [Google Scholar] [CrossRef]
- Chenal, A.; Iñaki Guijarro, J.; Raynal, B.; Delepierre, M.; Ladant, D. RTX calcium binding motifs are intrinsically disordered in the absence of calcium implication for protein secretion. J. Biol. Chem. 2009, 284, 1781–1789. [Google Scholar] [CrossRef]
- Ostolaza, H.; Soloaga, A.; Goni, F.M. The binding of divalent cations to Escherichia coli alpha-haemolysin. Eur. J. Biochem. 1995, 228, 39–44. [Google Scholar] [CrossRef] [PubMed]
- González-Bullón, D.; Uribe, K.B.; Largo, E.; Guembelzu, G.; García-Arribas, A.B.; Martín, C.; Ostolaza, H. Membrane Permeabilization by Bordetella Adenylate Cyclase Toxin Involves Pores of Tunable Size. Biomolecules 2019, 9, 183. [Google Scholar] [CrossRef] [PubMed]
- Döbereiner, A. The effects of calcium and other polyvalent cations on channel formation by Escherichia coli a-hemolysin in red blood cells and lipid bilayer membranes. Eur. J. Biochem. 1996, 240, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Knapp, O.; Maier, E.; Polleichtner, G.; Mašín, J.; Šebo, P.; Benz, R. Channel formation in model membranes by the adenylate cyclase toxin of Bordetella pertussis: Effect of calcium. Biochemistry 2003, 42, 8077–8084. [Google Scholar] [CrossRef]
- Martín, C.; Requero, M.; Masin, J.; Konopasek, I.; Goñi, F.M.; Sebo, P.; Ostolaza, H. Membrane restructuring by Bordetella pertussis adenylate cyclase toxin, a member of the RTX toxin family. J. Bacteriol. 2004, 186, 3760–3765. [Google Scholar]
- Moayeri, M.; Welch, R.A. Effects of temperature, time, and toxin concentration on lesion formation by the Escherichia coli hemolysin. Infect. Immun. 1994, 62, 4124–4134. [Google Scholar]
- Goñi, F.M.; Ostolaza, H. E. coli a-hemolysin: A membrane-active protein toxin. Braz. J. Med. Biol. Res. 1998, 31, 1019–1034. [Google Scholar] [CrossRef]
- Bakás, L.; Chanturiya, A.; Herlax, V.; Zimmerberg, J. Paradoxical lipid dependence of pores formed by the Escherichia coli a-hemolysin in planar phospholipid bilayer membranes. Biophys. J. 2006, 91, 3748–3755. [Google Scholar] [CrossRef]
- Brown, A.C.; Boesze-Battaglia, K.; Du, Y.; Stefano, F.P.; Kieba, I.R.; Epand, R.F.; Kakalis, L.; Yeagle, P.L.; Epand, R.M.; Lally, E.T. Aggregatibacter actinomycetemcomitans leukotoxin cytotoxicity occurs through bilayer destabilization. Cell. Microbiol. 2012, 14, 869–881. [Google Scholar] [CrossRef]
- Carbonetti, N.H. Pertussis toxin and adenylate cyclase toxin: Key virulence factors of Bordetella pertussis and cell biology tools. Future Microbiol. 2010, 5, 455–469. [Google Scholar] [CrossRef]
- Mattoo, S.; Cherry, J.D. Molecular pathogenesis, epidemiology, and clinical manifestations of respiratory infections due to Bordetella pertussis and other Bordetella subspecies. Clin. Microbiol. Rev. 2005, 18, 326–382. [Google Scholar] [CrossRef] [PubMed]
- Hewlett, E.L.; Urban, M.A.; Manclark, C.R.; Wolff, J. Extracytoplasmic adenylate cyclase of Bordetella pertussis. Proc. Natl. Acad. Sci. USA 1976, 73, 1926–1930. [Google Scholar] [CrossRef] [PubMed]
- Wolff, J.; Cook, G.H.; Goldhammer, A.R.; Berkowitz, S.A. Calmodulin activates prokaryotic adenylate cyclase. Proc. Natl. Acad. Sci. USA 1980, 77, 3841–3844. [Google Scholar] [CrossRef] [PubMed]
- Confer, D.L.; Eaton, J.W. Phagocyte impotence caused by an invasive bacterial adenylate cyclase. Science 1982, 217, 948–950. [Google Scholar] [CrossRef] [PubMed]
- Ladant, D.; Ullmann, A. Bordetella pertussis adenylate cyclase: A toxin with multiple talents. Trends Microbiol. 1999, 7, 172–176. [Google Scholar] [CrossRef]
- Hackett, M.; Guo, L.; Shabanowitz, J.; Hunt, D.F.; Hewlett, E.L. Internal lysine palmitoylation in adenylate cyclase toxin from Bordetella pertussis. Science 1994, 266, 433–435. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, S.A.; Goldhammer, A.R.; Hewlett, E.L.; Wolff, J. Activation of prokaryotic adenylate cyclase by calmodulin. Ann. N. Y. Acad. Sci. 1980, 356, 360. [Google Scholar] [CrossRef]
- Karst, J.C.; Barker, R.; Devi, U.; Swann, M.J.; Davi, M.; Roser, S.J.; Ladant, D.; Chenal, A. Identification of a region that assists membrane insertion and translocation of the catalytic domain of Bordetella pertussis CyaA toxin. J. Biol. Chem. 2012, 287, 9200–9212. [Google Scholar] [CrossRef]
- Rose, T.; Sebo, P.; Bellalou, J.; Ladant, D. Interaction of calcium with Bordetella pertussis adenylate cyclase toxin. Characterization of multiple calcium-binding sites and calcium-induced conformational changes. J. Biol. Chem. 1995, 270, 26370–26376. [Google Scholar] [CrossRef]
- Hewlett, E.L.; Gray, L.; Allietta, M.; Ehrmann, I.; Gordon, V.M.; Gray, M.C. Adenylate cyclase toxin from Bordetella pertussis. Conformational change associated with toxin activity. J. Biol. Chem. 1991, 266, 17503–17508. [Google Scholar]
- Hanski, E.; Farfel, Z. Bordetella pertussis invasive adenylate cyclase. Partial resolution and properties of its cellular penetration. J. Biol. Chem. 1985, 260, 5526–5532. [Google Scholar] [PubMed]
- Basler, M.; Masin, J.; Osicka, R.; Sebo, P. Pore-forming and enzymatic activities of Bordetella pertussis adenylate cyclase toxin synergize in promoting lysis of monocytes. Infect. Immun. 2006, 74, 2207–2214. [Google Scholar] [CrossRef] [PubMed]
- Hewlett, E.L.; Donato, G.M.; Gray, M.C. Macrophage cytotoxicity produced by adenylate cyclase toxin from Bordetella pertussis: More than just making cyclic AMP! Mol. Microbiol. 2006, 59, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Osicková, A.; Osicka, R.; Maier, E.; Benz, R.; Šebo, P. An amphipathic a-helix including glutamates 509 and 516 is crucial for membrane translocation of adenylate cyclase toxin and modulates formation and cation selectivity of its membrane channels. J. Biol. Chem. 1999, 274, 37644–37650. [Google Scholar] [PubMed]
- Powthongchin, B.; Angsuthanasombat, C. Effects on haemolytic activity of single proline substitutions in the Bordetella pertussis CyaA pore-forming fragment. Arch. Microbiol. 2009, 191, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Roderova, J.; Osickova, A.; Sukova, A.; Mikusova, G.; Fiser, R.; Sebo, P.; Osicka, R.; Masin, J. Residues 529 to 549 participate in membrane penetration and pore-forming activity of the Bordetella adenylate cyclase toxin. Sci. Rep. 2019, 9, 5758. [Google Scholar] [CrossRef] [PubMed]
- Juntapremjit, S.; Thamwiriyasati, N.; Kurehong, C.; Prangkio, P.; Shank, L.; Powthongchin, B.; Angsuthanasombat, C. Functional importance of the Gly cluster in transmembrane helix 2 of the Bordetella pertussis CyaA-hemolysin: Implications for toxin oligomerization and pore formation. Toxicon 2015, 106, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.C.; Lee, S.; Gray, L.S.; Zaretzky, F.R.; Otero, A.S.; Szabo, G.; Hewlett, E.L. Translocation-specific conformation of adenylate cyclase toxin from Bordetella pertussis inhibits toxin-mediated hemolysis. J. Bacteriol. 2001, 183, 5904–5910. [Google Scholar] [CrossRef] [PubMed]
- Osickova, A.; Masin, J.; Fayolle, C.; Krusek, J.; Basler, M.; Pospisilova, E.; Leclerc, C.; Osicka, R.; Sebo, P. Adenylate cyclase toxin translocates across target cell membrane without forming a pore. Mol. Microbiol. 2010, 75, 1550–1562. [Google Scholar] [CrossRef]
- Masin, J.; Osickova, A.; Sukova, A.; Fiser, R.; Halada, P.; Bumba, L.; Linhartova, I.; Osicka, R.; Sebo, P. Negatively charged residues of the segment linking the enzyme and cytolysin moieties restrict the membrane-permeabilizing capacity of adenylate cyclase toxin. Sci. Rep. 2016, 6, 29137. [Google Scholar] [CrossRef]
- Gray, M.; Szabo, G.; Otero, A.S.; Gray, L.; Hewlett, E. Distinct mechanisms for K+ efflux, intoxication, and hemolysis by Bordetella pertussis AC toxin. J. Biol. Chem. 1998, 273, 18260–18267. [Google Scholar] [CrossRef] [PubMed]
- Moldenhauer, H.; Díaz-Franulic, I.; González-Nilo, F.; Naranjo, D. Effective pore size and radius of capture for K+ ions in K-channels. Sci. Rep. 2016, 6, 19893. [Google Scholar] [CrossRef] [PubMed]
- Fuertes, G.; Giménez, D.; Esteban-Martín, S.; Sánchez-Muñoz, O.L.; Salgado, J. A lipocentric view of peptide-induced pores. Eur. Biophys. J. 2011, 40, 399–415. [Google Scholar] [CrossRef] [PubMed]
- Terrones, O.; Antonsson, B.; Yamaguchi, H.; Wang, H.; Liu, J.; Lee, R.M.; Herrmann, A.; Basañez, G. Lipidic pore formation by the concerted action of proapoptotic BAX and tBID. J. Biol. Chem. 2004, 279, 30081–30091. [Google Scholar] [CrossRef] [PubMed]
- Bleicken, S.; Landeta, O.; Landajuela, A.; Basañez, G.; García-Sáez, A.J. Proapoptotic Bax and Bak proteins form stable protein-permeable pores of tunable size. J. Biol. Chem. 2013, 288, 33241–33252. [Google Scholar] [CrossRef] [PubMed]
- Salvador-Gallego, R.; Mund, M.; Cosentino, K.; Schneider, J.; Unsay, J.; Schraermeyer, U.; Engelhardt, J.; Ries, J.; García-Sáez, A.J. Bax assembly into rings and arcs in apoptotic mitochondria is linked to membrane pores. EMBO J. 2016, 35, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Cannella, S.E.; Ntsogo Enguéné, V.Y.; Davi, M.; Malosse, C.; Sotomayor Pérez, A.C.; Chamot-Rooke, J.; Vachette, P.; Durand, D.; Ladant, D.; Chenal, A. Stability, structural and functional properties of a monomeric, calcium-loaded adenylate cyclase toxin, CyaA, from Bordetella pertussis. Sci. Rep. 2017, 7, 42065. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ostolaza, H.; González-Bullón, D.; Uribe, K.B.; Martín, C.; Amuategi, J.; Fernandez-Martínez, X. Membrane Permeabilization by Pore-Forming RTX Toxins: What Kind of Lesions Do These Toxins Form? Toxins 2019, 11, 354. https://doi.org/10.3390/toxins11060354
Ostolaza H, González-Bullón D, Uribe KB, Martín C, Amuategi J, Fernandez-Martínez X. Membrane Permeabilization by Pore-Forming RTX Toxins: What Kind of Lesions Do These Toxins Form? Toxins. 2019; 11(6):354. https://doi.org/10.3390/toxins11060354
Chicago/Turabian StyleOstolaza, Helena, David González-Bullón, Kepa B. Uribe, Cesar Martín, Jone Amuategi, and Xabier Fernandez-Martínez. 2019. "Membrane Permeabilization by Pore-Forming RTX Toxins: What Kind of Lesions Do These Toxins Form?" Toxins 11, no. 6: 354. https://doi.org/10.3390/toxins11060354
APA StyleOstolaza, H., González-Bullón, D., Uribe, K. B., Martín, C., Amuategi, J., & Fernandez-Martínez, X. (2019). Membrane Permeabilization by Pore-Forming RTX Toxins: What Kind of Lesions Do These Toxins Form? Toxins, 11(6), 354. https://doi.org/10.3390/toxins11060354