Recombinant Antibodies against Mycolactone

 , ,
, ,
Abstract
1. Introduction
2. Results
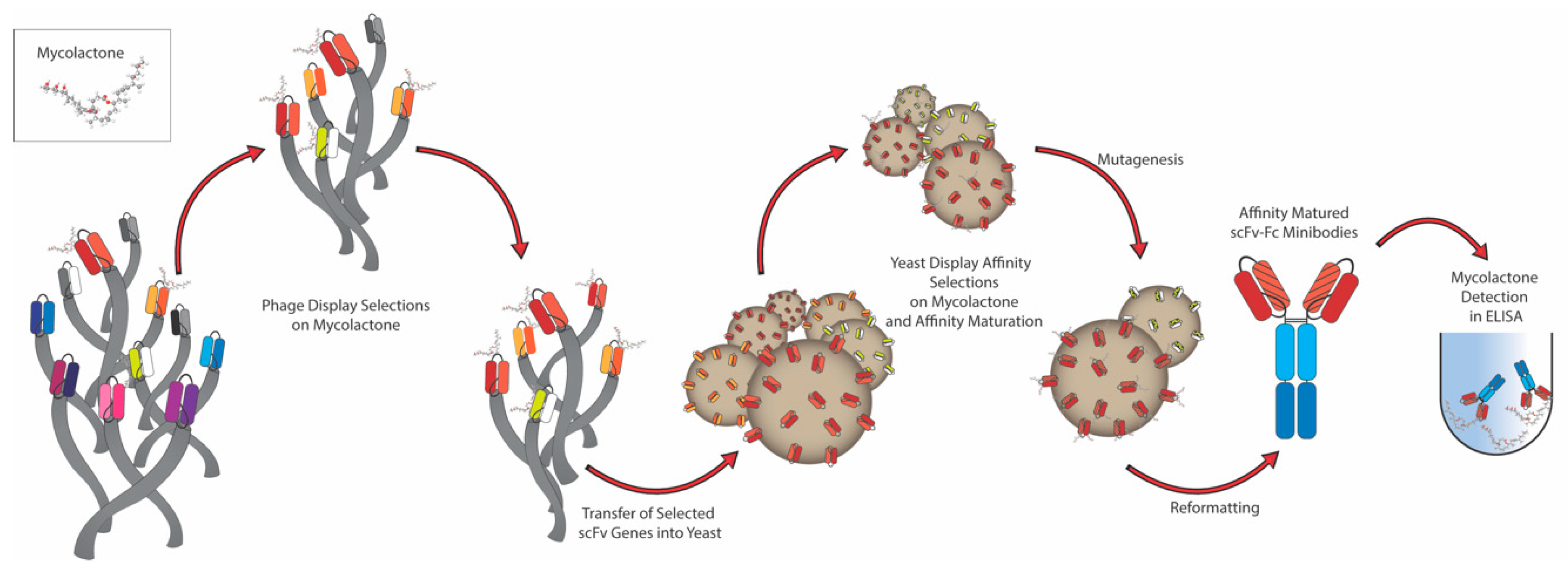
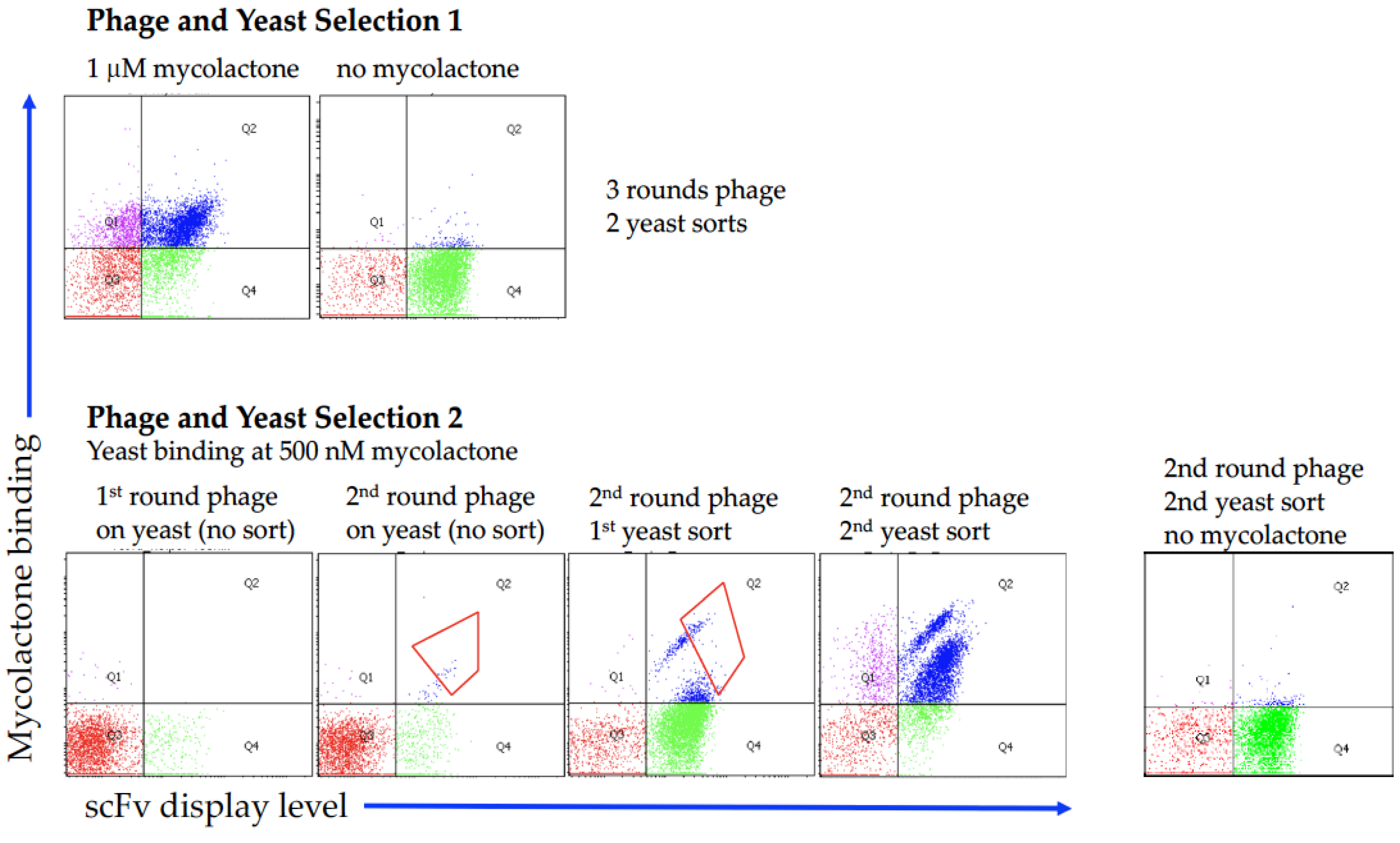
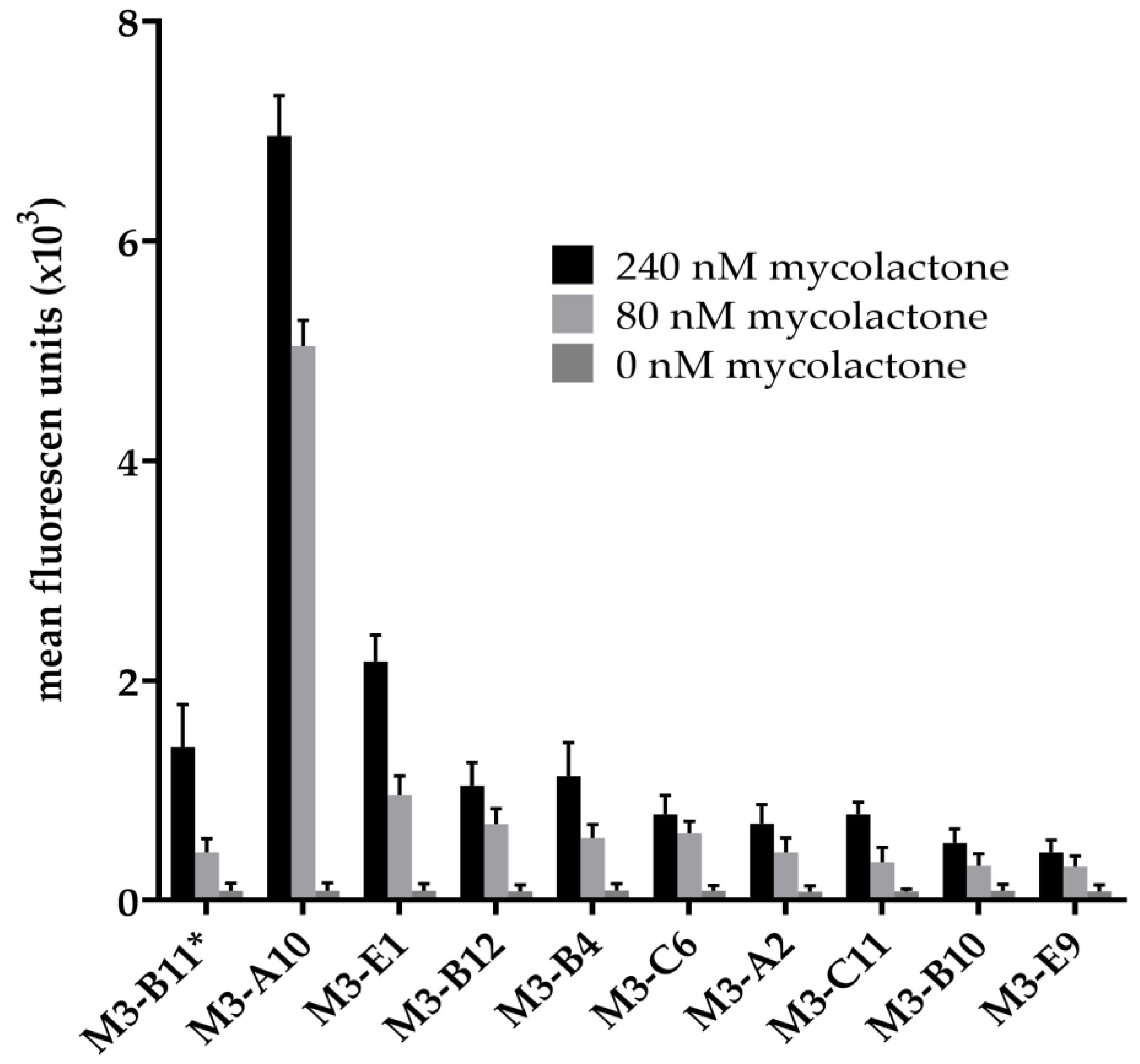
2.1. Antibody Selection
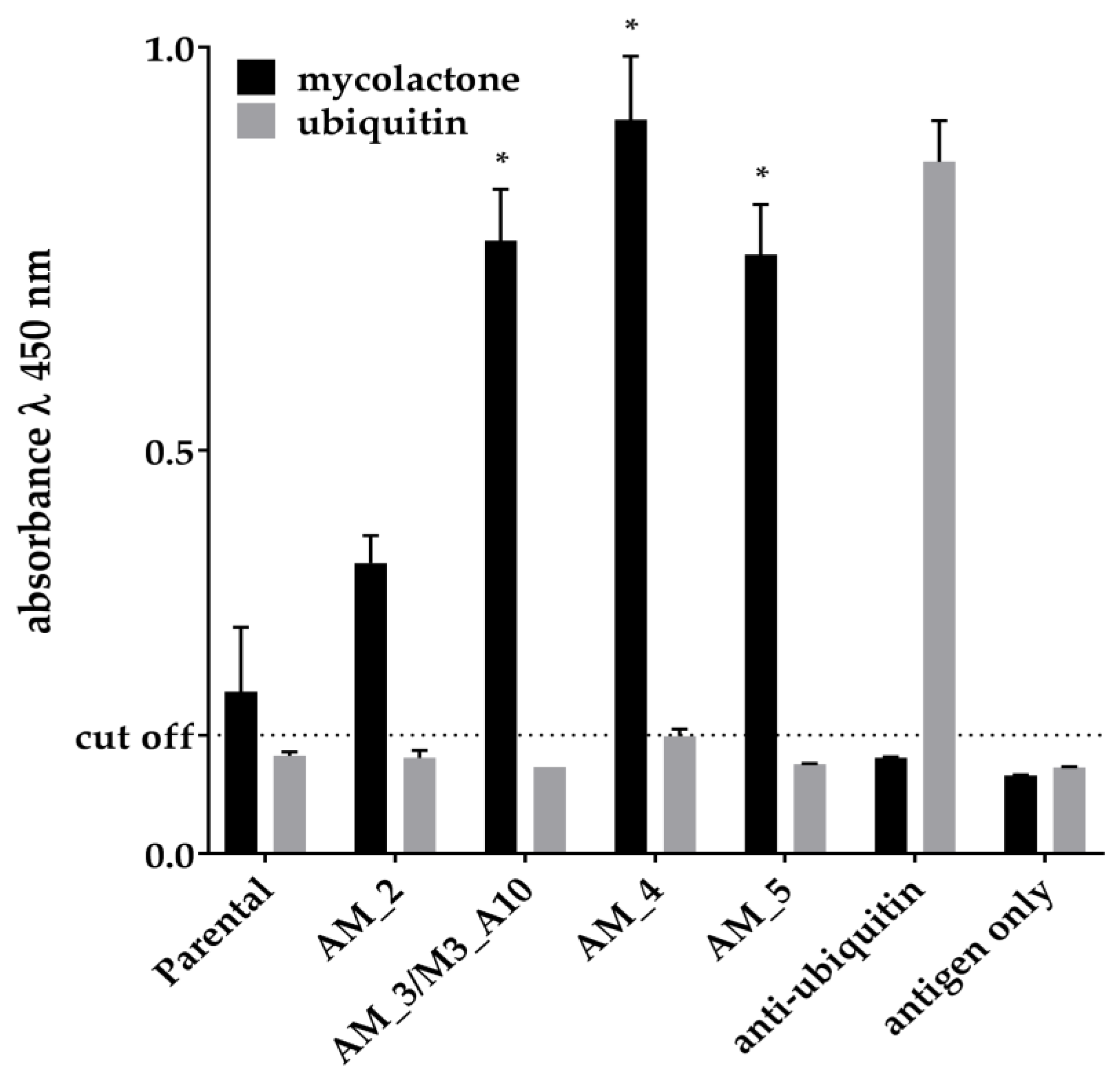
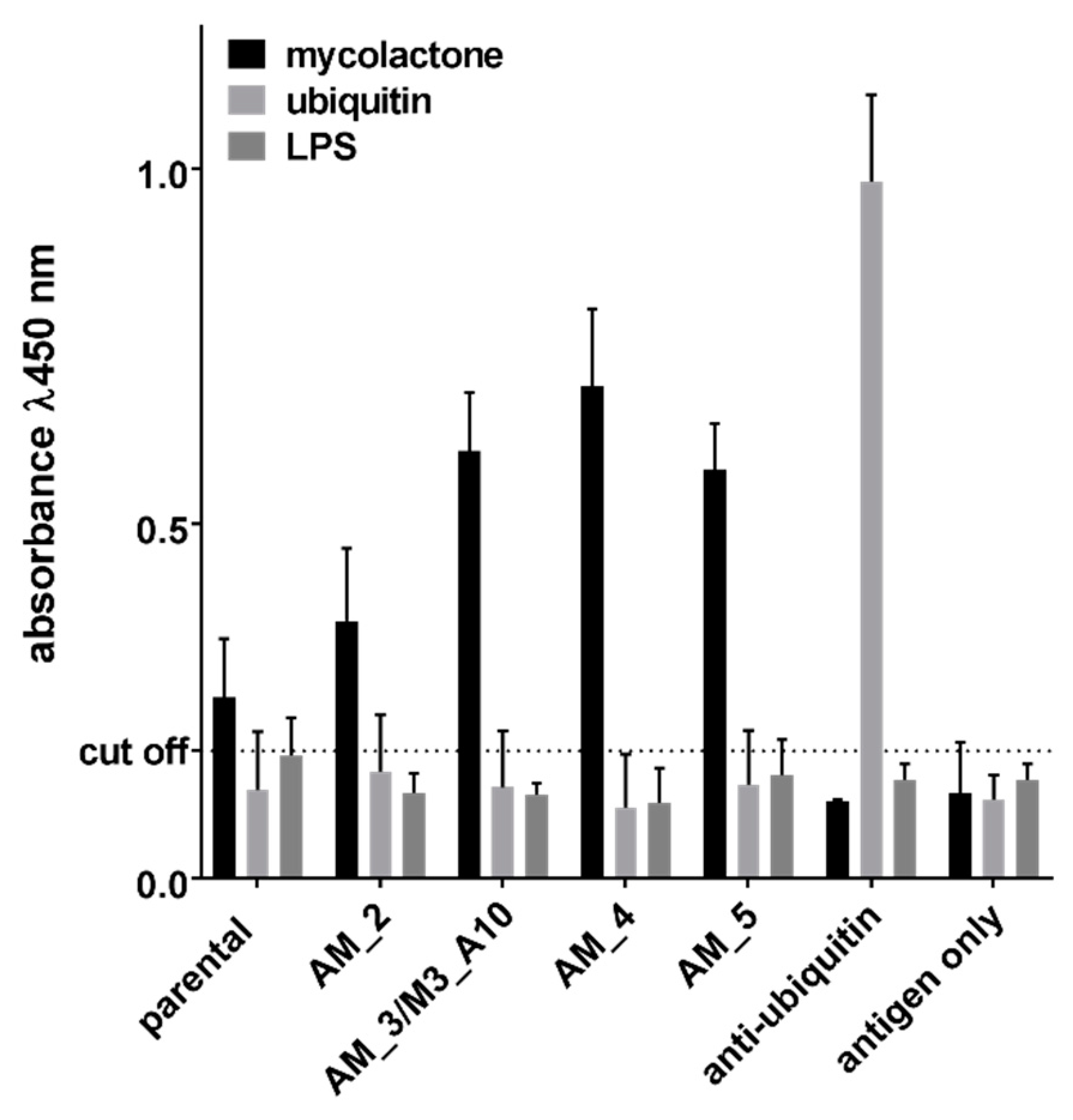
2.2. Affinity Maturation
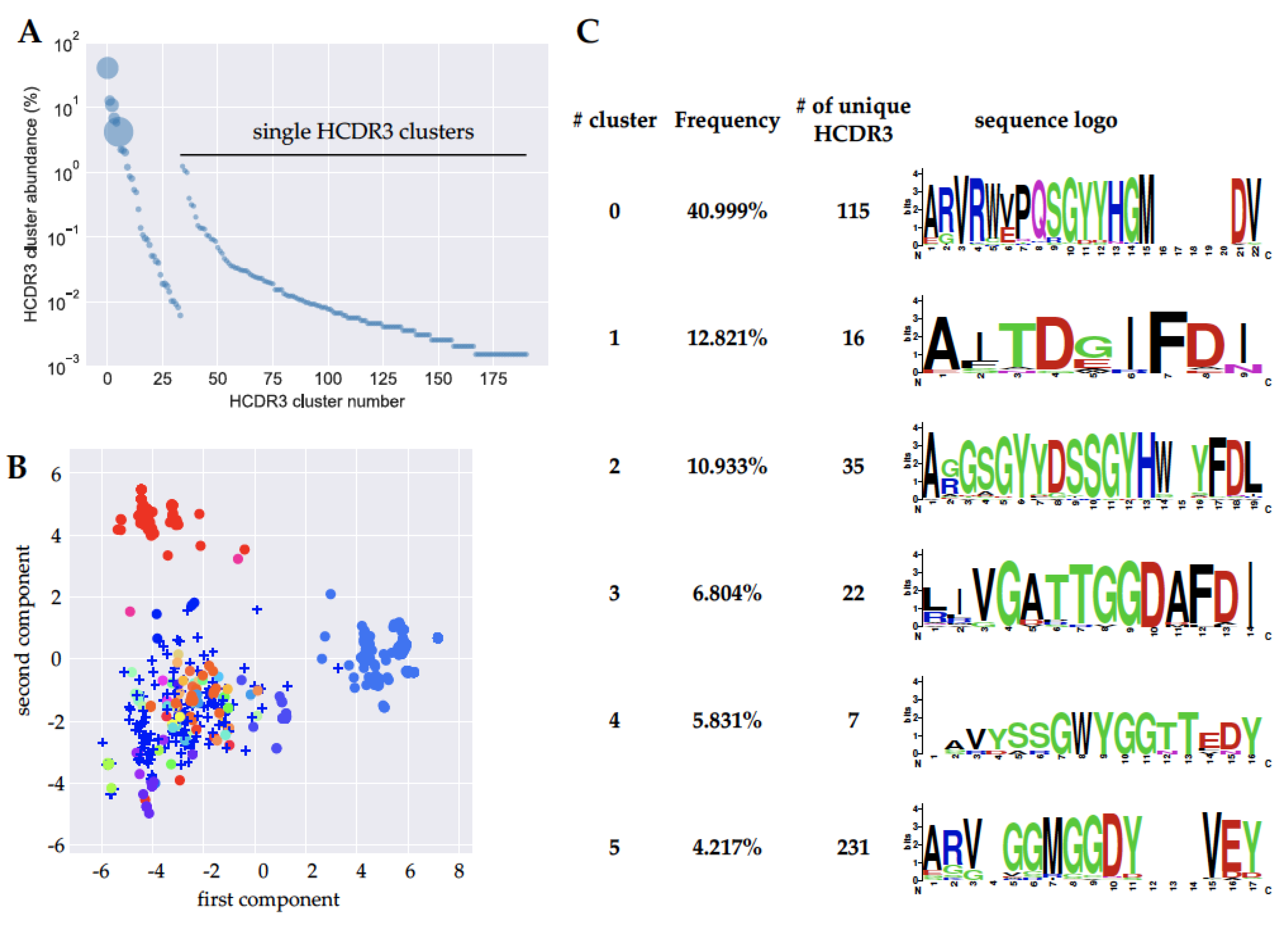
2.3. Next Generation Sequencing (NGS) Analysis
3. Discussion
4. Materials and Methods
4.1. Biotinylated Mycolactone
4.2. Phage Display Selections
4.3. Yeast Display and Sorting of scFvs
4.4. Affinity Maturation
4.5. Production of Monoclonal Antibodies (Yeast)
4.6. ELISA Screening of Antibodies
4.7. Next-Generation Sequencing
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Connor, D.H.; Lunn, H.F. Mycobacterium ulcerans infection (with comments on pathogenesis). Int. J. Lepr. 1965, 33, 698–709. [Google Scholar]
- Connor, D.H.; Lunn, H.F. Buruli Ulceration: A clincopathologic study of 38 Ugandans with Mycobacterium ulcerans ulceration. Arch. Pathol. 1966, 81, 183–199. [Google Scholar]
- Dobos, K.M.; Small, P.L.; Deslauriers, M.; Quinn, F.D.; King, C.H. Mycobacterium ulcerans cytotoxicity in an adipose cell model. Infect. Immun. 2001, 69, 7182–7186. [Google Scholar] [CrossRef]
- George, K.M.; Pascopella, L.; Welty, D.M.; Small, P.L. A Mycobacterium ulcerans toxin, mycolactone, causes apoptosis in guinea pig ulcers and tissue culture cells. Infect. Immun. 2000, 68, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Sarfo, F.S.; Converse, P.J.; Almeida, D.V.; Zhang, J.; Robinson, C.; Wansbrough-Jones, M.; Grosset, J.H. Microbiological, Histological, Immunological, and Toxin Response to Antibiotic Treatment in the Mouse Model of Mycobacterium ulcerans Disease. PLoS Negl. Trop. Dis. 2013, 7, e2101. [Google Scholar] [CrossRef] [PubMed]
- Schutte, D.; Umboock, A.; Pluschke, G. Phagocytosis of Mycobacterium ulcerans in the course of rifampicin and streptomycin chemotherapy in Buruli ulcer lesions. Br. J. Dermatol. 2009, 160, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Adusumilli, S.; Mve-Obiang, A.; Sparer, T.; Meyers, W.; Hayman, J.; Small, P.L.C. Mycobacterium ulcerans toxic macrolide, mycolactone modulates the host immune response and cellular location of M. ulcerans in vitro and in vivo. Cell. Microbiol. 2005, 7, 1295–1304. [Google Scholar] [CrossRef] [PubMed]
- Guenin-Macé, L.; Veyron-Churlet, R.; Thoulouze, M.-I.; Romet-Lemonne, G.; Hong, H.; Leadlay, P.F.; Danckaert, A.; Ruf, M.-T.; Mostowy, S.; Zurzolo, C.; et al. Mycolactone activation of Wiskott-Aldrich syndrome proteins underpins Buruli ulcer formation. J. Clin. Investig. 2013, 123, 1501–1512. [Google Scholar] [CrossRef]
- Hong, H.; Demangel, C.; Pidot, S.J.; Leadlay, P.F.; Stinear, T. Mycolactones: Immunosuppressive and cytotoxic polyketides produced by aquatic mycobacteria. Nat. Prod. Rep. 2008, 25, 447–454. [Google Scholar] [CrossRef]
- Phillips, R.; Sarfo, F.S.; Guenin-Mace, L.; Decalf, J.; Wansbrough-Jones, M.; Albert, M.L.; Demangel, C. Immunosuppressive signature of cutaneous Mycobacterium ulcerans infection in the peripheral blood of patients with buruli ulcer disease. J. Infect. Dis. 2009, 200, 1675–1684. [Google Scholar] [CrossRef]
- Simmonds, R.E.; Lali, F.V.; Smallie, T.; Small, P.L.; Foxwell, B.M. Mycolactone inhibits monocyte cytokine production by a posttranscriptional mechanism. J. Immunol. 2009, 182, 2194–2202. [Google Scholar] [CrossRef] [PubMed]
- Torrado, E.; Adusumilli, S.; Fraga, A.G.; Small, P.L.; Castro, A.G.; Pedrosa, J. Mycolactone-mediated inhibition of tumor necrosis factor production by macrophages infected with Mycobacterium ulcerans has implications for the control of infection. Infect. Immun. 2007, 75, 3979–3988. [Google Scholar] [CrossRef] [PubMed]
- Demangel, C.; High, S. Sec61 blockade by mycolactone: A central mechanism in Buruli ulcer disease. Biol. Cell 2018, 110, 237–248. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Joins Battle against a New Emerging Disease, Buruli Ulcer; Press Release WHO/88; World Health Organization: Geneva, Switzerland, 1997. [Google Scholar]
- World Health Organization. Buruli ulcer disease. Wkly. Epidemiol. Rec. 2003, 78, 163–168. [Google Scholar]
- Fyfe, J.A.; Lavender, C.J.; Handasyde, K.A.; Legione, A.R.; O’Brien, C.R.; Stinear, T.P.; Pidot, S.J.; Seemann, T.; Benbow, M.E.; Wallace, J.R.; et al. A Major Role for Mammals in the Ecology of Mycobacterium ulcerans. PLoS Negl. Trop. Dis. 2010, 4, e791. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.D.; Azuolas, J.; Lavender, C.J.; Wishart, E.; Stinear, T.P.; Hayman, J.A.; Brown, L.; Jenkin, G.A.; Fyfe, J.A. Mycobacterium ulcerans in mosquitoes captured during outbreak of Buruli ulcer, southeastern Australia. Emerg. Infect. Dis. 2007, 13, 1653–1660. [Google Scholar] [CrossRef]
- Coloma, J.N.; Navarrete-Franco, G.; Iribe, P.; Lopez-Cepeda, L.D. Ulcerative Cutaneous Mycobacteriosis Due to Mycobacterium ulcerans: Report of Two Mexican Cases. Int. J. Lepr. Other Mycobact. Dis. 2005, 73, 5–12. [Google Scholar] [CrossRef]
- Nakanaga, K.; Hoshino, Y.; Yotsu, R.R.; Makino, M.; Ishii, N. Nineteen Cases of Buruli Ulcer Diagnosed in Japan from 1980 to 2010. J. Clin. Microbiol. 2011, 49, 3829–3836. [Google Scholar] [CrossRef]
- Marion, E.; Eyangoh, S.; Yeramian, E.; Doannio, J.; Landier, J.; Aubry, J.; Fontanet, A.; Rogier, C.; Cassisa, V.; Cottin, J.; et al. Seasonal and regional dynamics of M. ulcerans transmission in environmental context: Deciphering the role of water bugs as hosts and vectors. PLoS Negl. Trop. Dis. 2010, 4, e731. [Google Scholar] [CrossRef]
- Maman, I.; Tchacondo, T.; Kere, A.B.; Piten, E.; Beissner, M.; Kobara, Y.; Kossi, K.; Badziklou, K.; Wiedemann, F.X.; Amekuse, K.; et al. Risk factors for Mycobacterium ulcerans infection (Buruli Ulcer) in Togo horizontal line a case-control study in Zio and Yoto districts of the maritime region. BMC Infect. Dis. 2018, 18, 48. [Google Scholar] [CrossRef]
- Converse, P.J.; Nuermberger, E.L.; Almeida, D.V.; Grosset, J.H. Treating Mycobacterium ulcerans disease (Buruli ulcer): From surgery to antibiotics, is the pill mightier than the knife? Future Microbiol. 2011, 6, 1185–1198. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Provisional Guidance on the Role of Specific Antibiotics in the Management of Mycobacterium ulcerans Disease (Buruli ulcer); World Health Organization: Geneva, Switzerland, 2004. [Google Scholar]
- Bentoucha, A.; Robert, J.; Dega, H.; Lounis, N.; Jarlier, V.; Grosset, J. Activities of new macrolides and fluoroquinolones against Mycobacterium ulcerans infection in mice. Antimicrob. Agents Chemother. 2001, 45, 3109–3112. [Google Scholar] [CrossRef] [PubMed]
- Dega, H.; Bentoucha, A.; Robert, J.; Jarlier, V.; Grosset, J. Bactericidal activity of rifampin-amikacin against Mycobacterium ulcerans in mice. Antimicrob. Agents Chemother. 2002, 46, 3193–3196. [Google Scholar] [CrossRef] [PubMed]
- Dega, H.; Robert, J.; Bonnafous, P.; Jarlier, V.; Grosset, J. Activities of several antimicrobials against Mycobacterium ulcerans infection in mice. Antimicrob. Agents Chemother. 2000, 44, 2367–2372. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chauty, A.; Ardant, M.F.; Adeye, A.; Euverte, H.; Guedenon, A.; Johnson, C.; Aubry, J.; Nuermberger, E.; Grosset, J. Promising clinical efficacy of streptomycin-rifampin combination for treatment of buruli ulcer (Mycobacterium ulcerans disease). Antimicrob. Agents Chemother. 2007, 51, 4029–4035. [Google Scholar] [CrossRef] [PubMed]
- Etuaful, S.; Carbonnelle, B.; Grosset, J.; Lucas, S.; Horsfield, C.; Phillips, R.; Evans, M.; Ofori-Adjei, D.; Klustse, E.; Owusu-Boateng, J.; et al. Efficacy of the combination rifampin-streptomycin in preventing growth of Mycobacterium ulcerans in early lesions of Buruli ulcer in humans. Antimicrob. Agents Chemother. 2005, 49, 3182–3186. [Google Scholar] [CrossRef] [PubMed]
- Herbinger, K.H.; Adjei, O.; Awua-Boateng, N.Y.; Nienhuis, W.A.; Kunaa, L.; Siegmund, V.; Nitschke, J.; Thompson, W.; Klutse, E.; Agbenorku, P.; et al. Comparative study of the sensitivity of different diagnostic methods for the laboratory diagnosis of Buruli ulcer disease. Clin. Infect. Dis. 2009, 48, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Wadagni, A.; Frimpong, M.; Phanzu, D.M.; Ablordey, A.; Kacou, E.; Gbedevi, M.; Marion, E.; Xing, Y.; Babu, V.S.; Phillips, R.O.; et al. Simple, Rapid Mycobacterium ulcerans Disease Diagnosis from Clinical Samples by Fluorescence of Mycolactone on Thin Layer Chromatography. PLoS Negl. Trop. Dis. 2015, 9, e0004247. [Google Scholar] [CrossRef]
- O’Brien, D.P.; Comte, E.; Serafini, M.; Ehounou, G.; Antierens, A.; Vuagnat, H.; Christinet, V.; Hamani, M.D.; du Cros, P. The urgent need for clinical, diagnostic, and operational research for management of Buruli ulcer in Africa. Lancet Infect. Dis. 2014, 14, 435–440. [Google Scholar] [CrossRef]
- Koczula, K.M.; Gallotta, A. Lateral flow assays. Essays Biochem. 2016, 60, 111–120. [Google Scholar] [CrossRef]
- Dangy, J.P.; Scherr, N.; Gersbach, P.; Hug, M.N.; Bieri, R.; Bomio, C.; Li, J.; Huber, S.; Altmann, K.H.; Pluschke, G. Antibody-Mediated Neutralization of the Exotoxin Mycolactone, the Main Virulence Factor Produced by Mycobacterium ulcerans. PLoS Negl. Trop. Dis. 2016, 10, e0004808. [Google Scholar] [CrossRef] [PubMed]
- Stanford, J.L.; Revill, W.D.; Gunthorpe, W.J.; Grange, J.M. The production and preliminary investigation of Burulin, a new skin test reagent for Mycobacterium ulcerans infection. J. Hyg. (Lond) 1975, 74, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Gooding, T.M.; Johnson, P.D.; Campbell, D.E.; Hayman, J.A.; Hartland, E.L.; Kemp, A.S.; Robins-Browne, R.M. Immune response to infection with Mycobacterium ulcerans. Infect. Immun. 2001, 69, 1704–1707. [Google Scholar] [CrossRef] [PubMed]
- Dobos, K.M.; Spotts, E.A.; Marston, B.J.; Horsburgh, C.R., Jr.; King, C.H. Serologic response to culture filtrate antigens of Mycobacterium ulcerans during Buruli ulcer disease. Emerg. Infect. Dis. 2000, 6, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, A.R.M.; Sidhu, S.; Dübel, S.; McCafferty, J. Beyond natural antibodies: The power of in vitro display technologies. Nat. Biotechnol. 2011, 29, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Huston, J.S.; Levinson, D.; Mudgett-Hunter, M.; Tai, M.S.; Novotny, J.; Margolies, M.N.; Ridge, R.J.; Bruccoleri, R.E.; Haber, E.; Crea, R.; et al. Protein engineering of antibody binding sites: Recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc. Natl. Acad. Sci. USA 1988, 85, 5879–5883. [Google Scholar] [CrossRef] [PubMed]
- McCafferty, J.; Griffiths, A.D.; Winter, G.; Chiswell, D.J. Phage antibodies: Filamentous phage displaying antibody variable domains. Nature 1990, 348, 552–554. [Google Scholar] [CrossRef] [PubMed]
- Marks, J.D.; Hoogenboom, H.R.; Bonnert, T.P.; McCafferty, J.; Griffiths, A.D.; Winter, G. By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J. Mol. Biol. 1991, 222, 581–597. [Google Scholar] [CrossRef]
- Boder, E.T.; Wittrup, K.D. Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 1997, 15, 553–557. [Google Scholar] [CrossRef]
- Feldhaus, M.J.; Siegel, R.W.; Opresko, L.K.; Coleman, J.R.; Feldhaus, J.M.; Yeung, Y.A.; Cochran, J.R.; Heinzelman, P.; Colby, D.; Swers, J.; et al. Flow-cytometric isolation of human antibodies from a nonimmune Saccharomyces cerevisiae surface display library. Nat. Biotechnol. 2003, 21, 163–170. [Google Scholar] [CrossRef]
- Bradbury, A.R.M.; Trinklein, N.D.; Thie, H.; Wilkinson, I.C.; Tandon, A.K.; Anderson, S.; Bladen, C.L.; Jones, B.; Aldred, S.F.; Bestagno, M.; et al. When monoclonal antibodies are not monospecific: Hybridomas frequently express additional functional variable regions. MAbs 2018. [Google Scholar] [CrossRef] [PubMed]
- Sblattero, D.; Bradbury, A. Exploiting recombination in single bacteria to make large phage antibody libraries. Nat. Biotechnol. 2000, 18, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Sblattero, D.; Lou, J.; Marzari, R.; Bradbury, A. In vivo recombination as a tool to generate molecular diversity in phage antibody libraries. J. Biotechnol. 2001, 74, 303–315. [Google Scholar] [CrossRef]
- Ferrara, F.; Naranjo, L.A.; Kumar, S.; Gaiotto, T.; Mukundan, H.; Swanson, B.; Bradbury, A.R. Using phage and yeast display to select hundreds of monoclonal antibodies: Application to antigen 85, a tuberculosis biomarker. PLoS ONE 2012, 7, e49535. [Google Scholar] [CrossRef] [PubMed]
- Kehoe, J.W.; Velappan, N.; Walbolt, M.; Rasmussen, J.; King, D.; Lou, J.; Knopp, K.; Pavlik, P.; Marks, J.D.; Bertozzi, C.R.; et al. Using phage display to select antibodies recognizing post-translational modifications independently of sequence context. Mol. Cell. Proteom. 2006, 5, 2350–2363. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.; Staquicini, F.I.; Ferrara, F.; Staquicini, D.I.; Sharma, G.; Tarleton, C.A.; Nguyen, H.; Naranjo, L.A.; Sidman, R.L.; Arap, W.; et al. Selection of phage-displayed accessible recombinant targeted antibodies (SPARTA): Methodology and applications. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Glanville, J.; D’Angelo, S.; Khan, T.A.; Reddy, S.T.; Naranjo, L.; Ferrara, F.; Bradbury, A.R. Deep sequencing in library selection projects: What insight does it bring? Curr. Opin. Struct. Biol. 2015, 33, 146–160. [Google Scholar] [CrossRef]
- Ferrara, F.; D’Angelo, S.; Gaiotto, T.; Naranjo, L.; Tian, H.; Graslund, S.; Dobrovetsky, E.; Hraber, P.; Lund-Johansen, F.; Saragozza, S.; et al. Recombinant renewable polyclonal antibodies. MAbs 2015, 7, 32–41. [Google Scholar] [CrossRef]
- Hanke, T.; Szawlowski, P.; Randall, R.E. Construction of solid matrix-antibody-antigen complexes containing simian immunodeficiency virus p27 using tag-specific monoclonal antibody and tag-linked antigen. J. Gen. Virol. 1992, 73 Pt 3, 653–660. [Google Scholar] [CrossRef]
- Randall, R.E.; Young, D.F.; Goswami, K.K.; Russell, W.C. Isolation and characterization of monoclonal antibodies to simian virus 5 and their use in revealing antigenic differences between human, canine and simian isolates. J. Gen. Virol. 1987, 68 Pt 11, 2769–2780. [Google Scholar] [CrossRef]
- Razai, A.; Garcia-Rodriguez, C.; Lou, J.; Geren, I.N.; Forsyth, C.M.; Robles, Y.; Tsai, R.; Smith, T.J.; Smith, L.A.; Siegel, R.W.; et al. Molecular evolution of antibody affinity for sensitive detection of botulinum neurotoxin type A. J. Mol. Biol. 2005, 351, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Angelini, A.; Chen, T.F.; de Picciotto, S.; Yang, N.J.; Tzeng, A.; Santos, M.S.; Van Deventer, J.A.; Traxlmayr, M.W.; Wittrup, K.D. Protein Engineering and Selection Using Yeast Surface Display. Methods Mol. Biol. 2015, 1319, 3–36. [Google Scholar] [CrossRef] [PubMed]
- Presta, L.G. Molecular engineering and design of therapeutic antibodies. Curr. Opin. Immunol. 2008, 20, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Harding, F.A.; Stickler, M.M.; Razo, J.; DuBridge, R.B. The immunogenicity of humanized and fully human antibodies: Residual immunogenicity resides in the CDR regions. MAbs 2010, 2, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Getts, D.R.; Getts, M.T.; McCarthy, D.P.; Chastain, E.M.; Miller, S.D. Have we overestimated the benefit of human(ized) antibodies? MAbs 2010, 2, 682–694. [Google Scholar] [CrossRef]
- Rajpal, A.; Beyaz, N.; Haber, L.; Cappuccilli, G.; Yee, H.; Bhatt, R.R.; Takeuchi, T.; Lerner, R.A.; Crea, R. A general method for greatly improving the affinity of antibodies by using combinatorial libraries. Proc. Natl. Acad. Sci. USA 2005, 102, 8466–8471. [Google Scholar] [CrossRef]
- Sela-Culang, I.; Kunik, V.; Ofran, Y. The structural basis of antibody-antigen recognition. Front. Immunol. 2013, 4, 302. [Google Scholar] [CrossRef]
- Briney, B.; Inderbitzin, A.; Joyce, C.; Burton, D.R. Commonality despite exceptional diversity in the baseline human antibody repertoire. Nature 2019. [Google Scholar] [CrossRef]
- Elhanati, Y.; Sethna, Z.; Marcou, Q.; Callan, C.G., Jr.; Mora, T.; Walczak, A.M. Inferring processes underlying B-cell repertoire diversity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370. [Google Scholar] [CrossRef]
- D’Angelo, S.; Ferrara, F.; Naranjo, L.; Erasmus, M.F.; Hraber, P.; Bradbury, A.R.M. Many Routes to an Antibody Heavy-Chain CDR3: Necessary, Yet Insufficient, for Specific Binding. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Chany, A.C.; Veyron-Churlet, R.; Tresse, C.; Mayau, V.; Casarotto, V.; Le Chevalier, F.; Guenin-Mace, L.; Demangel, C.; Blanchard, N. Synthetic variants of mycolactone bind and activate Wiskott-Aldrich syndrome proteins. J. Med. Chem. 2014, 57, 7382–7395. [Google Scholar] [CrossRef] [PubMed]
- Marks, J.D.; Bradbury, A. Selection of human antibodies from phage display libraries. Methods Mol. Biol. 2004, 248, 161–176. [Google Scholar] [PubMed]
- Boder, E.T.; Wittrup, K.D. Optimal screening of surface-displayed polypeptide libraries. Biotechnol. Prog. 1998, 14, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Boder, E.T.; Wittrup, K.D. Yeast surface display for directed evolution of protein expression, affinity, and stability. Methods Enzymol. 2000, 328, 430–444. [Google Scholar] [PubMed]
- Boder, E.T.; Midelfort, K.S.; Wittrup, K.D. Directed evolution of antibody fragments with monovalent femtomolar antigen-binding affinity. Proc. Natl. Acad. Sci. USA 2000, 97, 10701–10705. [Google Scholar] [CrossRef]
- Di Niro, R.; Ziller, F.; Florian, F.; Crovella, S.; Stebel, M.; Bestagno, M.; Burrone, O.; Bradbury, A.R.; Secco, P.; Marzari, R.; et al. Construction of miniantibodies for the in vivo study of human autoimmune diseases in animal models. BMC Biotechnol. 2007, 7, 46–55. [Google Scholar] [CrossRef]
- Wentz, A.E.; Shusta, E.V. Enhanced secretion of heterologous proteins from yeast by overexpression of ribosomal subunit RPP0. Biotechnol. Prog. 2008, 24, 748–756. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clone ID | LCDR3 | HCDR1 | HCDR2 | HCDR3 | Abundance after Selection 1 | Abundance after Selection 2 |
|---|---|---|---|---|---|---|
| M3_B11 | MQARQTPPT | GGTFSSYA | IIPIFGTA | ARVRWEPQSGYYHGMDVW | 100% | 18% |
| M3_A10 | MQARQTPPT | GGTFSSYA | IIPIFGTA | ARVRWVPQSGYYHGMDVW | 0% | 30% |
| M3_E1 | AAWDDSLNGPA | GYTFTSYG | YTFTSYG | ARVGGMGGDYVEYW | 0% | 20% |
| M3_B12 | SSYSSSSSYV | GGTFSSYA | IIPIFGTA | LIVGATTGGDAFDIW | 0% | 16% |
| M3_B4 | LLYYGGDWV | GGTFSSYA | IIPIFGTA | AAVGLDAFDIW | 0% | 4% |
| M3_C6 | MQGTHWPPT | GGTFSSYA | IIPIFGTA | AITDGIFDIW | 0% | 4% |
| M3_A2 | AAWDDRLNGVV | GGTFSSYA | IIPIFGTA | ARGSGYYDSSGYHWYFDLW | 0% | 2% |
| M3_C11 | SSYAGSNGSV | GGTFSSYA | IIPIFGTA | AVYSSGWYGGTTEDYW | 0% | 2% |
| M3_E9 | MQGTHWPPT | GGTFSSYA | IIPIFGTA | ARVAYYYGSGSYSFDYW | 0% | 2% |
| M3_B10 | SSYSSSSSYV | GGTFSSYA | IIPIFGTA | AAADYYDSSGYYYGGVEEHW | 0% | 2% |
| Clone ID | HCDR1 | HCDR2 | HCDR3 | Percentage Sequence Abundance | Yeast-Based Affinity | CI | ||
|---|---|---|---|---|---|---|---|---|
| Selection Output | 2 rds Affinity Maturation | 4 rds Affinity Maturation | ||||||
| parental | GGTFSSYA | IIPIFGTA | ARVRWEPQSGYYHGMDVW | 91% | 17% | 470 nM | 241–720 | |
| AM_1 | GGTFSSYA | IIPIFGTA | ARVRWEPRSGYYHGMDVW | 10% | 360 nM | 260–450 | ||
| AM_2 | GGTFSSYA | IIPIFGTA | ARVRWVPRSGYYHGMDVW | 7% | 345 nM | 181–676 | ||
| AM_3/M3_A10 | GGTFSSYA | IIPIFGTA | ARVRWVPQSGYYHGMDVW | 10% | 20% | 149 nM | 71–271 | |
| AM_4 | GGAFSRYA | IIPIFGTA | ARVRWVPQSGYYHGMDVW | 29% | 54% | 145 nM | 69–298 | |
| AM_5 | GGTFSRYA | IVPIFGTA | ARVRWVPQSGYYHGMDVW | 24% | 16% | 212 nM | 159–324 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naranjo, L.; Ferrara, F.; Blanchard, N.; Demangel, C.; D’Angelo, S.; Erasmus, M.F.; Teixeira, A.A.; Bradbury, A.R.M. Recombinant Antibodies against Mycolactone. Toxins 2019, 11, 346. https://doi.org/10.3390/toxins11060346
Naranjo L, Ferrara F, Blanchard N, Demangel C, D’Angelo S, Erasmus MF, Teixeira AA, Bradbury ARM. Recombinant Antibodies against Mycolactone. Toxins. 2019; 11(6):346. https://doi.org/10.3390/toxins11060346
Chicago/Turabian StyleNaranjo, Leslie, Fortunato Ferrara, Nicolas Blanchard, Caroline Demangel, Sara D’Angelo, M. Frank Erasmus, Andre A. Teixeira, and Andrew R.M. Bradbury. 2019. "Recombinant Antibodies against Mycolactone" Toxins 11, no. 6: 346. https://doi.org/10.3390/toxins11060346
APA StyleNaranjo, L., Ferrara, F., Blanchard, N., Demangel, C., D’Angelo, S., Erasmus, M. F., Teixeira, A. A., & Bradbury, A. R. M. (2019). Recombinant Antibodies against Mycolactone. Toxins, 11(6), 346. https://doi.org/10.3390/toxins11060346