1. Introduction
Staphylococcus aureus, which is a pervasive human pathogen, is a leading cause of life-threatening community and hospital-acquired infections world-wide. This Gram-positive bacterium is often associated with a range of diseases, from mild skin and soft tissue infections to invasive bacteremia, septic arthritis, endocarditis, and osteomyelitis [
1]. Moreover, recent widespread emergence of multi-drug resistant strains, specifically methicillin and vancomycin-resistance, have not only complicated the use of available treatment options, but have also considerably raised the economic burden that is associated with staphylococcal infections [
2,
3,
4]. Methicillin-resistant
S. aureus (MRSA) causes ~80,000 invasive infections and 11,000 deaths per year in the United States alone [
3].
S. aureus expresses a myriad of virulence factors, including cell surface attachment factors, capsular polysaccharides, enzymes, immune modulatory molecules, pore-forming toxins, and superantigens that aimed at establishing the infection or colonization as well as immune evasion [
5]. Among the pore-forming toxins,
S. aureus produces single component alpha hemolysin (Hla or α-toxin) and bicomponent pore-forming toxins (BCPFTs), Panton-Valentine leukocidin (PVL; composed of LukS-PV and LukF-PV), Leukocidin AB (LukAB), Leukocidin ED (LukED), and γ-hemolysins (HlgAB and HlgCB) [
6]. While Hla is secreted as a monomer and oligomerizes on the plasma membrane of target cells upon interaction with its specific cellular receptor ADAM10 [
7], the BCPFTs are produced from two distinct polypeptides, S (~32.4 kDa) and F (~34.6 kDa), which have β-barrel structures and hetero-oligomerize in a stepwise fashion with alternating S (LukS-PV, LukE, HlgA, HlgC, and LukA) and F (LukF-PV, LukD, HlgB, and LukB) subunits on the cell surface [
8]. Following oligomerization, structural rearrangements within the C-terminal stem domain promote membrane insertion, resulting in ion efflux, disruption of the host cell lipid bilayer, and ultimately cell death.
Among the BCPFTs, LukAB is the most recently identified and it is one of the most potent members of the leukocidin family that kills human neutrophils, macrophages, monocytes, and dendritic cells [
9,
10]. While the majority of the different BCPFTs exhibit a high sequence identity of 70–80% among the S and F components, LukA and LukB share a low sequence identity of 30–40% with the other leukotoxins [
6]. LukAB is also unique, in that it is secreted as a dimer in solution and it requires both S and F components for cell surface engagement in contrast to other BCPFTs [
11]. The crystal structure of LukAB from USA300, which is a predominant methicillin-resistant
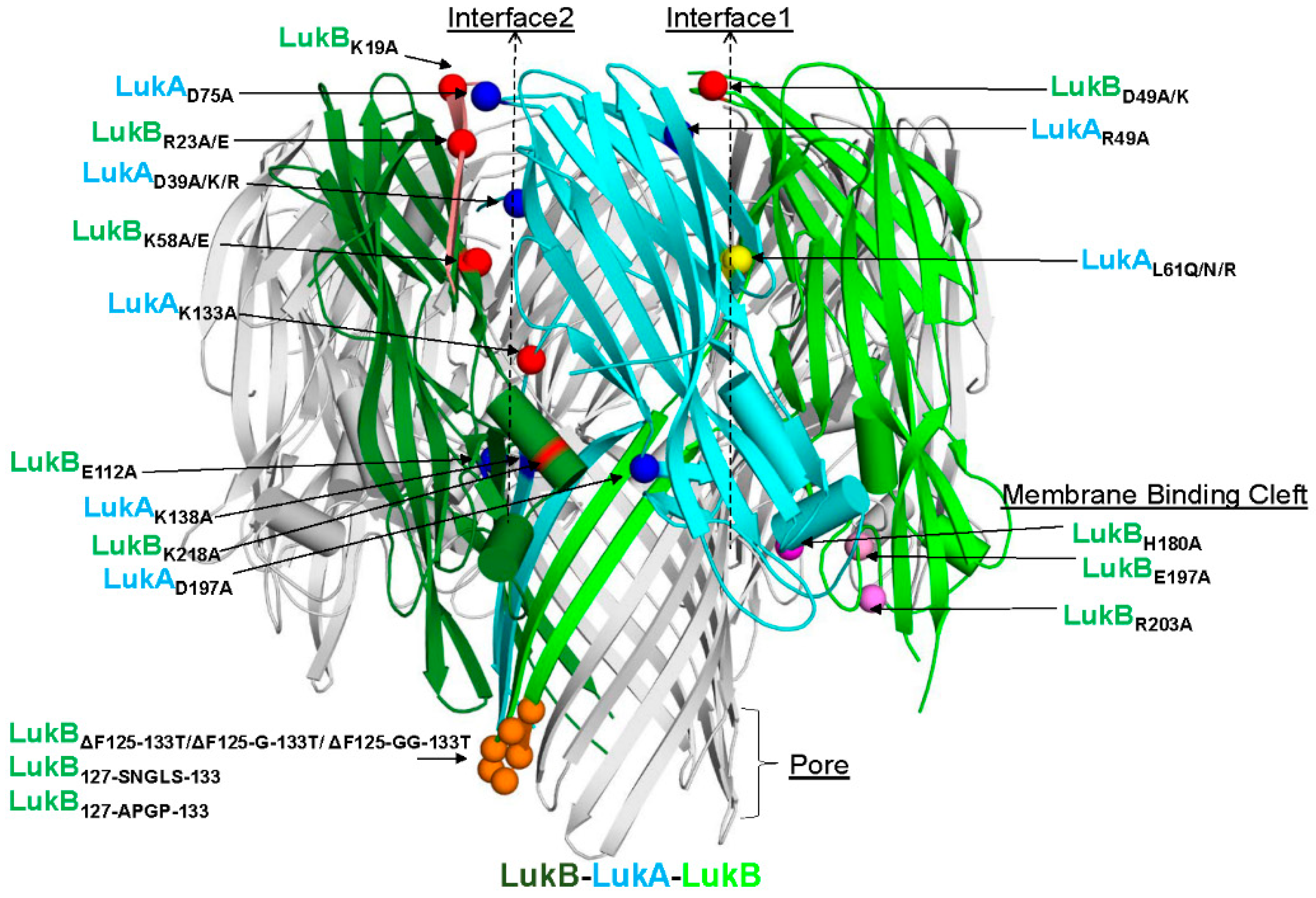
S. aureus (MRSA) strain circulating in the United States (US), revealed an octameric arrangement of four LukAB dimers with two unique interfaces 1 (intra-protomeric) and 2 (inter-protomeric), which primarily govern the cap and rim domains [
11]. Highly conserved residues among known LukAB variants that form salt bridges, a characteristic that is not found in other BCPFTs, hold these interfaces together. Of the two interfaces, residues within interface 1 were reported to be important for dimer formation, whilst interface 2 is important for octamerization. A surface-exposed residue E323 within the rim domain of LukA is critical to LukAB-mediated cytotoxicity by directly interacting with the integrin αM/β2 receptor (CD11b/CD18) on the surface of human neutrophils and monocytes (THP-1) [
11,
12,
13,
14]. In addition to the receptor binding properties, the rim domain harbors a high-affinity antibody epitope, which is also conserved among different LukAB sequence variants, and is suggestive of its role as an antigenic determinant as well as a site important for neutralization [
15]. LukAB production not only allows the bacteria to escape from phagocytic killing by human neutrophils, but also lyse monocytes in a CD11b-targeted manner [
9,
12,
13,
16]. Furthermore, LukAB, along with α-toxin, were identified as key players in host macrophage dysfunction enabling USA 300 biofilm formation, highlighting yet another role of LukAB in circumventing the immune-mediated clearance in the host [
17].
Highly virulent MRSA strains, like USA300, contribute to the clinical severity of SA infections by secreting a higher load of toxins to evade the innate immune system. Amongst other leukocidins, high titers to LukAB have been reported in acute and convalescent patients when compared to healthy controls [
18]. Additionally, a study reported high levels of functional antibodies against LukAB in patients with invasive
S. aureus infections as compared to healthy individuals or commercially available intravenous immunoglobulin (IVIG) [
19]. Altogether, these studies suggest LukAB as a key protagonist in SA-related disease pathogenesis, bacterial survival, and persistence, and therefore a potential vaccine target.
Here, iterative rounds of targeted single-amino acid and combination mutations within different functional domains of LukAB were designed in a structure-guided manner to identify the attenuated forms of LukAB. To this end, we first developed a novel production strategy that was based on co-expression from a single vector and a multistep purification process to generate tag-free, soluble dimers, and then functionally characterized them based on toxicity, thermostability, immunogenicity, and the ability to compete with cytotoxicity of wild type (WT) LukAB to identify several attenuated toxoids. Furthermore, we generated a polyclonal antibody against a selected attenuated LukAB mutant that demonstrated neutralizing activity towards supernatants from several laboratory and clinical strains, including USA300, which is responsible for the current CA-MRSA outbreak in the United States. Our findings further indicate that a multivalent toxoid vaccine targeting all the pore-forming toxins is needed to fully neutralize the cytotoxicity of different clinical strains of S. aureus toward neutrophils and monocytes.
3. Discussion
The evolution and emergence of MRSA strains has become a major challenge to global health. Among the diverse virulence factors that were secreted by SA to subvert the encountering host defense system, α-toxin/Hla and the bicomponent leukotoxins, such as LukAB, HlgAB, HlgCB, LukED, and PVL, are the most potent, because they specifically target and kill innate immune cells and disrupt biological barriers by lysing epithelial and endothelial cells [
24], as well as keratinocytes [
25]. Prior efforts toward vaccine development for
S. aureus have myopically focused on promoting the opsonophagocytic uptake of the bacteria and the subsequent killing by phagocytic cells, an approach that has been successful for several pathogens, such as
S. pneumoniae,
N. meningitidis, and
H. influenzae B. However, these efforts have failed to deliver an effective vaccine for
S. aureus and at least one of the experimental vaccines, Merck V710, led to increased mortality in vaccinated individuals who developed SA infection [
26], which suggested possible immunopathology. Several epidemiological studies indicate that the SA toxins are important vaccine targets [
27,
28,
29]. We have previously developed vaccine candidates for PVL subunits that elicit cross-reactivity to HlgAB, HlgCB, and LukED [
21]. LukAB plays a significant role in mediating SA virulence and its potency is most comparable to PVL [
8]. Unlike PVL, which is carried by phages and is only found in 5–15% clinical isolates, LukAB is chromosomally encoded and found in the majority of the isolates [
6]. However, LukAB is phylogenetically distant to these bicomponent toxins and anti-PVL antibodies are unable to neutralize this toxin. Here, we took a systematic and rational approach to design vaccine candidates for LukAB that are attenuated, stable, and highly immunogenic, and we identified several candidates for inclusion in a multivalent toxoid vaccine.
All of the bicomponent leukotoxins, except for LukAB, are secreted as S and F subunit monomers. Cell receptor binding is initiated by S subunit followed by the binding of F and oligomerization, which leads to pore formation [
6]. In contrast, LukAB is secreted as a stable dimer before engaging with the CD11b receptor molecules on the target cells following, which it hetero-oligomerizes into octomeric pore [
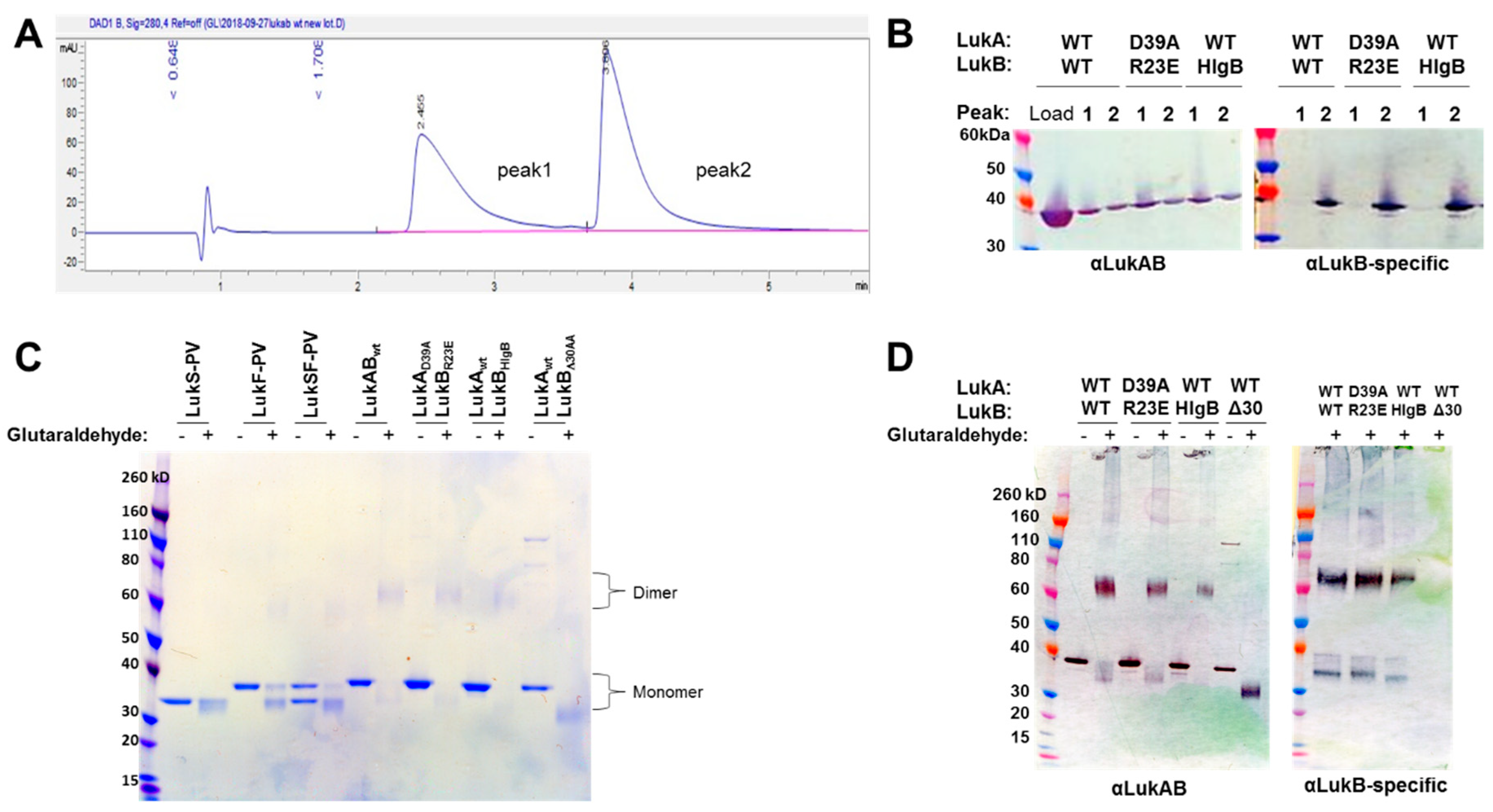
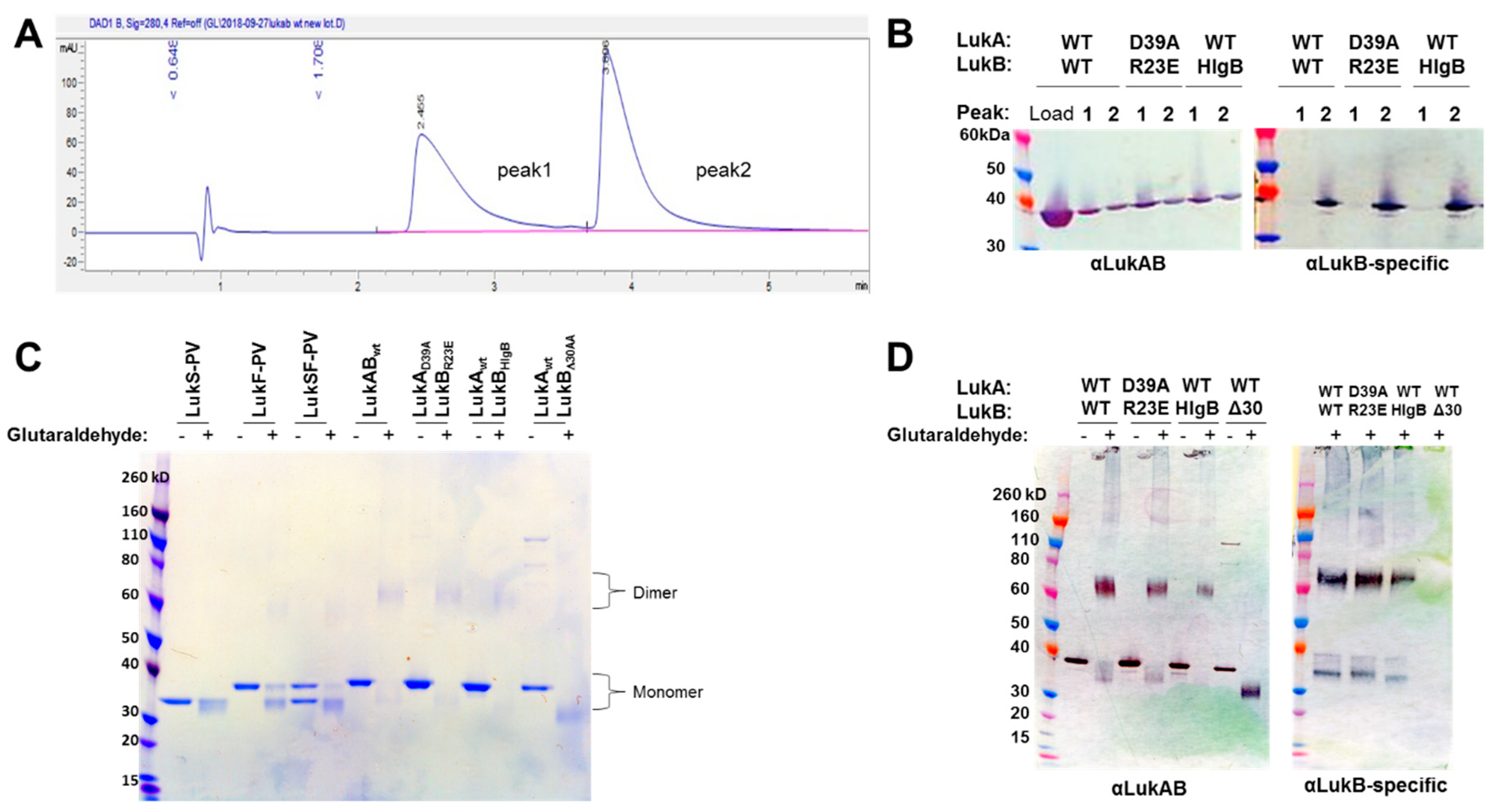
16]. In this report, we developed a process to express LukAB from a single vector (pETDuet) to purify the protein at high yield without the use of an affinity tag. Additionally, we were able to confirm that the produced LukAB contains both S and F components and are present as dimers in solution by using reverse-phase HPLC and glutaraldehyde cross-linking.
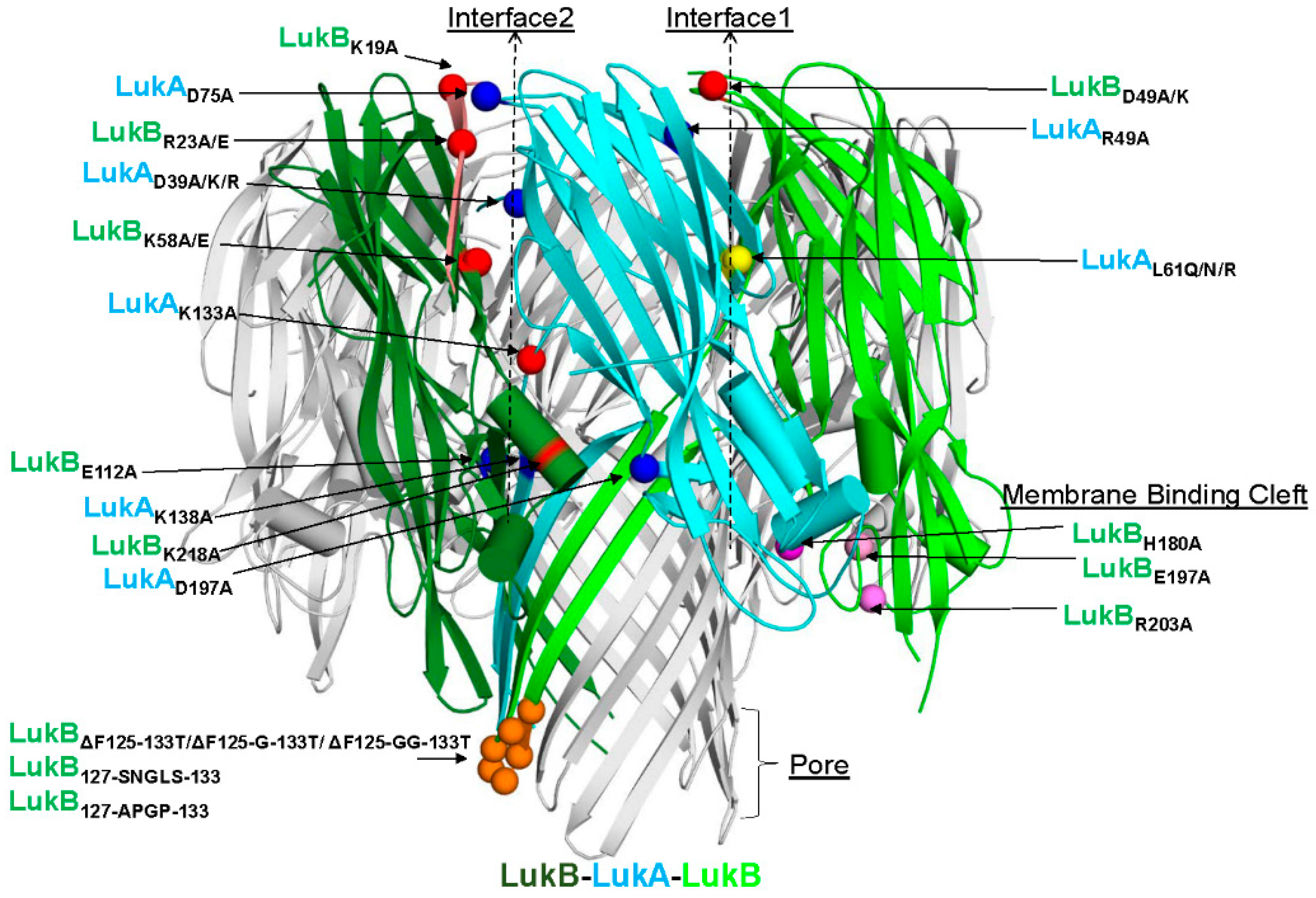
Badarau et al. solved the crystal structure of LukAB heterodimer in complex with a potent neutralizing antibody (ASN102) [
15], as well as the octameric LukAB [
11]. These studies identified 56 hydrogen bonds and four electrostatic interactions that hold the heterodimer together, as well as 34 hydrogen bonds and three electrostatic interactions that govern the formation of LukAB octamers. The authors also demonstrated that maintaining a stable dimer is critical for binding to neutralizing antibodies [
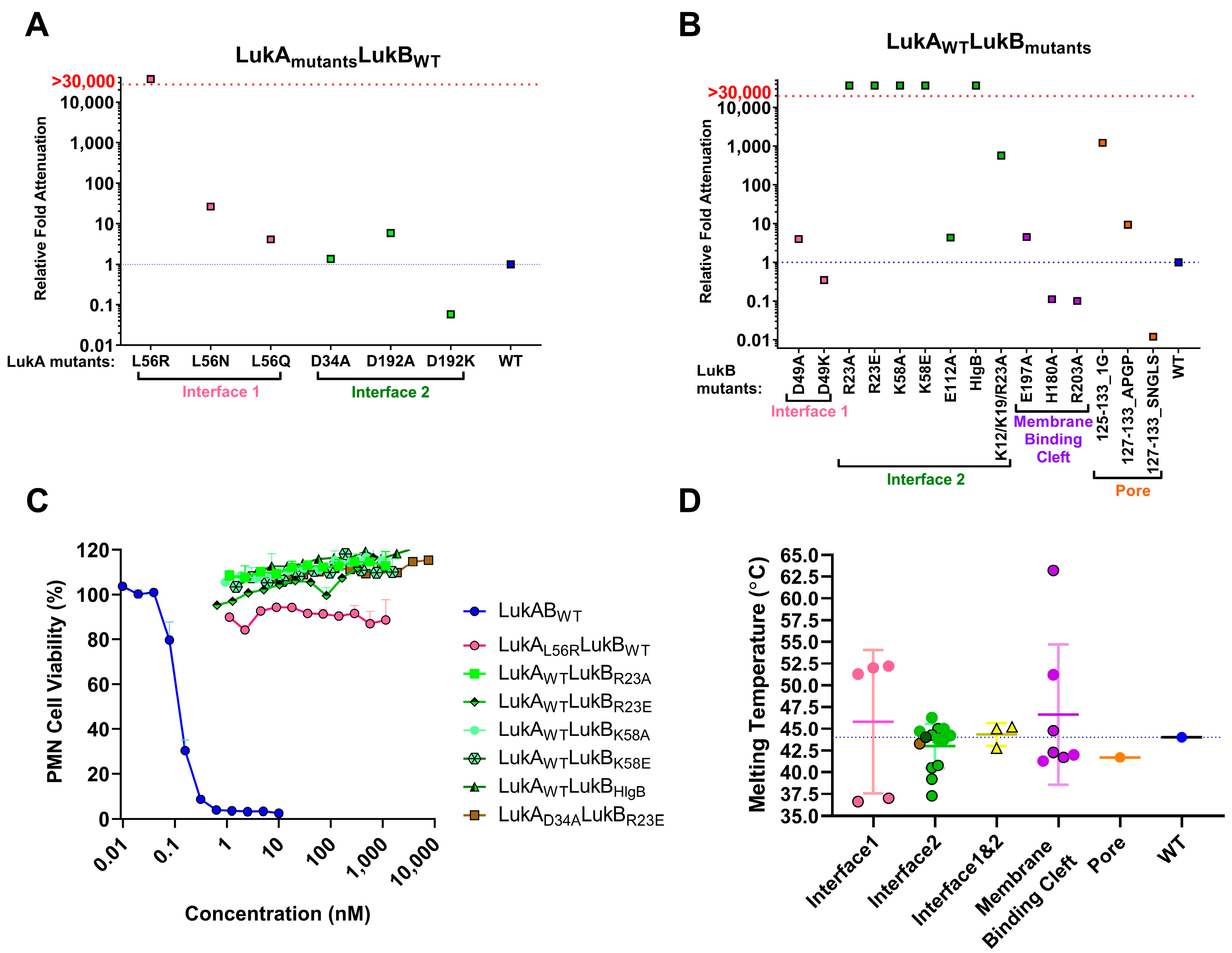
15]. Badarau et al. also generated several attenuated mutants, including double mutations in LukB consisting of R23A/E and K218A or LukA D75A and D197A in interface 2 (referred to as interface 1 in [
11]), which appear to interfere with octamerization. We undertook a broad screening strategy that is based on these important findings, and on the basis of LukAB sequence from USA300, to identify potential vaccine candidates that maintain LukAB structural integrity and immunogenicity, but lack toxicity. By making single and combination mutations, we were able to highlight residues in different, functional domains of the LukAB dimer that are imperative to mediating LukAB cytotoxicity. Additionally, targeting different domains on LukAB also allowed for biochemical, biophysical, and functional characterization, and delineated additional “hotspots” within LukAB dimer that act as key determinants of solubility, cytotoxicity, stability, and immunogenicity.
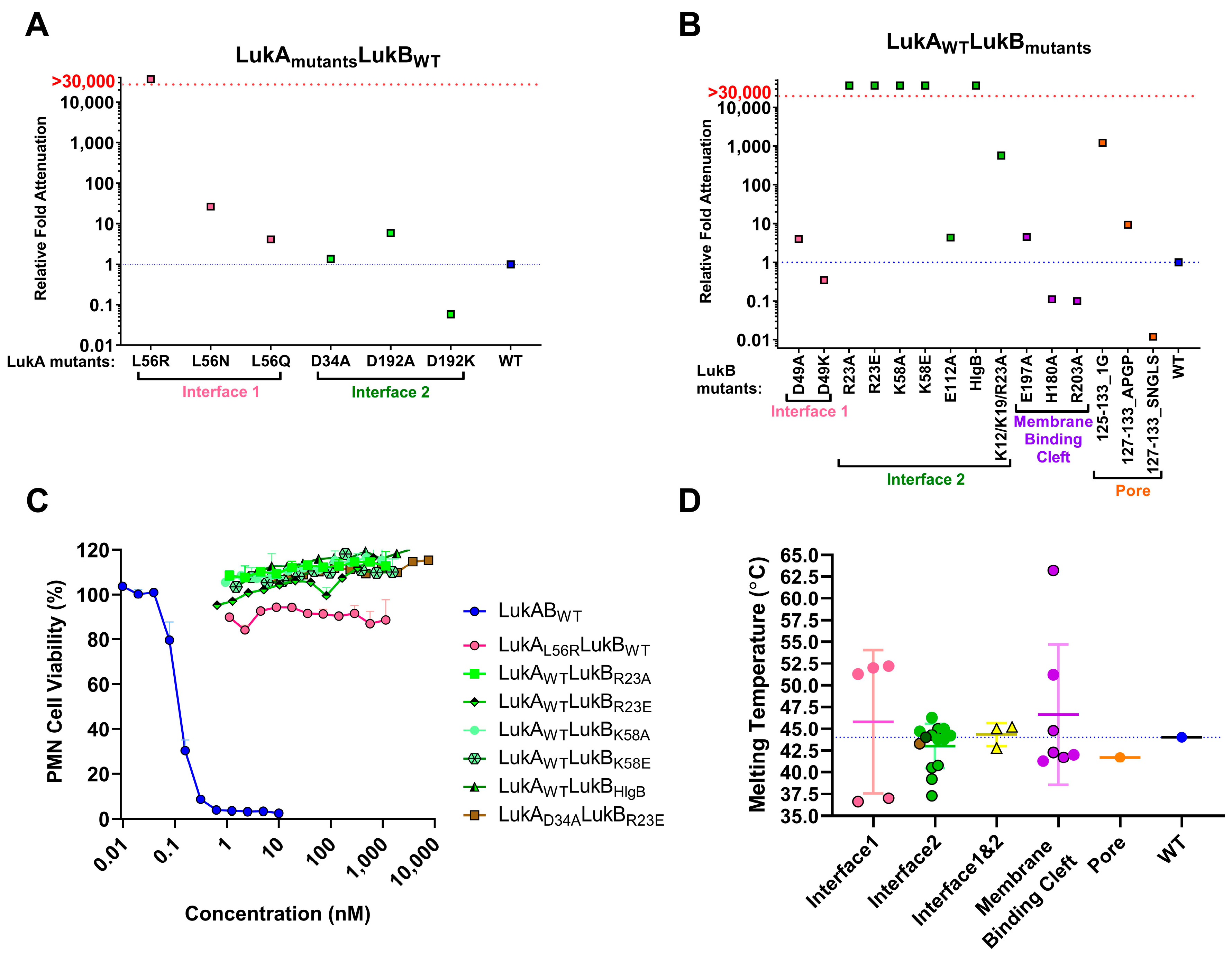
Of the electrostatic charges mediating protomer-protomer interactions (mediating heterodimer formation) and dimer-dimer interactions (mediating octamer formation), referred to here as interface 1 and 2, respectively, our data indicates that the residues within the salt bridges on interface 2 along with the bulky positively charged arginine and lysine residues at LukB N terminus are the key to mediating LukAB cytotoxicity. The electrostatic interactions that are part of the salt bridges within Interface 2, particularly participating LukB residues, which form a basic patch near the apical side of the cap domain, are crucial to cytotoxicity, as seen in this study. These residues have been previously shown to be important for LukAB octamerization and are known to be fully conserved between the LukAB sequence variants [
11]. In contrast, the salt bridge residues mutated within Interface 1 did not contribute to cytotoxicity as much. In our observation, substituting charged residues (K12/K19 and R23) or swapping of the first 30 residues of LukB N terminus with those of HlgB exhibited the highest impact on cytotoxic function. A previous report by DuMont et al. showed that the deletion of the first 33 residues of LukA does not affect copurification with LukB or heterodimer formation, but increases pore formation, thereby moderately increasing cytotoxicity [
12]. Our data indicates that, unlike LukA, the N terminus of LukB is indispensable for both heterodimer formation and cytotoxicity. While the N-terminal 30 residues of HlgB transplanted to LukB allow for heterodimer formation, it does not restore cytotoxicity, which is likely because of the loss of R23 that is critical for the salt bridge that is involved in dimer-dimer interaction. In contrast, the complete removal of those 30 residues resulted in the failure to express LukB. These observations are reminiscent of the functional properties of the amino latch that are found at the N terminus of α-toxin, where the residues control hemolytic activity and remain central to protomer-protomer interactions [
30]. Interestingly, the N terminal residues of LukB are unique to LukAB and they are not conserved among other S and F components or α-toxin alluding to a possibility that LukB N terminus may be functionally unique with putative roles in oligomerization and toxicity, requiring further investigation.
Mutations that were made to disrupt the buried hydrophobic pocket found within Interface 1, particularly L61, was found to be crucial to LukAB thermostability and attenuating toxicity; however, drastically hampering immunogenicity. As this residue is not surface exposed, the pocket near L61 is not an antigenic target, but it lowers immunogenicity, which is likely due to altered oligomeric status. The structures of LukAB and α-toxin show that L61 is nestled between β-strands that are part of the β-sandwich within the cap domain and the introduction of bulky mutations perturbs the hydrophobicity within this pocket affecting crucial protomer-protomer interactions that are sacred to stability, thereby reducing its ability to generate neutralizing antibodies.
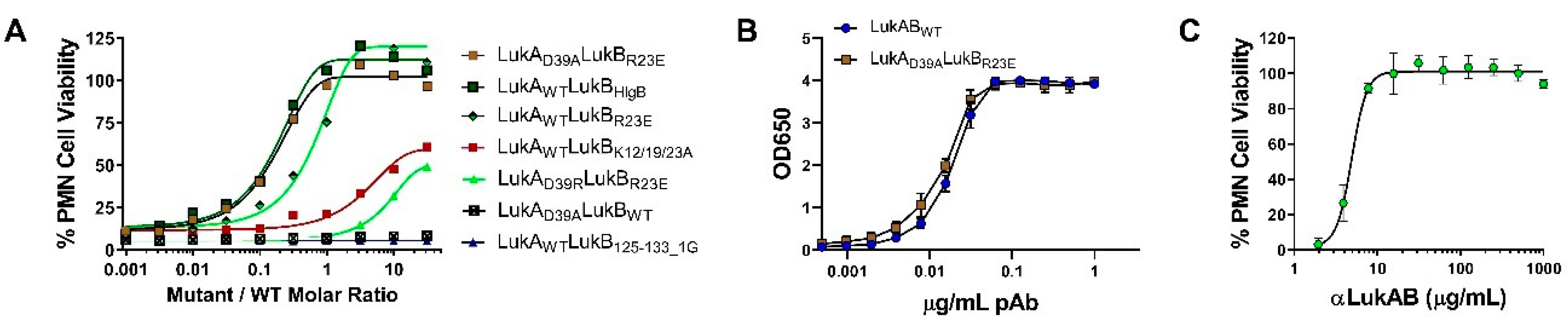
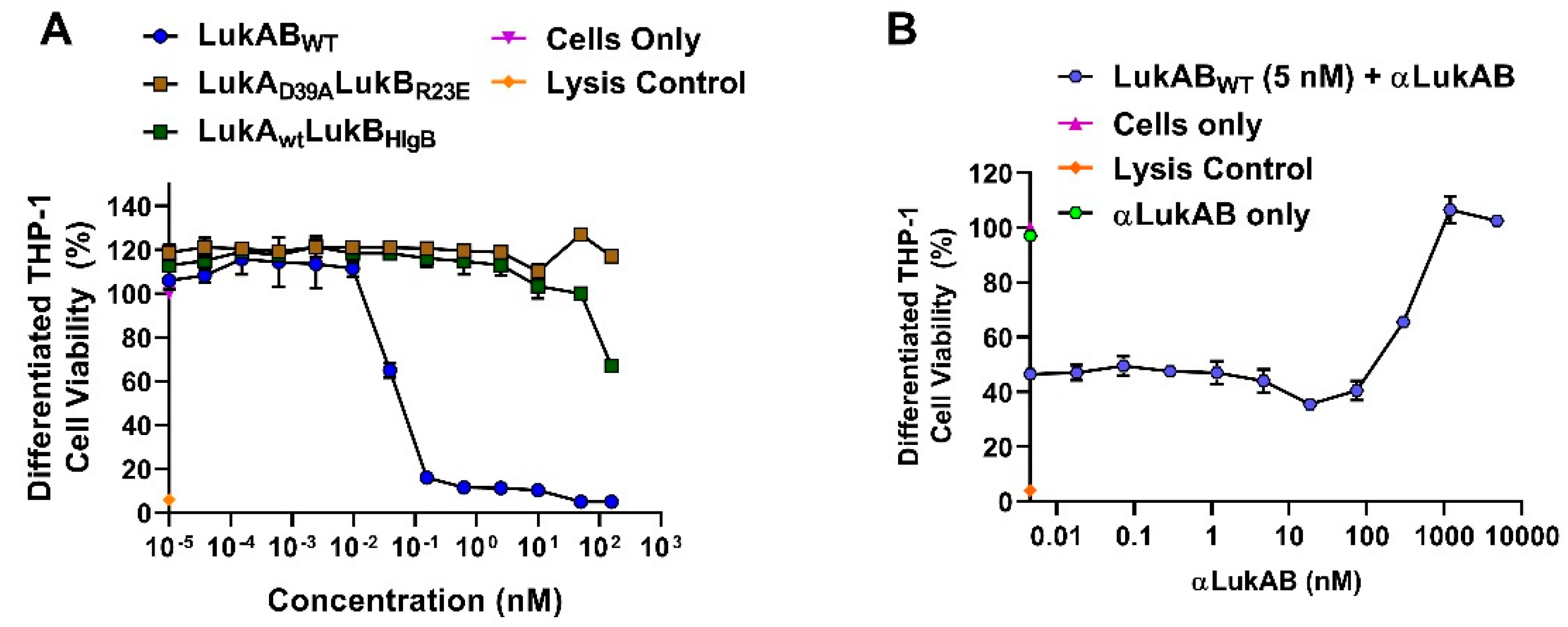
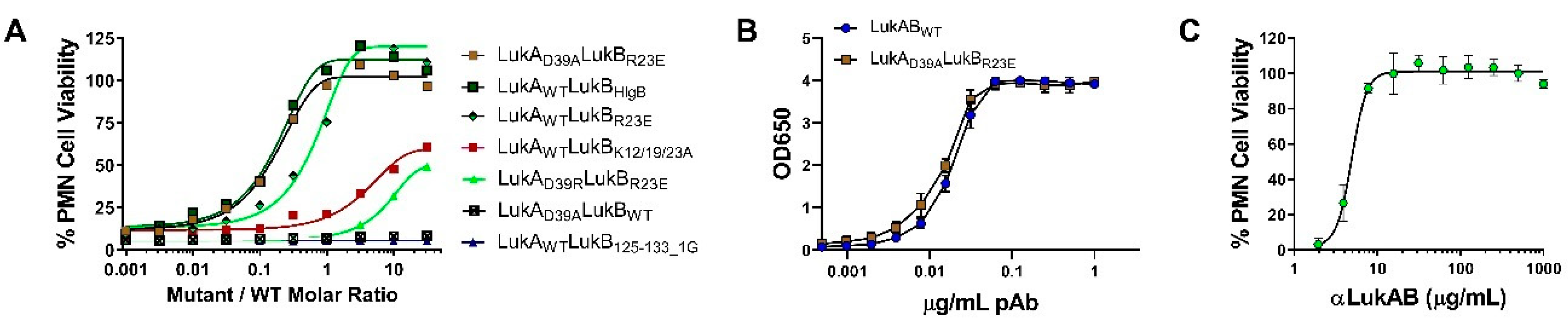
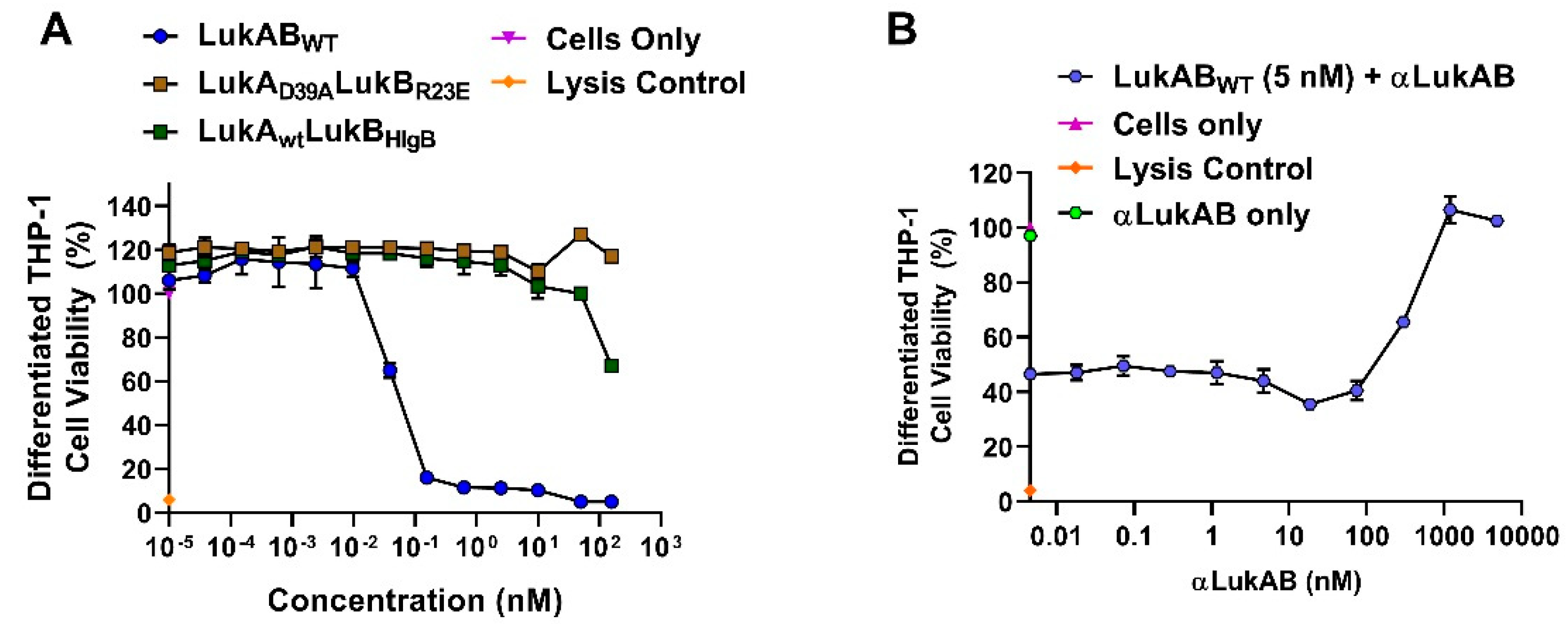
In our efforts, we have been successful in identifying a strong vaccine candidate in LukAD39ALukBR23E that satisfied biochemical and biophysical characterization, in addition to showing complete attenuation in PMNs. It is interesting that the second-best candidate, LukAwtLukBHlgB mutant, which exhibited the complete attenuation in PMN lytic activity, retained some toxicity, albeit at high concentrations in the THP-1 cells, suggesting that the substituted HlgB residues may be responsible for the relapsed cytotoxic effects. Interestingly, both of these mutants were able to compete with WT LukAB toxicity at a low molar ratio of below 1, which indicated that the competition cannot be entirely due to receptor binding by the mutant. The reversal of LukAB toxicity by these mutants may relate to the formation of defective mixed oligomeric structures, an observation that warrants further investigation. Moreover, with this study, we can show that the identified vaccine candidate generates antibodies that can reverse the toxicity and show protection in PMNs, as well THP-1 monocytes.
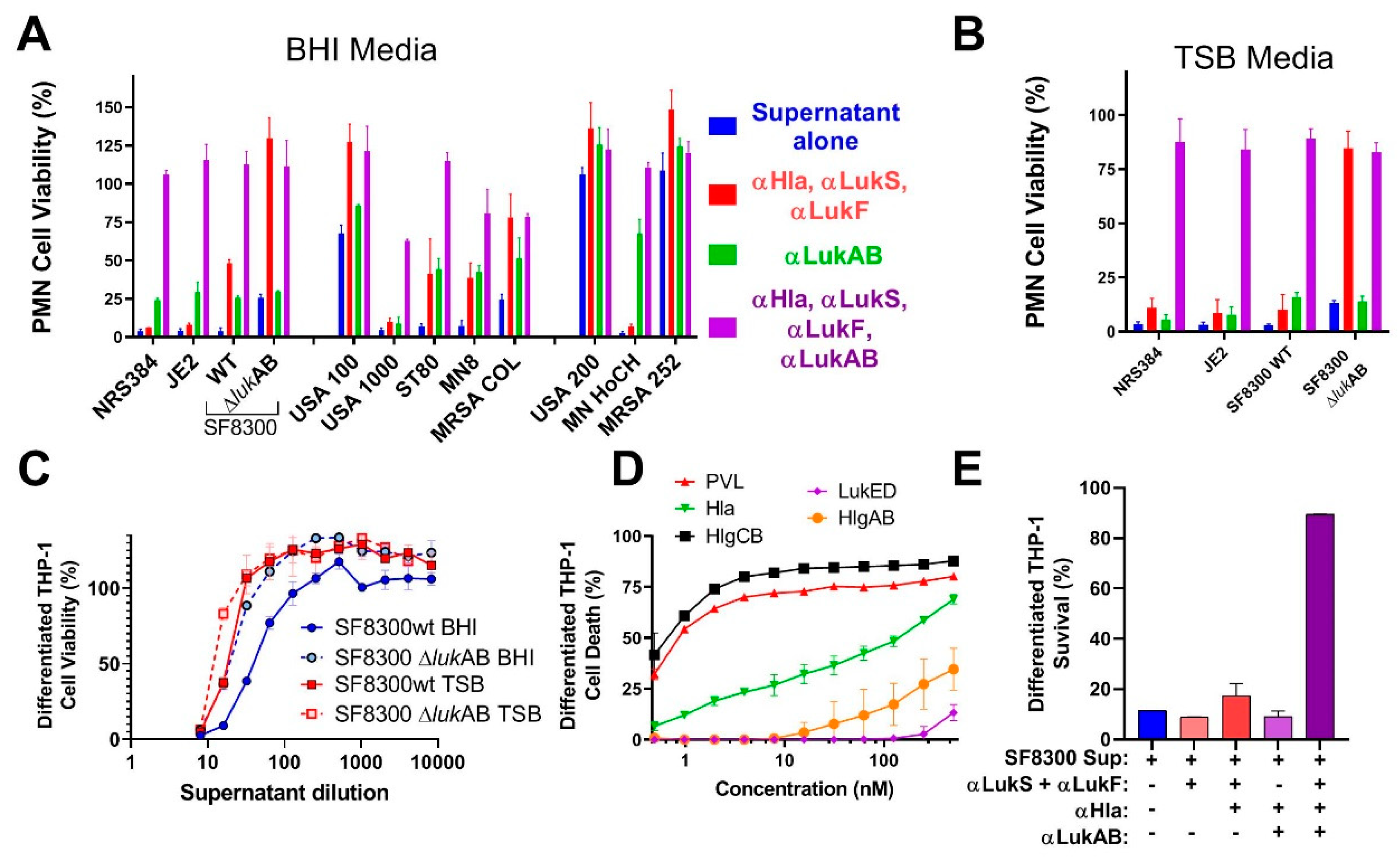
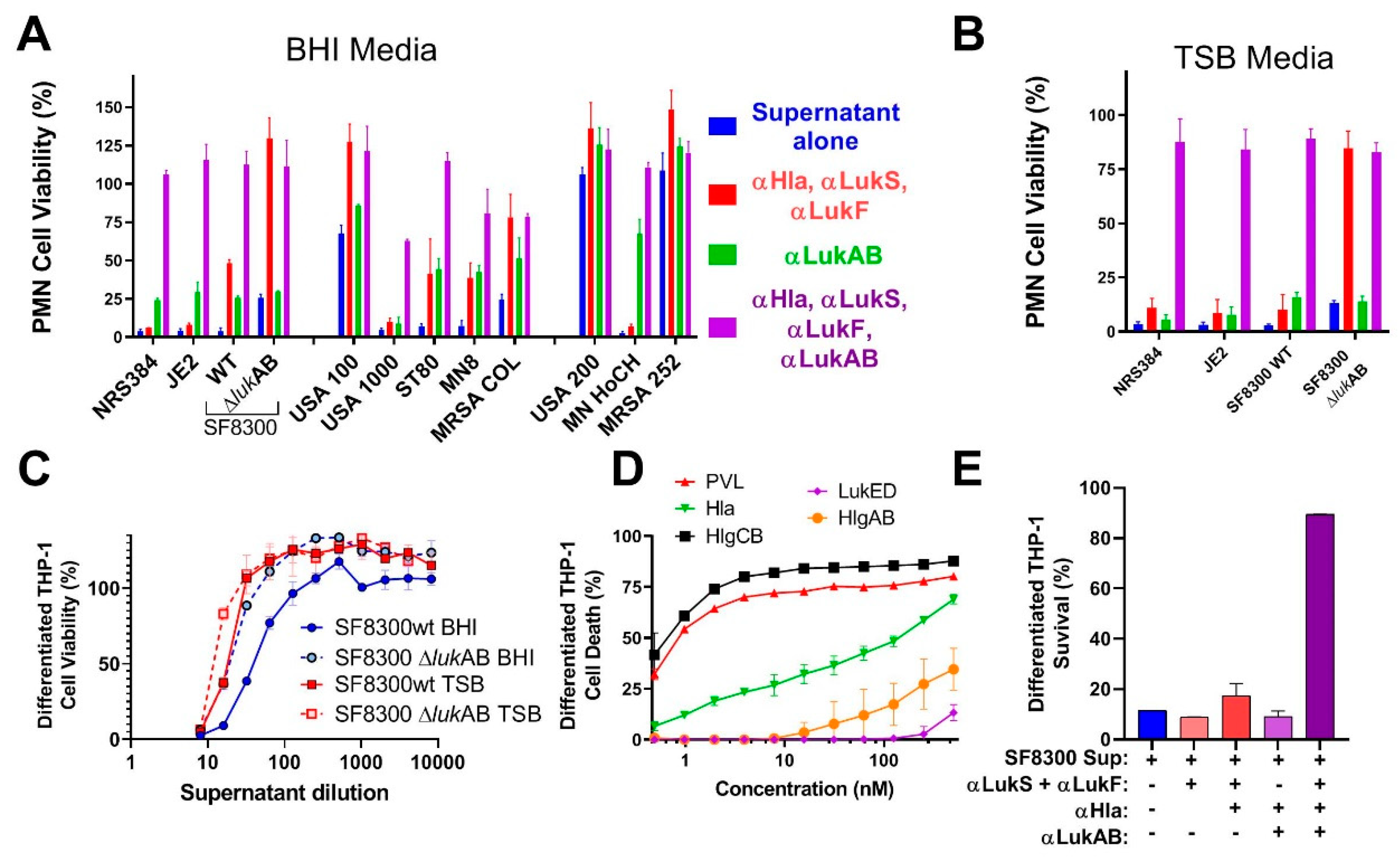
Previous studies showed the important role of LukAB in cell specific lysis of monocytes [
14] and dendritic cells (DC) [
31], as well as its role in macrophage dysfunction [
17]. However, these activities are also shared by some of the other pore-forming toxins. Most of the strains that were tested in our study showed clear synergism between polyclonal antibodies against LukAB and other pore-forming toxins. Cocktail polyclonal: αHla, LukS-PV, and LukF-PV (generated against three toxoid proteins) alone were unable to neutralize the toxicity of culture supernatants from most of the virulent strains in PMN lysis study. However, when this cocktail polyclonal was mixed with anti-LukAB polyclonal antibodies, 100% neutralization of culture supernatants was achieved, indicating the importance of neutralizing all the pore-forming toxins.
In summary, in this study, we have developed at least two vaccine candidates for LukAB, which is an important virulence factor of S. aureus. Our findings indicate that a multivalent approach targeting the related leukotoxins PVL, LukED, HlgAB, HlgCB, and the divergent LukAB, as well as the single component Hla is critical for protection against cytolytic activity of the most prevalent S. aureus strains. These data strongly support the development of a multivalent toxoid vaccine for S. aureus, which covers all major pore-forming toxins.
4. Materials and Methods
4.1. Generation of LukAB Wild-Type (WT) and Mutants in pET Duet and pET24a (+)
General methods that are used for bacterial culture have been described previously in detail (18,19). In this study, we optimized LukAB expression while using two different vectors: pET Duet-1 (Novagen), where LukA was cloned into the multiple cloning site 1 (MCS1) while using NcoI-HindIII, and LukB was cloned into MCS2 using NdeI-XhoI restriction sites within the same vector. In another system, LukA and LukB were expressed while using two different pET-24a(+) plasmids within the same E. coli cell. We compared LukAB WT expression, yield, and toxicity from these two systems. All of the mutants were expressed while using the latter system, where LukA and LukB were expressed using separate plasmids within the same E. coli. Towards this end, LukA (WT or mutants) was cloned into pET-24a(+) with a Kanamycin resistant marker within NdeI-XhoI sites. Similarly, LukB (WT/mutants) was inserted within NdeI-XhoI sites, but we replaced the inherent Kanamycin resistant cassette in pET-24a(+) with Ampicillin. The origin of replication of pET-24a(+) was replaced by p15a resulting in a recombinant pET24a(+)AmpRp15a LukB vector to increase the plasmid compatibility. This LukB plasmid (WT/mutant) was transformed into BL21(DE3) (NEB) containing pET24a(+) LukA plasmid (WT/mutant) and the colonies were selected on LB plates with 50 ug/mL of Kanamycin (Kan50) and 100 ug/mL of Ampicillin (Amp100). The genes for LukAB (USA300_TCH1516) WT and mutants were codon optimized prior to transformation by GenScript®.
4.2. Growth Media and Bacterial Strains
Overnight cultures of E. coli that were grown at 37 °C in LB Kan50Amp100 were expanded to 0.5 L in a shaking incubator (225 rpm), until they reached an OD OD600 of 0.5. The cultures were immediately chilled on ice for 10 min. with periodic shaking and then induced with 0.3 mM IPTG (Sigma, St.Louis, MO, USA) in a shaking incubator (225 rpm) overnight at 25 °C. The following day, the cells were harvested by centrifugation (14,000× g) and frozen at −80 °C. To lyse the cells, the pellet was resuspended in 3 mL of cell lysis buffer (20 mM Tris pH 8.0, 50 mM NaCl, 1 mM EDTA, 0.1% Triton X-100) per gram of cell paste and Hen egg lysozyme (Sigma, St. Louis, MO, USA) at 1 mg/mL final concentration prior to incubation at 37 °C for 30 min. The partially lysed cells were then cooled in a wet/dry ice ethanol bath, followed by sonication while using a microtip (10 × 10 s bursts with cooling between bursts, output 5, 50% duty). Lysis was confirmed by measuring the reduction of OD600 absorbance. Post lysis, the NaCl concentration was adjusted to 0.5 M and nucleic acid precipitation was carried out by the dropwise addition of 0.3–0.5% polyethyleneimine (PEI) while maintaining constant mixing. The PEI pellet was separated by centrifugation at 12,000 rpm in a Sorvall SS34 rotor and the supernatant containing the toxoid was subjected to ammonium sulphate (AmS04) precipitation. Towards this end, 0.472 g/mL of AmS04 powder was added to the PEI supernatant and then placed on a rotating mixer for 20 min. at room temperature. The resulting pellet that was obtained by centrifugation at 12,000× g for 30 min. at 4 °C was frozen at −80 °C until purification.
The AmS04 pellet was resuspended and buffer exchanged into 20 mM NaPi pH 6.5, 25 mM NaCl, 5% glycerol using a GE Healthcare PD10 desalting column. The mixture was then clarified by filtration using 0.8/0.2 µm Supor® low protein binding syringe filter (Pall Life Sciences, Port Washington, NY, USA). The toxoid from the resulting solution was purified while using a two-column purification approach. The first purification was carried over a 10 mL Poros 50 HS column using a 40-column volume (C.V.) gradient from 25 to 1000 mM NaCl in the phosphate buffer. The peak fractions were analyzed by standard SDS-PAGE analysis and accordingly pooled together for dialysis into 20 mM NaPi pH 6.8, 50 mM NaCl, 5% glycerol, the equilibration buffer for the second column—a 10 mL ceramic Hydroxyapatite (HTP) (BioRad, Hercules, CA, USA; Type 1 40 µm). The toxoid was eluted using a 40 C.V. gradient from 50 to 1000 mM NaCl in the phosphate buffer. The appropriate fractions were pooled together upon SDS-PAGE evaluation and then dialyzed into the final storage buffer (20 mM NaPi pH 7.4, 150 mM NaCl, 5% glycerol). All purified LukAB proteins (MW = ~72 kDa) were concentrated while using Amicon 3K MWCO Ultra 15, filtered through 0.8/0.2 µm low protein binding membrane, and stored at −80 °C prior to use.
4.3. Growth Media and Bacterial Strains
Table S2 lists the bacterial strains that were used in this study. The SA strains were grown in brain heart infusion broth/agar (BHI) and tryptic soy broth/agar (TSB) media at 37 °C, whichever appropriate. The overnight bacterial culture supernatants were normalized based on culture OD
600 absorbance. The next day, culture supernatants were filtered through 0.2 µm filter to sterilize the supernatants. Sterility was confirmed by culturing 100 μL of the filtered supernatants on BHI or TSA agar plates overnight.
4.4. Cell Culture Maintenance and Induction
The HL-60 cells (ATCC, Manassas, VA, USA) were cultured in RPMI 1640 (Gibco, Gaithersburg, MD, USA) supplemented with 16.4% heat inactivated fetal bovine serum (FBS), 4 mM L-glutamine, 82 U/mL each of penicillin, and streptomycin. Cells were passaged twice a week at a concentration of 6–8 × 105 cells/mL. 1 × 107 cells were seeded in 30 mL and grown in culture media with 1.5% dimethyl sulfoxide (DMSO) for seven days to differentiate cells into neutrophils. CD11b expression using flow cytometry confirmed induction.
4.5. PMN-based Cytotoxicity Assay
The induced HL-60 cells were harvested by centrifugation at 420×
g (Sorvall RT6000B rotor, ThermoFisher Scientific, Waltham, MA, USA) for 10 min. at 20 °C. Cells were washed and resuspended with phenol red-free RPMI 1640 (Gibco, Gaithersburg, MD, USA) supplemented with 2% FBS to a final concentration of 5 × 10
6 cells/mL. LukAB mutants (proteins) were serially diluted two-fold across 96-well plates (50 µL/well) and 100 µL of 5 × 10
6 cells/mL were added to each well. The plates were incubated at 37 °C, 5% CO
2, 95% humidity for 3 h. After 3 h, either XTT (Cell Signaling Technology) or CellTiter Glo (Promega) reagent was used to determine the cell viability and cytotoxicity [
21,
32]. When using XTT for readout, 50 µL of the activated XTT (50 μL electron coupling per 5 mL XTT) reagent was added to each well, and plate was returned to 37 °C, 5% CO
2, 95% humidity for 16–18 h. After incubation, the cells were pelleted by centrifugation at 3500 rpm 3565.9×
g (Sorvall RT6000B rotor, ThermoFisher Scientific, Waltham, MA, USA) for 3 min. The supernatants were transferred to 96-well ELISA plates and absorbance was read at 470 nm while using Spectramax 190 plate reader (Molecular Devices, San Jose, CA, USA) and Softmax 5.4.5 software (Molecular Devices, Waltham, MA, USA). When using CellTiter Glo, 50 μL of the reconstituted CellTiter Glo reagent was added to each well. The plate was shaken on an orbital shaker for 10–15 min., followed by measurement of luminescence (emission at 560 nm) using Cytation 5 imaging reader (Biotek, Winooski, VT, USA) and Gen5 2.09 software to determine cell viability. For the kinetic cytotoxicity studies, the replicates were incubated for 6 and 19 h, in addition to a 3-h incubation period. After incubation, the CellTiter Glo reagent was used to determine the cell viability.
4.6. Rabbit Polyclonal Antibody Generation
Rabbit polyclonal antibody generation for anti-HlaDM, LukSmut1, LukFmut9, and LukAD39ABR23E toxoid as immunogens were generated by Genscript® (Piscataway, NJ, USA) using >98% pure proteins as immunogens. Briefly, four New Zealand white rabbits were immunized per toxoid on day 0, 14, and 21 with 0.2 mg protein per rabbit, along with Freund’s Incomplete Adjuvant subcutaneously. The test bleeds and production bleeds were collected on day 21 and day 42. Hyperimmune sera were individually characterized for ELISA titer and TNA titers before pooling them together. The pooled serum was purified by Protein A affinity chromatography into total IgG and labeled, as follows: anti-Hla (IBT Cat: 1940-01 Rb pAb), anti-LukS- (IBT Cat: 1941-01 Rb pAb), anti-LukF-PV(IBT Cat: 1942-01 Rb pAb), and anti-LukAB (IBT Cat: 1944-02 Rb pAb). Full quality control (QC) were performed before use.
4.7. Toxin Neutralization Assay (TNA) in PMNs
The serum samples were serially diluted two-fold in RPMI across 96-well plates (25 µL/well). Twenty-five microliters of 2.5 nM LukAB toxin was added to each well. 100 µL of induced cells were prepared, as described above, at 5 × 10
6 cells/mL were added to each well. The plates were incubated at 37 °C, 5% CO
2, 95% humidity for 3 h, followed by XTT or CellTiter Glo readout to determine the cell viability and neutralization [
21,
32].
4.8. Reverse-TNA in PMNs
Select LukAB mutants were serially diluted from a starting concentration of 12 nM in RPMI semi-log across 96-well plates (25 µL/well). Polyclonal αLukAB at 100 µg/mL or RPMI was added to each well (12.5 µL/well), followed by incubation at RT for 10 min. Following incubation, 12.5 µL of 5 nM LukAB WT and 100 µL of 5 × 106 induced HL-60 cells/mL were added to each well. The plates were then incubated for 3 h at 37 °C, 5% CO2, 95% humidity, followed by CellTiter Glo readout determine cell viability and neutralization.
4.9. Differential Scanning Fluorimetry (DSF)
The proteins in the storage buffer (20 mM NaPi pH 7.4, 150 mM NaCl, 5% glycerol) were mixed with 2X SYPRO orange dye (Invitrogen, Carlsbad, CA, USA) in a 96-well hard shell plate with clear bottom (BIO-RAD, Hercules, CA, USA) and then placed into a thermal cycler, wherein the temperature scan rate was fixed at 0.5 °C/min over a range of 30–99 °C (21). The fluorescence intensities were plotted against temperature to get a sigmoidal curve and the melting temperatures (Tm) were calculated while using the first derivative. BSA (Pierce) was used as a control, which recorded a melting temperature of 66 ± 0 °C in 1 × PBS pH 7.4.
4.10. Cross-Linking with Glutaraldehyde
LukABWT and mutants at 50 ug/mL concentration were incubated with 0.25% glutaraldehyde (Sigma, St. Louis, MO, USA) for 2 min. at 37 °C in a final volume of 100 μL in 20 mM HEPES pH 7.5, 50 mM NaCl. The reaction was stopped by adding 10 μL of 1M Tris, pH 8.0 and 4 × LDS sample buffer, followed by SDS-PAGE and Western blot analysis while using αLukAB and rabbit pAb αLukB-specific (IBT Cat: 0313-001). αLukS-PV and αLukF-PV were used as the negative controls.
4.11. Reverse-Phase HPLC
LukAB (WT/Mutant; 100 μg) was injected into an AdvanceBio RP-mAb diphenyl column (Agilent 795975-944, Santa Clara, CA, USA, 4.6 × 100 mm, 3.5 micron) in a 1260 Infinity Quaternary instrument. A 30–90% gradient method was used consisting of 100% acetonitrile in Line A and 0.1% TFA (v/v) in Line B with a flow rate of 1.00 mL/min over 60 min. and a column temperature of 27 °C.
4.12. Animals and Immunizations
Female ICR (CD-1) mice, six weeks of age, were purchased from Envigo (US). The mice were maintained under pathogen-free conditions and fed laboratory chow and water ad libitum. All mouse work was conducted in accordance with protocols that were approved by institutional animal care and use committees (IACUC) of Integrated BioTherapeutics, where mouse studies were performed (approval date 28 February 2017; approval code: D17-00974). The mice were intramuscularly immunized (IM) three times two weeks apart with 10 µg of LukAB mutant in 50 µg of Al(OH)3. For serological analysis, the mice were test bled via retro-orbital (RO) route prior to and 10 days after the third and final immunization.
4.13. Enzyme-Linked Immunosorbent Assay (ELISA) for Determination of Serum Titers
Blood samples from mice were centrifuged in serum separator tubes and the serum samples were stored at −80 °C until further use in ELISA. Briefly, 96-well plates were coated with 300 ng/well of LukAB WT overnight at 4 °C. The plates were blocked with Starting Block (SB) (Thermo Scientific, Waltham, MA, USA) for one hour at room temperature (RT). Serum samples were diluted in a semi-log manner starting from 1:100 to 1:316,228 in a 96-well plate, using starting block buffer as the diluent. The plates were washed three times and sample dilutions were applied in 100 µL volume/well. The plates were incubated for 1h at RT and washed three times before applying the conjugate, goat anti-mouse IgG (H+L)- Horseradish Peroxidase (HRP) in SB. Plates were incubated for 1 h at RT, washed, as described above, and incubated with TMB (3,3′,5,5′-tetramethylbenzidine) for 30 min. to detect HRP activity. Optical density at 650 nm was measured while using a Versamax™ plate reader (Molecular Devices, Waltham, CA, USA). Data analysis for full dilution curves was performed using Softmax program and graphed in GraphPad Prism.
4.14. Cytotoxicity Assay in Human Monocytes and THP-1 Cells
Purified human monocytes (CD14+) up to 95.98% purity were purchased from BioIVT (San Carlos, CA, USA) and kept frozen in LiN2 until use. Acute monocytic leukemia THP-1 cells were purchased from ATCC (ATCC® TIB-202™) and were cultured at 4E5 cells/mL every 2–3 days at 37 °C + 5% CO2, as recommended in RPMI-1640, supplemented with 0.05 mM 2-mercaptoethanol and 10% FBS. To differentiate the THP-1 cells, 100 nM of Phorbol 12-myristate 13-acetate (PMA; Sigma, St. Louis, MO, USA) was added to THP-1 media and seeded at 4E5 cells/mL. The cells were allowed to differentiate for three days and analyzed for extent of cell adherence and surface marker for differentiation (i.e., CD11b) by flow cytometry. After three days, the media was replaced with non-PMA containing medium and allowed to rest for five days before use, feeding the cells with fresh THP-1 medium every two days.
To evaluate the cytotoxicity effects of purified LukAB (WT/mutants) or other toxins, such as α-toxin, LukSF, HlgAB, and HlgCB on differentiated THP-1 cells, the adherent cells were harvested by treatment with 1 × PBS + 0.05 mM EDTA at 37 °C for ~5 min. and immediately centrifuged at 420×
g (Sorvall RT6000B rotor) for 10 min. The cells were then washed twice in 1% (
w/
v) RPMI+Cas (Bacto BD, Franklin Lakes, NJ, USA) and pelleted by centrifugation. Proteins (12.5 µL) were serially diluted two-fold or in semi-log fashion, as indicated in 1% RPMI+ Cas and incubated with 75 µL cells (seeded at 1E5 cells/well in a 96-well plate) in a final volume of 125 µL for 1 h at 37 °C, 5% CO
2, 95% humidity, followed by CellTiter Glo readout to determine the cell viability and % lysis. For experiments with supernatants from culture filtrates, the supernatants (as listed in
Table S2) were normalized to OD
600 = 6 prior to serial dilution in RPMI+Cas. The incubation times were increased to 4 h at 37 °C, 5% CO
2, 95% humidity prior to CTG treatment and readout. For conditions with a polyclonal antibody, 12.5 µL of single or each antibody was added to the well, followed by incubation with purified protein or supernatant for 10 min. at RT prior to the addition of cells. Cells incubated with a final concentration of 0.2 or 0.4% Triton-X-100 in 1% RPMI+ Cas was used as the control for complete lysis.
4.15. Flow Cytometry Analysis
HL60, DMSO-treated HL-60, THP-1, and PMA-treated THP-1 cells were seeded at 1.5E5 cells/well on a clear 96-well plate. The cells were washed with 200 µL/well of PBS without Ca2+ and Mg2+, supplemented with 2% FBS (FACS buffer). For HL60 cells, CD11b-FITC (BDPharmingen; Cat: 562,793 Clone: ICRF44) was added to each well at 1:40 dilution (50 µL) in FACS buffer and incubated at RT, being covered from light, for 15 min. For THP-1 cells, a combination of CD11b and CD14-APC/Cy7 stains (Cat: 301,820 Clone: M5E2) were used to monitor CD11b upregulation in addition to macrophage differentiation. The cells were washed twice with FACS buffer at 1400 rpm for 5 min. each and LIVE/DEAD Fixable Near-IR Dead Cell stain (ThermoFisher Scientific, Waltham, MA, USA) was added to each well at a dilution of 1:500 (50 µL) and incubated on ice, covered from light, for 15 min. The cells were washed again and resuspended in 200 µL FACS buffer. Fluorescence measurements were acquired using either Guava flow cytometer (EMD Millipore, Burlington, MA, USA) or they were acquired at a Symphony A3 (BD Biosciences, Franklin Lakes, NJ USA). Data was analyzed with FlowJo software V10. Induction was considered to be successful if CD11b expression in viable cells was found to be ~70% or higher.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}