Neurotoxic Effect of Ethanolic Extract of Propolis in the Presence of Copper Ions is Mediated through Enhanced Production of ROS and Stimulation of Caspase-3/7 Activity




Abstract
1. Introduction
2. Results
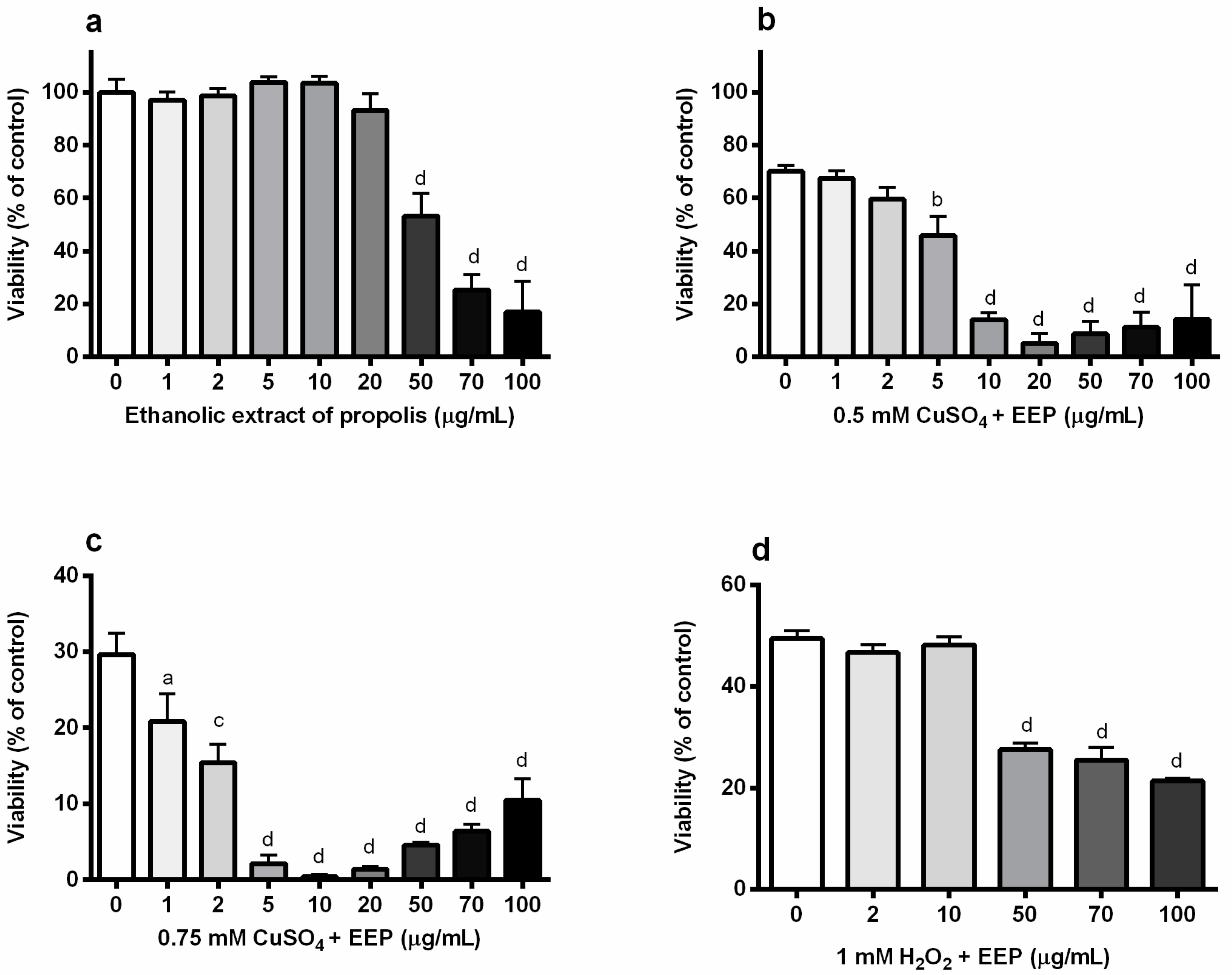
2.1. Propolis Exacerbated Copper-Induced Neuronal Injury in P19 Neuronal Cells
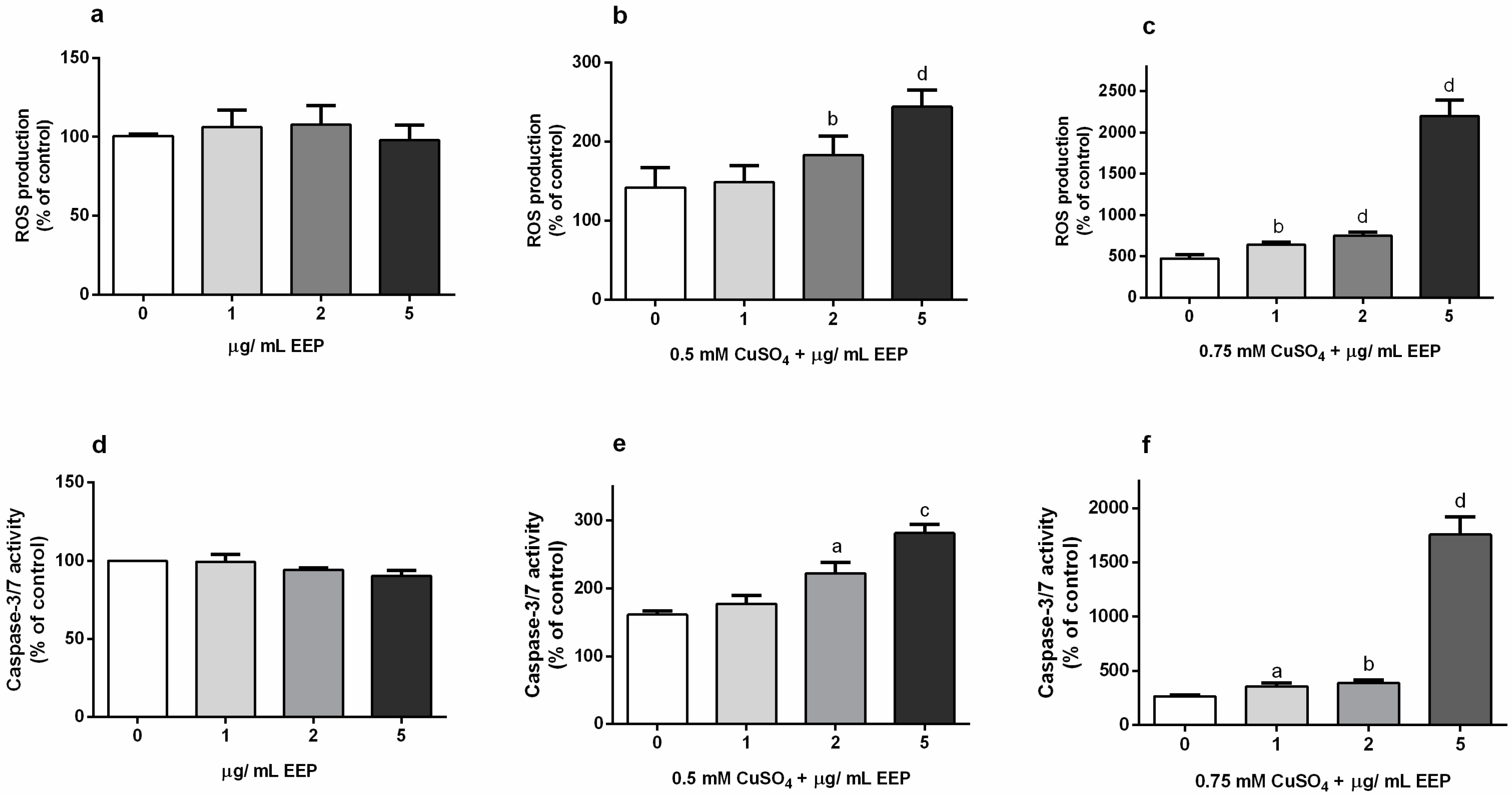
2.2. Propolis Promoted ROS Generation and Caspase-3/7-Activity in P19 Neurons Exposed to Excess Copper
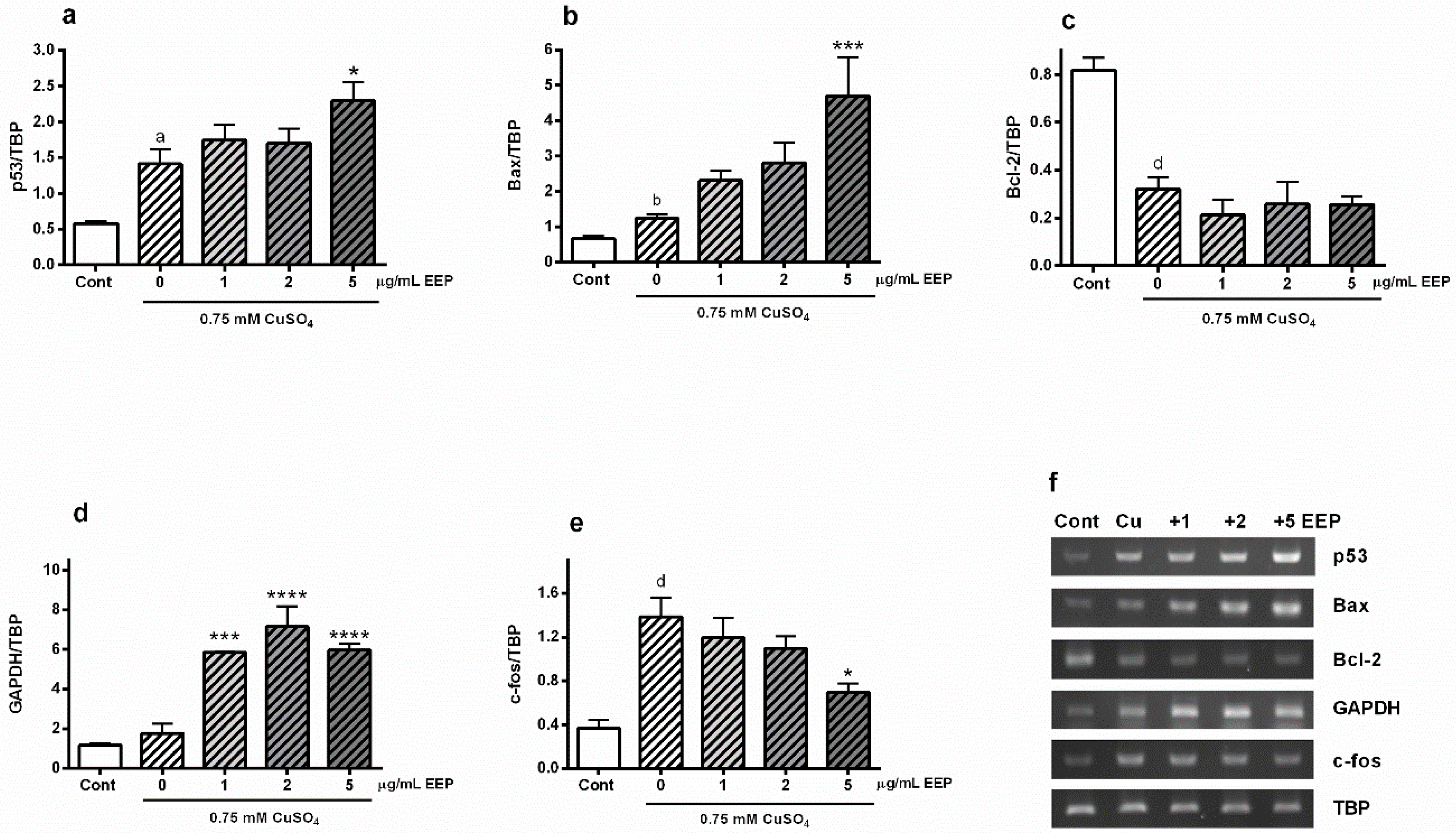
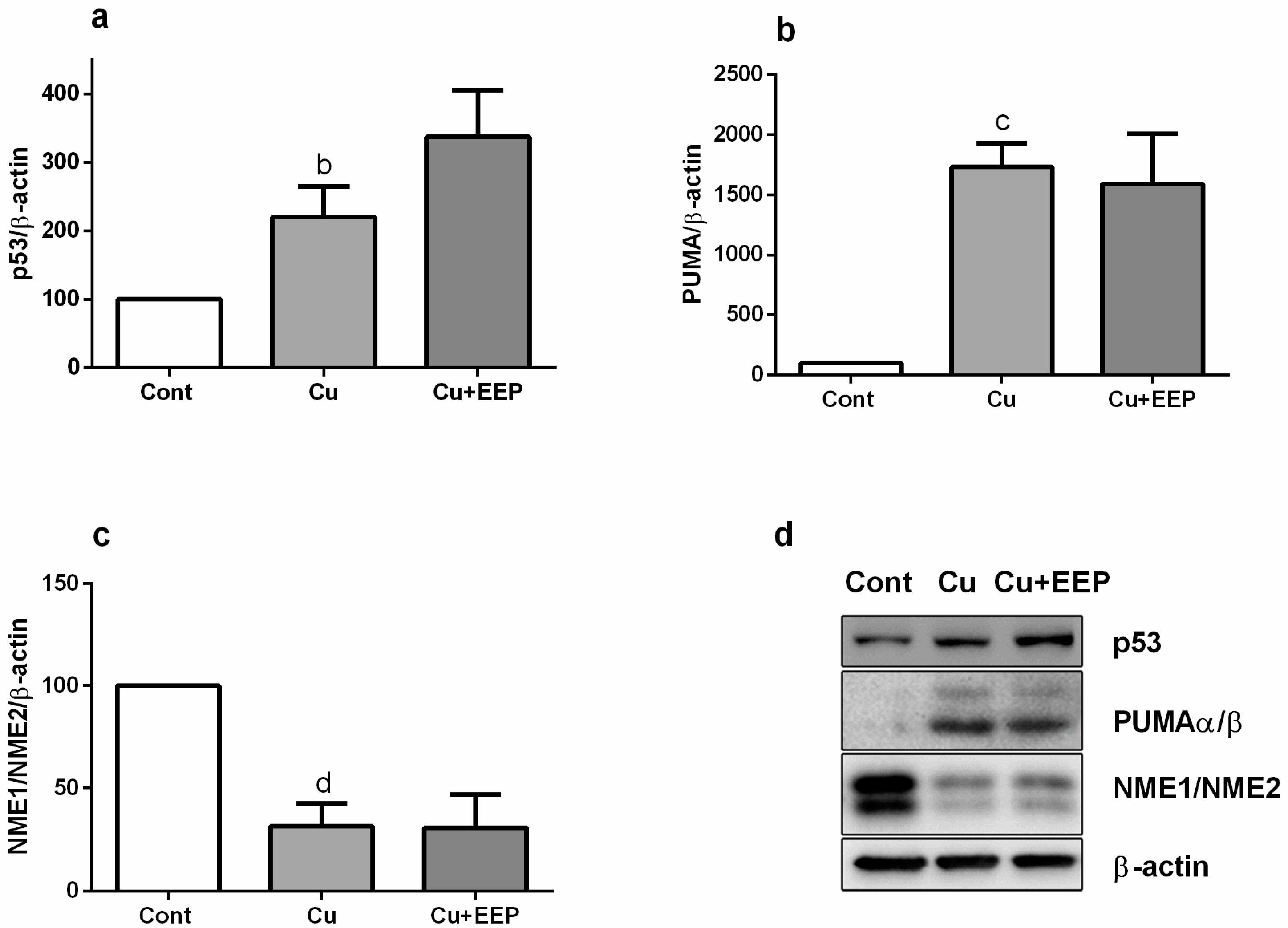
2.3. EEP Promoted Copper-Induced Up-Regulation of Bax and p53 Gene Expressions, Stimulated Transcription of GAPDH mRNA and Reduced Copper-Promoted Expression of c-fos Gene
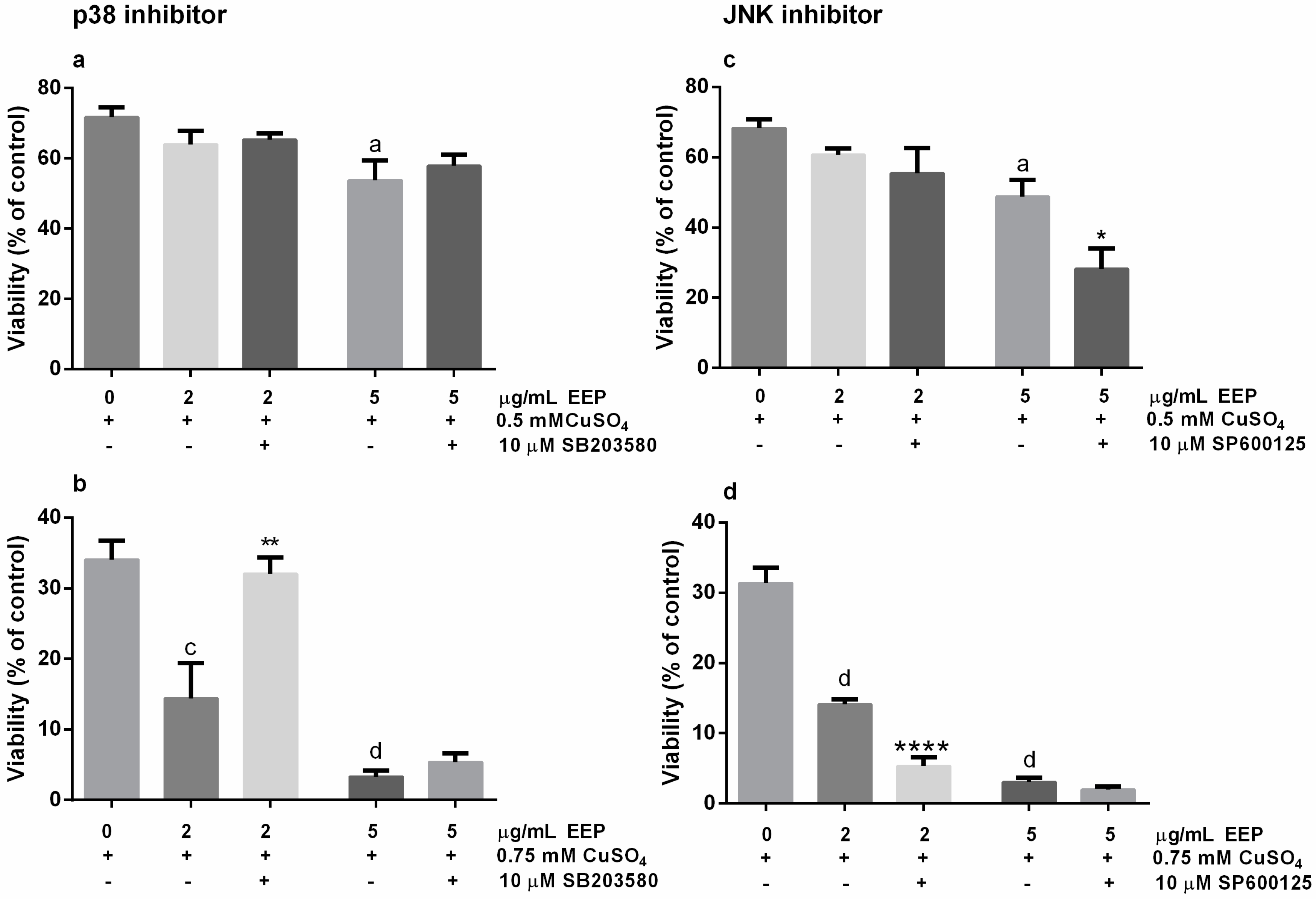
2.4. Neurotoxic Effects of EEP in The Presence of Excess Copper was Prevented by SB203580, The p38 Kinase Inhibitor, and Exacerbated by SP6000125, the JNK Pathway Inhibitor
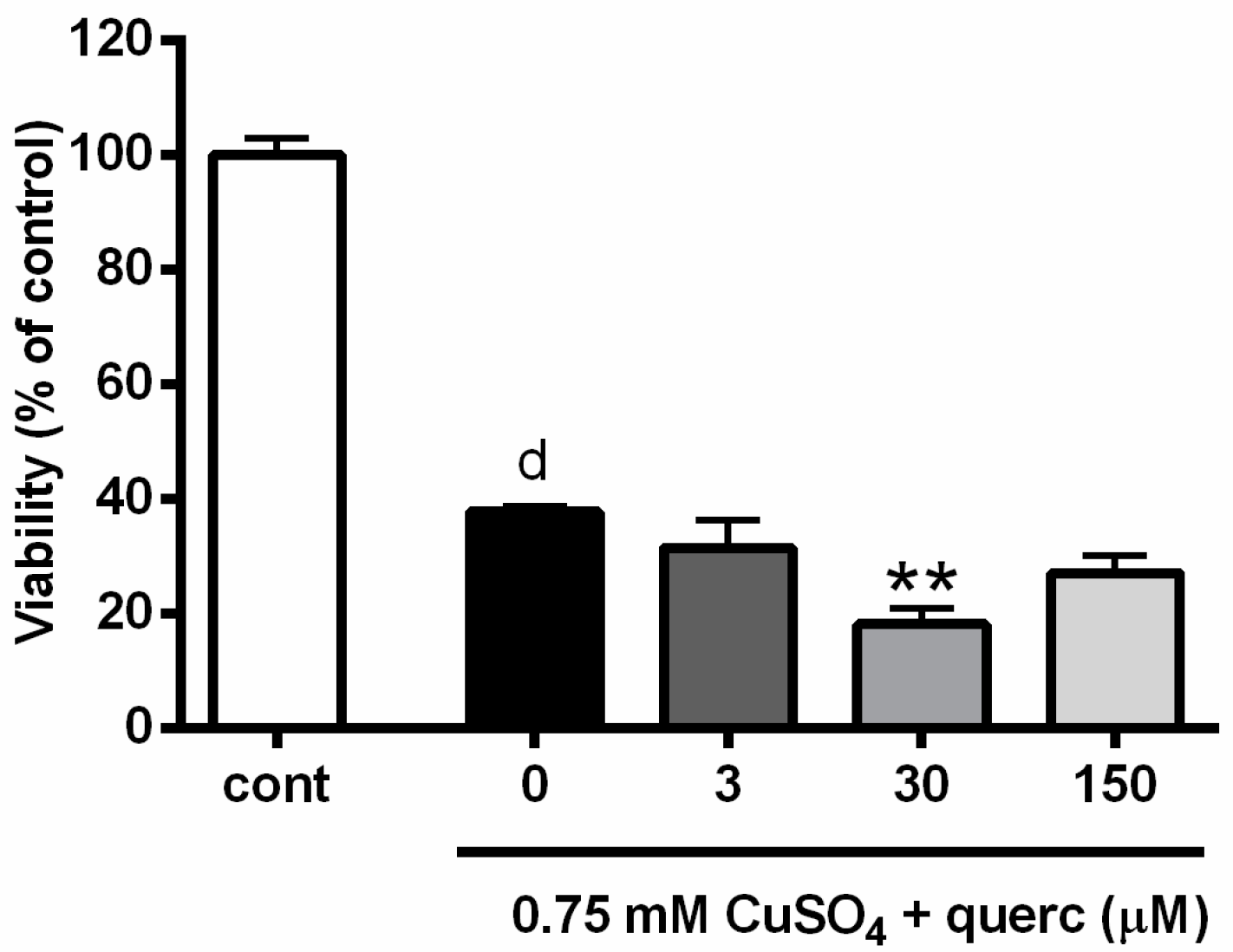
2.5. Quercetin Induced Similar Effects as Propolis and Promoted Copper-Induced Death of P19 Neuronal Cells
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Chemicals
5.2. P19 Cell Culturing and P19 Neuronal Differentiation
5.2.1. Neuronal Differentiation of P19 Cells
P19 Embryonal Body Formation (DIV 0-4)
P19 Neuronal Differentiation (DIV 4-8)
5.3. Drug Treatment
5.4. Assessment of Cell Death
5.5. Detection of ROS Levels
5.6. Determination of Caspase -3/7 Activity
5.7. Determination of mRNA Levels by Semi-Quantitative RT-PCR Method
5.8. Western Blot Analysis of p53, PUMA and NME1/2 Expression
5.9. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications. Oxid. Med. Cell Longev. 2017, 2017, 2525967. [Google Scholar] [CrossRef]
- Gaetke, L.M.; Chow-Johnson, H.S.; Chow, C.K. Copper: Toxicological relevance and mechanisms. Arch. Toxicol. 2014, 88, 1929–1938. [Google Scholar] [CrossRef]
- Jomova, K.M.; Valko, M. Advances in metal-induced oxidative stress and human disease. Toxicology 2011, 283, 65–87. [Google Scholar] [CrossRef] [PubMed]
- Jazvinscak Jembrek, M.; Vlainic, J.; Radovanovic, V.; Erhardt, J.; Orsolic, N. Effects of copper overload in P19 neurons: Impairment of glutathione redox homeostasis and crosstalk between caspase and calpain protease systems in ROS-induced apoptosis. Biometals 2014, 27, 1303–1322. [Google Scholar] [CrossRef] [PubMed]
- Letelier, M.E.; Sánchez-Jofré, S.; Peredo-Silva, L.; Cortés-Troncoso, J.; Aracena-Parks, P. Mechanisms underlying iron and copper ions toxicity in biological systems: Pro-oxidant activity and protein-binding effects. Chem. Biol. Interact. 2010, 188, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, M.; Inagawa, R.; Hosokawa, T.; Saito, T.; Kurasaki, M. Mechanism of apoptosis induced by copper in PC12 cells. Food Chem. Toxicol. 2008, 46, 2157–2164. [Google Scholar] [CrossRef]
- Paris, I.; Perez-Pastene, C.; Couve, E.; Caviedes, P.; Ledoux, S.; Segura-Aguilar, J. Copper dopamine complex induces mitochondrial autophagy preceding caspase-independent apoptotic cell death. J. Biol. Chem. 2009, 284, 13306–13315. [Google Scholar] [CrossRef]
- Ventriglia, M.; Bucossi, S.; Panetta, V.; Squitti, R. Copper in Alzheimer’s disease: A meta-analysis of serum, plasma, and cerebrospinal fluid studies. J. Alzheimers Dis. 2012, 30, 981–984. [Google Scholar] [CrossRef] [PubMed]
- Li, D.D.; Zhang, W.; Wang, Z.Y.; Zhao, P. Serum Copper, Zinc, and Iron Levels in Patients with Alzheimer’s Disease: A Meta-Analysis of Case-Control Studies. Front. Aging Neurosci 2017, 9, 300. [Google Scholar] [CrossRef]
- Martin, K.R.; Appel, C.L. Polyphenols as dietary supplements: A double-edged sword. Nutr. Diet. Suppl. 2010, 2, 1–12. [Google Scholar] [CrossRef]
- Azmi, A.S.; Sarkar, F.H.; Hadi, S.M. Pro-oxidant activity of dietary chemopreventive agents: An under-appreciated anti-cancer property. F1000Res 2013, 2, 135. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, Z.; Ren, Z.; Zhang, J.; Chuang, C.C.; Kandaswamy, E.; Zhou, T.; Zuo, L. Role of ROS and Nutritional Antioxidants in Human Diseases. Front. Physiol. 2018, 17, 477. [Google Scholar] [CrossRef]
- Orsolic, N.; Sirovina, D.; Gajski, G.; Garaj-Vrhovac, V.; Jazvinscak Jembrek, M.; Kosalec, I. Assessment of DNA damage and lipid peroxidation in diabetic mice: Effects of propolis and epigallocatechin gallate (EGCG). Mutat. Res. 2013, 757, 36–44. [Google Scholar] [CrossRef]
- Pasupuleti, V.R.; Sammugam, L.; Ramesh, N.; Gan, S.H. Honey, Propolis, and Royal Jelly: A Comprehensive Review of Their Biological Actions and Health Benefits. Oxid. Med. Cell Longev. 2017, 2017, 1259510. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Wu, Z.; Meng, J.; Zhu, A.; Zhong, X.; Wu, S.; Nakanishi, H. The Neuroprotective Effects of Brazilian Green Propolis on Neurodegenerative Damage in Human Neuronal SH-SY5Y Cells. Oxid. Med. Cell Longev. 2017, 2017, 7984327. [Google Scholar] [CrossRef]
- Shimazawa, M.; Chikamatsu, S.; Morimoto, N.; Mishima, S.; Nagai, H.; Hara, H. Neuroprotection by Brazilian Green Propolis against In vitro and In vivo Ischemic Neuronal Damage. Evid. Based Complement. Alternat. Med. 2005, 2, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Kalogeropoulos, N.; Konteles, S.J.; Troullidou, E.; Mourtzinos, I.; Karathanos, V.T. Chemical composition, antioxidant activity and antimicrobial properties of propolis extracts from Greece and Cyprus. Food Chem. 2009, 116, 452–461. [Google Scholar] [CrossRef]
- Franca, J.R.; De Luca, M.P.; Ribeiro, T.G.; Castilho, R.O.; Moreira, A.N.; Santos, V.R.; Faraco, A.A. Propolis--based chitosan varnish: Drug delivery, controlled release and antimicrobial activity against oral pathogen bacteria. BMC Complement Altern. Med. 2014, 14, 478. [Google Scholar] [CrossRef]
- Cregan, S.P.; MacLaurin, J.G.; Craig, C.G.; Robertson, G.S.; Nicholson, D.W.; Park, D.S.; Slack, R.S. Bax-dependent caspase-3 activation is a key determinant in p53-induced apoptosis in neurons. J. Neurosci 1999, 19, 7860–7869. [Google Scholar] [CrossRef]
- Jazvinscak Jembrek, M.; Radovanovic, V.; Vlainic, J.; Vukovic, L.; Hanzic, N. Neuroprotective effect of zolpidem against glutamate-induced toxicity is mediated via the PI3K/Akt pathway and inhibited by PK11195. Toxicology 2018, 406–407, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Steckley, D.; Karajgikar, M.; Dale, L.B.; Fuerth, B.; Swan, P.; Drummond-Main, C.; Poulter, M.O.; Ferguson, S.S.; Strasser, A.; Cregan, S.P. Puma is a dominant regulator of oxidative stress induced Bax activation and neuronal apoptosis. J. Neurosci. 2007, 27, 12989–12999. [Google Scholar] [CrossRef] [PubMed]
- Zhai, D.; Chin, K.; Wang, M.; Liu, F. Disruption of the nuclear p53-GAPDH complex protects against ischemia-induced neuronal damage. Mol. Brain 2014, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Oswald, M.C.W.; Garnham, N.; Sweeney, S.T.; Landgraf, M. Regulation of neuronal development and function by ROS. FEBS Lett. 2018, 592, 679–691. [Google Scholar] [CrossRef]
- Peuchant, E.; Bats, M.L.; Moranvillier, I.; Lepoivre, M.; Guitton, J.; Wendum, D.; Lacombe, M.L.; Moreau-Gaudry, F.; Boissan, M.; Dabernat, S. Metastasis suppressor NM23 limits oxidative stress in mammals by preventing activation of stress-activated protein kinases/JNKs through its nucleoside diphosphate kinase activity. FASEB J. 2017, 31, 1531–1546. [Google Scholar] [CrossRef]
- Cardaci, S.; Filomeni, G.; Rotilio, G.; Ciriolo, M.R. p38(MAPK)/p53 signalling axis mediates neuronal apoptosis in response to tetrahydrobiopterin-induced oxidative stress and glucose uptake inhibition: Implication for neurodegeneration. Biochem. J. 2010, 430, 439–451. [Google Scholar] [CrossRef]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef]
- Zheng, Y.Z.; Deng, G.; Liang, Q.; Chen, D.F.; Guo, R.; Lai, R.C. Antioxidant Activity of Quercetin and Its Glucosides from Propolis: A Theoretical Study. Sci. Rep. 2017, 7, 7543. [Google Scholar] [CrossRef]
- Jazvinscak Jembrek, M.; Vukovic, L.; Puhovic, J.; Erhardt, J.; Orsolic, N. Neuroprotective effect of quercetin against hydrogen peroxide-induced oxidative injury in P19 neurons. J. Mol. Neurosci. 2012, 47, 286–299. [Google Scholar] [CrossRef]
- Jazvinscak Jembrek, M.; Vlainic, J.; Cadez, V.; Segota, S. Atomic force microscopy reveals new biophysical markers for monitoring subcellular changes in oxidative injury: Neuroprotective effects of quercetin at the nanoscale. PLoS ONE 2018, 13, e0200119. [Google Scholar] [CrossRef]
- Swamy, M.; Norlina, W.; Azman, W.; Suhaili, D.; Sirajudeen, K.N.; Mustapha, Z.; Govindasamy, C. Restoration of glutamine synthetase activity, nitric oxide levels and amelioration of oxidative stress by propolis in kainic acid mediated excitotoxicity. Afr. J. Tradit. Complement. Altern. Med. 2014, 11, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Sawicka, D.; Car, H.; Borawska, M.H.; Nikliński, J. The anticancer activity of propolis. Folia. Histochem. Cytobiol. 2012, 50, 25–37. [Google Scholar] [CrossRef]
- Begnini, K.R.; Moura de Leon, P.M.; Thurow, H.; Schultze, E.; Campos, V.F.; Martins Rodrigues, F.; Borsuk, S.; Dellagostin, O.A.; Savegnago, L.; Roesch-Ely, M.; et al. Brazilian red propolis induces apoptosis-like cell death and decreases migration potential in bladder cancer cells. Evid. Based. Complement. Alternat. Med. 2014, 639856. [Google Scholar] [CrossRef] [PubMed]
- Frión-Herrera, Y.; Díaz-García, A.; Ruiz-Fuentes, J.; Rodríguez-Sánchez, H.; Sforcin, J.M. Brazilian green propolis induced apoptosis in human lung cancer A549 cells through mitochondrial-mediated pathway. J. Pharm. Pharmacol. 2015, 67, 1448–1456. [Google Scholar] [CrossRef]
- Salim, E.I.; Abd El-Magid, A.D.; Farara, K.M.; Maria, D.S. Antitumoral and Antioxidant Potential of Egyptian Propolis Against the PC3 Prostate Cancer Cell Line. Asian Pac. J. Cancer Prev. 2015, 16, 7641–7651. [Google Scholar] [CrossRef] [PubMed]
- Benkovic, V.; Knezevic, A.H.; Dikic, D.; Lisicic, D.; Orsolic, N.; Basic, I.; Kosalec, I.; Kopjar, N. Radioprotective effects of propolis and quercetin in gamma-irradiated mice evaluated by the alkaline comet assay. Phytomedicine 2008, 15, 851–858. [Google Scholar] [CrossRef] [PubMed]
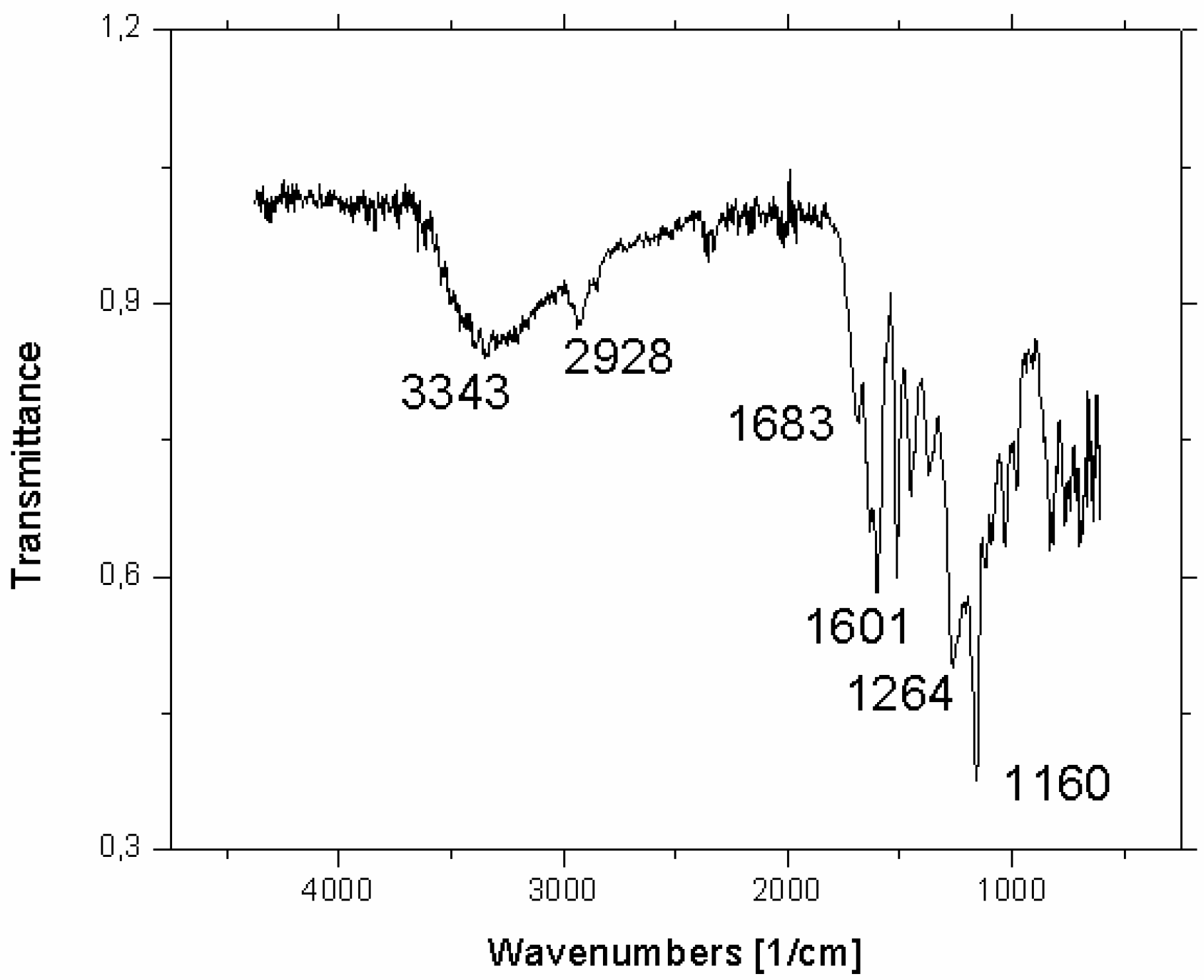
- Baranovic, G.; Segota, S. Infrared spectroscopy of flavones and flavonols. Reexamination of the hydroxyl and carbonyl vibrations in relation to the interactions of flavonoids with membrane lipids. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2018, 192, 473–486. [Google Scholar] [CrossRef]
- Boisard, S.; Le Ray, A.M.; Gatto, J.; Aumond, M.C.; Blanchard, P.; Derbré, S.; Flurin, C.; Richomme, P. Chemical composition, antioxidant and anti-AGEs activities of a French poplar type propolis. J. Agric. Food Chem. 2014, 62, 1344–1351. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.C.; Wang, Y.H.; Liou, C.C.; Lin, Y.C.; Huang, H.; Liu, Y.C. Induction of oxidative DNA damage by flavonoids of propolis: Its mechanism and implication about antioxidant capacity. Chem. Res. Toxicol. 2012, 25, 191–196. [Google Scholar] [CrossRef]
- Culmsee, C.; Mattson, M.P. p53 in neuronal apoptosis. Biochem. Biophys. Res. Commun. 2005, 331, 761–777. [Google Scholar] [CrossRef]
- Wong, H.K.; Fricker, M.; Wyttenbach, A.; Villunger, A.; Michalak, E.M.; Strasser, A.; Tolkovsky, A.M. Mutually exclusive subsets of BH3-only proteins are activated by the p53 and c-Jun N-terminal kinase/c-Jun signaling pathways during cortical neuron apoptosis induced by arsenite. Mol. Cell Biol. 2005, 25, 8732–8747. [Google Scholar] [CrossRef]
- Chatoo, W.; Abdouh, M.; Bernier, G. p53 pro-oxidant activity in the central nervous system: Implication in aging and neurodegenerative diseases. Antioxid. Redox. Signal. 2011, 15, 1729–1737. [Google Scholar] [CrossRef]
- Fricker, M.; O’Prey, J.; Tolkovsky, A.M.; Ryan, K.M. Phosphorylation of Puma modulates its apoptotic function by regulating protein stability. Cell Death Dis. 2010, 1, e59. [Google Scholar] [CrossRef]
- Aleyasin, H.; Cregan, S.P.; Iyirhiaro, G.; O’Hare, M.J.; Callaghan, S.M.; Slack, R.S.; Park, D.S. Nuclear factor-(kappa)B modulates the p53 response in neurons exposed to DNA damage. J. Neurosci. 2004, 24, 2963–2973. [Google Scholar] [CrossRef]
- Draganova-Filipova, M.N.; Georgieva, M.G.; Peycheva, E.N.; Miloshev, G.A.; Sarafian, V.S.; Peychev, L.P. Effects of propolis and CAPE on proliferation and apoptosis of McCoy-Plovdiv cell line. Folia Med. (Plovdiv) 2008, 50, 53–59. [Google Scholar]
- Chen, R.W.; Saunders, P.A.; Wei, H.; Li, Z.; Seth, P.; Chuang, D.M. Involvement of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and p53 in neuronal apoptosis: Evidence that GAPDH is upregulated by p53. J. Neurosci. 1999, 19, 9654–9662. [Google Scholar] [CrossRef]
- Wang, D.B.; Kinoshita, C.; Kinoshita, Y.; Morrison, R.S. p53 and mitochondrial function in neurons. Biochim. Biophys. Acta 2014, 1842, 1186–1197. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Chen, Y.; St Clair, D.K. ROS and p53: A versatile partnership. Free Radic. Biol. Med. 2008, 44, 1529–1535. [Google Scholar] [CrossRef]
- Xuan, H.; Li, Z.; Yan, H.; Sang, Q.; Wang, K.; He, Q.; Wang, Y.; Hu, F. Antitumor Activity of Chinese Propolis in Human Breast Cancer MCF-7 and MDA-MB-231 Cells. Evid. Based Complement. Alternat. Med. 2014, 2014, 280120. [Google Scholar] [CrossRef]
- Wu, F.; Wang, Z.; Gu, J.H.; Ge, J.B.; Liang, Z.Q.; Qin, Z.H. p38(MAPK)/p53-Mediated Bax induction contributes to neurons degeneration in rotenone-induced cellular and rat models of Parkinson’s disease. Neurochem. Int. 2013, 63, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Formisano, L.; Guida, N.; Laudati, G.; Boscia, F.; Esposito, A.; Secondo, A.; Di Renzo, G.; Canzoniero, L.M. Extracellular signal-related kinase 2/specificity protein 1/specificity protein 3/repressor element-1 silencing transcription factor pathway is involved in Aroclor 1254-induced toxicity in SH-SY5Y neuronal cells. J. Neurosci. Res. 2015, 93, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Guida, N.; Laudati, G.; Mascolo, L.; Valsecchi, V.; Sirabella, R.; Selleri, C.; Di Renzo, G.; Canzoniero, L.M.; Formisano, L. p38/Sp1/Sp4/HDAC4/BDNF Axis Is a Novel Molecular Pathway of the Neurotoxic Effect of the Methylmercury. Front. Neurosci. 2017, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Guida, N.; Valsecchi, V.; Laudati, G.; Serani, A.; Mascolo, L.; Molinaro, P.; Montuori, P.; Di Renzo, G.; Canzoniero, L.M.; Formisano, L. The miR206-JunD Circuit Mediates the Neurotoxic Effect of Methylmercury in Cortical Neurons. Toxicol. Sci. 2018, 163, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Bragado, P.; Armesilla, A.; Silva, A.; Porras, A. Apoptosis by cisplatin requires p53 mediated p38α MAPK activation through ROS generation. Apoptosis 2007, 12, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Liu, A.; Ming, X.; Deng, P.; Jiang, Y. UV-induced interaction between p38 MAPK and p53 serves as a molecular switch in determining cell fate. FEBS Lett. 2010, 584, 4711–4716. [Google Scholar] [CrossRef] [PubMed]
- Dewil, M.; dela Cruz, V.F.; Van Den Bosch, L.; Robberecht, W. Inhibition of p38 mitogen activated protein kinase activation and mutant SOD1(G93A)-induced motor neuron death. Neurobiol. Dis. 2007, 26, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Maher, P. How protein kinase C activation protects nerve cells from oxidative stress-induced cell death. J. Neurosci. 2001, 21, 2929–2938. [Google Scholar] [CrossRef]
- Zheng, L.; Ishii, Y.; Tokunaga, A.; Hamashima, T.; Shen, J.; Zhao, Q.L.; Ishizawa, S.; Fujimori, T.; Nabeshima, Y.; Mori, H.; et al. Neuroprotective effects of PDGF against oxidative stress and the signaling pathway involved. J. Neurosci. Res. 2010, 88, 1273–1284. [Google Scholar] [CrossRef] [PubMed]
- Aihara, K.; Kuroda, S.; Kanayama, N.; Matsuyama, S.; Tanizawa, K.; Horie, M. A neuron-specific EGF family protein, NELL2, promotes survival of neurons through mitogen-activated protein kinases. Brain Res. Mol. Brain Res. 2003, 116, 86–93. [Google Scholar] [CrossRef]
- Huang, S.; Zhang, C.P.; Wang, K.; Li, G.Q.; Hu, F.L. Recent advances in the chemical composition of propolis. Molecules 2014, 19, 19610–19632. [Google Scholar] [CrossRef]
- Tlak Gajger, I.T.; Pavlovic, I.; Bojic, M.; Kosalec, I.; Srecec, S.; Vlainic, T.; Vlainic, J. The Components Responsible for the Antimicrobial Activity of Propolis from Continental and Mediterranean Regions in Croatia. CJFS 2017, 35, 376–385. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Hirasawa, T.; Maruyama, Y.; Ishii, Y.; Ito, R.; Saito, K.; Umemura, T.; Nishikawa, A.; Nakazawa, H. Effect of interaction between phenolic compounds and copper ion on antioxidant and pro-oxidant activities. Toxicol. In Vitro 2011, 25, 1320–1327. [Google Scholar] [CrossRef] [PubMed]
- Boots, A.W.; Li, H.; Schins, R.P.; Duffin, R.; Heemskerk, J.W.; Bast, A.; Haenen, G.R. The quercetin paradox. Toxicol. Appl. Pharmacol. 2007, 222, 89–96. [Google Scholar] [CrossRef]
- Filipe, P.; Haigle, J.; Silva, J.N.; Freitas, J.; Fernandes, A.; Mazière, J.C.; Mazière, C.; Santus, R.; Morlière, P. Anti- and pro-oxidant effects of quercetin in copper-induced low density lipoprotein oxidation. Quercetin as an effective antioxidant against pro-oxidant effects of urate. Eur. J. Biochem. 2004, 271, 1991–1999. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence (5′ → 3′) | Product Length (bp) | Annealing Temp. (°C) | Number of Cycles |
|---|---|---|---|---|
| Bcl-2 | F: GGAGATCGTGATGAAGTACATAC R: CCTGAAGAGTTCCTCCACCACC | 373 | 58 | 27–28 |
| Bax | F: ATCGAGCAGGGAGGATGGCT R: CTTCCAGATGGTGAGCGAGG | 470 | 62 | 27–28 |
| c-fos | F: GAATGGTGAAGACCGTGTCAGG R: CGTTGCTGATGCTCTTGACTGG | 456 | 60 | 24–25 |
| p53 | F: AGAGACCGCCGTACAGAAGA R: CTGTAGCATGGGCATCCTTT | 231 | 62 | 31–32 |
| GAPDH | F: ACCACAGTCCATGCCATCAC R: TCCACCACCCTGTTGCTGTA | 452 | 60 | 19–20 |
| TBP | F: ACCCTTCACCAATGACTCCTATG R: ATGATGACTGCAGCAAATCGC | 190 | 60 | 26–27 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radovanović, V.; Vlainić, J.; Hanžić, N.; Ukić, P.; Oršolić, N.; Baranović, G.; Jazvinšćak Jembrek, M. Neurotoxic Effect of Ethanolic Extract of Propolis in the Presence of Copper Ions is Mediated through Enhanced Production of ROS and Stimulation of Caspase-3/7 Activity. Toxins 2019, 11, 273. https://doi.org/10.3390/toxins11050273
Radovanović V, Vlainić J, Hanžić N, Ukić P, Oršolić N, Baranović G, Jazvinšćak Jembrek M. Neurotoxic Effect of Ethanolic Extract of Propolis in the Presence of Copper Ions is Mediated through Enhanced Production of ROS and Stimulation of Caspase-3/7 Activity. Toxins. 2019; 11(5):273. https://doi.org/10.3390/toxins11050273
Chicago/Turabian StyleRadovanović, Vedrana, Josipa Vlainić, Nikolina Hanžić, Petra Ukić, Nada Oršolić, Goran Baranović, and Maja Jazvinšćak Jembrek. 2019. "Neurotoxic Effect of Ethanolic Extract of Propolis in the Presence of Copper Ions is Mediated through Enhanced Production of ROS and Stimulation of Caspase-3/7 Activity" Toxins 11, no. 5: 273. https://doi.org/10.3390/toxins11050273
APA StyleRadovanović, V., Vlainić, J., Hanžić, N., Ukić, P., Oršolić, N., Baranović, G., & Jazvinšćak Jembrek, M. (2019). Neurotoxic Effect of Ethanolic Extract of Propolis in the Presence of Copper Ions is Mediated through Enhanced Production of ROS and Stimulation of Caspase-3/7 Activity. Toxins, 11(5), 273. https://doi.org/10.3390/toxins11050273