Deoxynivalenol Modulates the Viability, ROS Production and Apoptosis in Prostate Cancer Cells

 ,
,  ,
, 
 ,
,
Abstract
1. Introduction
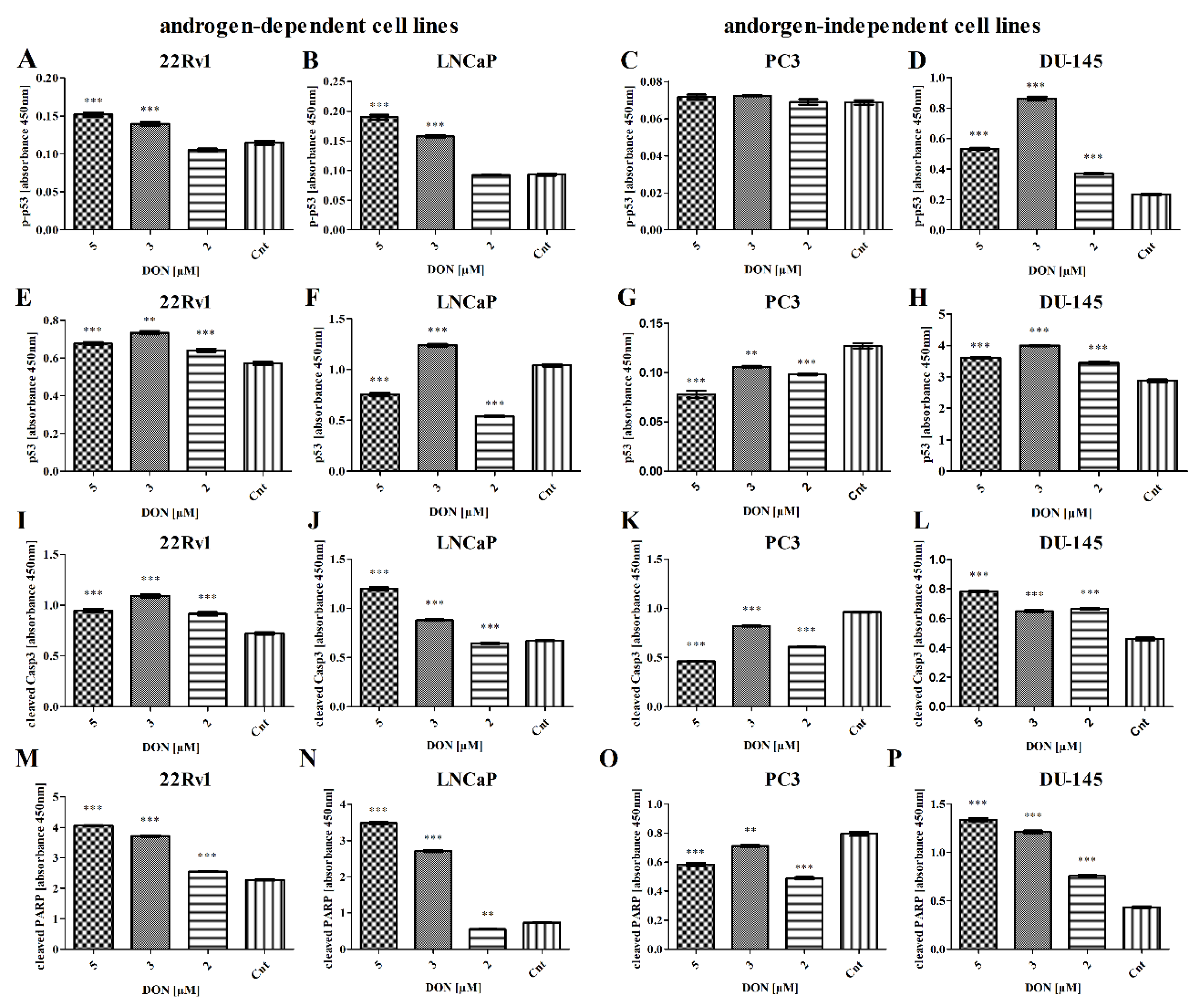
2. Results
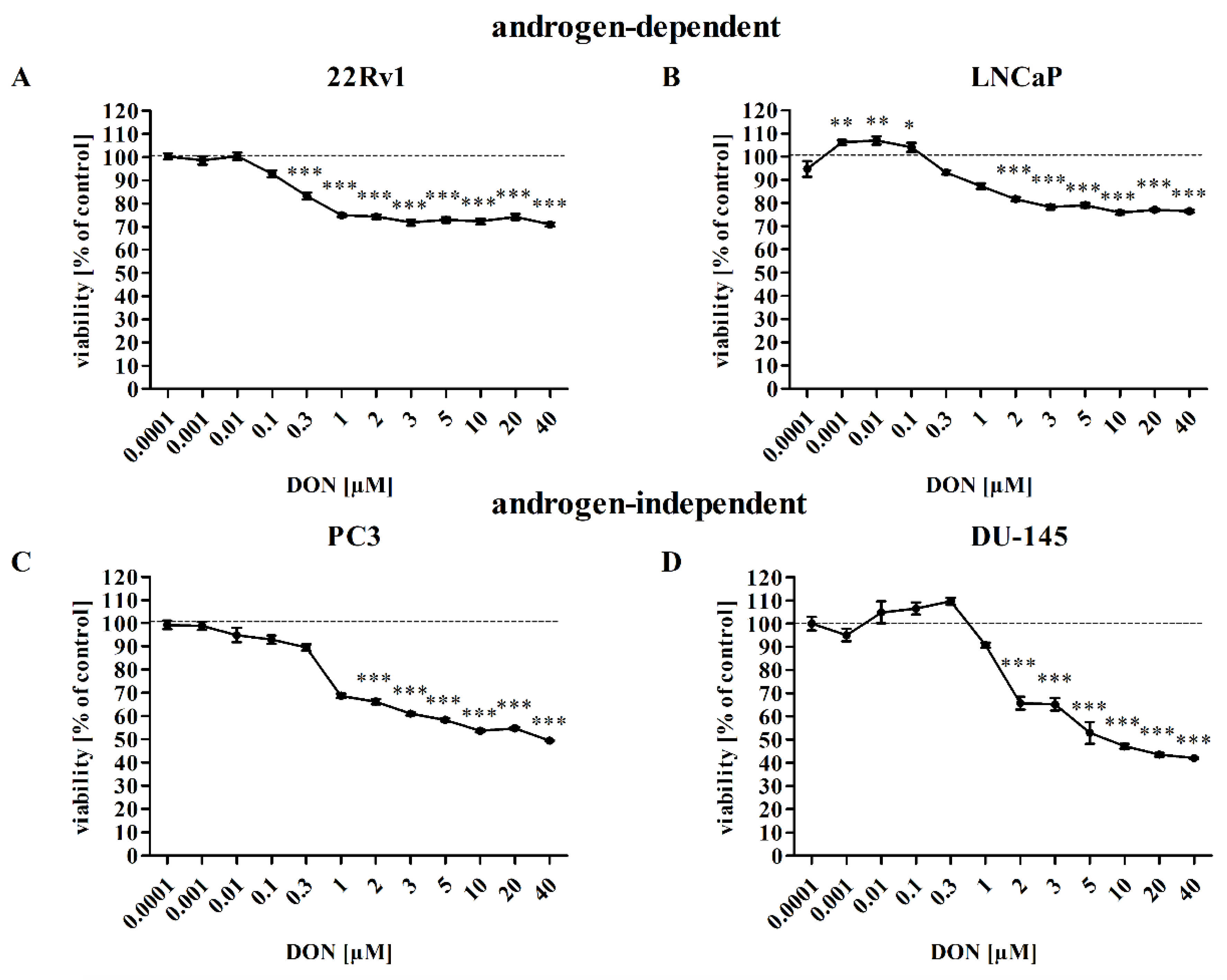
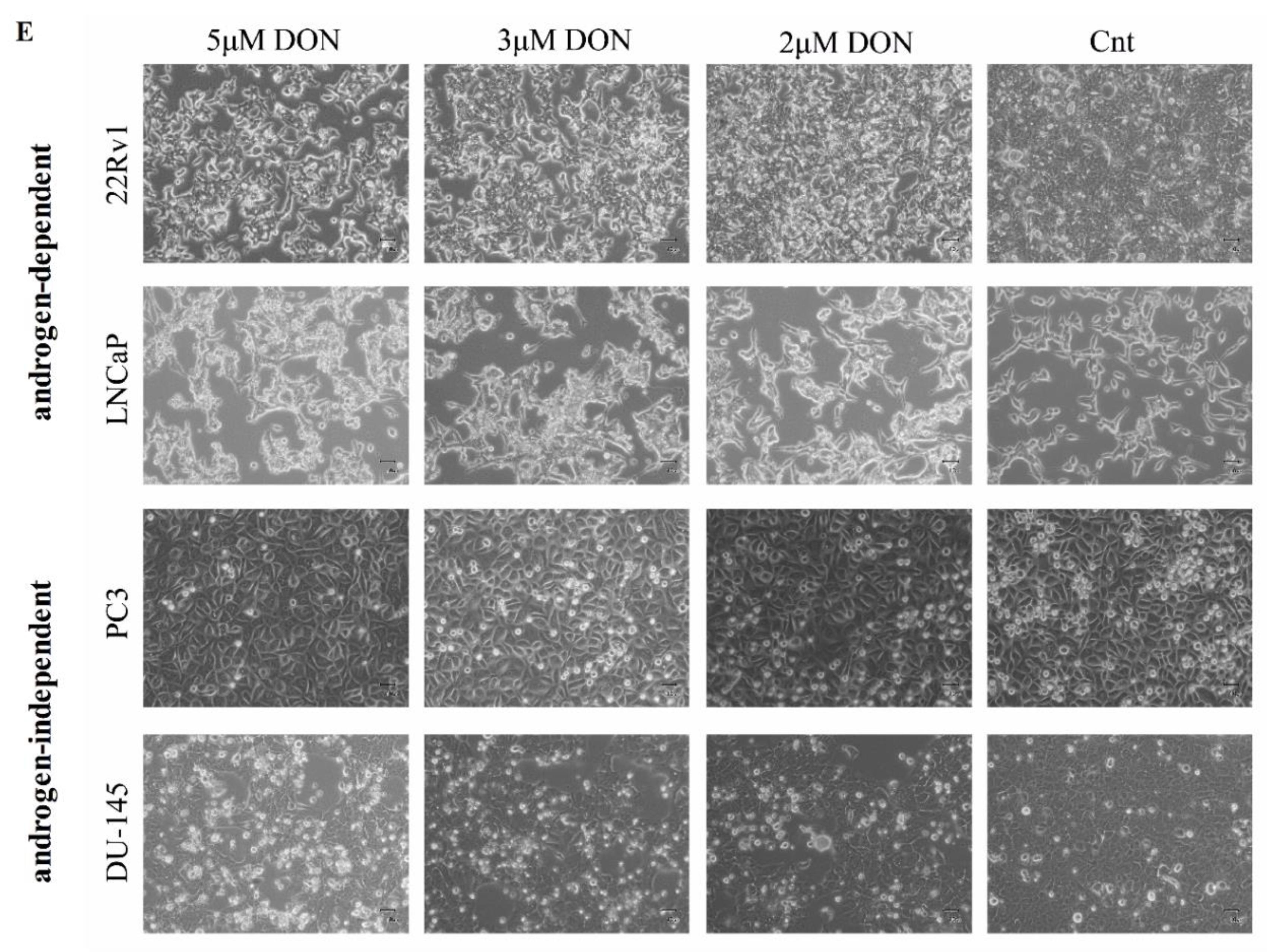
2.1. DON Decreases Viability of Prostate Cancer Cells
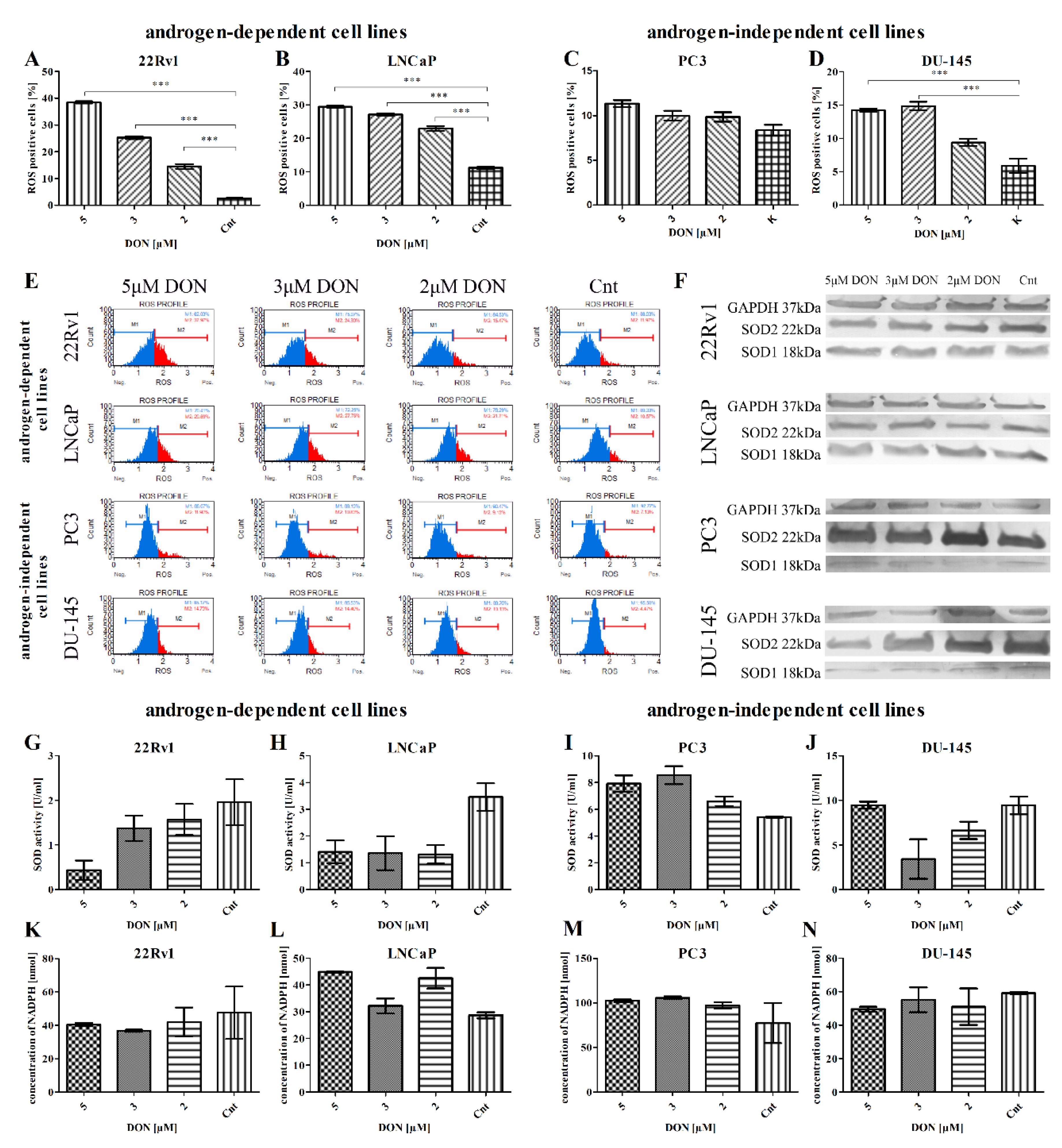
2.2. DON Induces ROS Production in PCa Cells
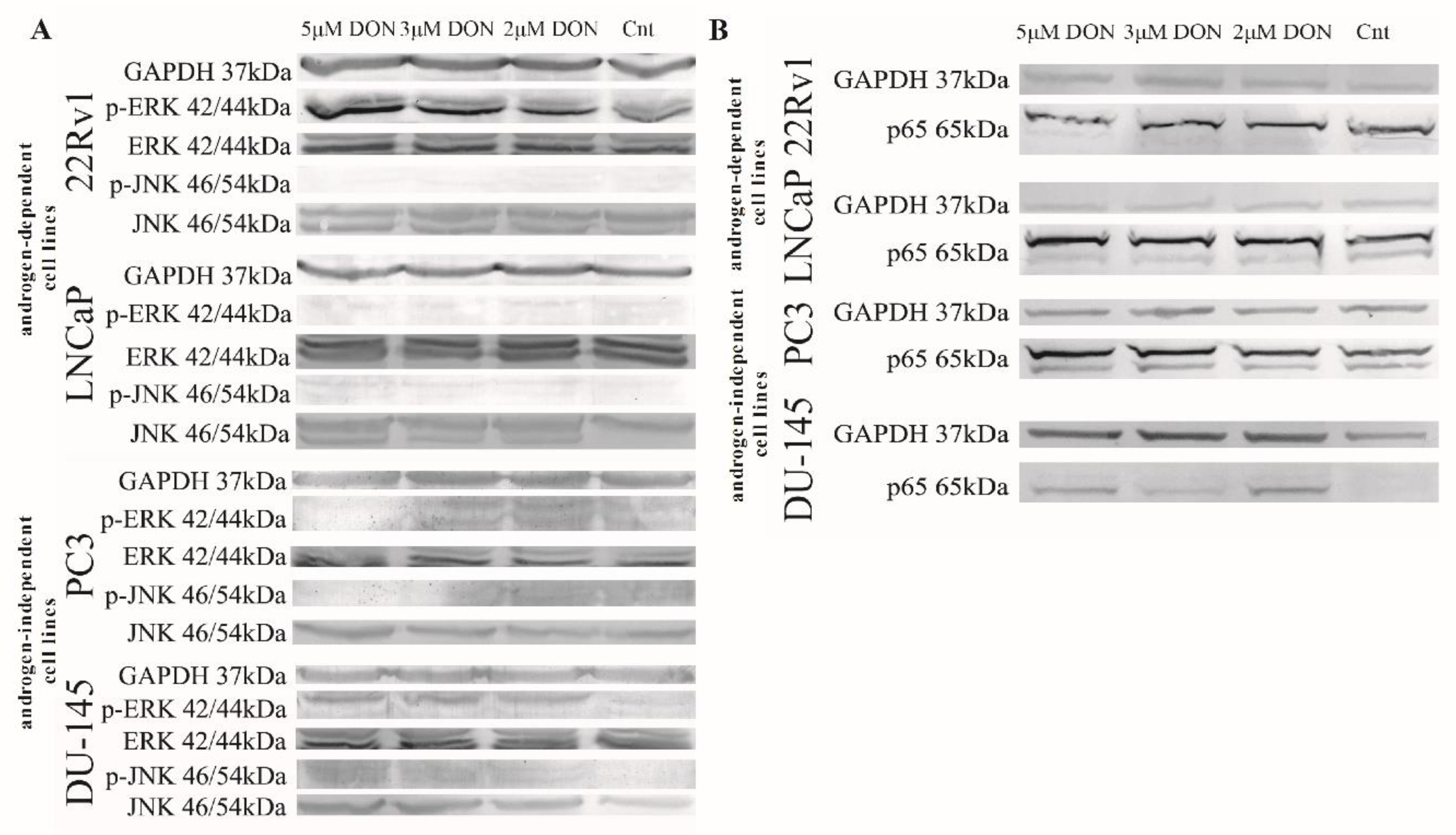
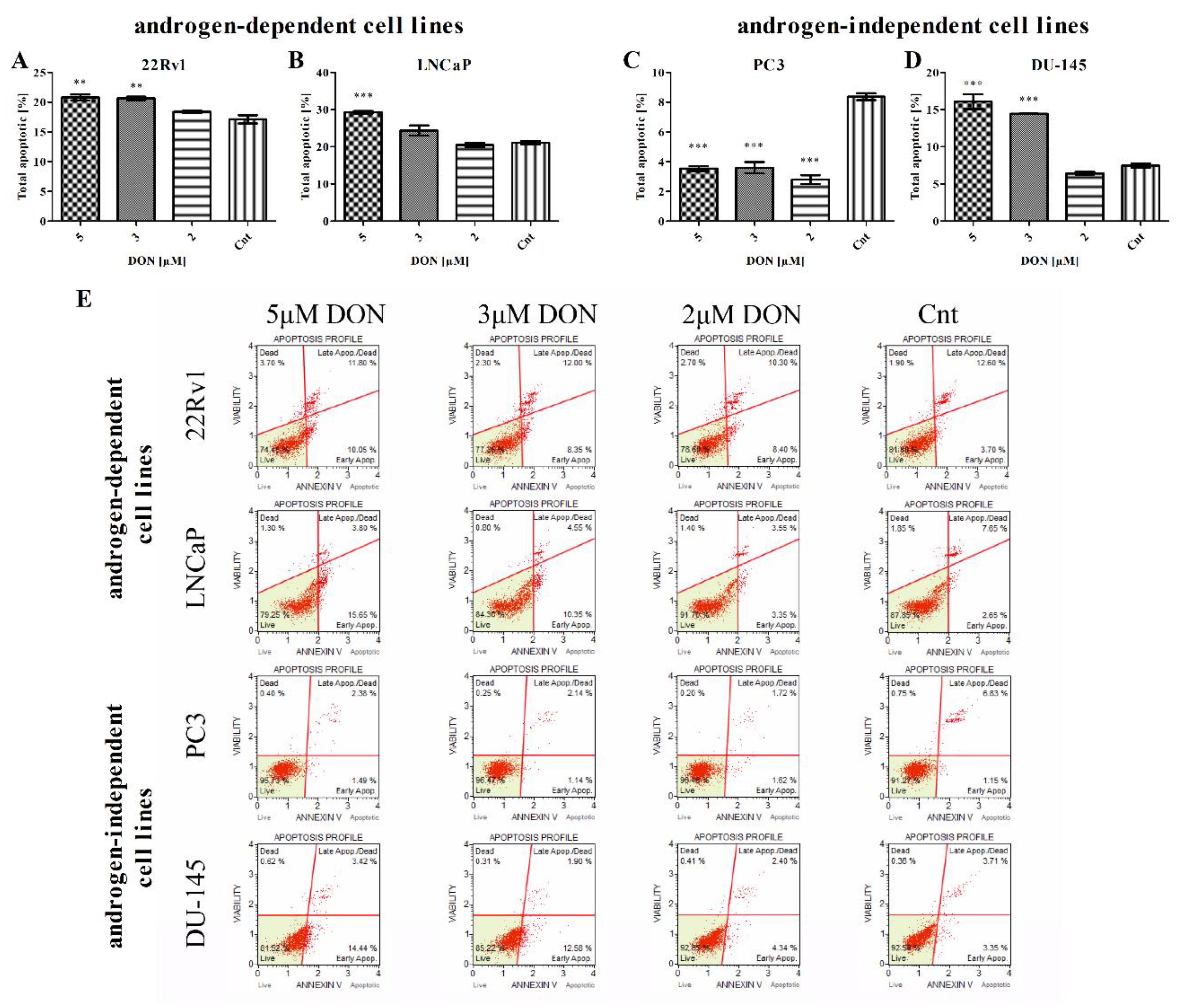
2.3. DON Induces Apoptosis in PCa Cells
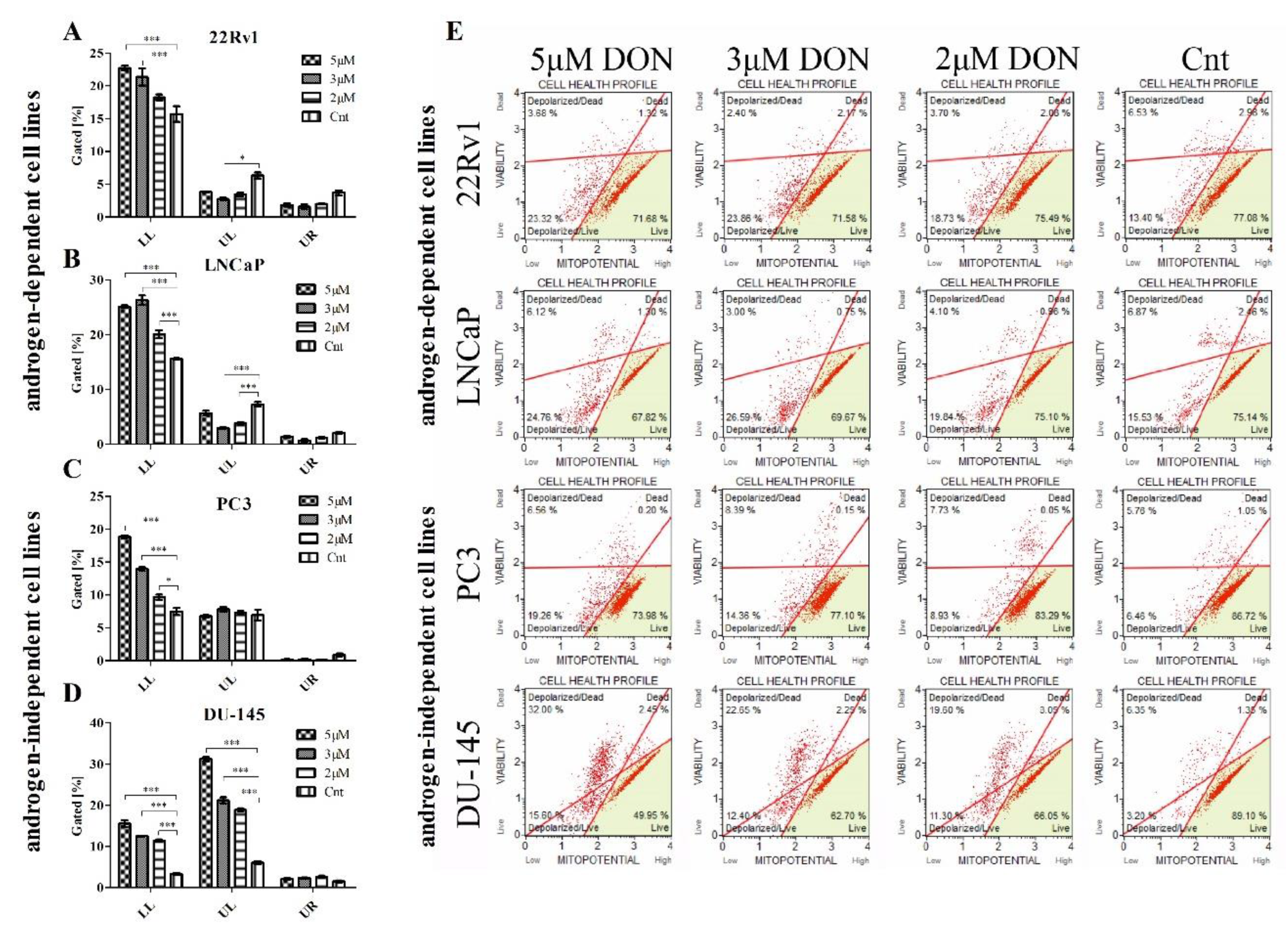
2.4. DON Modulates the Mitochondrial Potential in PCa Cells
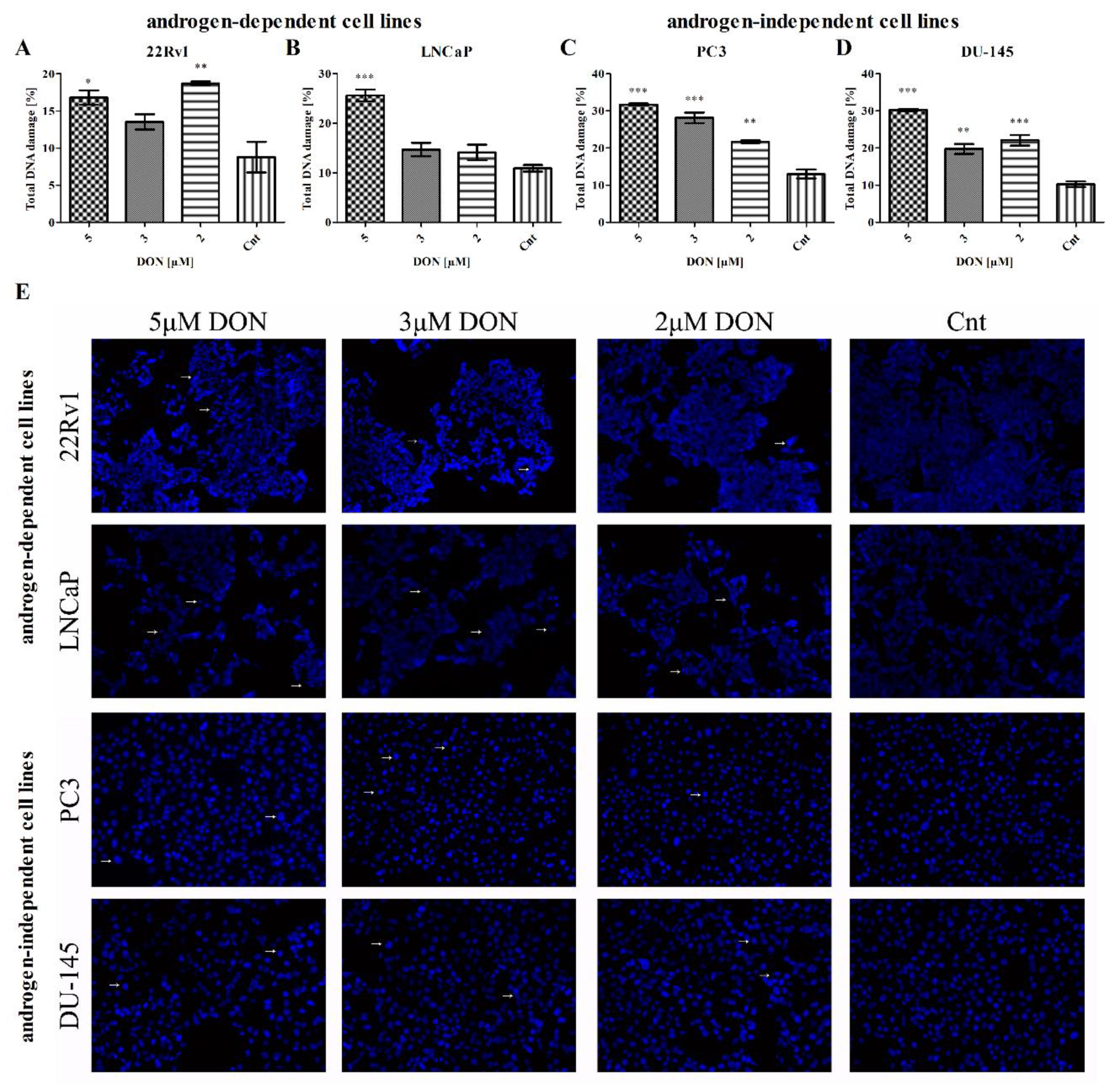
2.5. DON Induces DNA Damage in PCa Cells
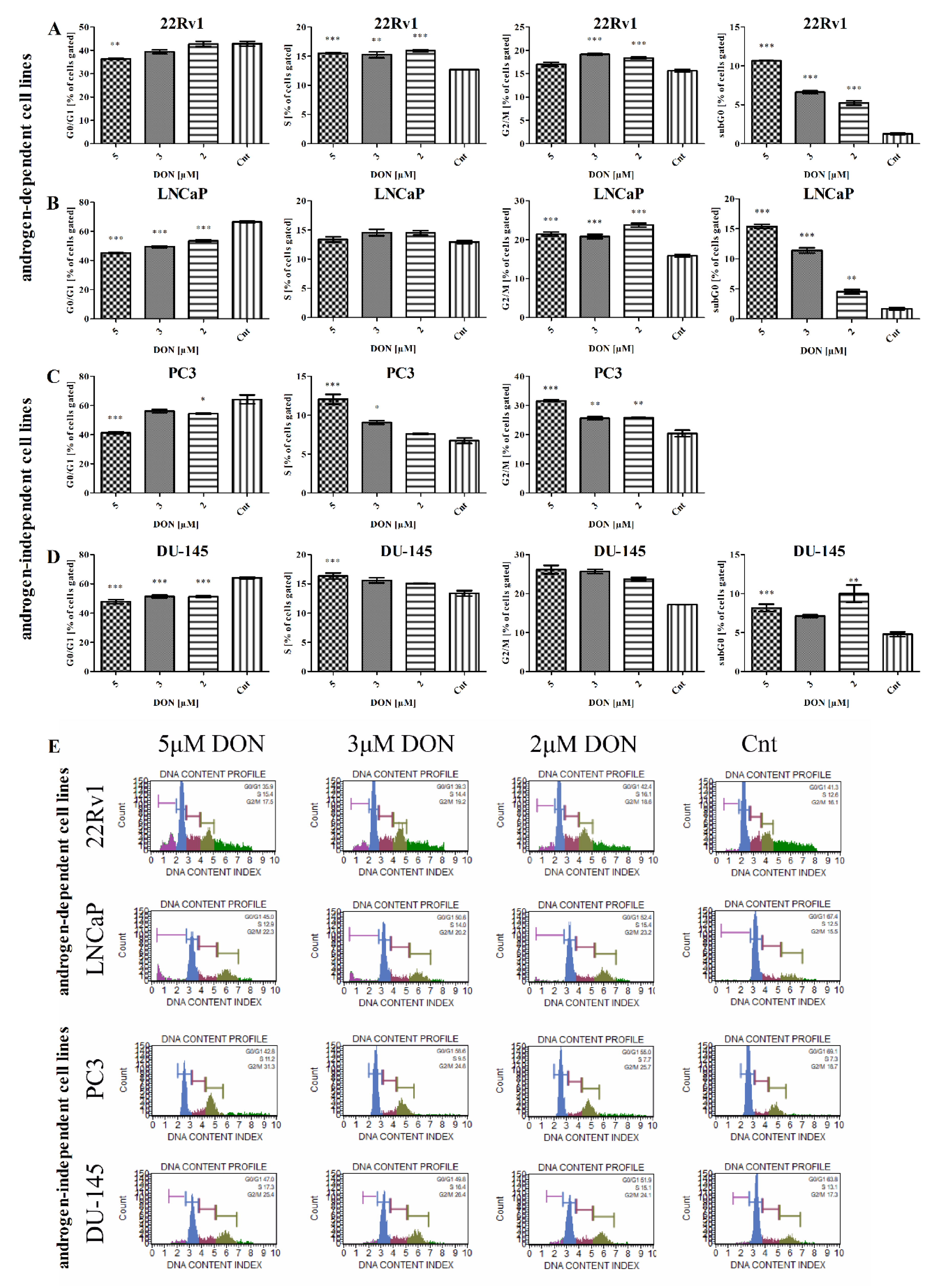
2.6. DON Modulates Cell Cycle in PCa Cells
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Cell Culture and DON Exposure
5.2. Cell Viability
5.3. Morphological Investigation
5.4. Cell Cycle Analysis
5.5. Analysis of Changes in Mitochondrial Transmembrane Potential
5.6. Annexin V and Dead Cell Double Staining Assay
5.7. Multi-Color DNA Damage
5.8. Morphology of Mitochondria and Nuclei
5.9. Evaluation of Reactive Oxygen Species (ROS) Levels
5.10. Measurement of Cu/Zn Superoxide Dismutase (SOD1) and Glutathione Peroxidase (GPx) Activity
5.11. Expression of Apoptotic Proteins
5.12. Real Time quantitative Polymerase Chain Reaction (RT-qPCR)
5.13. Western Blot Analysis
5.14. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Li, D.; Ye, Y.; Lin, S.; Deng, L.; Fan, X.; Zhang, Y.; Deng, X.; Li, Y.; Yan, H.; Ma, Y. Evaluation of deoxynivalenol-induced toxic effects on DF-1 cells in vitro: Cell-cycle arrest, oxidative stress, and apoptosis. Environ.Toxicol. Pharmacol. 2014, 37, 141–149. [Google Scholar] [CrossRef]
- Juan-García, A.; Taroncher, M.; Font, G.; Ruiz, M.-J. Micronucleus induction and cell cycle alterations produced by deoxynivalenol and its acetylated derivatives in individual and combined exposure on HepG2 cells. Food Chem. Toxicol. 2018, 118, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Dixit, S.; Dwivedi, P.D.; Pandey, H.P.; Das, M. Influence of temperature and pH on the degradation of deoxynivalenol (DON) in aqueous medium: Comparative cytotoxicity of DON and degraded product. Food Addit. Contam. Part A 2014, 31, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Urbanek, K.A.; Habrowska-Górczyńska, D.E.; Kowalska, K.; Stańczyk, A.; Domińska, K.; Piastowska-Ciesielska, A.W. Deoxynivalenol as potential modulator of human steroidogenesis. J. Appl. Toxicol. 2018, 38, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- The Commission of the European Communities. Commission Regulation (EC) No 401/2006 of 23 February 2006 laying down the methods of sampling and analysis for the official control of the levels of mycotoxins in foodstuffs. Off. J. Eur. Union 2006, 70, 12–34. [Google Scholar]
- Knutsen, H.K.; Alexander, J.; Barregård, L.; Bignami, M.; Brüschweiler, B.; Ceccatelli, S.; Cottrill, B.; Dinovi, M.; Grasl-Kraupp, B.; Hogstrand, C.; et al. Risks to human and animal health related to the presence of deoxynivalenol and its acetylated and modified forms in food and feed. EFSA J. 2017, 15, e04718. [Google Scholar]
- IARC Monographs on the Identifiaction of Carcinogenic Hazards of Humans. Available online: http://monographs.iarc.fr/ENG/Monographs/vol96/mono96.pdf (accessed on 7 May 2019).
- Pestka, J.J. Deoxynivalenol: Mechanisms of action, human exposure, and toxicological relevance. Arch. Toxicol. 2010, 84, 663–679. [Google Scholar] [CrossRef]
- Bradlaw, J.A.; Swentzel, K.C.; Alterman, E.; Hauswirth, J.W. Evaluation of purified 4-deoxynivalenol (vomitoxin) for unscheduled DNA synthesis in the primary rat hepatocyte-DNA repair assay. Food Chem. Toxicol. 1985, 23, 1063–1067. [Google Scholar] [CrossRef]
- Knasmüller, S.; Bresgen, N.; Kassie, F.; Mersch-Sundermann, V.; Gelderblom, W.; Zöhrer, E.; Eckl, P.M. Genotoxic effects of three Fusarium mycotoxins, fumonisin B1, moniliformin and vomitoxin in bacteria and in primary cultures of rat hepatocytes. Mutat. Res. 1997, 391, 39–48. [Google Scholar] [CrossRef]
- Mikami, O.; Yamamoto, S.; Yamanaka, N.; Nakajima, Y. Porcine hepatocyte apoptosis and reduction of albumin secretion induced by deoxynivalenol. Toxicology 2004, 204, 241–249. [Google Scholar] [CrossRef]
- Chung, Y.-J.; Yang, G.-H.; Islam, Z.; Pestka, J.J. Up-regulation of macrophage inflammatory protein-2 and complement 3A receptor by the trichothecenes deoxynivalenol and satratoxin G. Toxicology 2003, 186, 51–65. [Google Scholar] [CrossRef]
- Islam, Z.; Gray, J.S.; Pestka, J.J. p38 mitogen-activated protein kinase mediates IL-8 induction by the ribotoxin deoxynivalenol in human monocytes. Toxicol. Appl. Pharmacol. 2006, 213, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Baltriukiene, D.; Kalvelyte, A.; Bukelskiene, V. Induction of apoptosis and activation of JNK and p38 MAPK pathways in deoxynivalenol-treated cell lines. Altern. Lab. Anim. 2007, 35, 53–59. [Google Scholar] [CrossRef]
- Nasri, T.; Bosch, R.R.; ten Voorde, S.; Fink-Gremmels, J. Differential induction of apoptosis by type A and B trichothecenes in Jurkat T-lymphocytes. Toxicol. In Vitro 2006, 20, 832–840. [Google Scholar] [CrossRef]
- Pestka, J.J.; Yan, D.; King, L.E. Flow cytometric analysis of the effects of in vitro exposure to vomitoxin (deoxynivalenol) on apoptosis in murine T, B and IgA+ cells. Food Chem. Toxicol. 1994, 32, 1125–1136. [Google Scholar] [CrossRef]
- Zhou, H.R.; Harkema, J.R.; Yan, D.; Pestka, J.J. Amplified proinflammatory cytokine expression andtoxicity in mice coexposed to lipopolysaccharide and the trichothecene vomitoxin (deoxynivalenol). J. Toxicol. Environ. Health Part A 1999, 57, 115–136. [Google Scholar]
- Ma, Y.; Zhang, A.; Shi, Z.; He, C.; Ding, J.; Wang, X.; Ma, J.; Zhang, H. A mitochondria-mediated apoptotic pathway induced by deoxynivalenol in human colon cancer cells. Toxicol. In Vitro 2012, 26, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhu, C.; Ye, J.L.; Lv, Y.; Wang, L.; Chen, Z.; Jiang, Z. Yong Resveratrol protects porcine intestinal epithelial cells from deoxynivalenol induced damage via the Nrf2 signaling pathway. J. Agric. Food Chem. 2018. [Google Scholar] [CrossRef]
- Desport, J.-C.; Couratier, P. Stress oxydant et maladies neurodégénératives. Nutr. Clin. Métab. 2002, 16, 253–259. [Google Scholar] [CrossRef]
- Kaya, E.; Ozgok, Y.; Zor, M.; Eken, A.; Bedir, S.; Erdem, O.; Ebiloglu, T.; Ergin, G. Oxidative stress parameters in patients with prostate cancer, benign prostatic hyperplasia and asymptomatic inflammatory prostatitis: A prospective controlled study. Adv. Clin. Exp. Med. 2017, 26, 1095–1099. [Google Scholar] [CrossRef]
- Rezende, L.P.; Galheigo, M.R.U.; Landim, B.C.; Cruz, A.R.; Botelho, F.V.; Zanon, R.G.; Góes, R.M.; Ribeiro, D.L. Effect of glucose and palmitate enviro nment on proliferation and migration of PC3-prostate cancer cells. Cell Biol. Int. 2019, 43, 373–383. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef]
- Sprando, R.L.; Collins, T.F.X.; Black, T.N.; Olejnik, N.; Rorie, J.I.; Eppley, R.M.; Ruggles, D.I. Characterization of the effect of deoxynivalenol on selected male reproductive endpoints. Food Chem. Toxicol. 2005, 43, 623–635. [Google Scholar] [CrossRef]
- Fernández-Blanco, C.; Elmo, L.; Waldner, T.; Ruiz, M.-J. Cytotoxic effects induced by patulin, deoxynivalenol and toxin T2 individually and in combination in hepatic cells (HepG2). Food Chem. Toxicol. 2018, 120, 12–23. [Google Scholar] [CrossRef]
- Yuan, L.; Mu, P.; Huang, B.; Li, H.; Mu, H.; Deng, Y. EGR1 is essential for deoxynivalenol-induced G2/M cell cycle arrest in HepG2 cells via the ATF3ΔZip2a/2b-EGR1-p21 pathway. Toxicol. Lett. 2018, 299, 95–103. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, F. Cancer Research. Cancer Res. 2004, 63, 7689–7693. [Google Scholar]
- Pestka, J.J. Mechanisms of deoxynivalenol-induced gene expression and apoptosis. Food Addit. Contam. 2008, 25, 1128–1140. [Google Scholar] [CrossRef]
- Van Uden, P.; Kenneth, N.S.; Rocha, S. Regulation of hypoxia-inducible factor-1α by NF-κB. Biochem. J. 2008, 412, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yu, S.; Liu, N.; Xu, H.; Gong, Y.; Wu, Y.; Wang, P.; Su, X.; Liao, Y.; De Saeger, S.; et al. Transcription Factor FOXO3a Is a Negative Regulator of Cytotoxicity of Fusarium mycotoxin in GES-1 Cells. Toxicol. Sci. 2018, 166, 370–381. [Google Scholar] [CrossRef]
- Bensassi, F.; El Golli-Bennour, E.; Abid-Essefi, S.; Bouaziz, C.; Hajlaoui, M.R.; Bacha, H. Pathway of deoxynivalenol-induced apoptosis in human colon carcinoma cells. Toxicology 2009, 264, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Perdomo, H.A.G.; Zapata-Copete, J.A.; Sanchez, A. Molecular alterations associated with prostate cancer. Cent. Eur. J. Urol. 2018, 71, 168–176. [Google Scholar]
- Eze, U.A.; Huntriss, J.; Routledge, M.N.; Gong, Y.Y. Toxicological effects of regulated mycotoxins and persistent organochloride pesticides: In vitro cytotoxic assessment of single and defined mixtures on MA-10 murine Leydig cell line. Toxicol. In Vitro 2018, 48, 93–103. [Google Scholar] [CrossRef]
- Savard, C.; Nogues, P.; Boyer, A.; Chorfi, Y. Prevention of deoxynivalenol- and zearalenone-associated oxidative stress does not restore MA-10 Leydig cell functions. Toxicology 2016, 341–343, 17–27. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, S.; Zhang, M.; Yang, L.; Cheng, B.; Li, J.; Shan, A. Individual and combined effects of Fusarium toxins on apoptosis in PK15 cells and the protective role of N-acetylcysteine. Food Chem. Toxicol. 2018, 111, 27–43. [Google Scholar] [CrossRef]
- Osselaere, A.; Santos, R.; Hautekiet, V.; De Backer, P.; Chiers, K.; Ducatelle, R.; Croubels, S. Deoxynivalenol Impairs Hepatic and Intestinal Gene Expression of Selected Oxidative Stress, Tight Junction and Inflammation Proteins in Broiler Chickens, but Addition of an Adsorbing Agent Shifts the Effects to the Distal Parts of the Small Intestine. PLoS ONE 2013, 8, e69014. [Google Scholar] [CrossRef]
- Wu, Q.; Wang, X.; Nepovimova, E.; Miron, A.; Liu, Q.; Wang, Y.; Su, D.; Yang, H.; Li, L.; Kuca, K. Trichothecenes: Immunomodulatory effects, mechanisms, and anti-cancer potential. Arch. Toxicol. 2017, 91, 3737–3785. [Google Scholar] [CrossRef]
- Bensassi, F.; Gallerne, C.; Sharaf el Dein, O.; Hajlaoui, M.R.; Lemaire, C.; Bacha, H. In vitro investigation of toxicological interactions between the fusariotoxins deoxynivalenol and zearalenone. Toxicon 2014, 84, 1–6. [Google Scholar] [CrossRef]
- Bondy, G.S.; Coady, L.; Curran, I.; Caldwell, D.; Armstrong, C.; Aziz, S.A.; Nunnikhoven, A.; Gannon, A.M.; Liston, V.; Shenton, J.; et al. Effects of chronic deoxynivalenol exposure on p53 heterozygous and p53 homozygous mice. Food Chem. Toxicol. 2016, 96, 24–34. [Google Scholar] [CrossRef]
- Mishra, S.; Dwivedi, P.D.; Pandey, H.P.; Das, M. Role of oxidative stress in Deoxynivalenol induced toxicity. Food Chem. Toxicol. 2014, 72, 20–29. [Google Scholar] [CrossRef]
- Lei, Y.; Guanghui, Z.; Xi, W.; Yingting, W.; Xialu, L.; Fangfang, Y.; Goldring, M.B.; Xiong, G.; Lammi, M.J. Cellular responses to T-2 toxin and/or deoxynivalenol that induce cartilage damage are not specific to chondrocytes. Sci. Rep. 2017, 7, 2231. [Google Scholar] [CrossRef]
- Mishra, S.; Tewari, P.; Chaudhari, B.P.; Dwivedi, P.D.; Pandey, H.P.; Das, M. Deoxynivalenol induced mouse skin tumor initiation: Elucidation of molecular mechanisms in human HaCaT keratinocytes. Int. J. Cancer 2016, 139, 2033–2046. [Google Scholar] [CrossRef]
- Koul, H.K.; Pal, M.; Koul, S. Role of p38 MAP Kinase Signal Transduction in Solid Tumors. Genes Cancer 2013, 4, 342–359. [Google Scholar] [CrossRef]
- Smith, M.-C.; Madec, S.; Troadec, S.; Coton, E.; Hymery, N. Effects of fusariotoxin co-exposure on THP-1 human immune cells. Cell Biol. Toxicol. 2018, 34, 191–205. [Google Scholar] [CrossRef]
- Zhang, Z.-Q.; Wang, S.-B.; Wang, R.-G.; Zhang, W.; Wang, P.-L.; Su, X.-O. Phosphoproteome Analysis Reveals the Molecular Mechanisms Underlying Deoxynivalenol-Induced Intestinal Toxicity in IPEC-J2 Cells. Toxins 2016, 8, 270. [Google Scholar] [CrossRef]
- Springler, A.; Hessenberger, S.; Reisinger, N.; Kern, C.; Nagl, V.; Schatzmayr, G.; Mayer, E. Deoxynivalenol and its metabolite deepoxy-deoxynivalenol: Multi-parameter analysis for the evaluation of cytotoxicity and cellular effects. Mycotoxin Res. 2017, 33, 25–37. [Google Scholar] [CrossRef]
- Wang, X.; Fan, M.; Chu, X.; Zhang, Y.; Rahman, S.U.; Jiang, Y.; Chen, X.; Zhu, D.; Feng, S.; Li, Y.; et al. Deoxynivalenol induces toxicity and apoptosis in piglet hippocampal nerve cells via the MAPK signaling pathway. Toxicon 2018, 155, 1–8. [Google Scholar] [CrossRef]
- Kowalska, K.; Habrowska-Górczyńska, D.E.; Domińska, K.; Piastowska-Ciesielska, A.W. The dose-dependent effect of zearalenone on mitochondrial metabolism, plasma membrane permeabilization and cell cycle in human prostate cancer cell lines. Chemosphere 2017, 180, 455–466. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Androgen Dependency | Cell Line | Dose | SOD1 | SOD2 |
|---|---|---|---|---|
| Androgen-dependent cell lines | 22Rv1 | 5 µM DON | 0.8216 *** | 0.4710 |
| 3 µM DON | 0.9158 *** | 0.5009 | ||
| 2 µM DON | 0.9122 *** | 0.5504 | ||
| Cnt | 0.6205 | 0.5013 | ||
| LNCaP | 5 µM DON | 1.038 *** | 1.310 *** | |
| 3 µM DON | 1.009 *** | 0.7880 *** | ||
| 2 µM DON | 0.9110 ** | 0.5913 ** | ||
| Cnt | 0.5760 | 0.1688 | ||
| Androgen-independent cell lines | PC3 | 5 µM DON | 1.104 *** | 4.268 * |
| 3 µM DON | 1.208 *** | 3.135 | ||
| 2 µM DON | 1.241 *** | 3.310 | ||
| Cnt | 1.568 | 2.685 | ||
| DU-145 | 5 µM DON | 0.8172 *** | 1.030 *** | |
| 3 µM DON | 0.9245 *** | 1.087 *** | ||
| 2 µM DON | 1.061 * | 1.314 *** | ||
| Cnt | 1.216 | 2.214 |
| Androgen Dependency | Cell Line | Dose | RelA | FOXO3 | HIF1α |
|---|---|---|---|---|---|
| Androgen-dependent cell lines | 22Rv1 | 5 µM DON | 4.940 *** | 1.760 *** | 1.152 *** |
| 3 µM DON | 5.045 *** | 1.300 *** | 1.021 *** | ||
| 2 µM DON | 5.015 *** | 0.9127 *** | 0.8682 *** | ||
| Cnt | 0.8955 | 0.4464 | 0.2178 | ||
| LNCaP | 5 µM DON | 9.409 *** | 1.557 *** | 1.557 *** | |
| 3 µM DON | 7.976 *** | 1.075 *** | 1.073 *** | ||
| 2 µM DON | 7.424 *** | 1.010 *** | 1.012 *** | ||
| Cnt | 0.7651 | 0.2067 | 0.2066 | ||
| Androgen-independent cell lines | PC3 | 5 µM DON | 9.593 *** | 5.230 *** | 5.228 *** |
| 3 µM DON | 8.874 *** | 5.765 *** | 5.765 *** | ||
| 2 µM DON | 7.222 *** | 4.850 *** | 4.848 *** | ||
| Cnt | 3.803 | 3.245 | 3.246 | ||
| DU-145 | 5 µM DON | 6.836 *** | 1.499 *** | 1.784 *** | |
| 3 µM DON | 5.495 *** | 1.163 *** | 1.867 *** | ||
| 2 µM DON | 6.307 *** | 1.125 *** | 2.214 *** | ||
| Cnt | 2.17 | 0.4242 | 1.275 |
| Androgen Dependency | Cell Line | Dose | Casp3 | Bax |
|---|---|---|---|---|
| Androgen-dependent cell lines | 22Rv1 | 5 µM DON | 2.080 *** | 1.372 ** |
| 3 µM DON | 1.713 *** | 1.392 ** | ||
| 2 µM DON | 1.416 *** | 1.353 * | ||
| Cnt | 0.3414 | 0.8033 | ||
| LNCaP | 5 µM DON | 3.850 *** | 1.407 ** | |
| 3 µM DON | 2.927 *** | 1.242 * | ||
| 2 µM DON | 2.430 *** | 1.052 | ||
| Cnt | 0.4825 | 0.7100 | ||
| Androgen-independent cell lines | PC3 | 5 µM DON | 1.756 ** | 0.7383 * |
| 3 µM DON | 1.800 *** | 0.8233 | ||
| 2 µM DON | 1.687 * | 0.9817 | ||
| Cnt | 1.367 | 1.033 | ||
| DU-145 | 5 µM DON | 1.048 | 1.128 *** | |
| 3 µM DON | 0.8198 | 0.8817 | ||
| 2 µM DON | 1.036 | 1.170 *** | ||
| Cnt | 0.9308 | 0.6750 |
| Androgen Dependency | Cell Line | Dose | CCND1 | CDK4 | CDC2 | CCNB1 | CDKN1A |
|---|---|---|---|---|---|---|---|
| Androgen-dependent cell lines | 22Rv1 | 5 µM DON | 6.570 | 1.727 *** | 1.148 | 0.5915 | 1.928 *** |
| 3 µM DON | 8.919 *** | 1.489 ** | 0.8959 | 0.5901 | 2.017 *** | ||
| 2 µM DON | 11.33 *** | 1.441 ** | 0.8747 | 0.6257 | 1.914 *** | ||
| Cnt | 4.163 | 1.024 | 0.8164 | 0.3827 | 0.6593 | ||
| LNCaP | 5 µM DON | 1.669 *** | 1.001 *** | 1.303 ** | 0.9518 *** | 30.80 *** | |
| 3 µM DON | 1.338 *** | 0.9578 *** | 0.8296 | 0.5194 | 28.24 *** | ||
| 2 µM DON | 1.363 *** | 0.9136 *** | 1.208 * | 0.9653 *** | 22.53 *** | ||
| Cnt | 0.6477 | 0.5710 | 0.6072 | 0.2413 | 2.946 | ||
| Androgen-independent cell lines | PC3 | 5 µM DON | 8.594 *** | 1.639 *** | 2.603 *** | 1.007 | 1.071 |
| 3 µM DON | 8.000 *** | 1.909 *** | 2.948 ** | 0.8900 *** | 0.9634 | ||
| 2 µM DON | 7.236 ** | 1.781 *** | 2.932 ** | 0.8485 *** | 0.9790 | ||
| Cnt | 4.831 | 1.557 | 3.811 | 1.164 | 1.250 | ||
| DU-145 | 5 µM DON | 2.417 *** | 1.036 ** | 1.445 *** | 0.8115 *** | 4.855 *** | |
| 3 µM DON | 1.748 *** | 0.9628 ** | 1.707 *** | 1.178 | 3.503 *** | ||
| 2 µM DON | 2.102 *** | 1.003 ** | 1.928 * | 1.786 *** | 3.131 *** | ||
| Cnt | 0.7869 | 0.7160 | 1.121 | 1.235 | 0.9874 *** |
| Gene | Sequence (5′-3′) | Product Size (bp) |
|---|---|---|
| CCNB1 | For ACCTATGCTGGTGCCAGTG Rev GGCTTGGAGAGGCAGTA | 128 |
| CDC2 | For TTTTCAGAGCTTTGGGCACT Rev AGGCTTCCTGGTTTCCATTT | 100 |
| CDK4 | For TTACTGAGGCGACTGGAGG Rev GTCCTTAGGTCCTGGTCTACAT | 131 |
| CDKN1A | For GACAGATTTCTACCACTCCAA Rev CTGAGACTAAGGCAGAAGAGT | 134 |
| CCND1 | For TGTCCTACTACCGCCTCACACGCTTCCTCTCCAG Rev TCCTCTTCCTCCTCCTCGGCGGCCTTG | 160 |
| Casp3 | For GGAATATCCCTGGACAACAGTT Rev TTGCTGCATCGACATCTGT | 130 |
| Bax | For AGAGGTCTTTTTCCGAGTGGCAGC Rev TTCTGATCAGTTCCGGCACCTTG | 137 |
| SOD1 | For GCGTGGCCTAGCGAGTTAT Rev ACACCTTCACTGGTCCATTACT | 114 |
| SOD2 | For GGGTTGGCTTGGTTTCAATAAG Rev CTGAAGGTAGTAAGCGTGCTC | 136 |
| HIF-1α | For TTACTCATCCATGTGACCATGA Rev AGTTCTTCCTCGGCTAGTTAG | 140 |
| FOXO3 | For CAAGGATAAGGGCGACAG Rev GGTTGATGATCCACCAAGA | 131 |
| RelA | For GCACAGATACCACCAAGACC Rev TCAGCCTCATAGAAGCCATC | 157 |
| RPS17 | For AAGCGCGTGTGCGAGGAGATCG Rev TCGCTTCATCAGATGCGTGACATAACCTG | 87 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Habrowska-Górczyńska, D.E.; Kowalska, K.; Urbanek, K.A.; Domińska, K.; Sakowicz, A.; Piastowska-Ciesielska, A.W. Deoxynivalenol Modulates the Viability, ROS Production and Apoptosis in Prostate Cancer Cells. Toxins 2019, 11, 265. https://doi.org/10.3390/toxins11050265
Habrowska-Górczyńska DE, Kowalska K, Urbanek KA, Domińska K, Sakowicz A, Piastowska-Ciesielska AW. Deoxynivalenol Modulates the Viability, ROS Production and Apoptosis in Prostate Cancer Cells. Toxins. 2019; 11(5):265. https://doi.org/10.3390/toxins11050265
Chicago/Turabian StyleHabrowska-Górczyńska, Dominika Ewa, Karolina Kowalska, Kinga Anna Urbanek, Kamila Domińska, Agata Sakowicz, and Agnieszka Wanda Piastowska-Ciesielska. 2019. "Deoxynivalenol Modulates the Viability, ROS Production and Apoptosis in Prostate Cancer Cells" Toxins 11, no. 5: 265. https://doi.org/10.3390/toxins11050265
APA StyleHabrowska-Górczyńska, D. E., Kowalska, K., Urbanek, K. A., Domińska, K., Sakowicz, A., & Piastowska-Ciesielska, A. W. (2019). Deoxynivalenol Modulates the Viability, ROS Production and Apoptosis in Prostate Cancer Cells. Toxins, 11(5), 265. https://doi.org/10.3390/toxins11050265