Structural Insights to the Heterotetrameric Interaction between the Vibrio parahaemolyticus PirAvp and PirBvp Toxins and Activation of the Cry-Like Pore-Forming Domain

 ,
, 
Abstract
1. Introduction
2. Results and Discussion
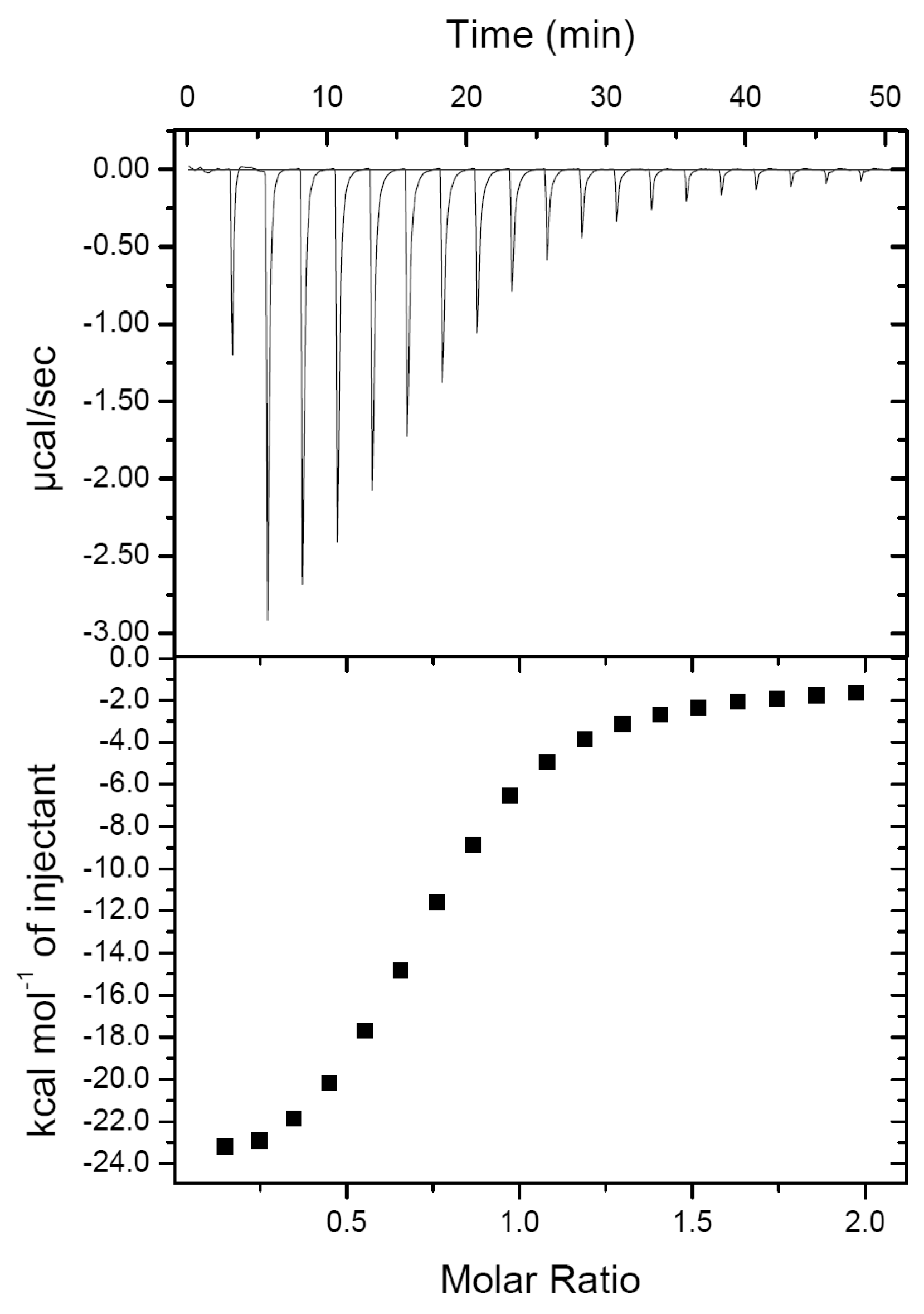
2.1. PirAvp and PirBvp Have a Low Binding Affinity
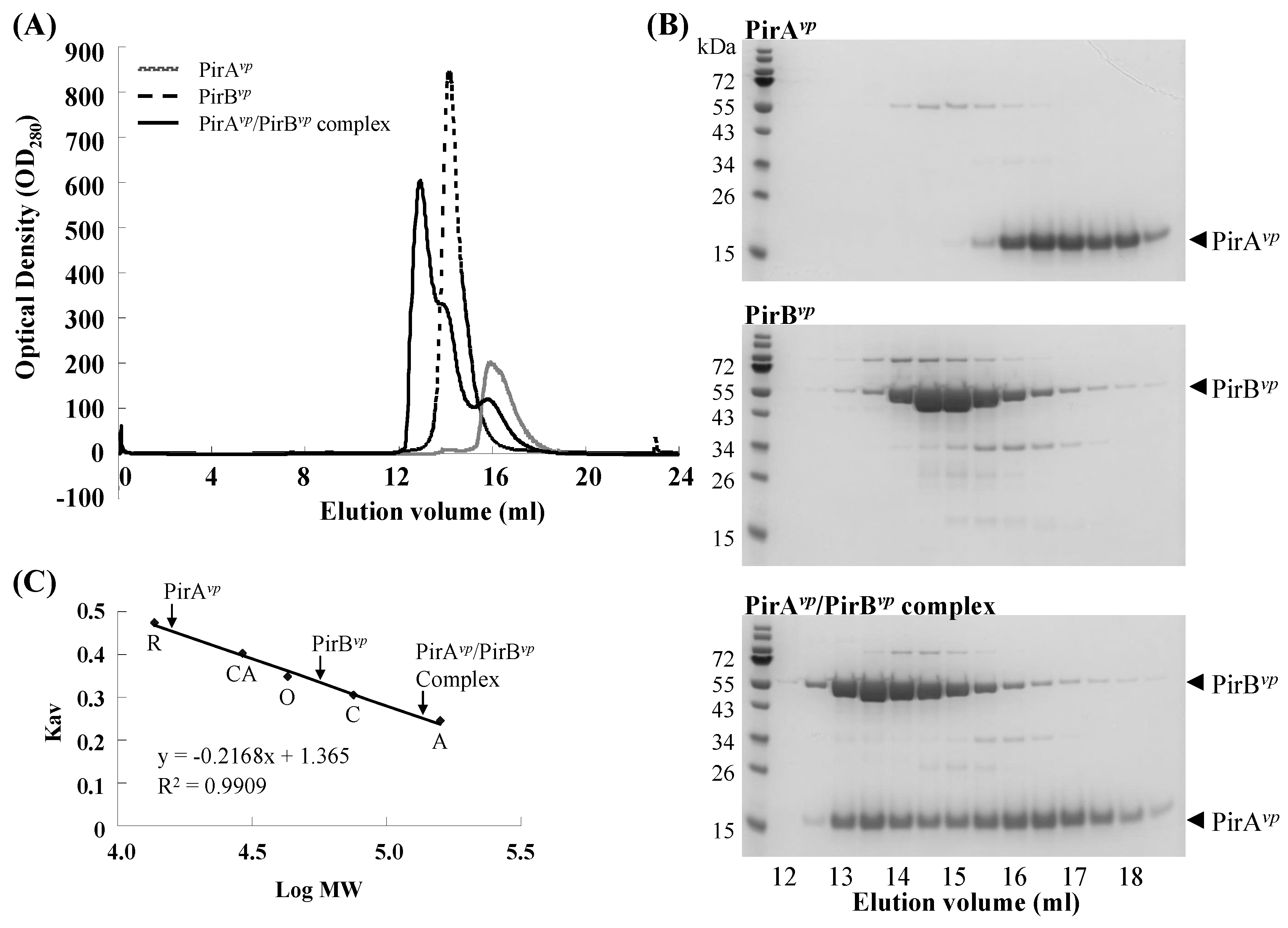
2.2. Calculation of the Native Molecular Weights of PirAvp, PirBvp and the PirAvp/PirBvp Complex by Gel Filtration
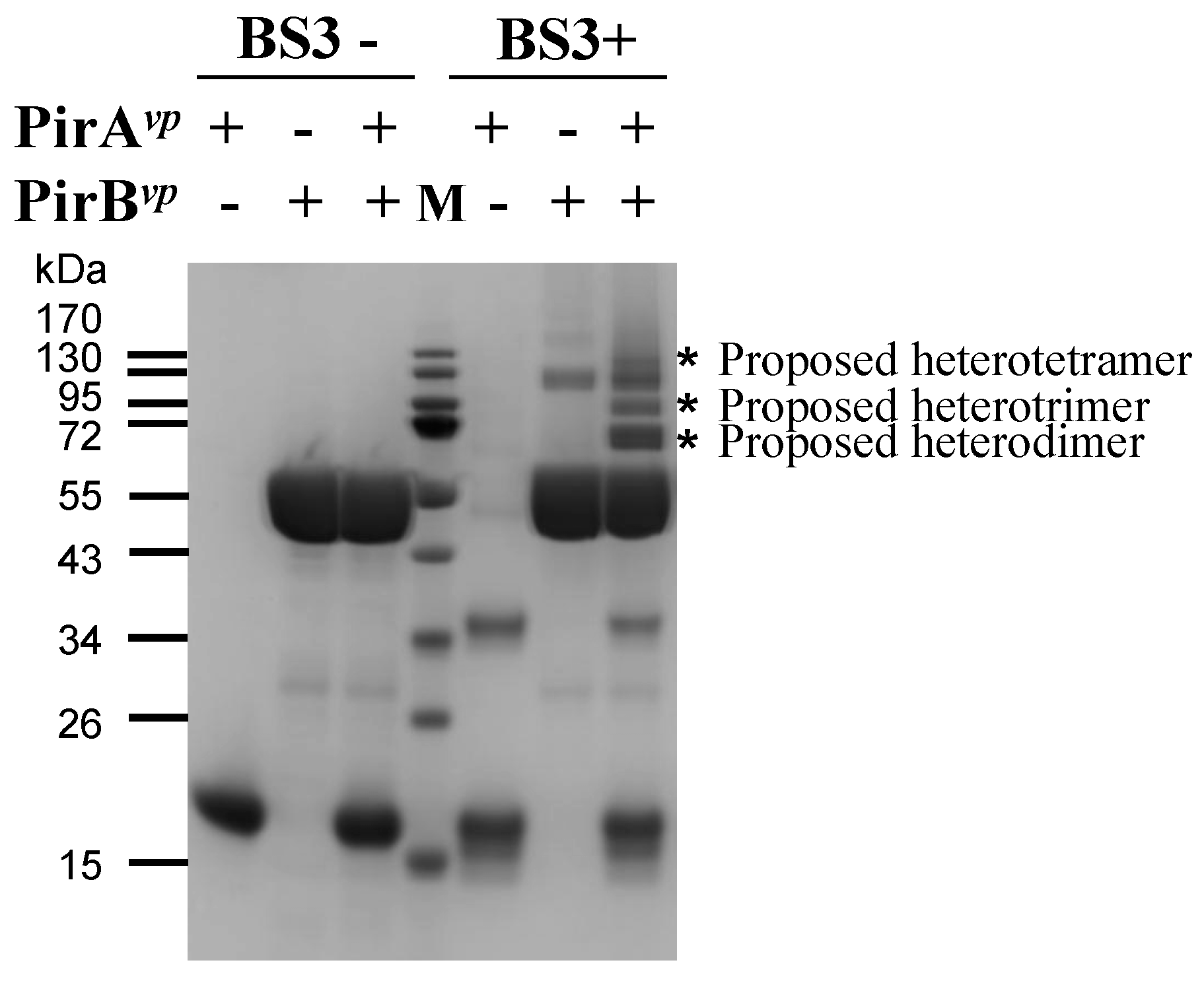
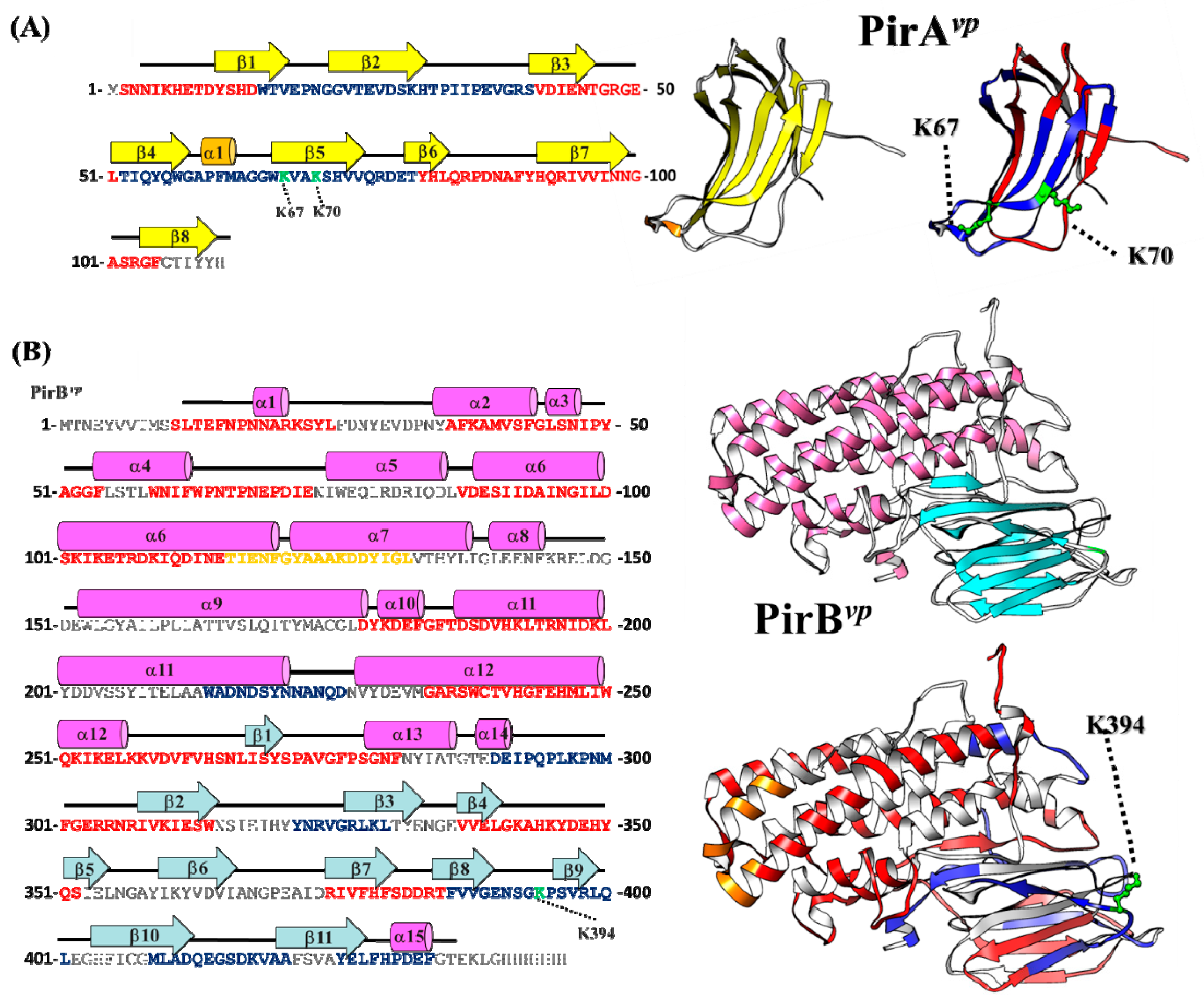
2.3. Determination of the Interface between PirAvp and PirBvp Using Cross-Linking Coupled Mass Spectrometry Analysis
2.4. Hydrogen-Deuterium Exchange (HDX) Coupled Mass Spectrometry Analysis of the PirAvp/PirBvp Interface
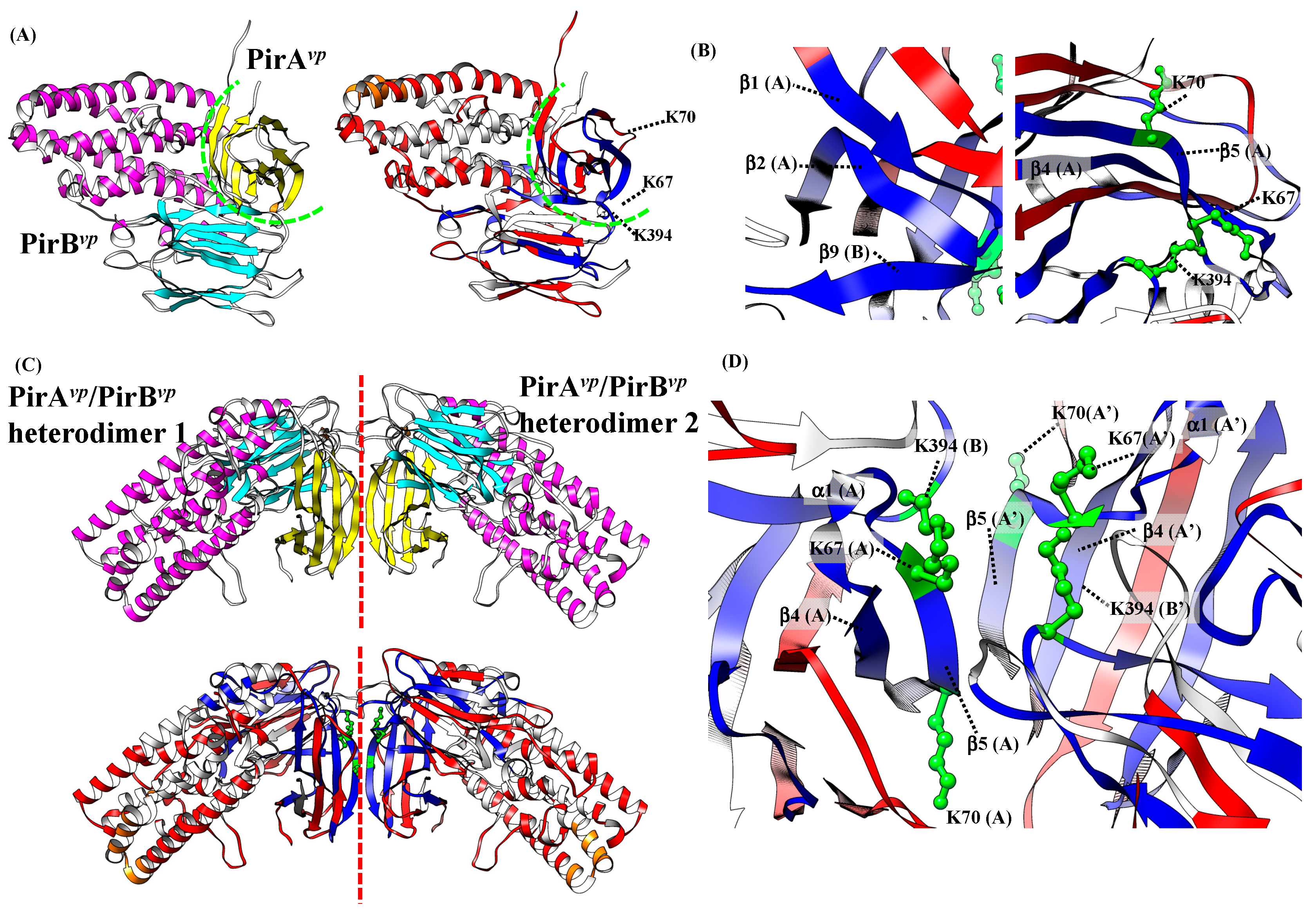
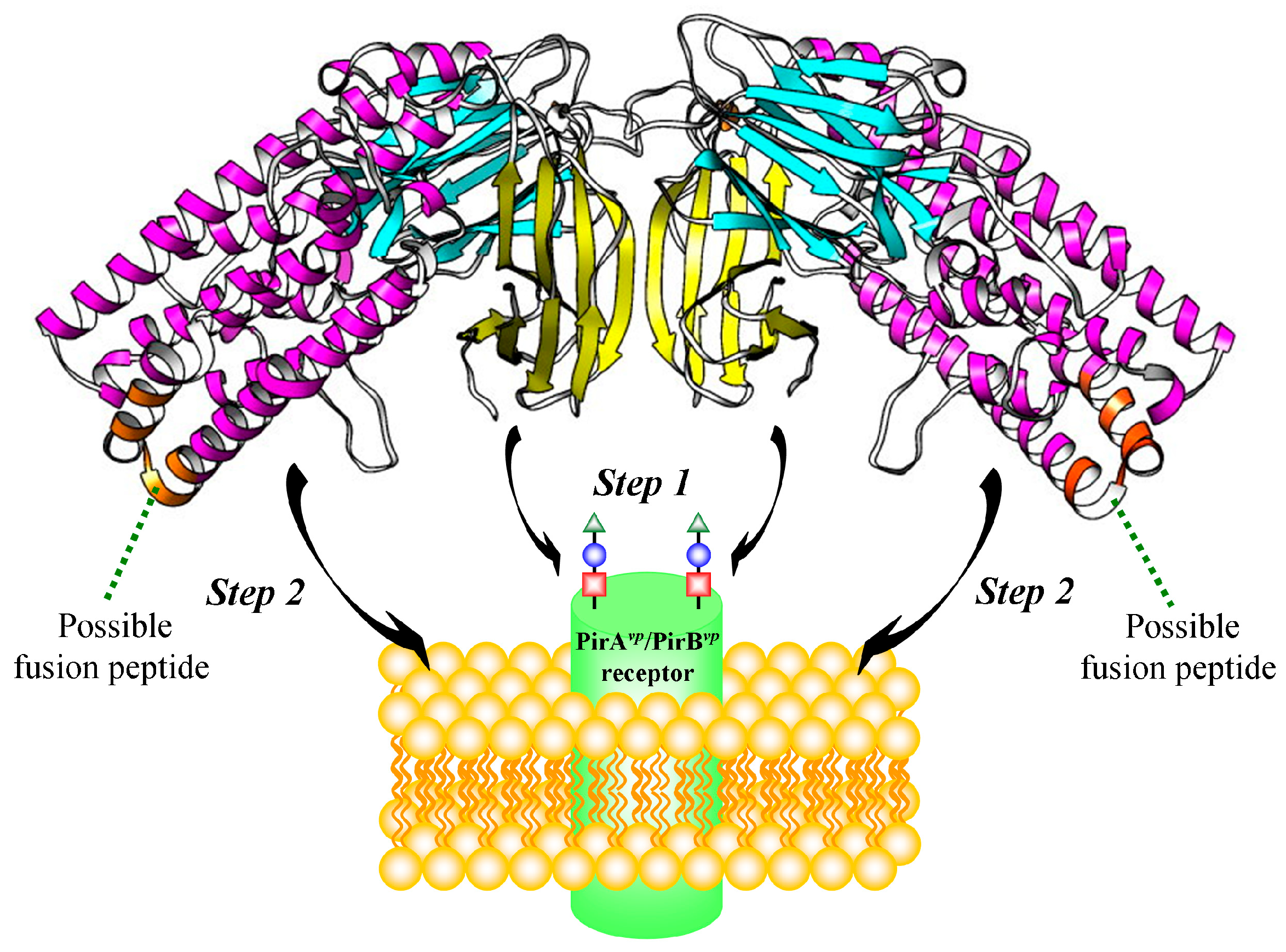
2.5. Proposed PirAvp/PirBvp Binding Model Using Cross-Linking Coupled Mass Spectrometry and HDX Analysis
3. Materials and Methods
3.1. Construction and Recombinant Protein Purification
3.2. Determination of the Binding Affinity between PirAvp and PirBvp by Isothermal Titration Calorimetry (ITC)
3.3. Determination of the Native Molecular Weights of PirAvp, PirBvp and PirAvp/PirBvp Complex by Using Gel Filtration
3.4. Determination of the Binding Stoichiometry of PirAvp and PirBvp by Densitometric Analysis
3.5. Cross-Linking Coupled Mass Spectrometry Analysis of PirAvp and PirBvp
3.6. Hydrogen-Deuterium Exchange (HDX) Mass Spectrometry Analysis of PirAvp and PirBvp
3.7. Peptide Identification and HDX Data Analysis
3.8. Molecular Docking Analysis of PirAvp/PirBvp Complex
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tran, L.; Nunan, L.; Redman, R.; Mohney, L.; Pantoja, C.; Fitzsimmons, K.; Lightner, D. Determination of the infectious nature of the agent of acute hepatopancreatic necrosis syndrome affecting penaeid shrimp. Dis. Aquat. Org. 2013, 105, 45–55. [Google Scholar] [CrossRef]
- Lightner, D.V.; Redman, R.M.; Pantoja, C.R.; Noble, B.L.; Tran, L. Early mortality syndrome affects shrimp in Asia. Glob. Aquac. Advocate 2012, 15, 40. [Google Scholar]
- Gomez-Gil, B.; Soto-Rodríguez, S.; Lozano, R.; Betancourt-Lozano, M. Draft genome sequence of Vibrio parahaemolyticus Strain M0605, which causes severe mortalities of shrimps in Mexico. Genome Announc. 2014, 2, e00055-14. [Google Scholar] [CrossRef] [PubMed]
- Soto-Rodriguez, S.A.; Gomez-Gil, B.; Lozano-Olvera, R.; Betancourt-Lozano, M.; Morales-Covarrubias, M.S. Field and experimental evidence of Vibrio parahaemolyticus as the causative agent of acute hepatopancreatic necrosis disease of cultured shrimp (Litopenaeus vannamei) in Northwestern Mexico. Appl. Environ. Microbiol. 2015, 81, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Sirikharin, R.; Taengchaiyaphum, S.; Sanguanrut, P.; Chi, T.D.; Mavichak, R.; Proespraiwong, P.; Nuangsaeng, B.; Thitamadee, S.; Flegel, T.W.; Sritunyalucksana, K. Characterization and PCR detection of binary, Pir-Like toxins from Vibrio parahaemolyticus isolates that cause acute hepatopancreatic necrosis disease (AHPND) in shrimp. PLoS ONE 2015, 10, e0126987. [Google Scholar] [CrossRef]
- Kondo, H.; Van, P.T.; Dang, L.T.; Hirono, I. Draft genome sequence of non-Vibrio parahaemolyticus acute hepatopancreatic necrosis disease strain KC13.17.5, isolated from diseased shrimp in Vietnam. Genome Announc. 2015, 3, e00978-15. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Wang, H.; Xie, G.; Zou, P.; Guo, C.; Liang, Y.; Huang, J. An isolate of Vibrio campbellii carrying the pirVP gene causes acute hepatopancreatic necrosis disease. Emerg. Microbes Infect. 2017, 6, e2. [Google Scholar] [CrossRef]
- Liu, L.; Xiao, J.; Xia, X.; Pan, Y.; Yan, S.; Wang, Y. Draft genome sequence of Vibrio owensii strain SH-14, which causes shrimp acute hepatopancreatic necrosis disease. Genome Announc. 2015, 3, e01395-e15. [Google Scholar] [CrossRef]
- Restrepo, L.; Bayot, B.; Arciniegas, S.; Bajaña, L.; Betancourt, I.; Panchana, F.; Muñoz, A.R. PirVP genes causing AHPND identified in a new Vibrio species (Vibrio punensis) within the commensal Orientalis clade. Sci. Rep. 2018, 8, 13080. [Google Scholar] [CrossRef]
- Lee, C.T.; Chen, I.T.; Yang, Y.T.; Ko, T.P.; Huang, Y.T.; Huang, J.Y.; Huang, M.F.; Lin, S.J.; Chen, C.Y.; Lin, S.S.; et al. The opportunistic marine pathogen Vibrio parahaemolyticus becomes virulent by acquiring a plasmid that expresses a deadly toxin. Proc. Natl. Acad. Sci. USA 2015, 112, 10798–10803. [Google Scholar] [CrossRef]
- Durán-Avelar, M.J.; Vázquez-Reyes, A.; González-Mercado, A.L.; Zambrano-Zaragoza, J.F.; Ayón-Pérez, M.F.; Agraz-Cibrián, J.M.; Gutiérrez-Franco, J.; Vibanco-Pérez, N. pirA- and pirB-like gene identification in Micrococcus luteus strains in Mexico. J. Fish Dis. 2018, 41, 1667–1673. [Google Scholar] [CrossRef]
- Lin, S.J.; Hsu, K.C.; Wang, H.C. Structural insights into the cytotoxic mechanism of Vibrio parahaemolyticus PirAvp and PirBvp toxins. Mar. Drugs 2017, 15, 373. [Google Scholar] [CrossRef]
- Rappsilber, J.; Siniossoglou, S.; Hurt, E.C.; Mann, M. A generic strategy to analyze the spatial organization of multi-protein complexes by cross-linking and mass spectrometry. Anal. Chem. 2000, 72, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Denison, C.; Kodadek, T. Toward a general chemical method for rapidly mapping multi-protein complexes. J. Proteome Res. 2004, 3, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Sharon, M.; Taverner, T.; Ambroggio, X.I.; Deshaies, R.J.; Robinson, C.V. Structural organization of the 19S proteasome lid: Insights from MS of intact complexes. PLoS. Biol. 2006, 4, e267. [Google Scholar] [CrossRef] [PubMed]
- Guilliam, T.A.; Bailey, L.J.; Brissett, N.C.; Doherty, A.J. PolDIP2 interacts with human PrimPol and enhances its DNA polymerase activities. Nucleic Acids Res. 2016, 44, 3317–3329. [Google Scholar] [CrossRef]
- Komolov, K.E.; Du, Y.; Duc, N.M.; Betz, R.M.; Rodrigues, J.P.G.L.M.; Leib, R.D.; Patra, D.; Skiniotis, G.; Adams, C.M.; Dror, R.O.; et al. Structural and Functional Analysis of a β2-Adrenergic Receptor Complex with GRK5. Cell 2017, 169, 407–421. [Google Scholar] [CrossRef]
- Weisz, D.A.; Liu, H.; Zhang, H.; Thangapandian, S.; Tajkhorshid, E.; Gross, M.L.; Pakrasi, H.B. Mass spectrometry-based crosslinking study shows that the Psb28 protein binds to cytochrome b559 in Photosystem II. Proc. Natl. Acad. Sci. USA 2017, 114, 2224–2229. [Google Scholar] [CrossRef]
- Paterson, Y.; Englander, S.W.; Roder, H. An antibody binding site on cytochrome c defined by hydrogen exchange and two-dimensional NMR. Science 1990, 249, 755–759. [Google Scholar] [CrossRef]
- Oganesyan, I.; Lento, C.; Wilson, D.J. Contemporary hydrogen deuterium exchange mass spectrometry. Methods 2018, 144, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Soberón, M.; Pardo, L.; Muñóz-Garay, C.; Sánchez, J.; Gómez, I.; Porta, H.; Bravo, A. Pore formation by Cry toxins. Adv. Exp. Med. Biol. 2010, 677, 127–142. [Google Scholar] [PubMed]
- Xu, C.; Wang, B.C.; Yu, Z.; Sun, M. Structural insights into Bacillus thuringiensis Cry, Cyt and parasporin toxins. Toxins 2014, 6, 2732–2770. [Google Scholar] [CrossRef]
- Jenkins, J.L.; Lee, M.K.; Valaitis, A.P.; Curtiss, A.; Dean, D.H. Bivalent sequential binding model of a Bacillus thuringiensis toxin to gypsy moth aminopeptidase N receptor. J. Biol. Chem. 2000, 275, 14423–14431. [Google Scholar] [CrossRef]
- Tinwongger, S.; Nochiri, Y.; Thawonsuwan, J.; Nozaki, R.; Kondo, H.; Awasthi, S.P.; Hinenoya, A.; Yamasaki, S.; Hirono, I. Virulence of acute hepatopancreatic necrosis disease PirAB-like relies on secreted proteins not on gene copy number. J. Appl. Microbiol. 2016, 121, 1755–1765. [Google Scholar] [CrossRef] [PubMed]
- Dal Peraro, M.; van der Goot, F.G. Pore-forming toxins: Ancient, but never really out of fashion. Nat. Rev. Microbiol. 2016, 14, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Qiu, J.; Zhang, Y.; Lu, C.; Dai, X.; Wang, J.; Li, H.; Wang, X.; Tan, W.; Luo, M.; et al. Oroxylin A inhibits hemolysis via hindering the self-assembly of α-hemolysin heptameric transmembrane pore. PLoS Comput. Biol. 2013, 9, e1002869. [Google Scholar] [CrossRef]
- Qiu, J.; Wang, D.; Zhang, Y.; Dong, J.; Wang, J.; Niu, X. Molecular modeling reveals the novel inhibition mechanism and binding mode of three natural compounds to staphylococcal α-hemolysin. PLoS ONE 2013, 8, e80197. [Google Scholar] [CrossRef]
- Foletti, D.; Strop, P.; Shaughnessy, L.; Hasa-Moreno, A.; Casas, M.G.; Russell, M.; Bee, C.; Wu, S.; Pham, A.; Zeng, Z.; et al. Mechanism of action and in vivo efficacy of a human-derived antibody against Staphylococcus aureus á-hemolysin. J. Mol. Biol. 2013, 425, 1641–1654. [Google Scholar] [CrossRef]
- Fernandes da Costa, S.P.; Savva, C.G.; Bokori-Brown, M.; Naylor, C.E.; Moss, D.S.; Basak, A.K.; Titball, R.W. Identification of a key residue for oligomerisation and pore-formation of Clostridium perfringens NetB. Toxins 2014, 6, 1049–1061. [Google Scholar] [CrossRef]
- Bokori-Brown, M.; Hall, C.A.; Vance, C.; Fernandes da Costa, S.P.; Savva, C.G.; Naylor, C.E.; Cole, A.R.; Basak, A.K.; Moss, D.S.; Titball, R.W. Clostridium perfringens epsilon toxin mutant Y30A-Y196A as a recombinant vaccine candidate against enterotoxemia. Vaccine 2014, 32, 2682–2687. [Google Scholar] [CrossRef]
- Lee, H.L.; Chiang, I.C.; Liang, S.Y.; Lee, D.Y.; Chang, G.D.; Wang, K.Y.; Lin, S.Y.; Shih, Y.L. Quantitative proteomics analysis reveals the min system of Escherichia coli modulates reversible protein association with the inner membrane. Mol. Cell Proteomics 2016, 15, 1572–1583. [Google Scholar] [CrossRef]
- Naveen, V.; Hsiao, C.D. NrdR Transcription regulation: Global proteome analysis and its role in Escherichia coli viability and virulence. PLoS ONE 2016, 11, e0157165. [Google Scholar] [CrossRef]
- Xu, H.; Freitas, M.A. MassMatrix: A database search program for rapid characterization of proteins and peptides from tandem mass spectrometry data. Proteomics 2009, 9, 1548–1555. [Google Scholar] [CrossRef]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ΔH (kcal/mol) | ΔG (kcal/mol) | Kd (μM) | N |
|---|---|---|---|
| −25.69 ± 1.42 | −7.01 ± 0.10 | 7.33 ± 1.20 | 0.74 ± 0.01 |
| Protein | Theoretical MW (kDa) | Estimated MW (kDa) |
|---|---|---|
| PirAvp (N-His10) | 15.19 | 15.75 |
| PirBvp (C-His6) | 51.13 | 56.12 |
| PirAvp/PirBvp complex | 132.59 | 136.08 |
| Crosslinked Lysine Residues | PP/PP2/PPtag Score | MW (obs) (Da) | MW (Da) | Assigned Peptide Sequence | |
|---|---|---|---|---|---|
| PirAvp Peptide (Chain A) | PirBvp Peptide (Chain B) | ||||
| PirAvpK67-PirBvpK394 | 29.8/15.4/4.5 | 2749.3991 | 2749.4212 | GAPFMAGGWK(67) | TFVVGENSGK(394)PSVRL |
| PirAvpK70-PirBvpK394 | 61.7/20.4/11.1 | 2637.4449 | 2637.4471 | VAK(70)SHVVQR | TFVVGENSGK(394)PSVR |
| 33.0/18.4/9.6 | 2637.4466 | 2637.4471 | VAK(70)SHVVQR | TFVVGENSGK(394)PSVR | |
| 34.5/14.9/5.2 | 2638.4289 | 2638.4505 | VAK(70)SHVVQR | TFVVGENSGK(394)PSVR | |
| 34.4/14.6/4.4 | 2638.4332 | 2638.4505 | VAK(70)SHVVQR | TFVVGENSGK(394)PSVR | |
| (A) | ||||
| Identified Peptide Sequences Derived from PirAvp | Deuterium Incorporation Fold | Classification | ||
| 10 s | 40 s | 80 s | ||
| 2-SNNIKHETDYSHD-14 | 1.2 | 1.0 | 1.0 | Not involved in binding |
| 15-WTVEPNGGVTEVDSKHTPIIPEVGRS-40 | Involved in binding; in the center of the interface or deep within the complex | |||
| 15-WTVEPNGGVTEVDSKHTPIIPEVG-38 | 1.6 | 1.6 | 1.4 | |
| 15-WTVEPNGGVTEVDSKHTPIIPEVGRSVD-42 | 1.5 | 1.5 | 1.4 | |
| 26-VDSKHTPIIPEVGRSVD-42 | 1.7 | 1.8 | 1.5 | |
| 41-VDIENTGRGEL-51 | 1.0 | 1.0 | 1.0 | Not involved in binding |
| 52-TIQYQWGAPFMAGGWKVAKSHVVQRDET-79 | Involved in binding; edge of the interface or near the surface of the complex | |||
| 52-TIQYQWGAPFMAGGWKVAKSHVVQRDET-79 | 1.2 | 1.2 | 1.2 | |
| 66-WKVAKSHVVQRDET-79 | 1.2 | 1.2 | 1.2 | |
| 80-YHLQRPDNAF-89 | 1.1 | 1.1 | 1.2 | Not involved in binding |
| 89-FYHQRIVVINNGASRGF-105 | Not involved in binding | |||
| 89-FYHQRIVVINNGASRG-104 | 1.1 | 1.2 | 1.1 | |
| 90-YHQRIVVINNGASRGF-105 | 1.1 | 1.2 | 1.1 | |
| (B) | ||||
| Identified Peptide Sequences Derived from PirBvp | Deuterium Incorporation Fold | Classification | ||
| 10 s | 40 s | 80 s | ||
| 11-SLTEFNPNNARKSYL-25 | 0.9 | 1.0 | 1.0 | Not involved in binding |
| 36-AFKAMVSFGLSNIPYAGGF-54 | Not involved in binding | |||
| 36-AFKAMVSF-43 | 1.1 | 1.0 | 1.0 | |
| 41-VSFGLSNIPYAGGF-54 | 1.0 | 1.1 | 1.2 | |
| 59-WNIFWPNTPNEPDIE-73 | 1.2 | 0.8 | 0.8 | Not involved in binding |
| 87-VDESIIDAINGILDSKIKETRDKIQDINE-115 | Not involved in binding | |||
| 87-VDESIIDAINGIL-99 | 1.5 | - | 1.7 | |
| 100-DSKIKETRDKIQDINE-115 | 1.1 | 1.1 | 1.2 | |
| 116-TIENFGYAAAKDDYIGL-132 | 1.0 | 0.8 | 0.7 | Exposed after complex formation |
| 178-DYKDEFGFTDSDVHKLTRNIDKL-200 | Not involved in binding | |||
| 178-DYKDEFGFTDSDVHKLTRNIDKL-200 | 1.0 | 1.1 | 1.2 | |
| 185-FTDSDVHKLTRNIDKL-200 | 1.0 | 1.1 | 1.4 | |
| 214-WADNDSYNNANQD-226 | 1.1 | 1.4 | 1.4 | Involved in binding; in the center of the interface or deep within the complex |
| 234-GARSWCTVHGFEHMLIWQKIKELKKVDVFVHSNLISYSPAVGFPSGNF-281 | Not involved in binding | |||
| 234-GARSWCTVHGFEHM-247 | 1.0 | 0.9 | 0.9 | |
| 248-LIWQKIKELKKVDVFVHSNL-267 | 0.9 | 1.0 | 1.1 | |
| 268-ISYSPAVGFPSGNF-281 | 1.1 | 1.1 | 1.2 | |
| 290-DEIPQPLKPNM-300 | 1.4 | 1.4 | 1.3 | Involved in binding; in the center of the interface or deep within the complex |
| 301-FGERRNRIVKIESW-314 | 1.1 | 1.1 | 1.1 | Not involved in binding |
| 322-YNRVGRLKL-330 | 1.3 | 1.6 | 1.8 | Involved in binding; in the center of the interface or deep within the complex |
| 337-VVELGKAHKYDEHYQS-352 | 0.8 | 1.0 | 1.1 | Not involved in binding |
| 375-RIVFHFSDDRT-385 | 0.9 | 0.9 | 0.9 | Not involved in binding |
| 386-FVVGENSGKPSVRLQL-401 | 1.1 | 1.3 | 1.3 | Involved in binding; edge of the interface or near the surface of the complex |
| 409-MLADQEGSDKVAA-421 | Involved in binding; in the center of the interface or deep within the complex | |||
| 409-MLADQEGSDKVAA-421 | 1.3 | 1.9 | 1.8 | |
| 410-LADQEGSDKVAA-421 | 1.5 | 1.8 | 1.9 | |
| 426-YELFHPDEF-434 | 1.5 | 1.2 | 1.0 | Involved in binding; in the center of the interface or deep within the complex |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, S.-J.; Chen, Y.-F.; Hsu, K.-C.; Chen, Y.-L.; Ko, T.-P.; Lo, C.-F.; Wang, H.-C.; Wang, H.-C. Structural Insights to the Heterotetrameric Interaction between the Vibrio parahaemolyticus PirAvp and PirBvp Toxins and Activation of the Cry-Like Pore-Forming Domain. Toxins 2019, 11, 233. https://doi.org/10.3390/toxins11040233
Lin S-J, Chen Y-F, Hsu K-C, Chen Y-L, Ko T-P, Lo C-F, Wang H-C, Wang H-C. Structural Insights to the Heterotetrameric Interaction between the Vibrio parahaemolyticus PirAvp and PirBvp Toxins and Activation of the Cry-Like Pore-Forming Domain. Toxins. 2019; 11(4):233. https://doi.org/10.3390/toxins11040233
Chicago/Turabian StyleLin, Shin-Jen, Yi-Fan Chen, Kai-Cheng Hsu, Yun-Ling Chen, Tzu-Ping Ko, Chu-Fang Lo, Han-Ching Wang, and Hao-Ching Wang. 2019. "Structural Insights to the Heterotetrameric Interaction between the Vibrio parahaemolyticus PirAvp and PirBvp Toxins and Activation of the Cry-Like Pore-Forming Domain" Toxins 11, no. 4: 233. https://doi.org/10.3390/toxins11040233
APA StyleLin, S.-J., Chen, Y.-F., Hsu, K.-C., Chen, Y.-L., Ko, T.-P., Lo, C.-F., Wang, H.-C., & Wang, H.-C. (2019). Structural Insights to the Heterotetrameric Interaction between the Vibrio parahaemolyticus PirAvp and PirBvp Toxins and Activation of the Cry-Like Pore-Forming Domain. Toxins, 11(4), 233. https://doi.org/10.3390/toxins11040233