The Dual Prey-Inactivation Strategy of Spiders—In-Depth Venomic Analysis of Cupiennius salei

 ,
,
Abstract
1. Introduction
2. Results and Discussion
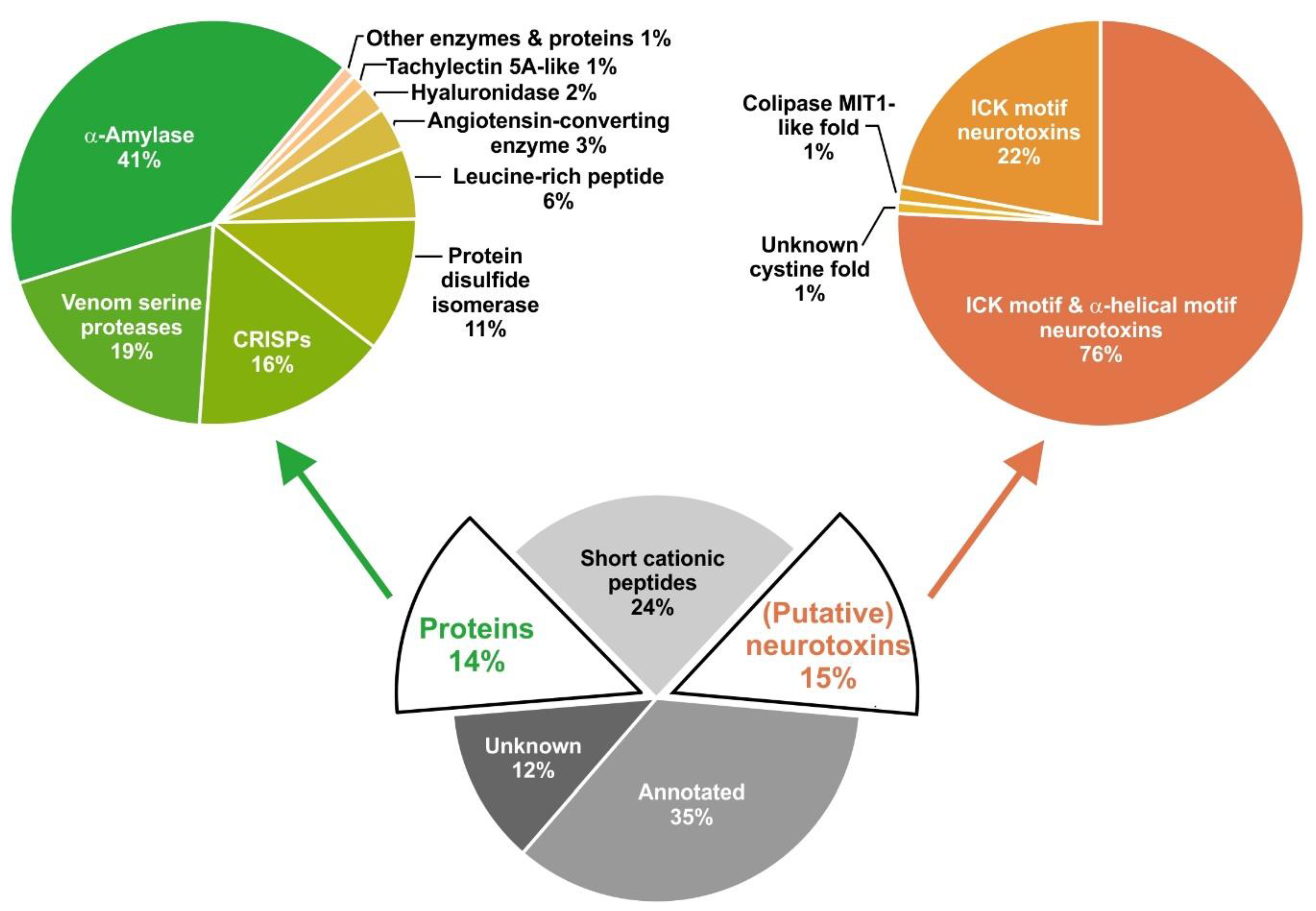
2.1. Overview of Venom Gland Composition
2.2. Most Abundant Proteins in the Venom Gland Transcriptome and in the Venom of C. salei
2.3. Proteins of the Protein- and Peptide-Processing Machinery
2.3.1. Signal Peptidase (SPase)
2.3.2. Protein Disulfide-Isomerase (PDI)
2.3.3. Venom Serine Proteases (VSPs)
2.3.4. Carboxypeptidase A-Like Protein (CPA)
2.3.5. Peptidylglycine α-Amidating Monooxygenase (PAM)
2.4. Recruited and Neofunctionalized Proteins
2.4.1. α-Amylase (α-AMY)
2.4.2. Cysteine–Rich Secretory Proteins (CRISPs)
2.4.3. Angiotensin-Converting Enzyme (ACE)
2.4.4. Hyaluronidase (HYAL)
2.4.5. Phospholipase A2 (PLA2)
2.4.6. Cystatin (CST)
2.4.7. Kunitz Domain-Containing Protein (KCP)
2.4.8. Thyroglobulin Type-1 Domain-Like Protein (TT1LP)
2.4.9. Insulin-Like Growth Factor-Binding Protein-Related Protein 1 (IGFBP-rP1)
2.5. Proteins Belonging to the Innate Immune System
2.5.1. Tachylectin 5A-Like Protein (TL5A)
2.5.2. Leucine-Rich Repeat Domain-Containing Protein (LRR)
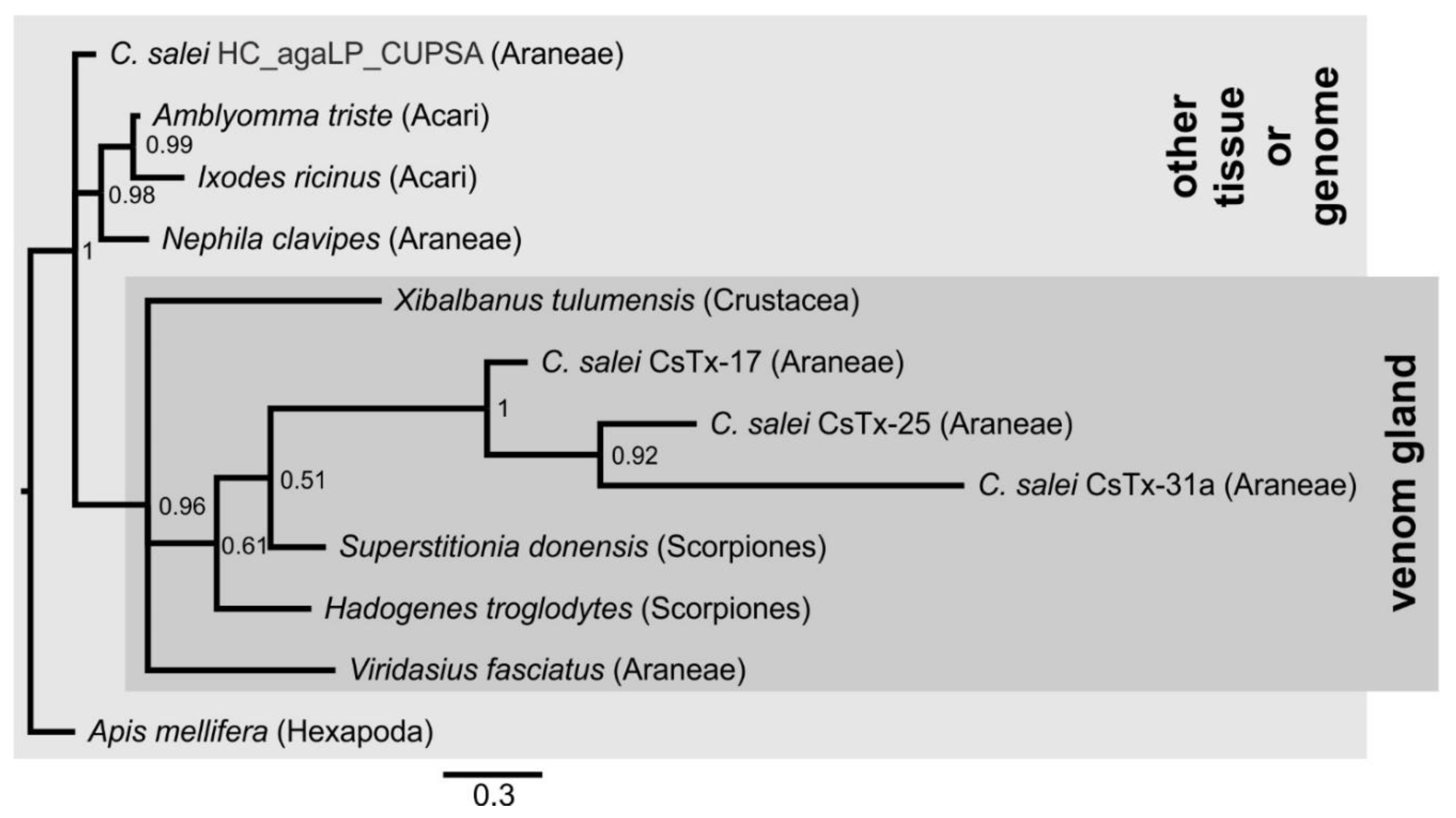
2.6. Cysteine-Containing (Putative) Neurotoxins
2.7. Signal Peptides and Pro-peptides of (Putative) Neurotoxins
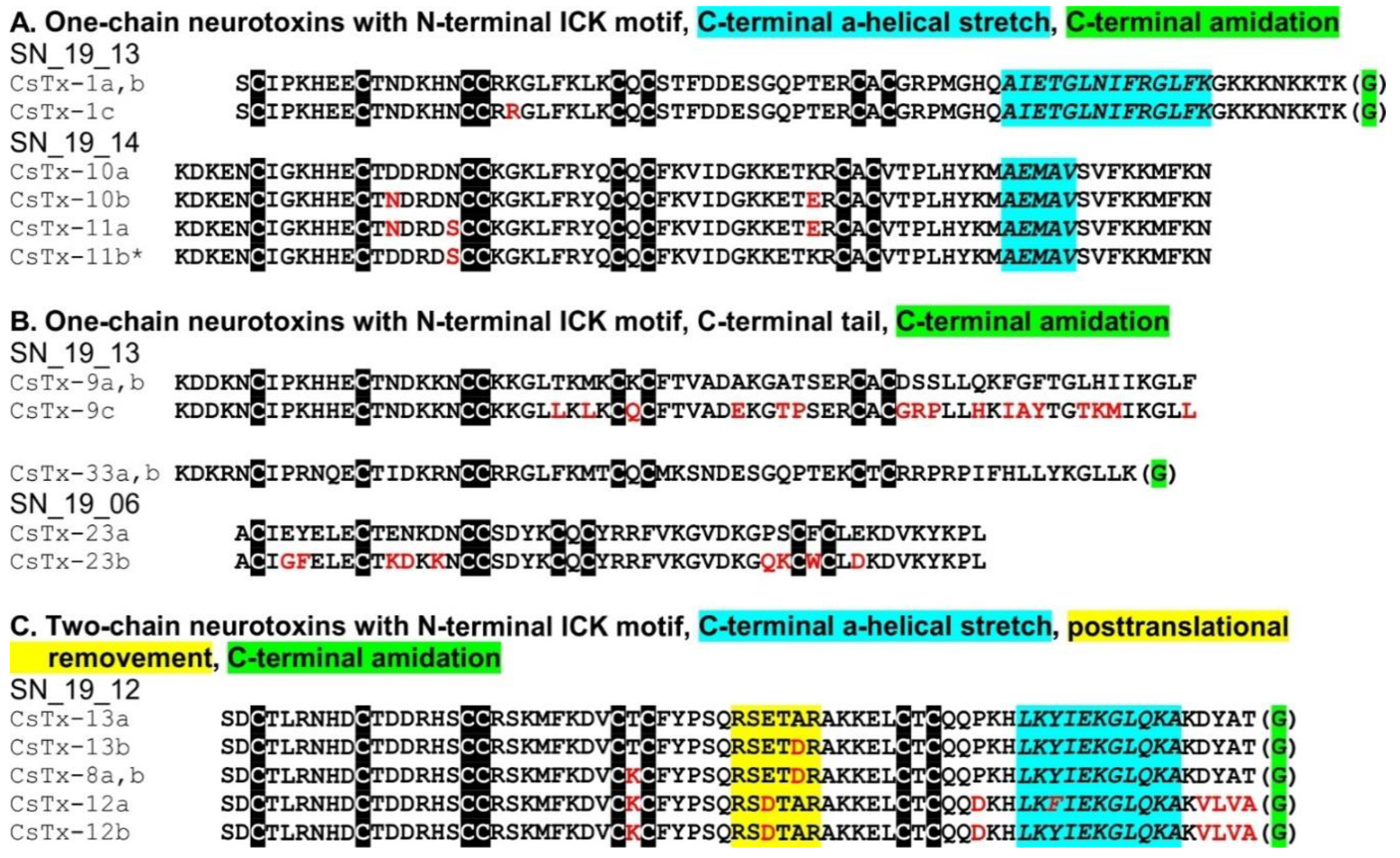
2.8. C-terminal Modifications of Mature (Putative) Neurotoxins
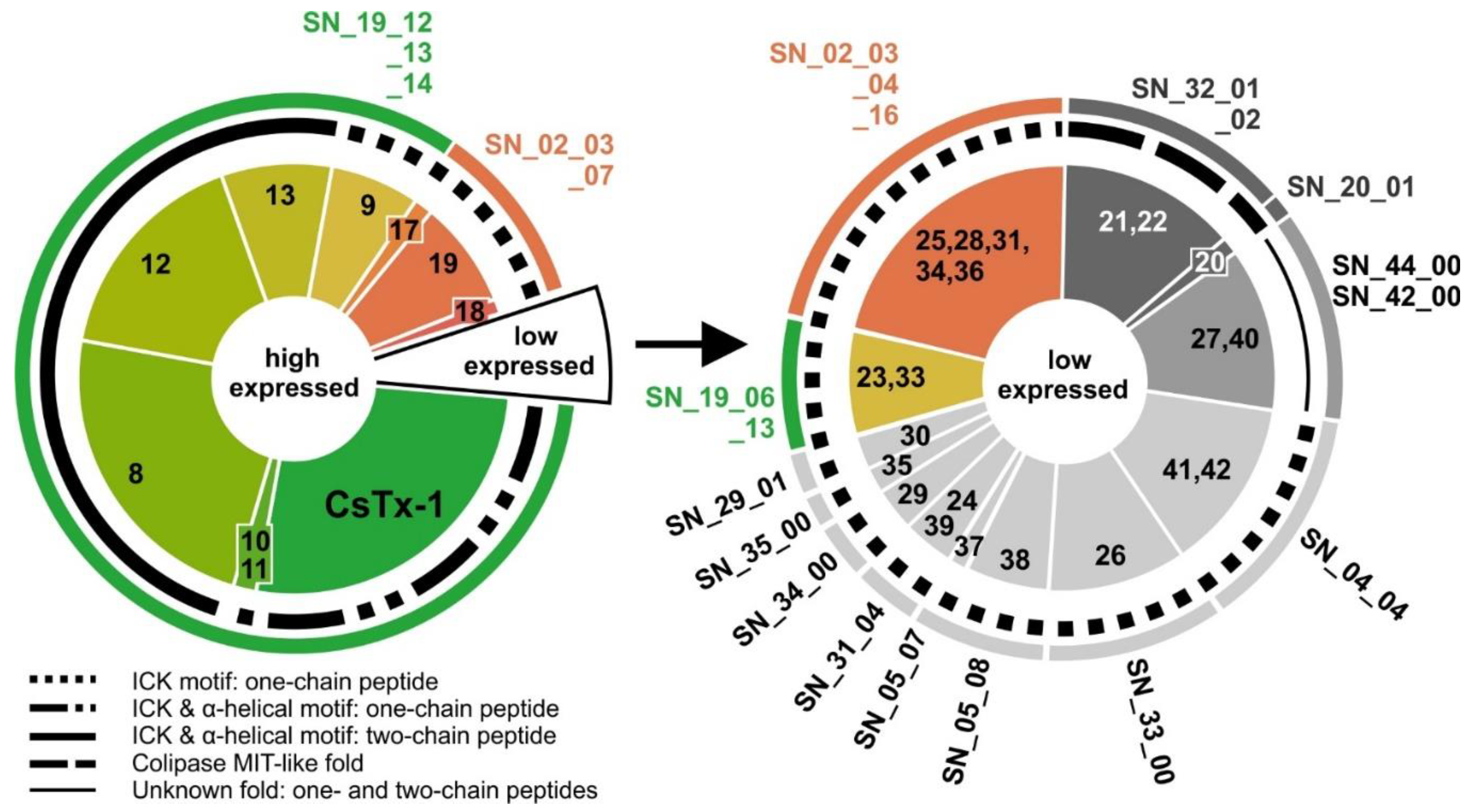
2.9. SN_19 Family
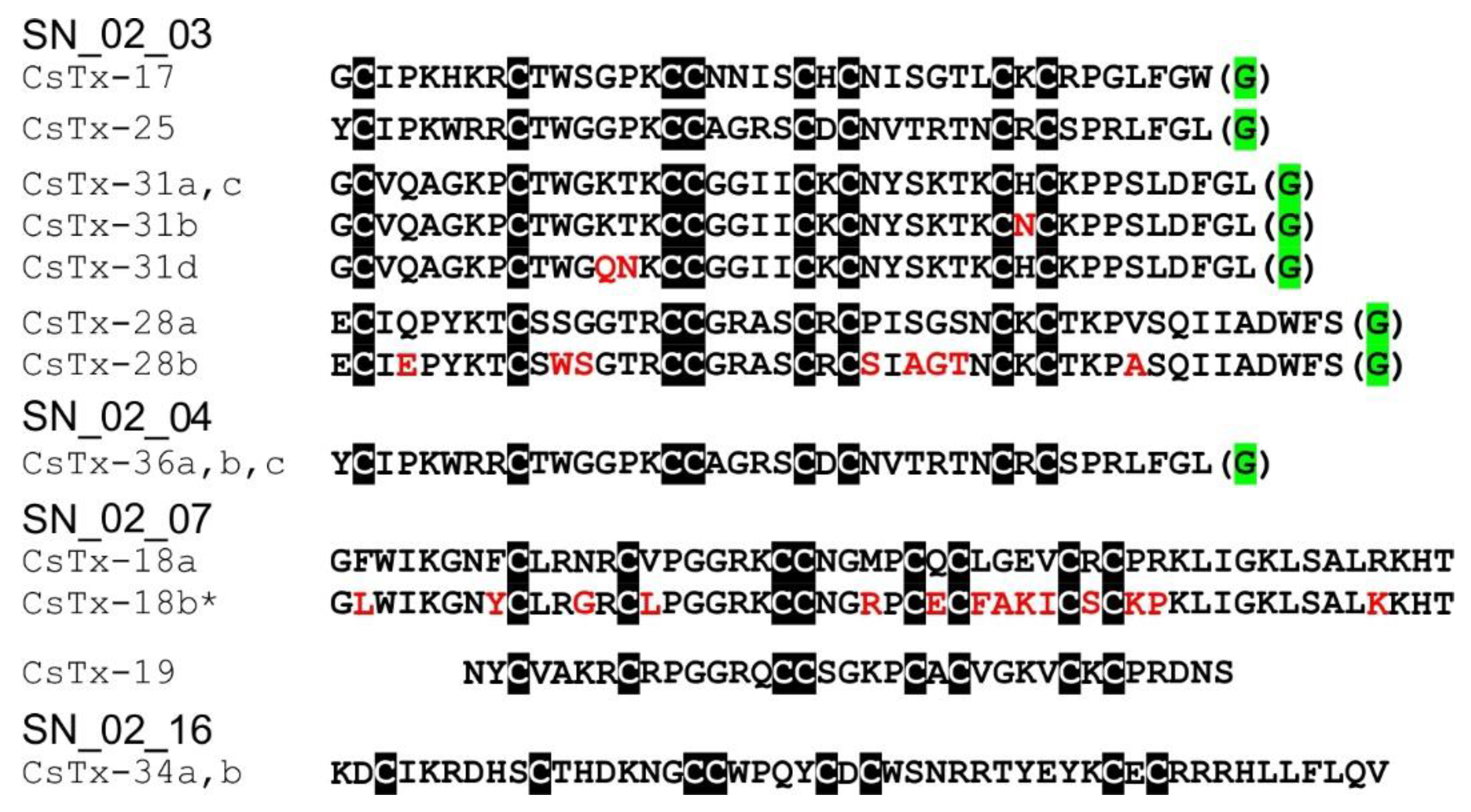
2.10. SN_02 Family
2.11. Low Abundant (Putative) Neurotoxins
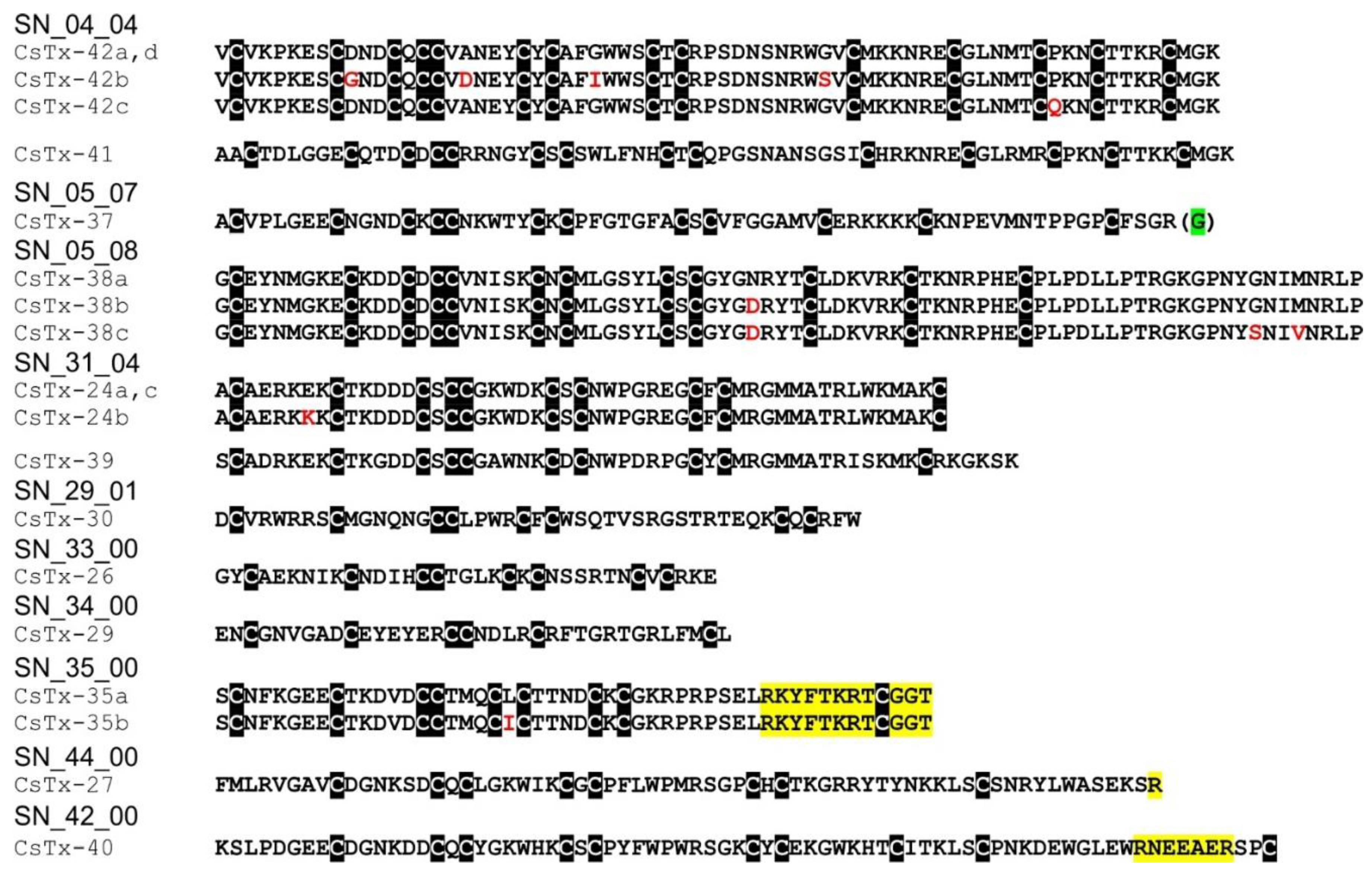
2.12. SN_04, SN_05, and SN_31 Family
2.13. SN_34, SN_29, SN_33, SN_19, and SN_35 Family
2.14. SN_42 and SN_44 Family
2.15. SN_20 and SN_32 Family
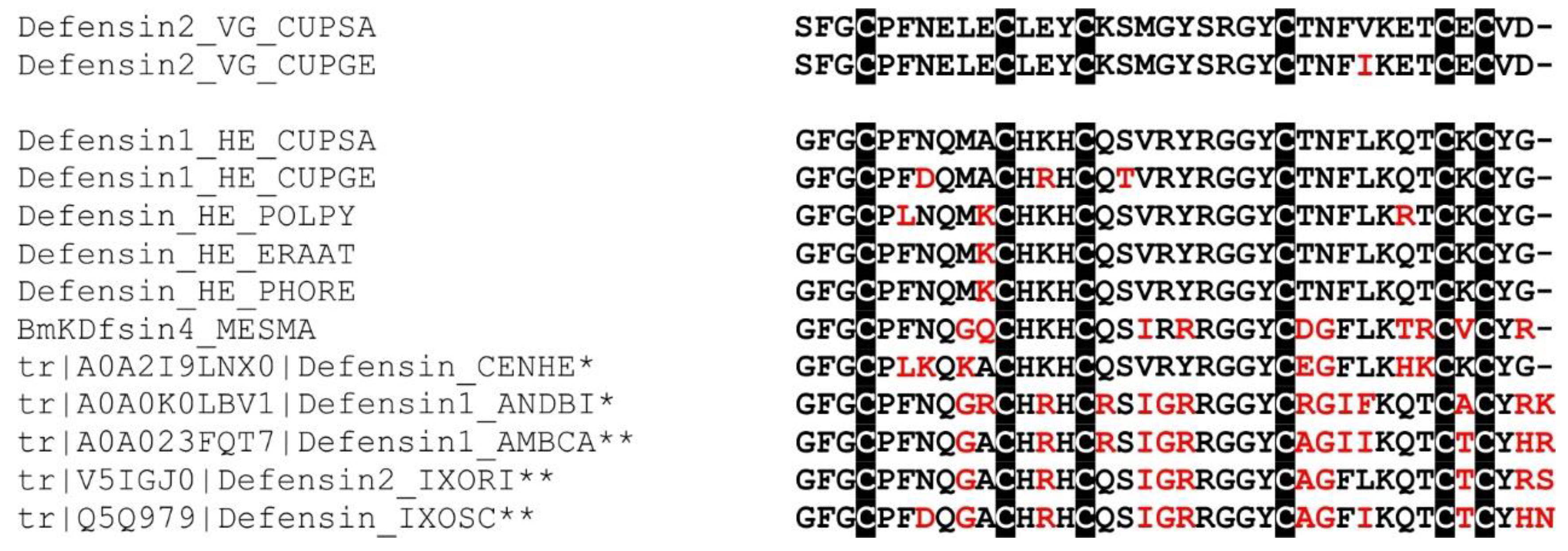
2.16. Defensin-Like Peptide
2.17. Proteomics
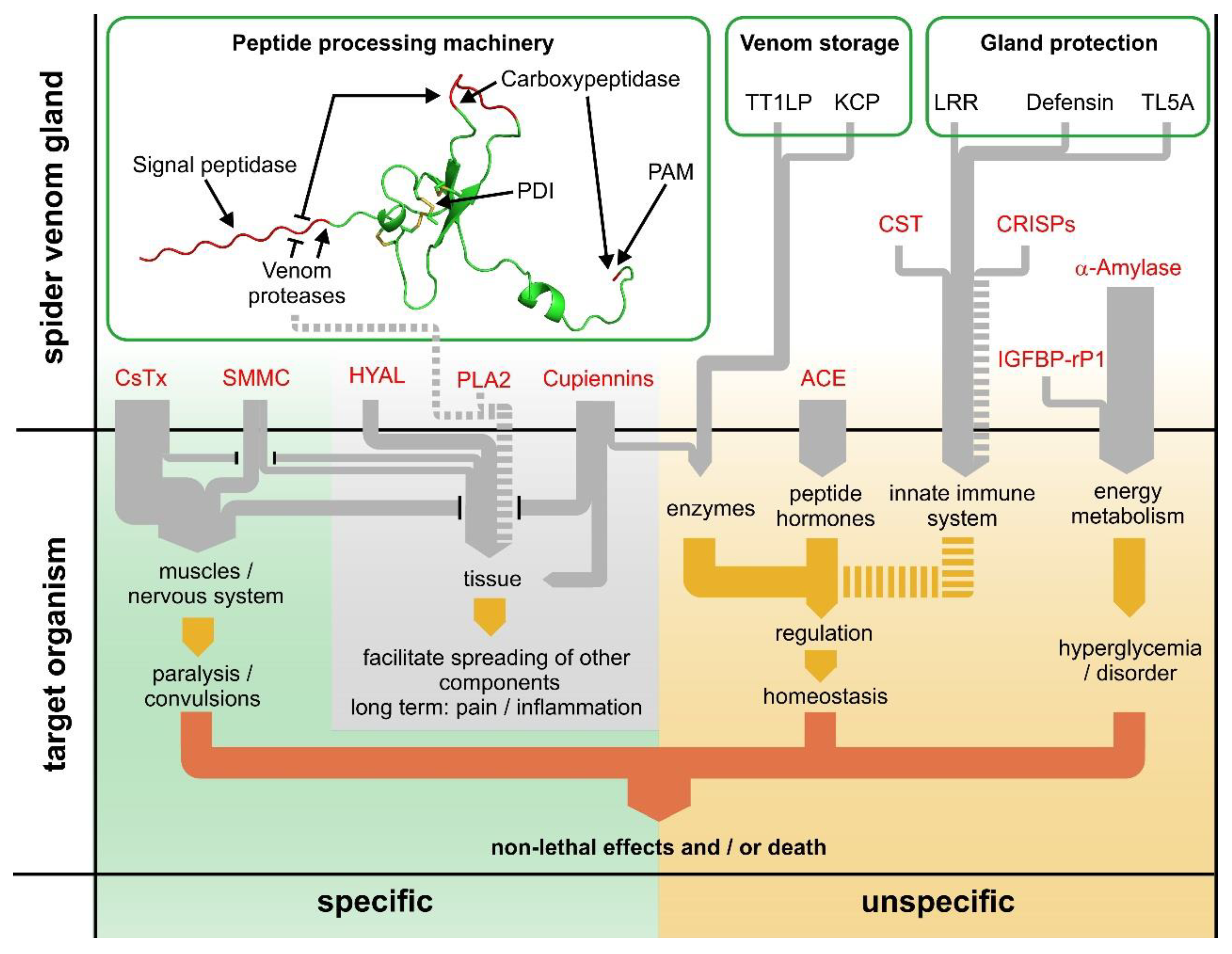
2.18. The Dual Prey-Inactivation Strategy of Spiders
3. Conclusions
4. Materials and Methods
4.1. Spider Maintenance, Venom Collection, and cDNA Libraries
4.2. Transcriptome Analysis
4.3. Tandem Mass Spectrometry
4.4. Tandem Mass Spectrometry Data Analysis
4.5. Sequence Analysis
4.6. Sequence Deposition
4.7. Proteomic Data Deposition
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- World Spider Catalog. Version 19.5. Natural History Museum Bern. Available online: http://wsc.nmbe.ch (accessed on 11 September 2018).
- Wise, D.H. Spiders in Ecological Webs; Cambridge University Press: Cambridge, MA, USA, 1993. [Google Scholar]
- Kuhn-Nentwig, L.; Stöcklin, R.; Nentwig, W. Venom composition and strategies in spiders: Is everything possible? Adv. Insect Physiol. 2011, 40, 1–86. [Google Scholar]
- Saez, N.J.; Senff, S.; Jensen, J.E.; Er, S.Y.; Herzig, V.; Rash, L.D.; King, G.F. Spider-venom peptides as therapeutics. Toxins 2010, 2, 2851–2871. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.J.; Herzig, V.; King, G.F.; Alewood, P.F. The insecticidal potential of venom peptides. Cell. Mol. Life Sci. 2013, 70, 3665–3693. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Duan, Z.; Yu, Y.; Liu, Z.; Liu, Z.; Liang, S. The venom gland transcriptome of Latrodectus tredecimguttatus revealed by deep sequencing and cDNA library analysis. PLoS ONE 2013, 8, e81357. [Google Scholar] [CrossRef] [PubMed]
- Oldrati, V.; Koua, D.; Allard, P.M.; Hulo, N.; Arrell, M.; Nentwig, W.; Lisacek, F.; Wolfender, J.L.; Kuhn-Nentwig, L.; Stocklin, R. Peptidomic and transcriptomic profiling of four distinct spider venoms. PLoS ONE 2017, 12, e0172966. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, Y.; He, Q.; Liu, J.; Luo, J.; Zhu, L.; Lu, S.; Huang, P.; Chen, X.; Zeng, X.; et al. Toxin diversity revealed by a transcriptomic study of Ornithoctonus huwena. PLoS ONE 2014, 9, e100682. [Google Scholar] [CrossRef]
- Biner, O.; Trachsel, C.; Moser, A.; Kopp, L.; Langenegger, N.; Kämpfer, U.; von Ballmoos, C.; Nentwig, W.; Schürch, S.; Schaller, J.; et al. Isolation, N-glycosylations and function of a hyaluronidase-like enzyme from the venom of the spider Cupiennius salei. PLoS ONE 2015, 10, e0143963. [Google Scholar] [CrossRef]
- Pedroso, A.; Matioli, S.R.; Murakami, M.T.; Pidde-Queiroz, G.; Tambourgi, D.V. Adaptive evolution in the toxicity of a spider’s venom enzymes. BMC Evol. Biol. 2015, 15, 290. [Google Scholar] [CrossRef]
- Diniz, M.R.V.; Paiva, A.L.B.; Guerra-Duarte, C.; Nishiyama, M.Y.J.; Mudadu, M.A.; Oliveira, U.; Borges, M.H.; Yates, J.R.; Junqueira-de-Azevedo, I.L. An overview of Phoneutria nigriventer spider venom using combined transcriptomic and proteomic approaches. PLoS ONE 2018, 13, e0200628. [Google Scholar] [CrossRef]
- Kuhn-Nentwig, L.; Schaller, J.; Schürch, S.; Nentwig, W. Venom of Cupiennius salei. In Spider Venoms; Gopalakrishnakone, P., Ed.; Springer Science + Business Media: Dordrecht, The Netherlands, 2016; pp. 47–70. [Google Scholar]
- Trachsel, C.; Siegemund, D.; Kämpfer, U.; Kopp, L.S.; Bühr, C.; Grossmann, J.; Luthi, C.; Cunningham, M.; Nentwig, W.; Kuhn-Nentwig, L.; et al. Multicomponent venom of the spider Cupiennius salei: A bioanalytical investigation applying different strategies. FEBS J. 2012, 279, 2683–2694. [Google Scholar] [CrossRef]
- Barth, F. A Spider’s World; Springer: Berlin, Germany, 2002. [Google Scholar]
- Kuhn-Nentwig, L.; Fedorova, I.M.; Lüscher, B.P.; Kopp, L.S.; Trachsel, C.; Schaller, J.; Vu, X.L.; Seebeck, T.; Streitberger, K.; Nentwig, W.; et al. A Venom-derived neurotoxin, CsTx-1, from the spider Cupiennius salei exhibits cytolytic activities. J. Biol. Chem. 2012, 287, 25640–25649. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Malli, H.; Kuhn-Nentwig, L.; Imboden, H.; Moon, M.J.; Wyler, T. Immunocytochemical localization and secretion process of the toxin CSTX-1 in the venom gland of the wandering spider Cupiennius salei (Araneae: Ctenidae). Cell Tissue Res. 2000, 299, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Kuhn-Nentwig, L.; Largiader, C.R.; Streitberger, K.; Chandru, S.; Baumann, T.; Kämpfer, U.; Schaller, J.; Schürch, S.; Nentwig, W. Purification, cDNA structure and biological significance of a single insulin-like growth factor-binding domain protein (SIBD-1) identified in the hemocytes of the spider Cupiennius salei. Insect Biochem. Mol. Biol. 2011, 41, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Auclair, S.M.; Bhanu, M.K.; Kendall, D.A. Signal peptidase I: Cleaving the way to mature proteins. Protein Sci. 2012, 21, 13–25. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J.; Finn, R. Twenty years of the MEROPS database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2016, 44, D343–D350. [Google Scholar] [CrossRef]
- Ali Khan, H.; Mutus, B. Protein disulfide isomerase a multifunctional protein with multiple physiological roles. Front. Chem. 2014, 2, 70. [Google Scholar] [CrossRef] [PubMed]
- Logunov, D.V. A new species of the genus Alopecosa Simon, 1885 (Araneae, Lycosidae) from south-east Kazakhstan. Arthropoda Selecta 2015, 22, 163–169. [Google Scholar]
- Veenstra, J.A. Mono- and dibasic proteolytic cleavage sites in insect neuroendocrine peptide precursors. Arch. Insect Biochem. Physiol. 2000, 43, 49–63. [Google Scholar] [CrossRef]
- Kozlov, S.A.; Grishin, E.V. The universal algorithm of maturation for secretory and excretory protein precursors. Toxicon 2007, 49, 721–726. [Google Scholar] [CrossRef]
- Langenegger, N.; Koua, D.; Schürch, S.; Heller, M.; Nentwig, W.; Kuhn-Nentwig, L. Identification of a precursor processing protease from the spider Cupiennius salei essential for venom neurotoxin maturation. J. Biol. Chem. 2018, 293, 2079–2090. [Google Scholar] [CrossRef] [PubMed]
- Wullschleger, B.; Kuhn-Nentwig, L.; Tromp, J.; Kämpfer, U.; Schaller, J.; Schürch, S.; Nentwig, W. CSTX-13, a highly synergistically acting two-chain neurotoxic enhancer in the venom of the spider Cupiennius salei (Ctenidae). Proc. Natl. Acad. Sci. USA 2004, 101, 11251–11256. [Google Scholar] [CrossRef] [PubMed]
- Kuhn-Nentwig, L.; Schaller, J.; Kämpfer, U.; Imboden, H.; Malli, H.; Nentwig, W. A lysine rich C-terminal tail is directly involved in the toxocity of CSTX-1, a neurotoxic peptide from the venom of the spider Cupiennius salei. Arch. Insect Biochem. Physiol. 2000, 44, 101–111. [Google Scholar] [CrossRef]
- Kozlov, S.A.; Vassilevski, A.A.; Feofanov, A.V.; Surovoy, A.Y.; Karpunin, D.V.; Grishin, E.V. Latarcins, antimicrobial and cytolytic peptides from the venom of the spider Lachesana tarabaevi (Zodariidae) that exemplify biomolecular diversity. J. Biol. Chem. 2006, 281, 20983–20992. [Google Scholar] [CrossRef] [PubMed]
- Ouafik, L.H.; Stoffers, D.A.; Campbell, T.A.; Johnson, R.C.; Bloomquist, B.T.; Mains, R.E.; Eipper, B.A. The multifunctional peptidylglycine alpha-amidating monooxygenase gene: Exon/intron organization of catalytic, processing, and routing domains. Mol. Endocrinol. 1992, 6, 1571–1584. [Google Scholar] [PubMed]
- Undheim, E.A.; Grimm, L.L.; Low, C.F.; Morgenstern, D.; Herzig, V.; Zobel-Thropp, P.; Pineda, S.S.; Habib, R.; Dziemborowicz, S.; Fry, B.G.; et al. Weaponization of a hormone: Convergent recruitment of hyperglycemic hormone into the venom of arthropod predators. Structure 2015, 23, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Fry, B.G.; Roelants, K.; Champagne, D.E.; Scheib, H.; Tyndall, J.D.; King, G.F.; Nevalainen, T.J.; Norman, J.A.; Lewis, R.J.; Norton, R.S.; et al. The toxicogenomic multiverse: Convergent recruitment of proteins into animal venoms. Annu. Rev. Genom. Hum. Genet. 2009, 10, 483–511. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhuo, X.; Zhao, K.; Zheng, T.; Han, Y.; Yuan, C.; Zhang, Q. Transcriptome profiles reveal the crucial roles of hormone and sugar in the bud dormancy of Prunus mume. Sci. Rep. 2018, 8, 5090. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Satyanarayana, T. Bacterial and archaeal alpha-amylases: Diversity and amelioration of the desirable characteristics for industrial applications. Front. Microbiol. 2016, 7, 1129. [Google Scholar] [CrossRef] [PubMed]
- Avwioroko, O.J.; Anigboro, A.A.; Unachukwu, N.N.; Tonukari, N.J. Isolation, identification and in silico analysis of alpha-amylase gene of Aspergillus niger strain CSA35 obtained from cassava undergoing spoilage. Biochem. Biophys. Rep. 2018, 14, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Boehlke, C.; Zierau, O.; Hannig, C. Salivary amylase—The enzyme of unspecialized euryphagous animals. Arch. Oral Biol. 2015, 60, 1162–1176. [Google Scholar] [CrossRef] [PubMed]
- DeLay, B.; Mamidala, P.; Wijeratne, A.; Wijeratne, S.; Mittapalli, O.; Wang, J.; Lamp, W. Transcriptome analysis of the salivary glands of potato leafhopper, Empoasca fabae. J. Insect Physiol. 2012, 58, 1626–1634. [Google Scholar] [CrossRef] [PubMed]
- Mommsen, T.P. Comparison of digestive α-amylases from two species of spiders (Tegenaria atrica and Cupiennius salei). J. Comp. Physiol. 1978, 127, 355–361. [Google Scholar] [CrossRef]
- Fuzita, F.J.; Pinkse, M.W.; Patane, J.S.; Verhaert, P.D.; Lopes, A.R. High throughput techniques to reveal the molecular physiology and evolution of digestion in spiders. BMC Genom. 2016, 17, 716. [Google Scholar] [CrossRef] [PubMed]
- Lipovsek, S.; Novak, T.; Janzekovic, F.; Leitinger, G. Changes in the midgut diverticula in the harvestmen Amilenus aurantiacus (Phalangiidae, Opiliones) during winter diapause. Arthropod Struct. Dev. 2015, 44, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Smrz, J.; Kovac, L.; Mikes, J.; Lukesova, A. Microwhip scorpions (Palpigradi) feed on heterotrophic cyanobacteria in Slovak caves—A curiosity among Arachnida. PLoS ONE 2013, 8, e75989. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Wang, S.; Wang, S.G.; Wang, H.J.; Zhang, J.Y.; Cui, S.Y. Invertebrate trehalose-6-phosphate synthase gene: Genetic architecture, biochemistry, physiological function, and potential applications. Front. Physiol. 2018, 9, 30. [Google Scholar] [CrossRef]
- Gronke, S.; Clarke, D.F.; Broughton, S.; Andrews, T.D.; Partridge, L. Molecular evolution and functional characterization of Drosophila insulin-like peptides. PLoS Genet. 2010, 6, e1000857. [Google Scholar] [CrossRef]
- Kerekes, E.; Kokai, E.; Paldy, F.S.; Dombradi, V. Functional analysis of the glycogen binding subunit CG9238/Gbs-70E of protein phosphatase 1 in Drosophila melanogaster. Insect Biochem. Mol. Biol. 2014, 49, 70–79. [Google Scholar] [CrossRef]
- Suren-Castillo, S.; Abrisqueta, M.; Maestro, J.L. FoxO is required for the activation of hypertrehalosemic hormone expression in cockroaches. Biochim. Biophys. Acta 2014, 1840, 86–94. [Google Scholar] [CrossRef]
- Post, S.; Karashchuk, G.; Wade, J.D.; Sajid, W.; De Meyts, P.; Tatar, M. Drosophila insulin-like peptides DILP2 and DILP5 differentially stimulate cell signaling and glycogen phosphorylase to regulate longevity. Front. Endocrinol. 2018, 9, 245. [Google Scholar] [CrossRef]
- Graham, P.; Pick, L. Drosophila as a model for diabetes and diseases of insulin resistance. Curr. Top. Dev. Biol. 2017, 121, 397–419. [Google Scholar]
- Milne, T.J.; Abbenante, G.; Tyndall, J.D.; Halliday, J.; Lewis, R.J. Isolation and characterization of a cone snail protease with homology to CRISP proteins of the pathogenesis-related protein superfamily. J. Biol. Chem. 2003, 278, 31105–31110. [Google Scholar] [CrossRef]
- Hansson, K.; Thamlitz, A.M.; Furie, B.; Furie, B.C.; Stenflo, J. A single gamma-carboxyglutamic acid residue in a novel cysteine-rich secretory protein without propeptide. Biochemistry 2006, 45, 12828–12839. [Google Scholar] [CrossRef]
- Qian, J.; Guo, Z.Y.; Chi, C.W. Cloning and isolation of a Conus cysteine-rich protein homologous to Tex31 but without proteolytic activity. Acta Biochim. Biophys. Sin. 2008, 40, 174–181. [Google Scholar] [CrossRef]
- Assumpcao, T.C.; Ma, D.; Schwarz, A.; Reiter, K.; Santana, J.M.; Andersen, J.F.; Ribeiro, J.M.; Nardone, G.; Yu, L.L.; Francischetti, I.M. Salivary antigen-5/CAP family members are Cu2+-dependent antioxidant enzymes that scavenge O(2)(-). and inhibit collagen-induced platelet aggregation and neutrophil oxidative burst. J. Biol. Chem. 2013, 288, 14341–14361. [Google Scholar] [CrossRef]
- Cajado-Carvalho, D.; Kuniyoshi, A.K.; Duzzi, B.; Iwai, L.K.; Oliveira, U.C.; Junqueira de Azevedo, I.L.; Kodama, R.T.; Portaro, F.V. Insights into the hypertensive effects of Tityus serrulatus scorpion venom: Purification of an angiotensin-converting enzyme-like peptidase. Toxins 2016, 8, 348. [Google Scholar] [CrossRef]
- de Oliveira, U.C.; Nishiyama, M.Y.J.; Dos Santos, M.B.V.; Santos-da-Silva, A.P.; Chalkidis, H.M.; Souza-Imberg, A.; Candido, D.M.; Yamanouye, N.; Dorce, V.A.C.; Junqueira-de-Azevedo, I.L.M. Proteomic endorsed transcriptomic profiles of venom glands from Tityus obscurus and T. serrulatus scorpions. PLoS ONE 2018, 13, e0193739. [Google Scholar] [CrossRef]
- Hernandez-Vargas, M.J.; Gil, J.; Lozano, L.; Pedraza-Escalona, M.; Ortiz, E.; Encarnacion-Guevara, S.; Alagon, A.; Corzo, G. Proteomic and transcriptomic analysis of saliva components from the hematophagous reduviid Triatoma pallidipennis. J. Proteom. 2017, 162, 30–39. [Google Scholar] [CrossRef]
- Yan, H.Y.; Mita, K.; Zhao, X.; Tanaka, Y.; Moriyama, M.; Wang, H.; Iwanaga, M.; Kawasaki, H. The angiotensin-converting enzyme (ACE) gene family of Bombyx mori. Gene 2017, 608, 58–65. [Google Scholar] [CrossRef]
- Yamagishi, T.; Endo, H.; Fukumura, K.; Nagata, S.; Hayakawa, T.; Adegawa, S.; Kasubuchi, M.; Sato, R. Glucose, some amino acids and a plant secondary metabolite, chlorogenic acid induce the secretion of a regulatory hormone, tachykinin-related peptide, from the silkworm midgut. Peptides 2018, 106, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.L.; Otwinowski, Z.; Gelb, M.H.; Sigler, P.B. Crystal structure of bee-venom phospholipase A2 in a complex with a transition-state analogue. Science 1990, 250, 1563–1566. [Google Scholar] [CrossRef] [PubMed]
- Shipolini, R.A.; Callewaert, G.L.; Cottrell, R.C.; Vernon, C.A. The amino-acid sequence and carbohydrate content of phospholipase A2 from bee venom. Eur. J. Biochem. 1974, 48, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Valdez-Cruz, N.A.; Segovia, L.; Corona, M.; Possani, L.D. Sequence analysis and phylogenetic relationship of genes encoding heterodimeric phospholipases A2 from the venom of the scorpion Anuroctonus phaiodactylus. Gene 2007, 396, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Kordis, D.; Turk, V. Phylogenomic analysis of the cystatin superfamily in eukaryotes and prokaryotes. BMC Evol. Biol. 2009, 9, 266. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.Y.; Tan, C.H.; Chanhome, L.; Tan, N.H. Comparative venom gland transcriptomics of Naja kaouthia (monocled cobra) from Malaysia and Thailand: Elucidating geographical venom variation and insights into sequence novelty. PeerJ 2017, 5, e3142. [Google Scholar] [CrossRef]
- Turk, V.; Stoka, V.; Turk, D. Cystatins: Biochemical and structural properties, and medical relevance. Front. Biosci. 2008, 13, 5406–5420. [Google Scholar] [CrossRef]
- Salat, J.; Paesen, G.C.; Rezacova, P.; Kotsyfakis, M.; Kovarova, Z.; Sanda, M.; Majtan, J.; Grunclova, L.; Horka, H.; Andersen, J.F.; et al. Crystal structure and functional characterization of an immunomodulatory salivary cystatin from the soft tick Ornithodoros moubata. Biochem. J. 2010, 429, 103–112. [Google Scholar] [CrossRef]
- Choo, Y.M.; Lee, K.S.; Yoon, H.J.; Qiu, Y.; Wan, H.; Sohn, M.R.; Sohn, H.D.; Jin, B.R. Antifibrinolytic role of a bee venom serine protease inhibitor that acts as a plasmin inhibitor. PLoS ONE 2012, 7, e32269. [Google Scholar] [CrossRef]
- Zhao, R.; Dai, H.; Qiu, S.; Li, T.; He, Y.; Ma, Y.; Chen, Z.; Wu, Y.; Li, W.; Cao, Z. SdPI, the first functionally characterized Kunitz-type trypsin inhibitor from scorpion venom. PLoS ONE 2011, 6, e27548. [Google Scholar] [CrossRef]
- Santibanez-Lopez, C.E.; Ontano, A.Z.; Harvey, M.S.; Sharma, P.P. Transcriptomic analysis of pseudoscorpion venom reveals a unique cocktail dominated by enzymes and protease inhibitors. Toxins 2018, 10, 207. [Google Scholar] [CrossRef]
- Wan, H.; Lee, K.S.; Kim, B.Y.; Zou, F.M.; Yoon, H.J.; Je, Y.H.; Li, J.; Jin, B.R. A spider-derived Kunitz-type serine protease inhibitor that acts as a plasmin inhibitor and an elastase inhibitor. PLoS ONE 2013, 8, e53343. [Google Scholar] [CrossRef]
- Soares, T.S.; Watanabe, R.M.; Tanaka-Azevedo, A.M.; Torquato, R.J.; Lu, S.; Figueiredo, A.C.; Pereira, P.J.; Tanaka, A.S. Expression and functional characterization of boophilin, a thrombin inhibitor from Rhipicephalus (Boophilus) microplus midgut. Vet. Parasitol. 2012, 187, 521–528. [Google Scholar] [CrossRef]
- Tsujimoto, H.; Kotsyfakis, M.; Francischetti, I.M.; Eum, J.H.; Strand, M.R.; Champagne, D.E. Simukunin from the salivary glands of the black fly Simulium vittatum inhibits enzymes that regulate clotting and inflammatory responses. PLoS ONE 2012, 7, e29964. [Google Scholar] [CrossRef]
- Corral-Rodriguez, M.A.; Macedo-Ribeiro, S.; Barbosa Pereira, P.J.; Fuentes-Prior, P. Tick-derived Kunitz-type inhibitors as antihemostatic factors. Insect Biochem. Mol. Biol. 2009, 39, 579–595. [Google Scholar] [CrossRef]
- Peigneur, S.; Billen, B.; Derua, R.; Waelkens, E.; Debaveye, S.; Beress, L.; Tytgat, J. A bifunctional sea anemone peptide with Kunitz type protease and potassium channel inhibiting properties. Biochem. Pharmacol. 2011, 82, 81–90. [Google Scholar] [CrossRef]
- Santibanez-Lopez, C.E.; Cid-Uribe, J.I.; Batista, C.V.; Ortiz, E.; Possani, L.D. Venom gland transcriptomic and proteomic analyses of the enigmatic scorpion Superstitionia donensis (Scorpiones: Superstitioniidae), with insights on the evolution of its venom components. Toxins 2016, 8, 367. [Google Scholar] [CrossRef]
- Anatriello, E.; Ribeiro, J.M.; de Miranda-Santos, I.K.; Brandao, L.G.; Anderson, J.M.; Valenzuela, J.G.; Maruyama, S.R.; Silva, J.S.; Ferreira, B.R. An insight into the sialotranscriptome of the brown dog tick, Rhipicephalus sanguineus. BMC Genom. 2010, 11, 450. [Google Scholar] [CrossRef]
- Mihelic, M.; Turk, D. Two decades of thyroglobulin type-1 domain research. Biol. Chem. 2007, 388, 1123–1130. [Google Scholar] [CrossRef]
- Chandler, J.C.; Gandhi, N.S.; Mancera, R.L.; Smith, G.; Elizur, A.; Ventura, T. Understanding insulin endocrinology in decapod Crustacea: Molecular modelling characterization of an insulin-binding protein and insulin-like peptides in the eastern spiny lobster, Sagmariasus verreauxi. Int. J. Mol. Sci. 2017, 18, 1832. [Google Scholar] [CrossRef]
- Mulenga, A.; Blandon, M.; Khumthong, R. The molecular basis of the Amblyomma americanum tick attachment phase. Exp. Appl. Acarol. 2007, 41, 267–287. [Google Scholar] [CrossRef]
- Gokudan, S.; Muta, T.; Tsuda, R.; Koori, K.; Kawahara, T.; Seki, N.; Mizunoe, Y.; Wai, S.N.; Iwanaga, S.; Kawabata, S. Horseshoe crab acetyl group-recognizing lectins involved in innate immunity are structurally related to fibrinogen. Proc. Natl. Acad. Sci. USA 1999, 96, 10086–10091. [Google Scholar] [CrossRef]
- Kawabata, S.; Beisel, H.G.; Huber, R.; Bode, W.; Gokudan, S.; Muta, T.; Tsuda, R.; Koori, K.; Kawahara, T.; Seki, N.; et al. Role of tachylectins in host defense of the Japanese horseshoe crab Tachypleus tridentatus. Adv. Exp. Med. Biol. 2001, 484, 195–202. [Google Scholar]
- Maeda, H.; Kurisu, K.; Miyata, T.; Kusakisako, K.; Galay, R.L.; Rio, T.M.; Mochizuki, M.; Fujisaki, K.; Tanaka, T. Identification of the Babesia-responsive leucine-rich repeat domain-containing protein from the hard tick Haemaphysalis longicornis. Parasitol. Res. 2015, 114, 1793–1802. [Google Scholar] [CrossRef]
- Koua, D.; Kuhn-Nentwig, L. Spider neurotoxins, short linear cationic peptides and venom protein classification improved by an automated competition between exhaustive profile HMM classifiers. Toxins 2017, 9, 245. [Google Scholar] [CrossRef]
- King, G.F.; Gentz, M.C.; Escoubas, P.; Nicholson, G.M. A rational nomenclature for naming peptide toxins from spiders and other venomous animals. Toxicon 2008, 52, 264–276. [Google Scholar] [CrossRef]
- Norton, R.S.; Pallaghy, P.K. The cystine knot structure of ion channel toxins and related polypeptides. Toxicon 1998, 36, 1573–1583. [Google Scholar] [CrossRef]
- Wang, X.; Connor, M.; Smith, R.; Maciejewski, M.W.; Howden, M.E.; Nicholson, G.M.; Christie, M.J.; King, G.F. Discovery and characterization of a family of insecticidal neurotoxins with a rare vicinal disulfide bridge. Nat. Struct. Biol. 2000, 7, 505–513. [Google Scholar]
- Zhu, S.; Peigneur, S.; Gao, B.; Umetsu, Y.; Ohki, S.; Tytgat, J. Experimental conversion of a defensin into a neurotoxin: Implications for origin of toxic function. Mol. Biol. Evol. 2014, 31, 546–559. [Google Scholar] [CrossRef]
- Lavergne, V.; Alewood, P.F.; Mobli, M.; King, G.F. The structural universe of disulfide-rich venom peptides. In Venoms to Drugs; King, G.F., Ed.; The Royal Society of Chemistry: London, UK, 2015; pp. 37–79. [Google Scholar]
- Kopp, L. Biochemical and Mass Spectrometric Analysis of Venom Components, Venom Gland, and Hemocytes of the Spider Cupiennius salei. Ph.D. Thesis, The University of Bern, Bern, Switzerland, 2013. [Google Scholar]
- Wen, S.; Wilson, D.T.; Kuruppu, S.; Korsinczky, M.L.; Hedrick, J.; Pang, L.; Szeto, T.; Hodgson, W.C.; Alewood, P.F.; Nicholson, G.M. Discovery of an MIT-like atracotoxin family: Spider venom peptides that share sequence homology but not pharmacological properties with AVIT family proteins. Peptides 2005, 26, 2412–2426. [Google Scholar] [CrossRef]
- Sforca, M.L.; Oyama, S.; Canduri, F.; Lorenzi, C.C.B.; Pertinhez, T.A.; Konno, K.; Souza, B.M.; Palma, M.S.; RuggieroNeto, J.; Azevedo, W.F.; et al. How C-terminal carboxyamidation alters the biological activity of peptides from the venom of the eumenine solitary wasp. Biochemistry 2004, 43, 5608–5617. [Google Scholar] [CrossRef]
- Kubista, H.; Mafra, R.A.; Chong, Y.; Nicholson, G.M.; Beirao, P.S.; Cruz, J.S.; Boehm, S.; Nentwig, W.; Kuhn-Nentwig, L. CSTX-1, a toxin from the venom of the hunting spider Cupiennius salei, is a selective blocker of L-type calcium channels in mammalian neurons. Neuropharmacology 2007, 52, 1650–1662. [Google Scholar] [CrossRef]
- Vassilevski, A.A.; Fedorova, I.M.; Maleeva, E.E.; Korolkova, Y.V.; Efimova, S.S.; Samsonova, O.V.; Schagina, L.V.; Feofanov, A.V.; Magazanik, L.G.; Grishin, E.V. Novel class of spider toxin: Active principle from the yellow sac spider Cheiracanthium punctorium venom is a unique two-domain polypeptide. J. Biol. Chem. 2010, 285, 32293–32302. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, J.; Tang, X.; Wang, F.; Jiang, L.; Xiong, X.; Wang, M.; Rong, M.; Liu, Z.; Liang, S. Transcriptome analysis of the venom glands of the Chinese wolf spider Lycosa singoriensis. Zoology 2010, 113, 10–18. [Google Scholar] [CrossRef]
- Oparin, P.B.; Nadezhdin, K.D.; Berkut, A.A.; Arseniev, A.S.; Grishin, E.V.; Vassilevski, A.A. Structure of purotoxin-2 from wolf spider: Modular design and membrane-assisted mode of action in arachnid toxins. Biochem. J. 2016, 473, 3113–3126. [Google Scholar] [CrossRef]
- Kuhn-Nentwig, L.; Nentwig, W. The cytotoxic mode of action of the venom of Cupiennius salei (Ctenidae). In Spider Ecophysiology; Nentwig, W., Ed.; Springer: Heidelberg, Germany, 2013; pp. 217–228. [Google Scholar]
- Borges, M.H.; de Lima, M.E.; Stankiewicz, M.; Pelhate, M.; do Nascimento Cordeiro, M.; Beirao, P.S. Structural and functional diversity in the venom of spiders of the genus Phoneutria. In Animal Toxins: State of the Art; de Lima, M.E., Ed.; Editora UFMG: Belo Horizonte, Brazil, 2009; pp. 291–311. [Google Scholar]
- Cordeiro Mdo, N.; de Figueiredo, S.G.; Valentim Ado, C.; Diniz, C.R.; von Eickstedt, V.R.; Gilroy, J.; Richardson, M. Purification and amino acid sequences of six Tx3 type neurotoxins from the venom of the Brazilian ‘armed’ spider Phoneutria nigriventer (Keys). Toxicon 1993, 31, 35–42. [Google Scholar] [CrossRef]
- Pluzhnikov, K.; Vassilevski, A.; Korolkova, Y.; Fisyunov, A.; Iegorova, O.; Krishtal, O.; Grishin, E. Omega-Lsp-IA, a novel modulator of P-type Ca2+ channels. Toxicon 2007, 50, 993–1004. [Google Scholar] [CrossRef]
- Bickmeyer, U.; Rossler, W.; Wiegand, H. Omega aga-toxin IVA blocks high-voltage-activated calcium channel currents in cultured pars intercerebralis neurosecretory cells of adult Locusta migratoria. Neurosci. Lett. 1994, 181, 113–116. [Google Scholar] [CrossRef]
- Adams, M.E.; Mintz, I.M.; Reily, M.D.; Thanabal, V.; Bean, B.P. Structure and properties of omega-agatoxin IVB, a new antagonist of P-type calcium channels. Mol. Pharmacol. 1993, 44, 681–688. [Google Scholar]
- Sturm, S.; Ramesh, D.; Brockmann, A.; Neupert, S.; Predel, R. Agatoxin-like peptides in the neuroendocrine system of the honey bee and other insects. J. Proteom. 2016, 132, 77–84. [Google Scholar] [CrossRef]
- von Reumont, B.M.; Blanke, A.; Richter, S.; Alvarez, F.; Bleidorn, C.; Jenner, R.A. The first venomous crustacean revealed by transcriptomics and functional morphology: Remipede venom glands express a unique toxin cocktail dominated by enzymes and a neurotoxin. Mol. Biol. Evol. 2014, 31, 48–58. [Google Scholar] [CrossRef]
- Kozlov, S.; Malyavka, A.; McCutchen, B.; Lu, A.; Schepers, E.; Herrmann, R.; Grishin, E. A novel strategy for the identification of toxinlike structures in spider venom. Proteins 2005, 59, 131–140. [Google Scholar] [CrossRef]
- Martin-Moutot, N.; Mansuelle, P.; Alcaraz, G.; Dos Santos, R.G.; Cordeiro, M.N.; De Lima, M.E.; Seagar, M.; Van Renterghem, C. Phoneutria nigriventer toxin 1: A novel, state-dependent inhibitor of neuronal sodium channels that interacts with micro conotoxin binding sites. Mol. Pharmacol. 2006, 69, 1931–1937. [Google Scholar] [CrossRef]
- Cassola, A.C.; Jaffe, H.; Fales, H.M.; Afeche, S.C.; Magnoli, F.; Cipolla-Neto, J. Omega-phonetoxin-IIA: A calcium channel blocker from the spider Phoneutria nigriventer. Pflügers Arch. 1998, 436, 545–552. [Google Scholar] [CrossRef]
- Dos Santos, R.G.; Van Renterghem, C.; Martin-Moutot, N.; Mansuelle, P.; Cordeiro, M.N.; Diniz, C.R.; Mori, Y.; De Lima, M.E.; Seagar, M. Phoneutria nigriventer omega-phonetoxin IIA blocks the Cav2 family of calcium channels and interacts with omega-conotoxin-binding sites. J. Biol. Chem. 2002, 277, 13856–13862. [Google Scholar] [CrossRef]
- Kozlov, S.A.; Lazarev, V.N.; Kostryukova, E.S.; Selezneva, O.V.; Ospanova, E.A.; Alexeev, D.G.; Govorun, V.M.; Grishin, E.V. Comprehensive analysis of the venom gland transcriptome of the spider Dolomedes fimbriatus. Sci. Data 2014, 1, 140023. [Google Scholar] [CrossRef]
- Venema, V.J.; Swiderek, K.M.; Lee, T.D.; Hathaway, G.M.; Adams, M.E. Antagonism of synaptosomal calcium channels by subtypes of omega-agatoxins. J. Biol. Chem. 1992, 267, 2610–2615. [Google Scholar]
- Kabanova, N.V.; Vassilevski, A.A.; Rogachevskaja, O.A.; Bystrova, M.F.; Korolkova, Y.V.; Pluzhnikov, K.A.; Romanov, R.A.; Grishin, E.V.; Kolesnikov, S.S. Modulation of P2X3 receptors by spider toxins. Biochim. Biophys. Acta 2012, 1818, 2868–2875. [Google Scholar] [CrossRef]
- Undheim, E.A.; Sunagar, K.; Herzig, V.; Kely, L.; Low, D.H.; Jackson, T.N.; Jones, A.; Kurniawan, N.; King, G.F.; Ali, S.A.; et al. A proteomics and transcriptomics investigation of the venom from the barychelid spider Trittame loki (brush-foot trapdoor). Toxins 2013, 5, 2488–2503. [Google Scholar] [CrossRef]
- Cheng, T.C.; Long, R.W.; Wu, Y.Q.; Guo, Y.B.; Liu, D.L.; Peng, L.; Li, D.Q.; Yang, D.W.; Xu, X.; Liu, F.X.; et al. Identification and characterization of toxins in the venom gland of the Chinese bird spider, Haplopelma hainanum, by transcriptomic analysis. Insect Sci. 2016, 23, 487–499. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Huang, Y.; He, Q.Z.; Luo, J.; Zhu, L.; Lu, S.S.; Liu, J.Y.; Huang, P.F.; Zeng, X.Z.; Liang, S.P. Structural and functional diversity of peptide toxins from tarantula Haplopelma hainanum (Ornithoctonus hainana) venom revealed by transcriptomic, peptidomic, and patch clamp approaches. J. Biol. Chem. 2015, 290, 26471–26472. [Google Scholar] [CrossRef] [PubMed]
- Satake, H.; Villegas, E.; Oshiro, N.; Terada, K.; Shinada, T.; Corzo, G. Rapid and efficient identification of cysteine-rich peptides by random screening of a venom gland cDNA library from the hexathelid spider Macrothele gigas. Toxicon 2004, 44, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Chong, Y.; Hayes, J.L.; Sollod, B.; Wen, S.; Wilson, D.T.; Hains, P.G.; Hodgson, W.C.; Broady, K.W.; King, G.F.; Nicholson, G.M. The omega-atracotoxins: Selective blockers of insect M-LVA and HVA calcium channels. Biochem. Pharmacol. 2007, 74, 623–638. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zhang, Y.; Hu, W.; Xu, D.; Tao, H.; Yang, X.; Li, Y.; Jiang, L.; Liang, S. Molecular diversification of peptide toxins from the tarantula Haplopelma hainanum (Ornithoctonus hainana) venom based on transcriptomic, peptidomic, and genomic analyses. J. Proteome Res. 2010, 9, 2550–2564. [Google Scholar] [CrossRef] [PubMed]
- Pocock, J.M.; Venema, V.J.; Adams, M.E. Omega-agatoxins differentially block calcium channels in locust, chick and rat synaptosomes. Neurochem. Int. 1992, 20, 263–270. [Google Scholar] [CrossRef]
- Santos, A.D.; Imperial, J.S.; Chaudhary, T.; Beavis, R.C.; Chait, B.T.; Hunsperger, J.P.; Olivera, B.M.; Adams, M.E.; Hillyard, D.R. Heterodimeric structure of the spider toxin omega-agatoxin IA revealed by precursor analysis and mass spectrometry. J. Biol. Chem. 1992, 267, 20701–20705. [Google Scholar] [PubMed]
- Pineda, S.S.; Chaumeil, P.A.; Kunert, A.; Kaas, Q.; Thang, M.W.C.; Li, L.; Nuhn, M.; Herzig, V.; Saez, N.J.; Cristofori-Armstrong, B.; et al. ArachnoServer 3.0: An online resource for automated discovery, analysis and annotation of spider toxins. Bioinformatics 2017, 34, 1074–1076. [Google Scholar] [CrossRef]
- Boisbouvier, J.; Albrand, J.P.; Blackledge, M.; Jaquinod, M.; Schweitz, H.; Lazdunski, M.; Marion, D. A structural homologue of colipase in black mamba venom revealed by NMR floating disulphide bridge analysis. J. Mol. Biol. 1998, 283, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Morales, R.A.; Daly, N.L.; Vetter, I.; Mobli, M.; Napier, I.A.; Craik, D.J.; Lewis, R.J.; Christie, M.J.; King, G.F.; Alewood, P.F.; et al. Chemical synthesis and structure of the prokineticin Bv8. Chembiochem 2010, 11, 1882–1888. [Google Scholar] [CrossRef]
- Szeto, T.H.; Wang, X.H.; Smith, R.; Connor, M.; Christie, M.J.; Nicholson, G.M.; King, G.F. Isolation of a funnel-web spider polypeptide with homology to mamba intestinal toxin 1 and the embryonic head inducer Dickkopf-1. Toxicon 2000, 38, 429–442. [Google Scholar] [CrossRef]
- Rokyta, D.R.; Ward, M.J. Venom-gland transcriptomics and venom proteomics of the black-back scorpion (Hadrurus spadix) reveal detectability challenges and an unexplored realm of animal toxin diversity. Toxicon 2017, 128, 23–37. [Google Scholar] [CrossRef]
- Garcia, G.R.; Gardinassi, L.G.; Ribeiro, J.M.; Anatriello, E.; Ferreira, B.R.; Moreira, H.N.; Mafra, C.; Martins, M.M.; Szabo, M.P.; de Miranda-Santos, I.K.; et al. The sialotranscriptome of Amblyomma triste, Amblyomma parvum and Amblyomma cajennense ticks, uncovered by 454-based RNA-seq. Parasit Vectors 2014, 7, 430. [Google Scholar] [CrossRef]
- Baumann, T.; Kuhn-Nentwig, L.; Largiadèr, C.R.; Nentwig, W. Expression of defensins in non-infected araneomorph spiders. Cell. Mol. Life Sci. 2010, 67, 2643–2651. [Google Scholar] [CrossRef]
- Meng, L.; Xie, Z.; Zhang, Q.; Li, Y.; Yang, F.; Chen, Z.; Li, W.; Cao, Z.; Wu, Y. Scorpion potassium channel-blocking defensin highlights a functional link with neurotoxin. J. Biol. Chem. 2016, 291, 7097–7106. [Google Scholar] [CrossRef]
- Ward, M.J.; Ellsworth, S.A.; Rokyta, D.R. Venom-gland transcriptomics and venom proteomics of the Hentz striped scorpion (Centruroides hentzi; Buthidae) reveal high toxin diversity in a harmless member of a lethal family. Toxicon 2018, 142, 14–29. [Google Scholar] [CrossRef]
- Zhang, L.; Shi, W.; Zeng, X.C.; Ge, F.; Yang, M.; Nie, Y.; Bao, A.; Wu, S.; E, G. Unique diversity of the venom peptides from the scorpion Androctonus bicolor revealed by transcriptomic and proteomic analysis. J. Proteom. 2015, 128, 231–250. [Google Scholar] [CrossRef]
- Kotsyfakis, M.; Schwarz, A.; Erhart, J.; Ribeiro, J.M. Tissue- and time-dependent transcription in Ixodes ricinus salivary glands and midguts when blood feeding on the vertebrate host. Sci. Rep. 2015, 5, 9103. [Google Scholar] [CrossRef]
- Wullschleger, B.; Nentwig, W.; Kuhn-Nentwig, L. Spider venom: Enhancement of venom efficacy mediated by different synergistic strategies in Cupiennius salei. J. Exp. Biol. 2005, 208, 2115–2121. [Google Scholar] [CrossRef]
- Kuhn-Nentwig, L.; Müller, J.; Schaller, J.; Walz, A.; Dathe, M.; Nentwig, W. Cupiennin 1, a new family of highly basic antimicrobial peptides in the venom of the spider Cupiennius salei (Ctenidae). J. Biol. Chem. 2002, 277, 11208–11216. [Google Scholar] [CrossRef]
- Kuhn-Nentwig, L.; Willems, J.; Seebeck, T.; Shalaby, T.; Kaiser, M.; Nentwig, W. Cupiennin 1a exhibits a remarkably broad, non-stereospecific cytolytic activity on bacteria, protozoan parasites, insects, and human cancer cells. Amino Acids 2011, 40, 69–76. [Google Scholar] [CrossRef][Green Version]
- Pukala, T.L.; Doyle, J.R.; Llewellyn, L.E.; Kuhn-Nentwig, L.; Apponyi, M.A.; Separovic, F.; Bowie, J.H. Cupiennin1a, an antimicrobial peptide from the venom of the neotropical wandering spider Cupiennius salei, also inhibits the formation of nitric oxide by neuronal nitric oxide synthase. FEBS Lett. 2007, 274, 1778–1784. [Google Scholar] [CrossRef]
- Casewell, N.R.; Wüster, W.; Vonk, F.J.; Harrison, R.A.; Fry, B.G. Complex cocktails: The evolutionary novelty of venoms. Trends Ecol. Evol. 2013, 28, 219–229. [Google Scholar] [CrossRef]
- Santibanez-Lopez, C.E.; Cid-Uribe, J.I.; Zamudio, F.Z.; Batista, C.V.F.; Ortiz, E.; Possani, L.D. Venom gland transcriptomic and venom proteomic analyses of the scorpion Megacormus gertschi Diaz-Najera, 1966 (Scorpiones: Euscorpiidae: Megacorminae). Toxicon 2017, 133, 95–109. [Google Scholar] [CrossRef]
- Kuhn-Nentwig, L.; Schaller, J.; Nentwig, W. Purification of toxic peptides and the amino acid sequence of CSTX-1 from the multicomponent venom of Cupiennius salei (Araneae: Ctenidae). Toxicon 1994, 32, 287–302. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Combet, C.; Blanchet, C.; Geourjon, C.; Deleage, G. NPS@: Network protein sequence analysis. Trends Biochem. Sci. 2000, 25, 147–150. [Google Scholar] [CrossRef]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Braga-Lagache, S.; Buchs, N.; Iacovache, M.I.; Zuber, B.; Jackson, C.B.; Heller, M. Robust label-free, quantitative profiling of circulating plasma microparticle (MP) associated proteins. Mol. Cell. Proteom. 2016, 15, 3640–3652. [Google Scholar] [CrossRef]
- Kou, Q.; Xun, L.; Liu, X. TopPIC: A software tool for top-down mass spectrometry-based proteoform identification and characterization. Bioinformatics 2016, 32, 3495–3497. [Google Scholar] [CrossRef]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef]
- Eng, J.K.; Hoopmann, M.R.; Jahan, T.A.; Egertson, J.D.; Noble, W.S.; MacCoss, M.J. A deeper look into Comet—Implementation and features. J. Am. Soc. Mass Spectrom. 2015, 26, 1865–1874. [Google Scholar] [CrossRef]
- Craig, R.; Beavis, R.C. A method for reducing the time required to match protein sequences with tandem mass spectra. Rapid Commun. Mass Spectrom. 2003, 17, 2310–2316. [Google Scholar] [CrossRef]
- Kim, S.; Pevzner, P.A. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 2014, 5, 5277. [Google Scholar] [CrossRef]
- Tabb, D.L.; Fernando, C.G.; Chambers, M.C. MyriMatch: Highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J. Proteome Res. 2007, 6, 654–661. [Google Scholar] [CrossRef]
- Choi, H.; Ghosh, D.; Nesvizhskii, A.I. Statistical validation of peptide identifications in large-scale proteomics using the target-decoy database search strategy and flexible mixture modeling. J. Proteome Res. 2008, 7, 286–292. [Google Scholar] [CrossRef]
- Deutsch, E.W.; Mendoza, L.; Shteynberg, D.; Farrah, T.; Lam, H.; Tasman, N.; Sun, Z.; Nilsson, E.; Pratt, B.; Prazen, B.; et al. A guided tour of the trans-proteomic pipeline. Proteomics 2010, 10, 1150–1159. [Google Scholar] [CrossRef]
- Shteynberg, D.; Deutsch, E.W.; Lam, H.; Eng, J.K.; Sun, Z.; Tasman, N.; Mendoza, L.; Moritz, R.L.; Aebersold, R.; Nesvizhskii, A.I. iProphet: Multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol. Cell. Proteom. 2011, 10, M111.007690. [Google Scholar] [CrossRef]
- Nesvizhskii, A.I.; Keller, A.; Kolker, E.; Aebersold, R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003, 75, 4646–4658. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | TPM | Transcripts/Precursors (n)/(n) | Length Signal Peptide (aa) | Length Mature Protein (aa) | Molecular Mass * (kDa) (pI) ** | InterProScan ID Predicted Functional Role | Proteomic Coverage [%] [Trypsin/Chymo-trypsin] |
|---|---|---|---|---|---|---|---|
| Signal peptidase catalytic subunit (SPase) | 7.97 | 1/1 | 177 | 20.0 (9.21) | IPR001733 Specific peptidase activity | [-/-] | |
| Protein disulfide isomerase (PDI) | 14,970.57 | 3/2 | 16 | 479 | 54.2 (4.77) 54.1 (4.81) | IPR005792 PDI family Isomerase activity | [81.6/60.2] [80.8/56.0] |
| Venom serine protease PQM protease (VSP1_a) A0A2I5YNR5 A0A2I5YNR1 | 10,970.57 | 3/2 | 18 | 260 | 28.0 (7.03) 28.0 (7.66) | IPR001314 IPR001254 PQM serine protease | [91.1/81.8] [91.1/91.1] |
| Venom serine protease 1_b1/b2 PQM protease (VSP1_b) | 12,251.37 | 2/2 | 18 | 261 | 28.3 (5.84) 28.3 (6.12) | IPR001314 IPR001254 Serine protease | [86.8/84.3] [86.8/77.9] |
| Venom serine protease 2 (VSP2) | 3476.51 | 3/1 | 16 | 281 | 31.0 (8.03) | IPR001314 IPR001254 Serine protease | [85.2/71.0] |
| Carboxypeptidase A-like protein (CPA) | 72.70 | 2/1 | 18 | 454 | 53.1 (6.55) | IPR000834 IPR008969 Carboxypeptidase A | [89.4/66.1] |
| Peptidylglycine alpha-amidating monooxygenase (PAM) | 153.60 | 1/1 | 25 | 328 | 36.9 (5.13) | IPR000720 C-terminal amidating activity | [82.4/79.9] |
| α-amylase (α-AMY) | 57,070.97 | >1 | 21 | 510 | 57.6 (6.61) | IPR006046) Alpha amylase | [92.2/89.3] |
| Cysteine-rich secretory protein (CRISP1) | 19,808.52 | 2/2 | 26 | 418 | 46.6 (7.54) 46.5 (7.54) | IPR001283 IPR002413 CAP domain IPR014044 | [94.1/93.2] [94.1/93.2] |
| Cysteine-rich secretory protein (CRISP2) | 2121.07 | 1/1 | 19 | 396 | 44.0 (5.93) | IPR001283 IPR002413 CAP domain IPR014044 | [90.8/82.2] |
| Angiotensin-converting enzyme (ACE) | 4795.97 | 4/4 | 20 | 614 | 70.9 (5.82) | IPR033591 Metallocarboxy-peptidase activity | [91.3/86.8] [91.3/86.8] [91.3/86.8] [91.3/86.8] |
| Hyaluronidase-like (HYAL) tr_A0A0S4JYH2 | 3040.68 | 1/1 | 16 | 378 | 43.9 (8.56) | IPR018155 O-glycosyl-hydrolase activity | [93.7/85.0] |
| Phospholipase A2 (PLA2) | 60.17 | 1/1 | 20 | 150 | 16.9 (5.04) | IPR001211 Cell membrane lysis | [62.9/47.6] |
| Cystatin (CST) | 559.42 | 3/2 | 20 | 116 | 13.2 (8.80) 13.2 (6.90) | IPR027214 Cysteine-type endopeptidase inhibitor activity | [50.7/49.3] [60.3/49.3] |
| Kunitz domain-containing protein (KCP) | 88.19 | 2/2 | 16 | 185 | 20.6 (4.22) 20.5 (4.22) | IPR002223 Serine-type endopeptidase inhibitor activity | [60.7/44.3] [60.7/27.9] |
| TT1LP Thyroglobulin type-1-like protein (TT1LP) | 392.26 | 4/2 | 18 | 133 | 14.9 (4.42) 14.9 (4.42) | IPR00716 | [90.1/84.1] [88.1/77.5] |
| Insulin-like growth factor-binding protein-related protein 1 (IGFBP-rP1) G4V4G1 | 142.67 | 1/1 | 17 | 244 | 26.7 (5.00) | IPR000867 IGFBP-like IPR002350 Kazal_dom IPR007110 Ig-like_domain | [11.9/-] |
| Tachylectin 5A-like (TL5A) | 1615.50 | 2/2 | 18 | 286 | 32.4 (5.93) 32.4 (5.93) | IPR002181 Innate immunity | [71.7/53.9] [71.7/53.9] |
| Leucine-rich repeat protein (LRR) | 7994.70 | 2/1 | 19 | 297 | 34.1 (5.29) | IPR032675Leucine-rich repeat domain; innate immunity | [83.5/78.2] |
| SN_Peptide Family_Group | Cupiennius salei Neurotoxin | Arachnoserver Nomenclature | Mature Toxins N | Cysteine Framework |
|---|---|---|---|---|
| Neurotoxins exhibiting N-terminal ICK fold and C-terminal α-helix | ||||
| SN_19_13 SN_19_14 SN_19_12 | CsTx-1a,b,c CsTx-10a,b CsTx-11a,b CsTx-13a,b CsTx-12a,b CsTx-8a,b | Omega-ctenitoxin-Cs1a,,c U5-ctenitoxin-Cs1a,b U6-ctenitoxin-Cs1a,b U2-ctenitoxin-Cs1a,b U3-ctenitoxin-Cs1a,b U4-ctenitoxin-Cs1a,b | 2 [1] 2 [2] 2 * [1] 1 [1] 2 [2] 1 [1] |  |
| Neurotoxins exhibiting ICK fold | ||||
| SN_19_13 | CsTx-9a,b,c CsTx-33a,b | U1-ctenitoxin-Cs1a,b,c U7-ctenitoxin-Cs1a,b | 2 [2] 1 [1] | #C6C6CC8C1C13C1C# |
| SN_19_06 | CsTx-23a,b | U8-ctenitoxin-Cs1a,b | 2 [2] | #C6C6CC4C1C13C1C# |
| SN_02_03 | CsTx-17 CsTx-31a,b,c,d CsTx-25 CsTx-28a,b | U9-ctenitoxin-Cs1 U10-ctenitoxin-Cs1a,b,c,d U11-ctenitoxin-Cs1 U12-ctenitoxin-Cs1a,b | 1 [1] 3 [2] 1 [1] 2 [2] | #C6C6CC4C1C6C1C# |
| SN_02_04 | CsTx-36a,b,c | U13-ctenitoxin-Cs1a,b,c | 1 [1] | #C6C4CC4C1C6C1C# |
| SN_02_07 | CsTx-18a,b CsTx-19 | U15-ctenitoxin-Cs1a,b U16-ctenitoxin-Cs1 | 2 * [1] 1 [1] | #C4C6CC4C1C4C1C# |
| SN_02_16 | CsTx-34a,b | U14-ctenitoxin-Cs1a,b | 1 [1] | #C6C6CC4C1C10C1C# |
| SN_33_00 | CsTx-26 | U17-ctenitoxin-Cs1 | 1 [1] | #C6C4CC4C1C6C1C# |
| SN_29_01 | CsTx-30 | U18-ctenitoxin-Cs1 | 1 [1] | #C6C6CC4C1C15C1C# |
| SN_34_00 | CsTx-29 | U19-ctenitoxin-Cs1 | 1 [1] |  |
| SN_35_00 | CsTx-35a,b | U20-ctenitoxin-Cs1a,b | 2 [2] ** |  |
| SN_04_04 | CsTx-42a,b,c,d CsTx-41 | U21-ctenitoxin-Cs1a,b,c,d U22-ctenitoxin-Cs1 | 3 [2] 1 [1] |  |
| SN_05_07 SN_05_08 | CsTx-37 CsTx-38a,b,c | U23-ctenitoxin-Cs1 U24-ctenitoxin-Cs1a,b,c | 1 [1] 3 [3] |  |
| SN_31_04 | CsTx-24a,b,c CsTx-39 | U25-ctenitoxin-Cs1a,b,c U26-ctenitoxin-Cs1 | 2 [1] 1 [1] |  |
| Unknown cystine fold | ||||
| SN_44_00 | CsTx-27 | U27-ctenitoxin-Cs1 | 1 [1] | #C6C1C6C1C10C1C13C# |
| SN_42_00 | CsTx-40 | U28-ctenitoxin-Cs1 | 1 [1] | #C6C1C6C1C10C1C7C5C19C |
| Colipase MIT1-like fold | ||||
| SN_20_01 | CsTx-20a,b | U29-ctenitoxin-Cs1a,b | 2 [2] |  |
| SN_32_01 SN_32_02 | CsTx-21a,b,c,d, e,f,g CsTx-22a,b,c | U30-ctenitoxin-Cs1a,b,c,d,e,f,g U31-ctenitoxin-Cs1a,b,c | 6 [5] 3 [3] |  |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuhn-Nentwig, L.; Langenegger, N.; Heller, M.; Koua, D.; Nentwig, W. The Dual Prey-Inactivation Strategy of Spiders—In-Depth Venomic Analysis of Cupiennius salei. Toxins 2019, 11, 167. https://doi.org/10.3390/toxins11030167
Kuhn-Nentwig L, Langenegger N, Heller M, Koua D, Nentwig W. The Dual Prey-Inactivation Strategy of Spiders—In-Depth Venomic Analysis of Cupiennius salei. Toxins. 2019; 11(3):167. https://doi.org/10.3390/toxins11030167
Chicago/Turabian StyleKuhn-Nentwig, Lucia, Nicolas Langenegger, Manfred Heller, Dominique Koua, and Wolfgang Nentwig. 2019. "The Dual Prey-Inactivation Strategy of Spiders—In-Depth Venomic Analysis of Cupiennius salei" Toxins 11, no. 3: 167. https://doi.org/10.3390/toxins11030167
APA StyleKuhn-Nentwig, L., Langenegger, N., Heller, M., Koua, D., & Nentwig, W. (2019). The Dual Prey-Inactivation Strategy of Spiders—In-Depth Venomic Analysis of Cupiennius salei. Toxins, 11(3), 167. https://doi.org/10.3390/toxins11030167