Evaluation of Mycotoxin Screening Tests in a Verification Study Involving First Time Users

 ,
, 
 ,
,  ,
, 

Abstract
:1. Introduction
2. Results
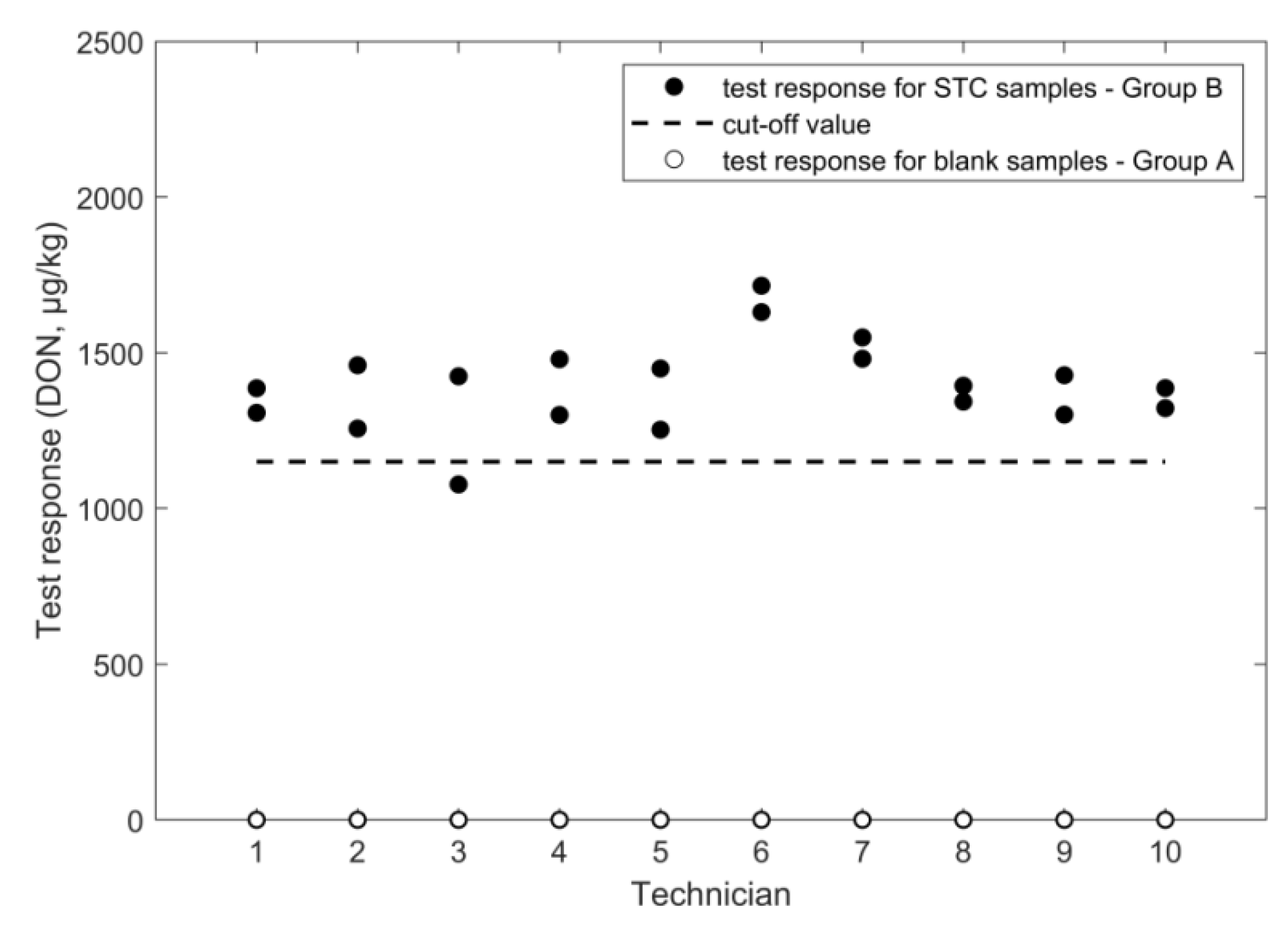
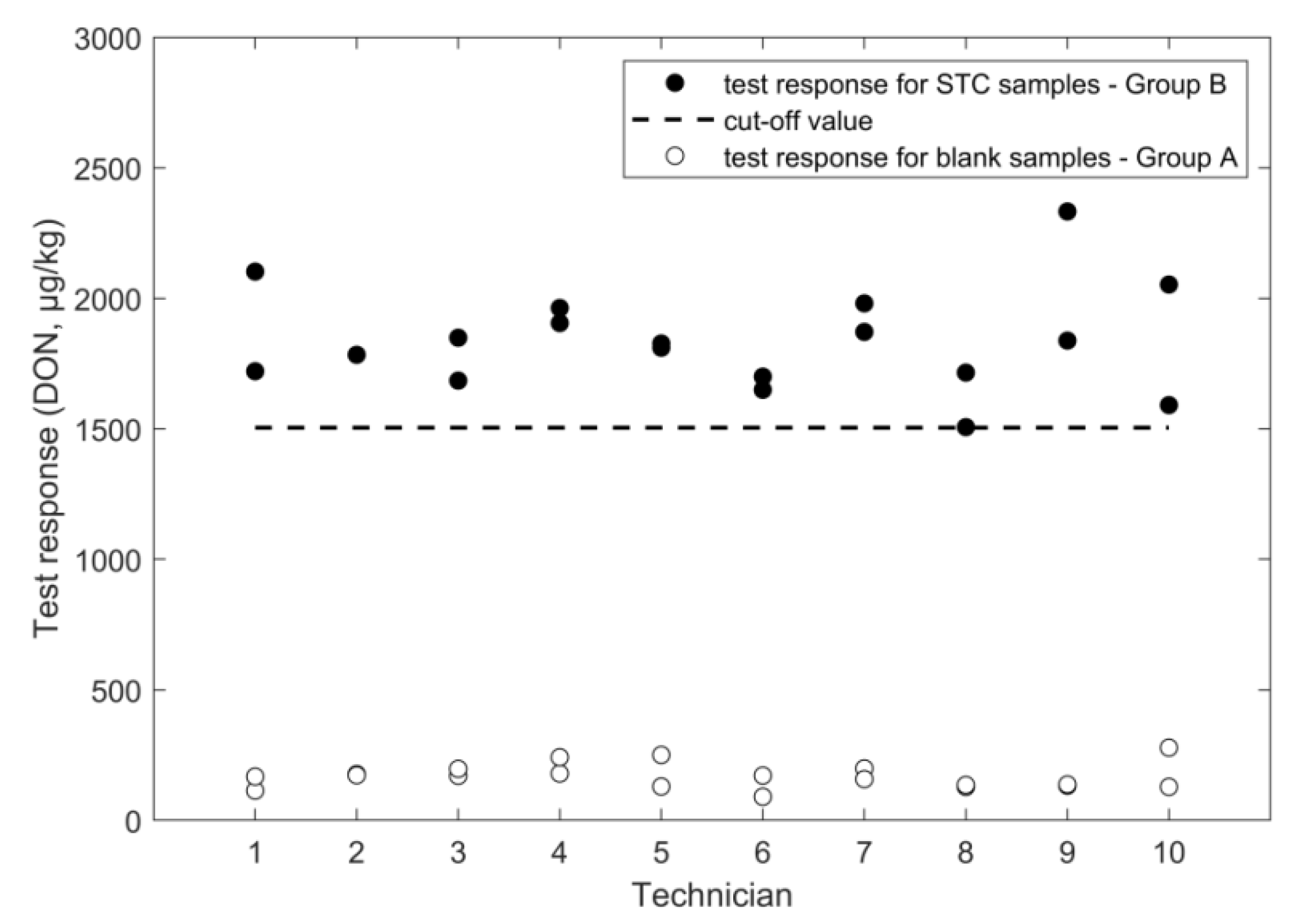
2.1. Determination of Deoxynivalenol
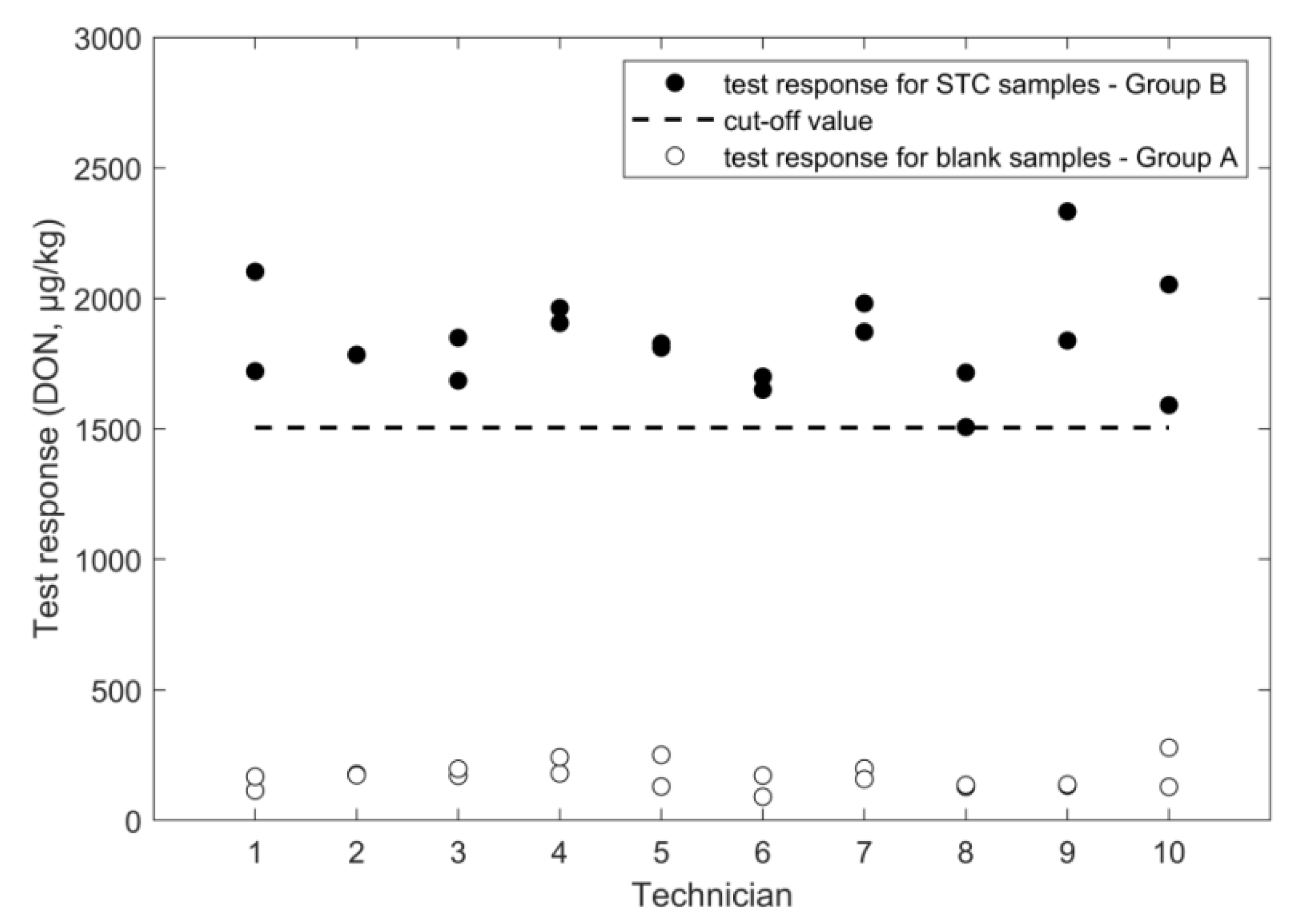
2.1.1. FPIA
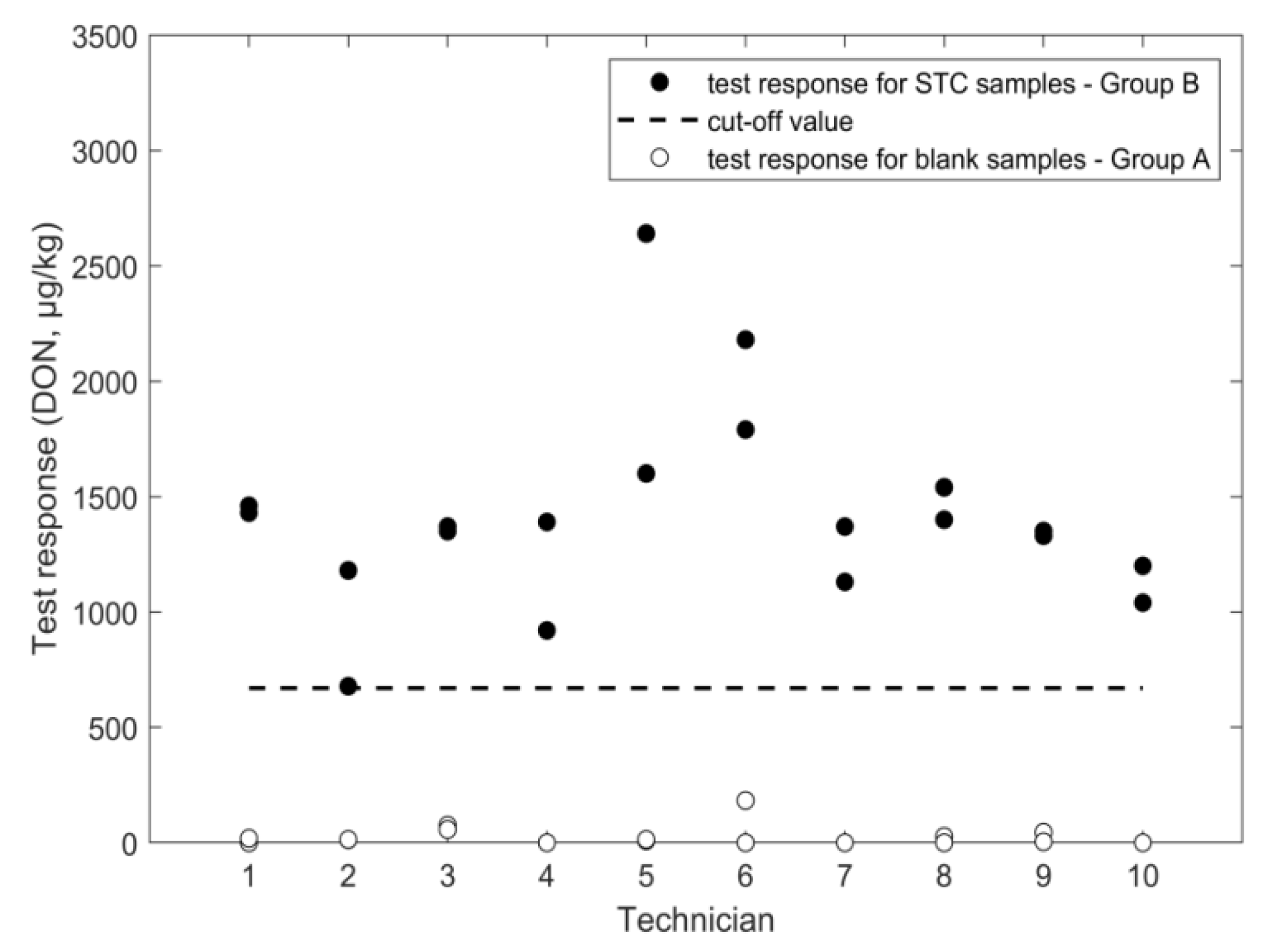
2.1.2. ELISA
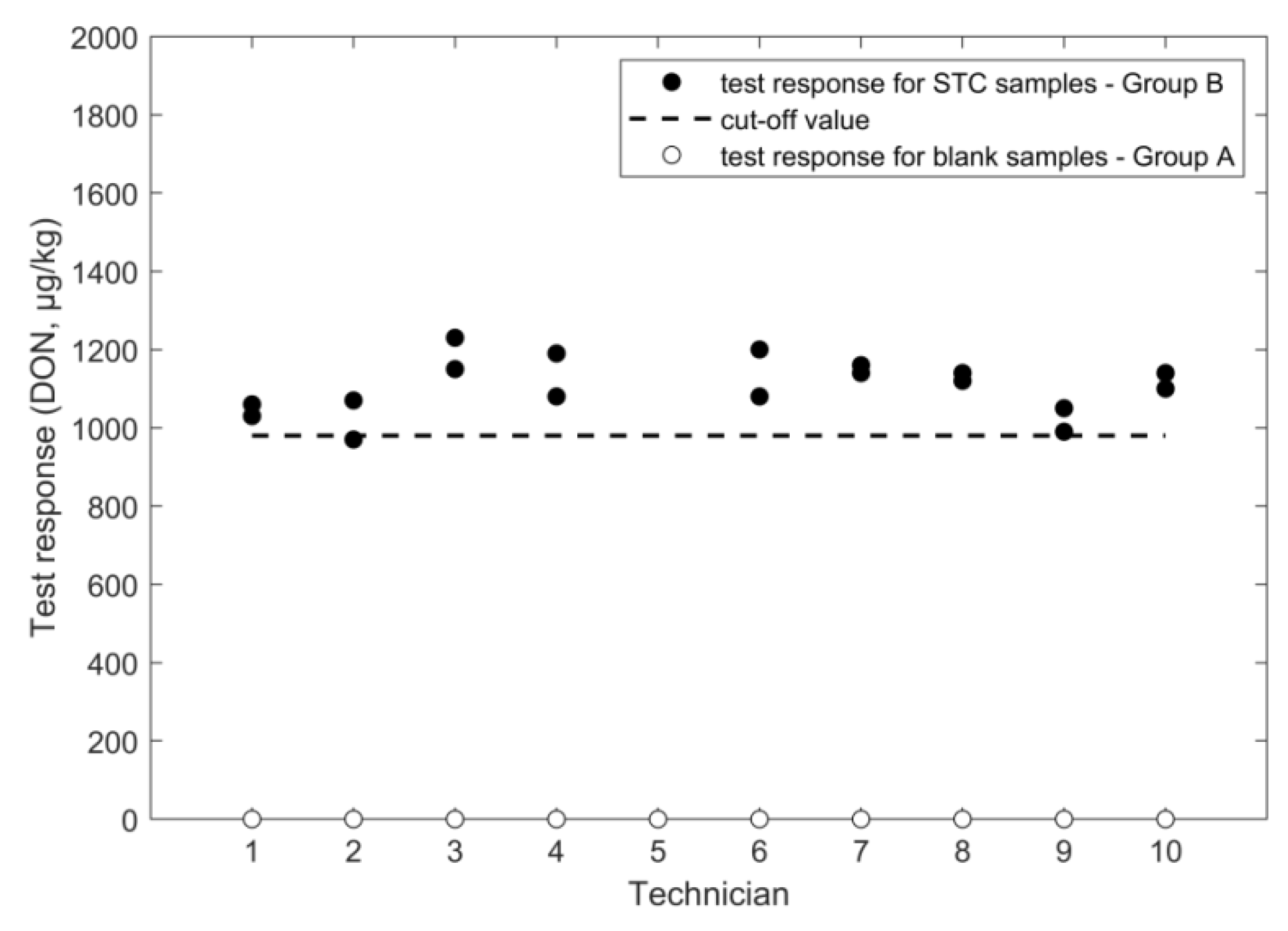
2.1.3. LFD
2.1.4. LC-HRMS
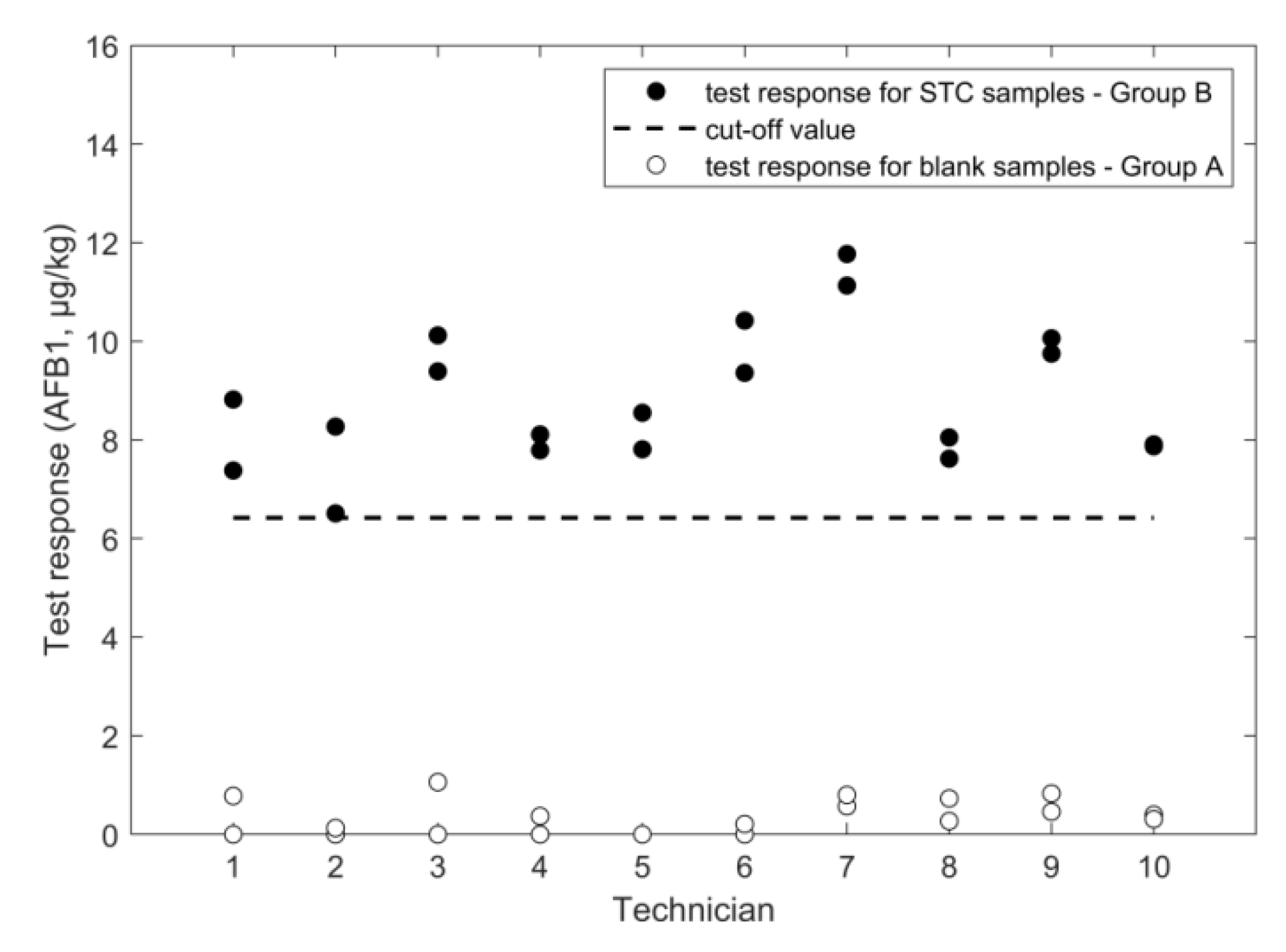
2.2. Determination of Aflatoxin B1
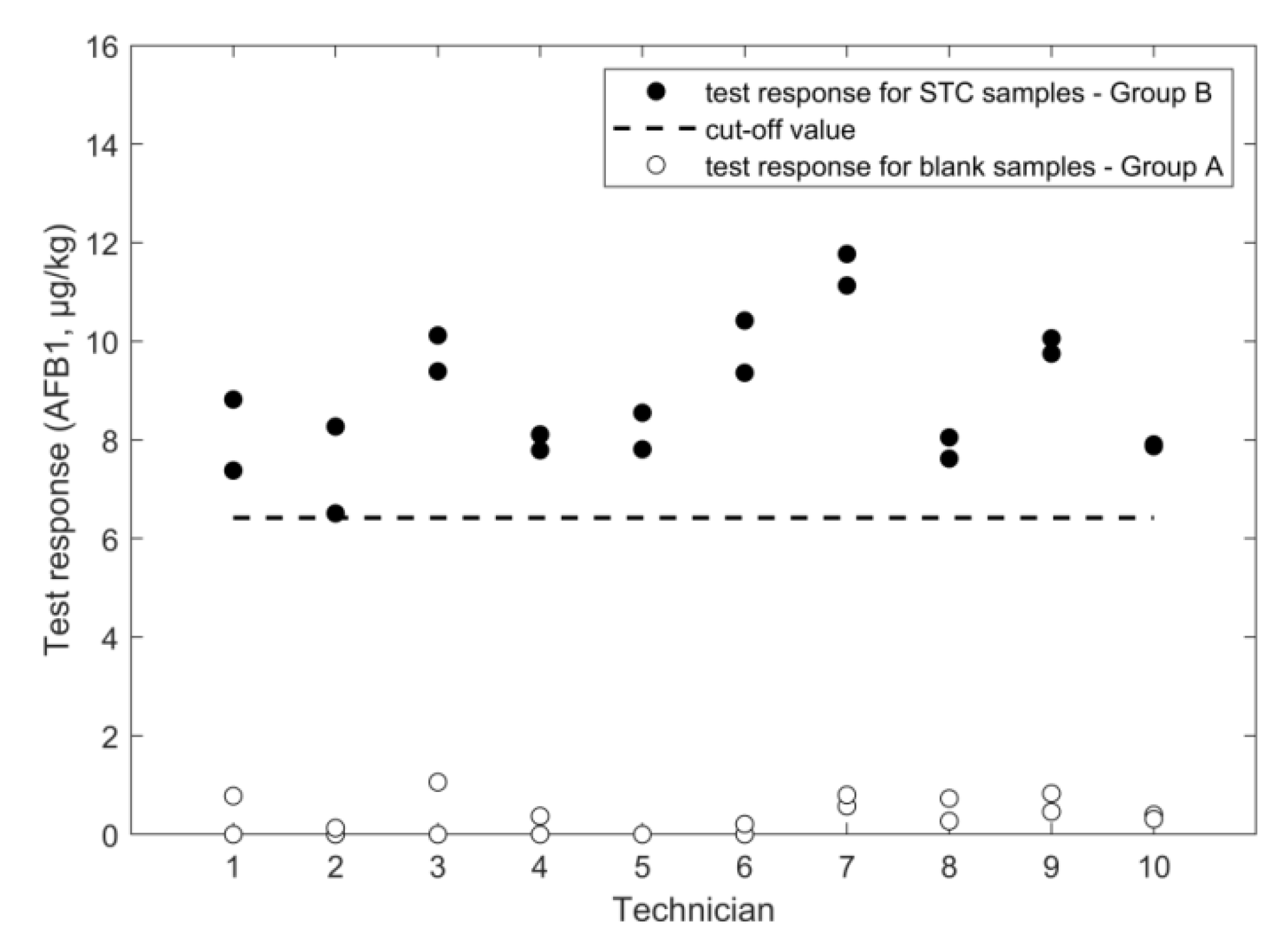
2.2.1. LFD
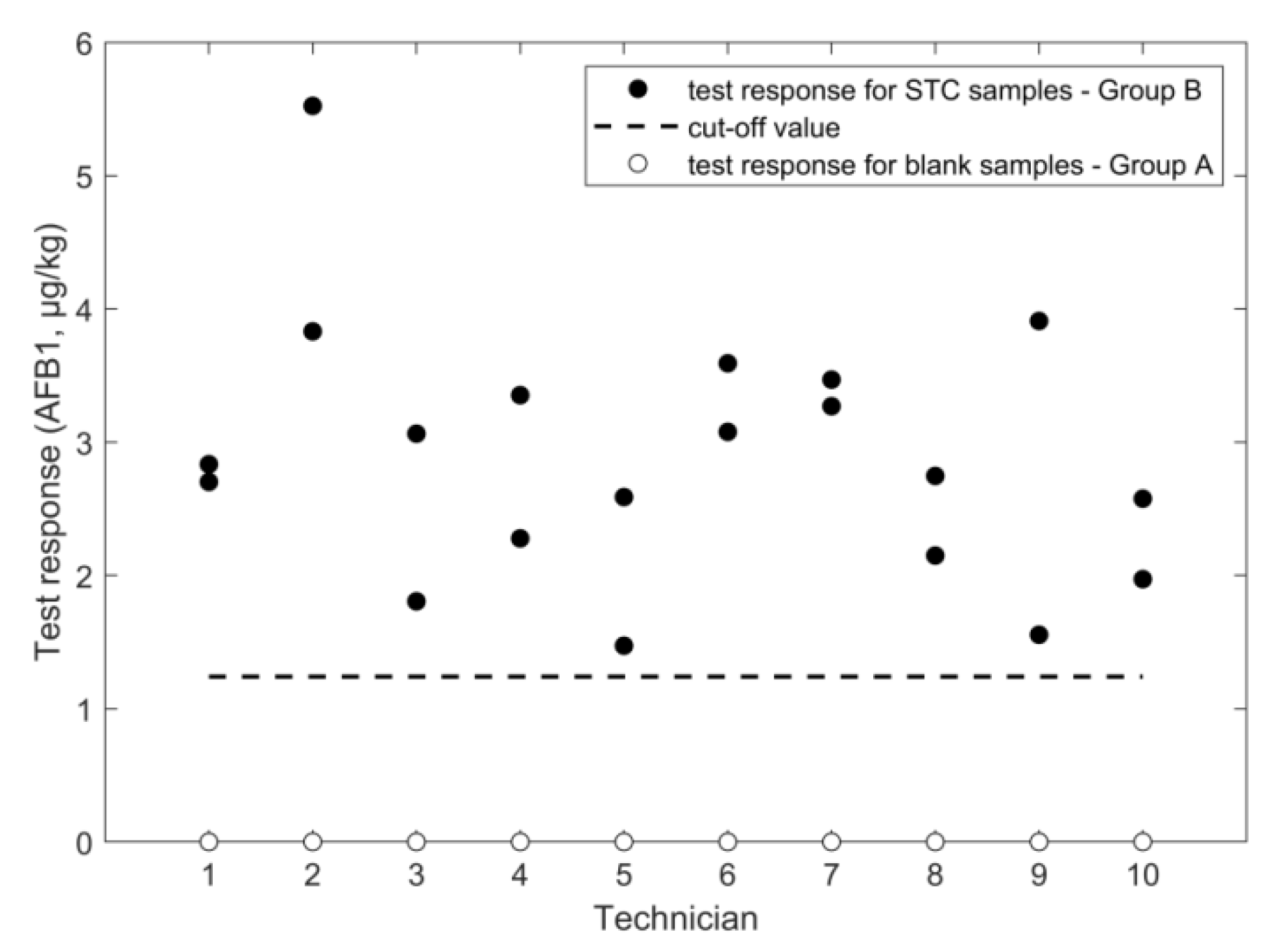
2.2.2. LC-HRMS
3. Discussion
Practicability of the Screening Methods
4. Conclusions
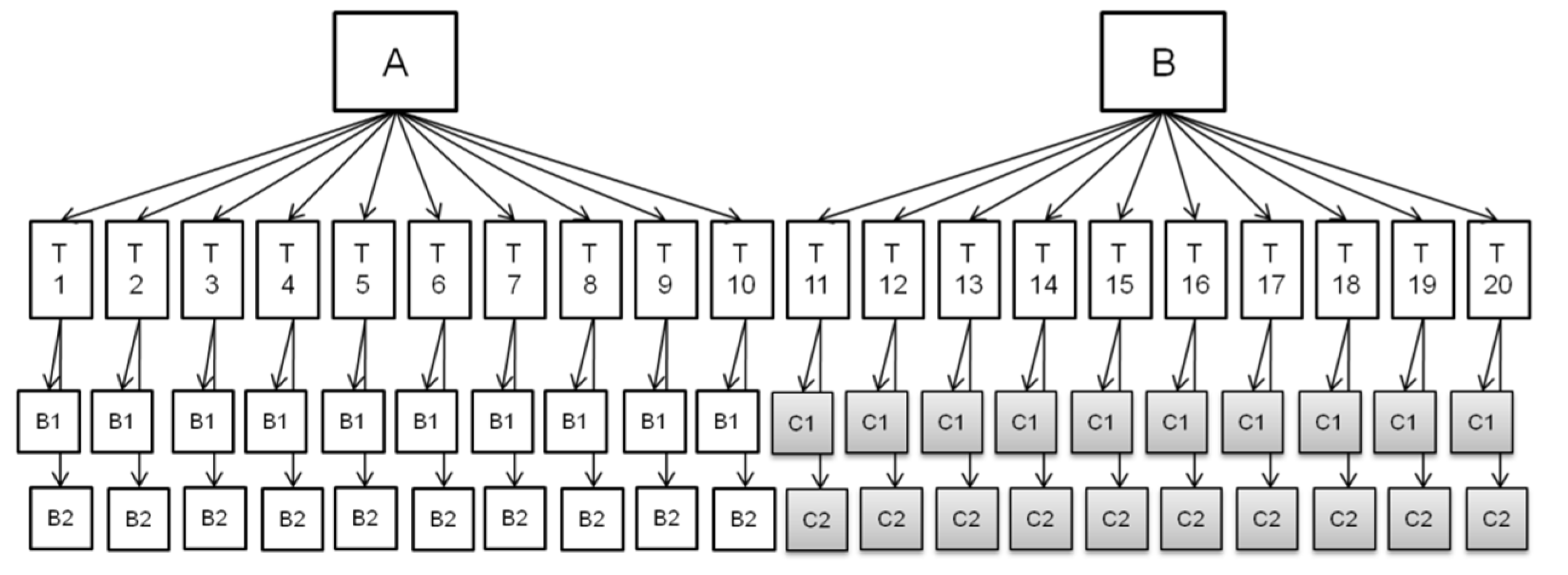
5. Materials and Methods
5.1. Test Materials
5.2. Description of the Analytical Methods
5.2.1. FPIA—Deoxynivalenol in Wheat
5.2.2. ELISA—Deoxynivalenol in Wheat
5.2.3. LFD—Deoxynivalenol in Wheat
5.2.4. LFD—Aflatoxin B1 in Maize
5.2.5. LC-HRMS—Deoxynivalenol and Aflatoxin B1 in Wheat
- CSample = mass fraction of the target analyte in the sample expressed in µg/kg;
- RSample = response (area) of peak in extracted ion chromatogram using the exact m/z ± 5 ppm of the protonated molecule;
- R(IS/STC) = response (area) of peak in extracted ion chromatogram using the exact m/z ± 5 ppm of the protonated molecule of the 13C-label added at the STC level;
- C(IS/STC) = STC of 13C-label in the extract, expressed in the corresponding µg/kg equivalent in sample
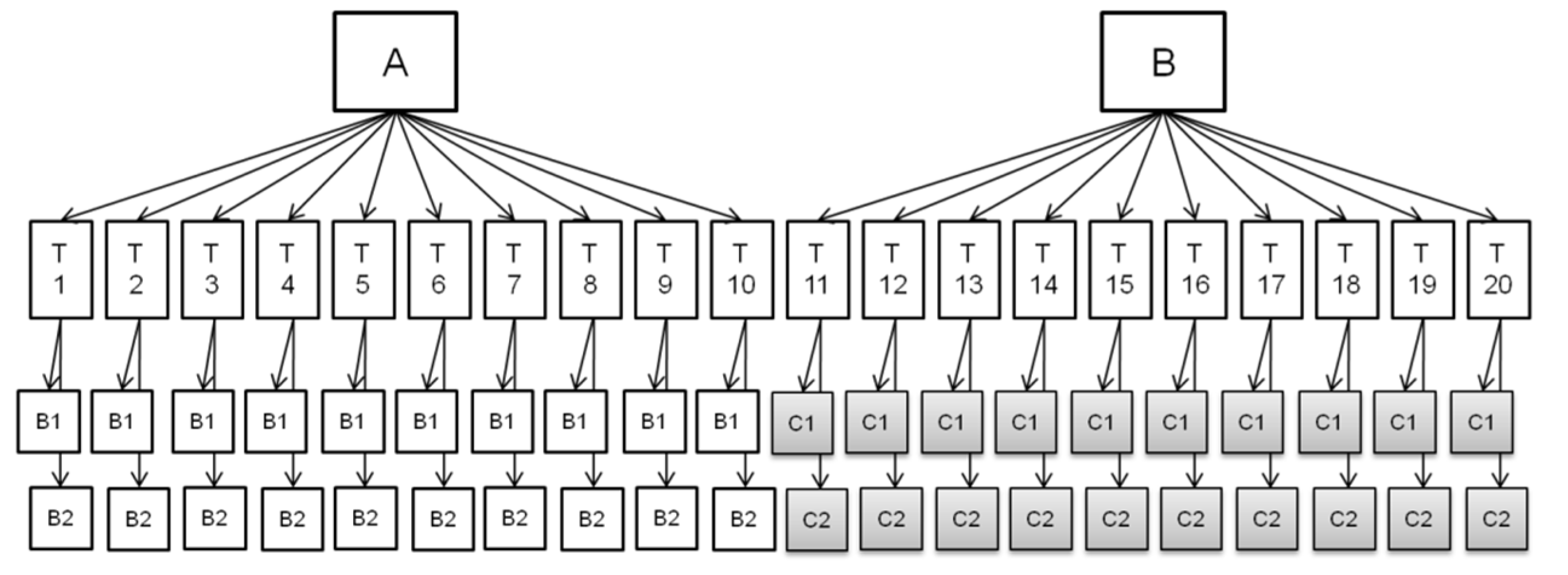
5.3. Set Up of the Validation Exercise
5.4. Statistics Applied
Author Contributions
Funding
Acknowledgments
- The test kit providers: r-biopharm AG, Vicam a Waters Business
- The training course assistants of the Institute of Sciences of Food Productions of the National Research Council of Italy: Cervellieri S., Ciasca B., Cortese M., D’Ascanio V., Gambacorta L., Greco D., and Guarducci N.
- All training course participants and instructors
Conflicts of Interest
References
- Lebesi, D.; Dimakou, C.; Alldrick, A.J.; Oreopoulou, V. Rapid test methods: A versatile tool to assist food-safety management. Qual. Assur. Saf. Crops Foods 2010, 2, 173–181. [Google Scholar] [CrossRef]
- European Commission. Commission Decision 2002/657/EC of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and interpretation of results. Off. J. Eur. Commun. 2002, L221, 8–36. [Google Scholar]
- Goryacheva, I.Y.; Rusanova, T.Y.; Burmistrova, N.A.; De Saeger, S. Immunochemical methods for the determination of mycotoxins. J. Anal. Chem. 2009, 64, 768–785. [Google Scholar] [CrossRef]
- Shephard, G.S. Current Status of Mycotoxin Analysis: A Critical Review. J. AOAC Int. 2016, 99, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Urusov, A.E.; Zherdev, A.V.; Dzantiev, B.B. Immunochemical Methods of Mycotoxin Analysis. Appl. Biochem. Microbiol. 2010, 46, 253–266. [Google Scholar] [CrossRef]
- Anfossi, L.; Baggiani, C.; Giovannoli, C.; D’Arco, G.; Giraudi, G. Lateral-flow immunoassays for mycotoxins and phycotoxins: A review. Anal. Bioanal. Chem. 2013, 405, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Powers, S.; Dai, S.Y. Using commercial immunoassay kits for mycotoxins: ‘joys and sorrows’? World Mycotox. J. 2014, 7, 417–430. [Google Scholar] [CrossRef]
- Maragos, C.M. Fluorescence Polarization Immunoassay of Mycotoxins: A Review. Toxins 2009, 1, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Lippolis, V.; Maragos, C.M. Fluorescence polarisation immunoassays for rapid, accurate and sensitive determination of mycotoxins. World Mycotox. J. 2014, 7, 479–489. [Google Scholar] [CrossRef]
- Berthiller, F.; Brera, C.; Iha, M.H.; Krska, R.; Lattanzio, V.M.T.; MacDonald, S.; Malone, R.J.; Maragos, C.; Solfrizzo, M.; Stranska-Zachariasova, M.; et al. Developments in mycotoxin analysis: An update for 2015–2016. World Mycotox. J. 2017, 10, 5–29. [Google Scholar] [CrossRef]
- Ciasca, B.; Pascale, M.; Altieri, V.G.; Longobardi, F.; Suman, M.; Catellani, D.; Lattanzio, V.M.T. In-house validation and small-scale collaborative study to evaluate analytical performances of multimycotoxin screening methods based on liquid chromatography–high-resolution mass spectrometry: Case study on Fusarium toxins in wheat. J. Mass Spectrom. 2018, 53, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Malachová, A.; Sulyok, M.; Beltrán, E.; Berthiller, F.; Krska, R. Optimization and validation of a quantitative liquid chromatography-tandem mass spectrometric method covering 295 bacterial and fungal metabolites including all regulated mycotoxins in four model food matrices. J. Chromatogr. A 2014, 1362, 145–156. [Google Scholar]
- European Commission. Commission Regulation (EU) No 519/2014 of 16 May 2014 amending Regulation (EC) No 401/2006 as regards methods of sampling of large lots, spices and food supplements, performance criteria for T-2, HT-2 toxin and citrinin and screening methods of analysis. Off. J. Eur. Union 2014, L147, 29–43. [Google Scholar]
- ALACC Guide. How to Meet ISO 17025 Requirements for Method Verification. Available online: http://www.aoac.org/aoac_prod_imis/AOAC_Docs/LPTP/alacc_guide_2008.pdf#page=6&zoom=auto,-169,634 (accessed on 12 December 2018).
- Lippolis, V.; Pascale, M.; Visconti, A. Optimization of a Fluorescence Polarization Immunoassay for Rapid Quantification of Deoxynivalenol in Durum Wheat–Based Products. J. Food Prot. 2006, 69, 2712–2719. [Google Scholar] [CrossRef] [PubMed]
- R-Biopharm, RIDASCREEN®FAST DON Certificate. Available online: https://food.r-biopharm.com/wp-content/uploads/sites/2/2017/12/r5901-r5902-fast-don-17-07-18.pdf (accessed on 19 February 2019).
- European Commission, Health Consumers Directorate-General. Multimethod for the screening of aflatoxin B1, deoxynivalenol, fumonisin B1 and B2, ochratoxin A, HT-2 and T-2 toxins and zearalenone in foodstuffs, excluding foods for infants and young children, by HPLC-MS/MS. In Mandate for Standardisation Addressed to CEN for Methods for Mycotoxin Analysis in Food; European Commission: Brussels, Belgium, 2013; Available online: http://ec.europa.eu/growth/tools-databases/mandates/index.cfm?fuseaction=search.detail&id=528 (accessed on 19 February 2019).
- Lattanzio, V.M.T.; Guarducci, N.; Powers, S.; Ciasca, B.; Pascale, M.; von Holst, C. Validation of a lateral flow immunoassay for the rapid determination of aflatoxins in maize by solvent free extraction. Anal. Meth. 2018, 10, 123–130. [Google Scholar] [CrossRef]
- Lattanzio, V.M.T.; Ciasca, B.; Powers, S.; von Holst, C. Validation of screening methods according to Regulation 519/2014/EU. Determination of deoxynivalenol in wheat by lateral flow immunoassay: A case study. Trends Anal. Chem. 2016, 76, 137–144. [Google Scholar] [CrossRef]
- Horwitz, W. Evaluation of Analytical Methods Used for Regulation of Foods and Drugs. Anal. Chem. 1982, 54, 67A–76A. [Google Scholar] [CrossRef]
- Lippolis, V.; Pascale, M.; Valenzano, S.; Visconti, A. Comparison of slurry mixing and dry milling in laboratory sample preparation for determination of ochratoxin A and deoxynivalenol in wheat. J. AOAC Int. 2012, 95, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, T.B. Sampling foods for mycotoxins. Food Addit. Contam. 2006, 23, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Breidbach, A. A Greener, Quick and Comprehensive Extraction Approach for LC-MS of Multiple Mycotoxins. Toxins 2017, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Mol, H.J.; Plaza-Bolaños, O.; Zomer, P.; de Rijk, T.C.; Stolker, A.A.M.; Mulder, P.P.J. Toward a generic extraction method for simultaneous determination of pesticides, mycotoxins, plant toxins, and veterinary drugs in feed and food matrixes. Anal. Chem. 2008, 80, 9450–9459. [Google Scholar] [CrossRef] [PubMed]
- Valenzano, S.; Lippolis, V.; Pascale, M.; De Marco, A.; Maragos, C.M.; Suman, M.; Visconti, A. Determination of deoxynivalenol in wheat bran and whole-wheat flour by fluorescence polarization immunoassay. Food Anal. Meth. 2014, 7, 806–813. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Set | Validation Parameters | DON | AFB1 | ||||
|---|---|---|---|---|---|---|---|
| LFD Wheat | ELISA Wheat | FPIA Wheat | LC-HRMS Wheat | LFD Maize | LC-HRMS Wheat | ||
| negative samples | mean response (µg/kg) | 0 | 23 | 168 | n.a. | 0.3 | n.a. |
| SD repeatability (µg/kg) | n.a. | 43 | 52 | n.a. | 0.3 | n.a. | |
| SD intermediate (µg/kg) | n.a. | 43 | 52 | n.a. | 0.3 | n.a. | |
| rate of false suspect (%) | <0.1% | <0.1% | <0.1% | <0.1% | <0.1% | <0.1% | |
| positive samples | STC(µg/kg) | 1600 | 1600 | 1600 | 1600 | 10.6 | 2.0 |
| mean response (µg/kg) | 1106 | 1417 | 1833 | 1397 | 8.8 | 2.9 | |
| SD repeatability (µg/kg) | 53 | 301 | 186 | 117 | 0.6 | 0.8 | |
| SD intermediate (µg/kg) | 72 | 429 | 190 | 142 | 1.4 | 1.0 | |
| RSD repeatability (%) | 4.8 | 21 | 10 | 8.4 | 7.2 | 28 | |
| RSD intermediate (%) | 6.5 | 30 | 10 | 10 | 16 | 33 | |
| cut off (µg/kg) | 981 | 674 | 1504 | 1151 | 6.4 | 1.2 | |
| Mycotoxin | Present Training Course | Previous Studies | ||||
|---|---|---|---|---|---|---|
| Validation Parameters | Study Design | Guidelines | Validation Parameters | Reference | ||
| DON | FPIA/ wheat | STC: 1600 µg/kg intermediate precision TC: 10% repeatability: 10% cut-off: 1504 µg/kg false suspects for blanks: <0.1% | single laboratory | CEN/TR 13505 | STC: 1750 µg/kg recovery: 97% repeatability: 4.1% limit of detection: 80 µg/kg | Lippolis et al. [15] |
| ELISA/ wheat | STC: 1600 µg/kg intermediate precision TC: 30% repeatability: 21% cut-off: 670 µg/kg false suspects for blanks: <0.1% | single laboratory | AOAC performance tested | STC: 1000 µg/kg recovery: 100% repeatability: 22% limit of quantification: 167 µg/kg | r-biopharm [16] | |
| LFD/ wheat | STC: 1600 µg/kg intermediate precision TC: 6.5% repeatability: 4.8% cut-off: 980 µg/kg false suspects for blanks: <0.1% | single laboratory | 519/2014/EC | STC: 1600 µg/kg intermediate precision: 13% repeatability: 12% cut-off: 1410 µg/kg false suspects for blanks: <0.1% | unpublished results | |
| LC-HRMS/ wheat | STC: 1600 µg/kg intermediate precision TC: 10% repeatability: 8.4% cut-off: 1151 µg/kg relative cut off: 0.71 false suspects for blanks: <0.1% | inter-laboratory | 519/2014/EC | STC: 250 µg/kg reproducibility: 11% repeatability: 3.4% cut-off: 184 µg/kg relative cut off: 0.74 false suspects for blanks: <0.1% | prEN 17279:2018 [17] | |
| AFB1 | LFD/ maize | STC: 10.6 µg/kg intermediate precision TC: 16% repeatability: 7.2% cut-off: 6.4 µg/kg false suspects for blanks: <0.1% | single laboratory | 519/2014/EC | STC: 4 µg/kg intermediate precision*: 29% repeatability: 21% cut-off: 2.18 µg/kg false suspects for blanks: 8% | Lattanzio et al. [18] |
| LC-HRMS/ wheat | STC: 2 µg/kg intermediate precision TC: 33% repeatability: 28% cut-off: 1.23 µg/kg false suspects for blanks: <0.1% | inter-laboratory | 519/2014/EC | STC: 2 µg/kg reproducibility: 25% repeatability: 19% cut-off: 1.08 µg/kg false suspects for blanks: <0.1% | prEN 17279:2018 [17] | |
| Mycotoxin/Matrix | Assay | Sample Size | Extraction | Additional Steps | Analysis | Calibration Curve | Results |
|---|---|---|---|---|---|---|---|
| DON/wheat | FPIA | 25 g | 100 mL PBS solution 2 min high speed blending | extract filtration through paper filter and glass microfiber filter | pipette: 830 µL PBS solution 80 µL antibody 120 µL filtered extract read the blank add 25 µL tracer result reading | provided by the TC organizers | evaluated trough an excel file*(input data: fluorescence polarization of the test sample) |
| ELISA | 5 g | 100 mL water 3 min manual shaking | extract filtration through paper filter | pipette: 50 µL filtered extract 50 µL conjugate 50 µL antibody 250 µL wash buffer 100 µL substrate/chromogen 100 µL stop solution result reading | each technician made its own calibration | evaluated trough a software * (input data: absorbance of the calibration standards and the test sample) | |
| LFD | 1 g | 15 mL buffer * 3 min manual shaking | 100 µL onto the test strip 5 min test strip development reading by smartphone | uploaded through bar code | provided by the smartphone app * | ||
| AFB1/maize | LFD | 5 g | 25 mL buffer * 2 min vortexing | extract filtration through paper filter | 100 µL onto the test strip 5 min test strip development reading by optical reader | uploaded through a bar code | provided by the reader |
| DON/wheat AFB1/wheat | LC-HRMS | 5 g | 10 mL water + 10 mL acetonitrile 30 min shaking | 5 g magnesium sulfate centrifugation | 200 µL acetonitrile extract 20 µL 13C-IS solution 180 µL water inject into LC-HRMS | One point internal calibration (13C-IS addition) | evaluated trough an excel file * (input data: peak area of the mycotoxin in the test sample and peak area of the relevant 13C-IS) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lattanzio, V.M.T.; von Holst, C.; Lippolis, V.; De Girolamo, A.; Logrieco, A.F.; Mol, H.G.J.; Pascale, M. Evaluation of Mycotoxin Screening Tests in a Verification Study Involving First Time Users. Toxins 2019, 11, 129. https://doi.org/10.3390/toxins11020129
Lattanzio VMT, von Holst C, Lippolis V, De Girolamo A, Logrieco AF, Mol HGJ, Pascale M. Evaluation of Mycotoxin Screening Tests in a Verification Study Involving First Time Users. Toxins. 2019; 11(2):129. https://doi.org/10.3390/toxins11020129
Chicago/Turabian StyleLattanzio, Veronica M. T., Christoph von Holst, Vincenzo Lippolis, Annalisa De Girolamo, Antonio F. Logrieco, Hans G. J. Mol, and Michelangelo Pascale. 2019. "Evaluation of Mycotoxin Screening Tests in a Verification Study Involving First Time Users" Toxins 11, no. 2: 129. https://doi.org/10.3390/toxins11020129
APA StyleLattanzio, V. M. T., von Holst, C., Lippolis, V., De Girolamo, A., Logrieco, A. F., Mol, H. G. J., & Pascale, M. (2019). Evaluation of Mycotoxin Screening Tests in a Verification Study Involving First Time Users. Toxins, 11(2), 129. https://doi.org/10.3390/toxins11020129