Inclusion of a Furin Cleavage Site Enhances Antitumor Efficacy against Colorectal Cancer Cells of Ribotoxin α-Sarcin- or RNase T1-Based Immunotoxins

 ,
,
Abstract
1. Introduction
2. Results
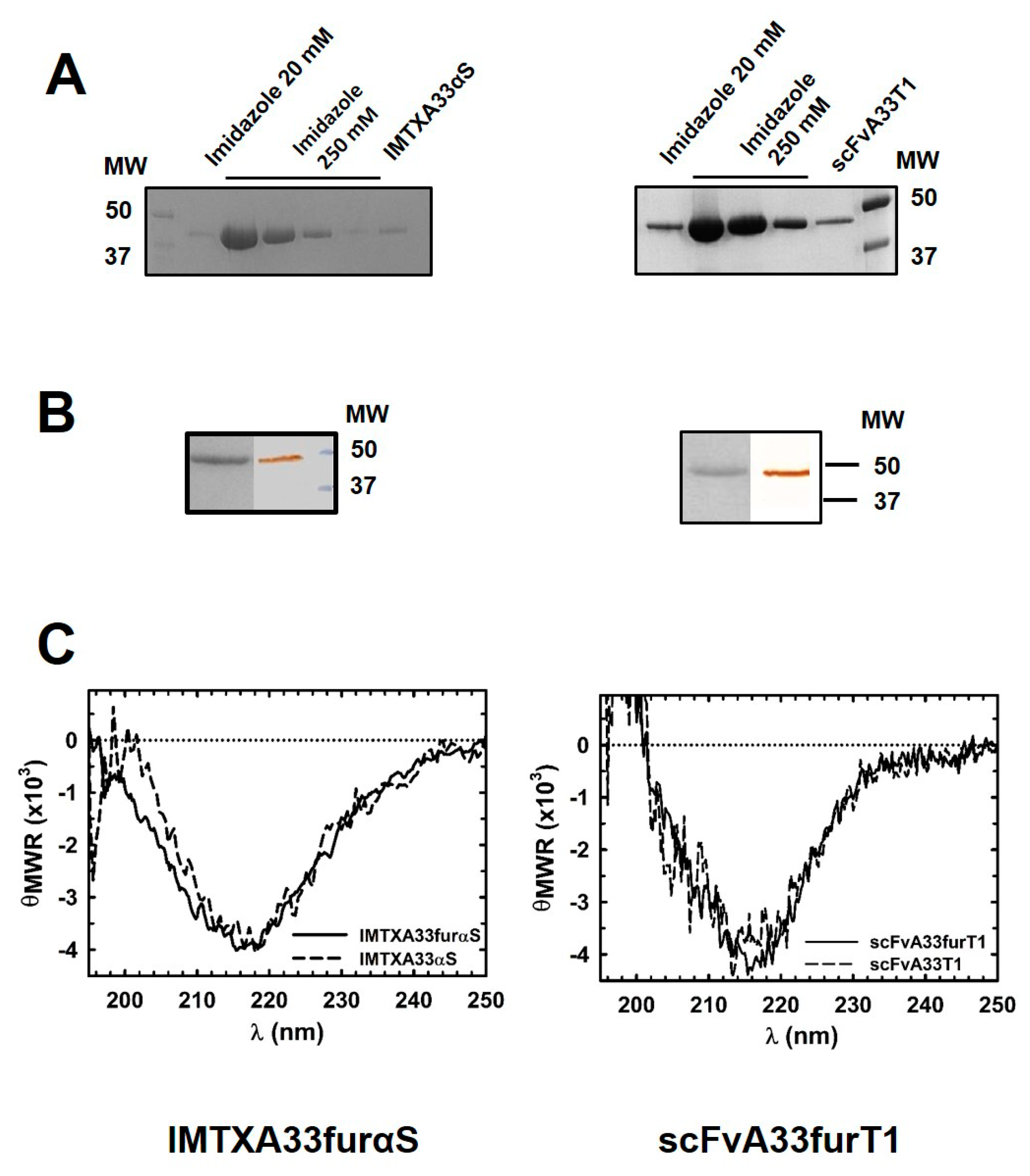
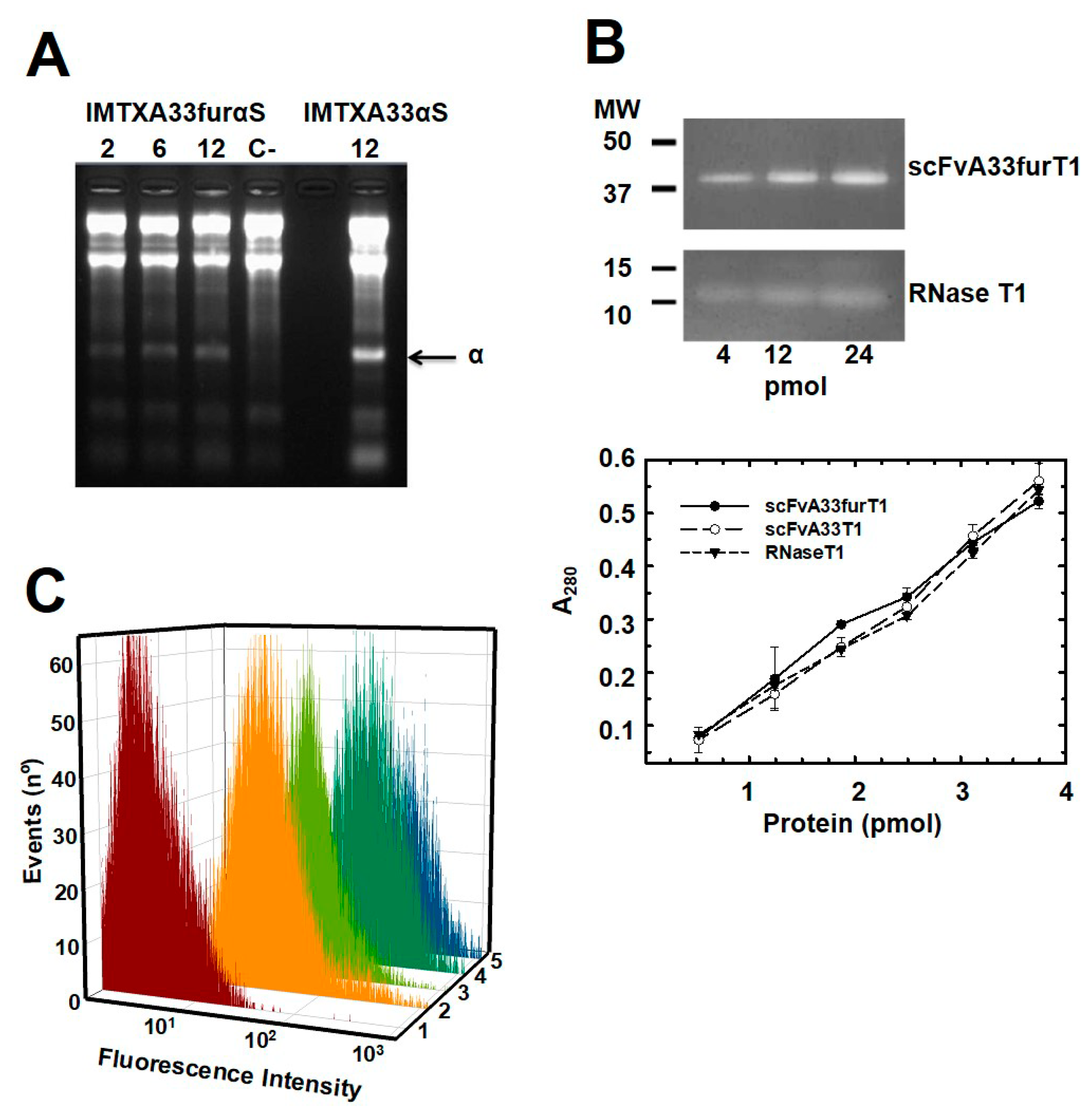
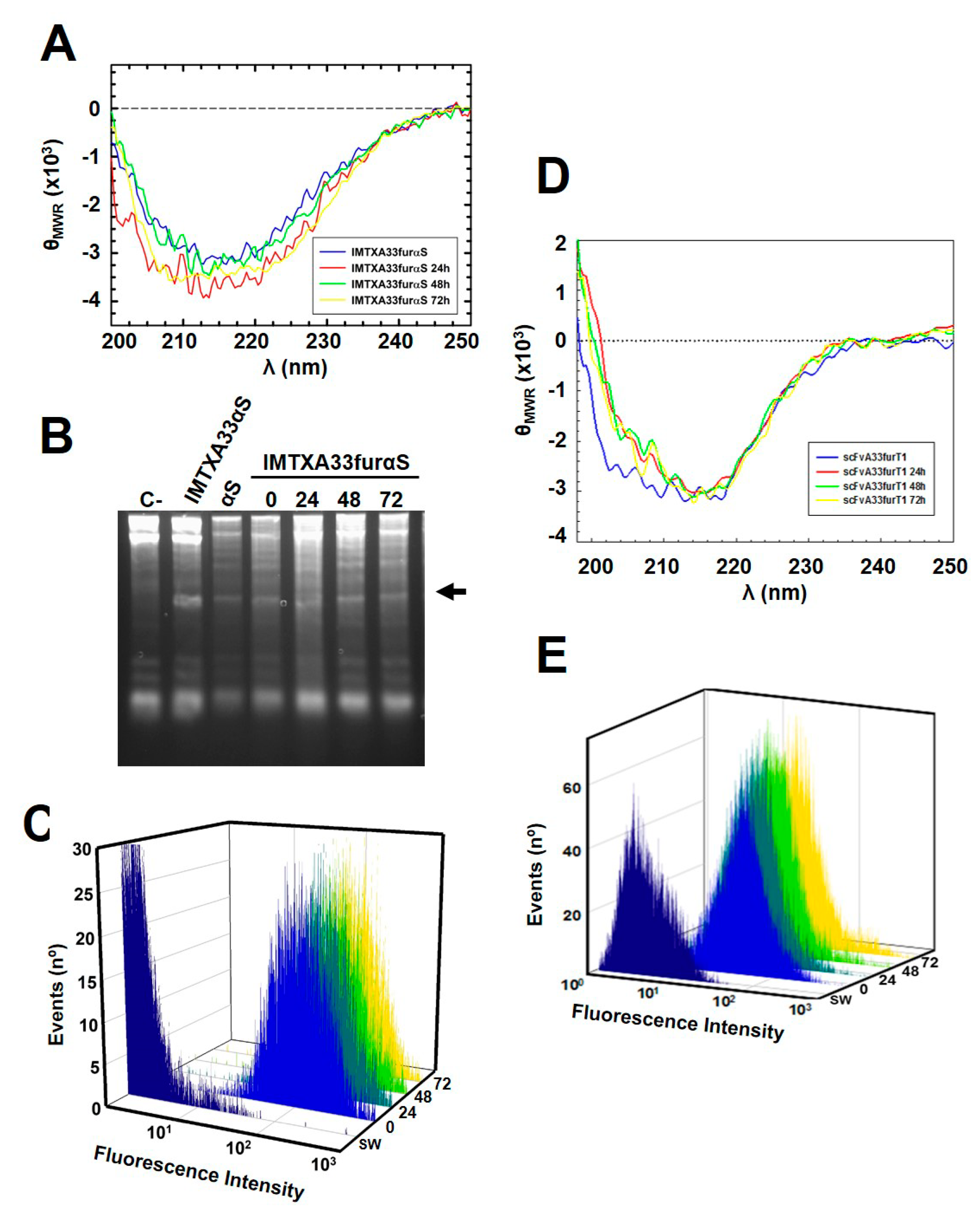
2.1. IMTXA33furαS and scFVA33furT1 Variants Were Purified as Fully Functional Immunoconjugates
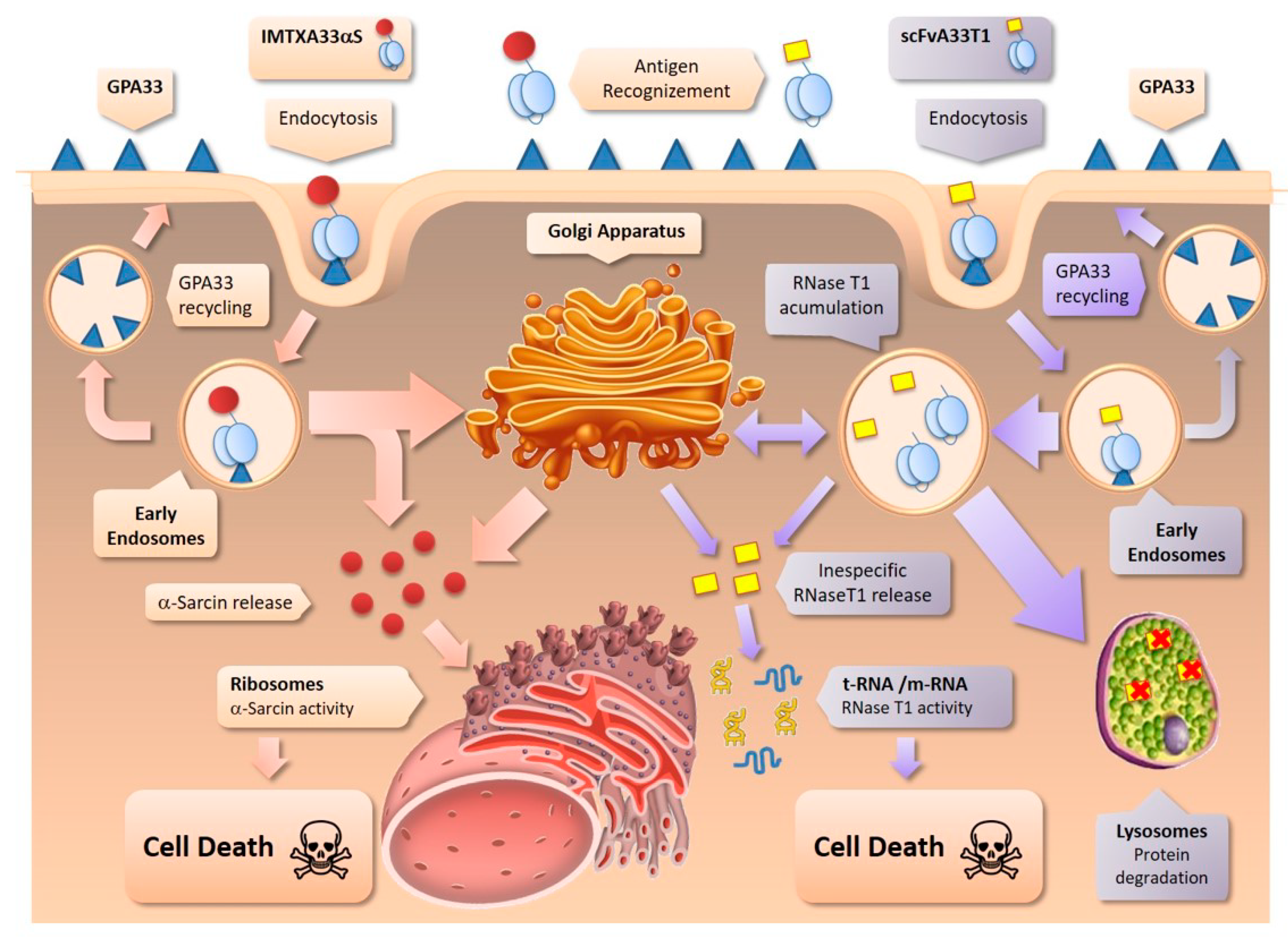
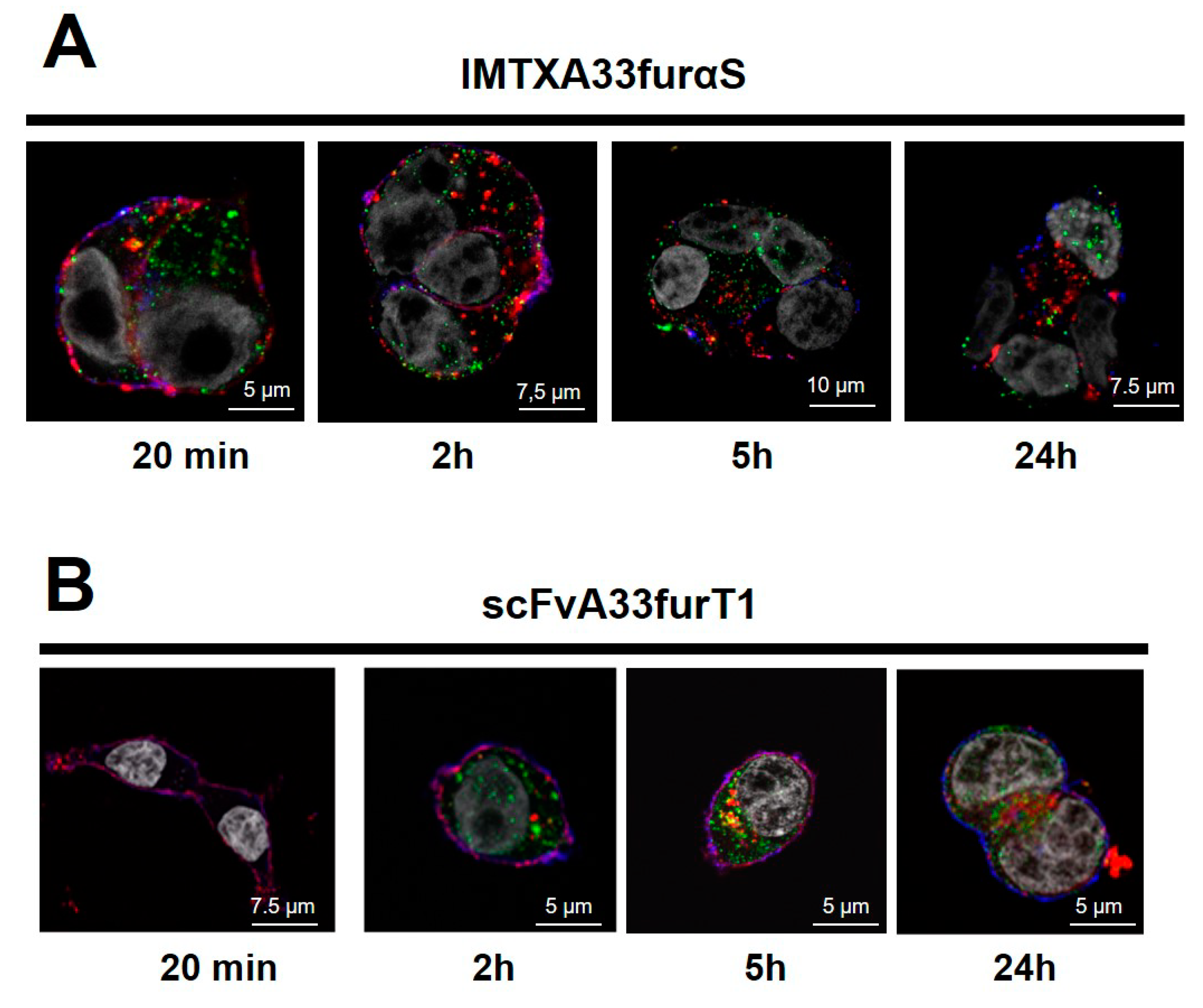
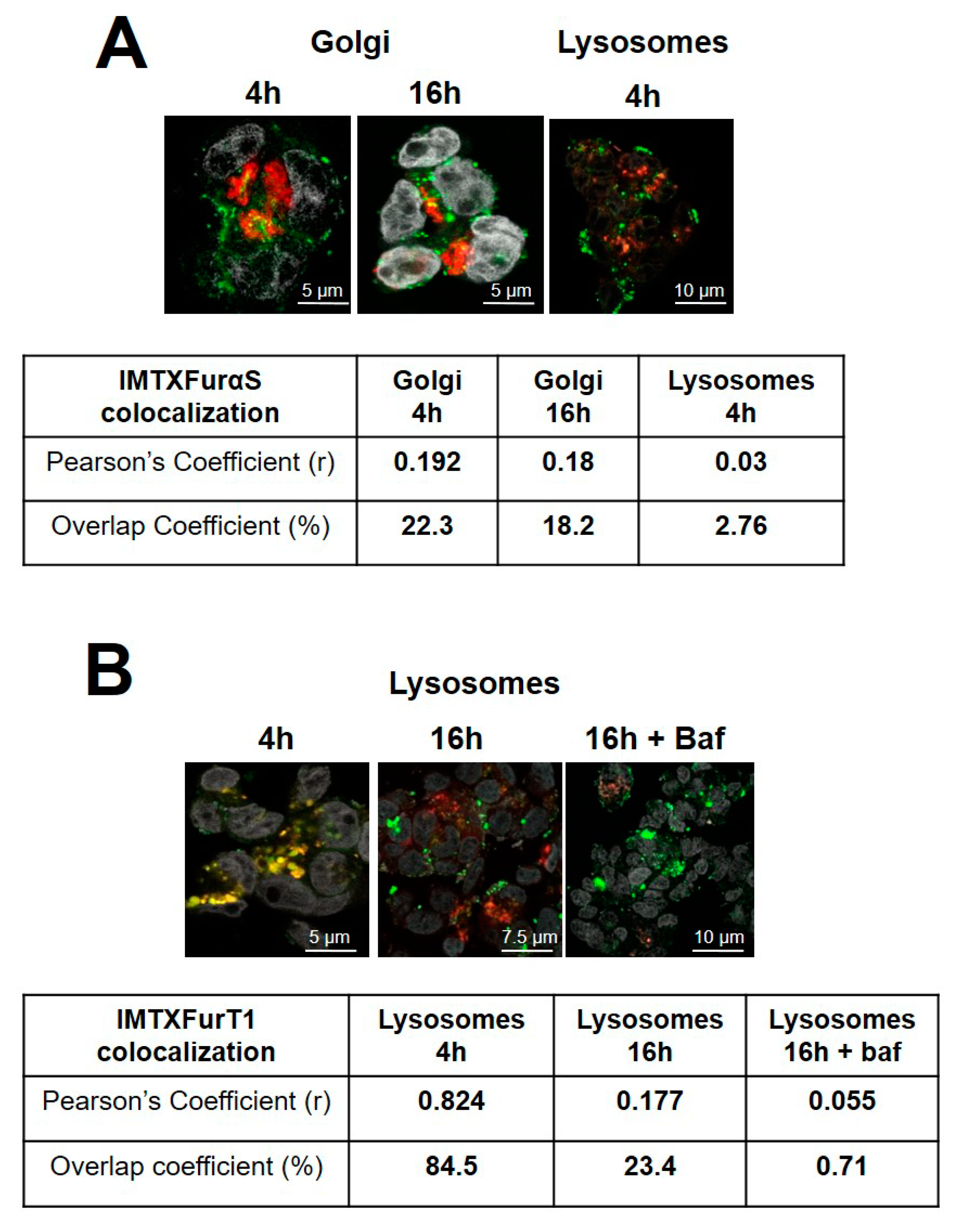
2.2. IMTXA33furαS and scFVA33furT1 Follow Different Intracellular Pathway
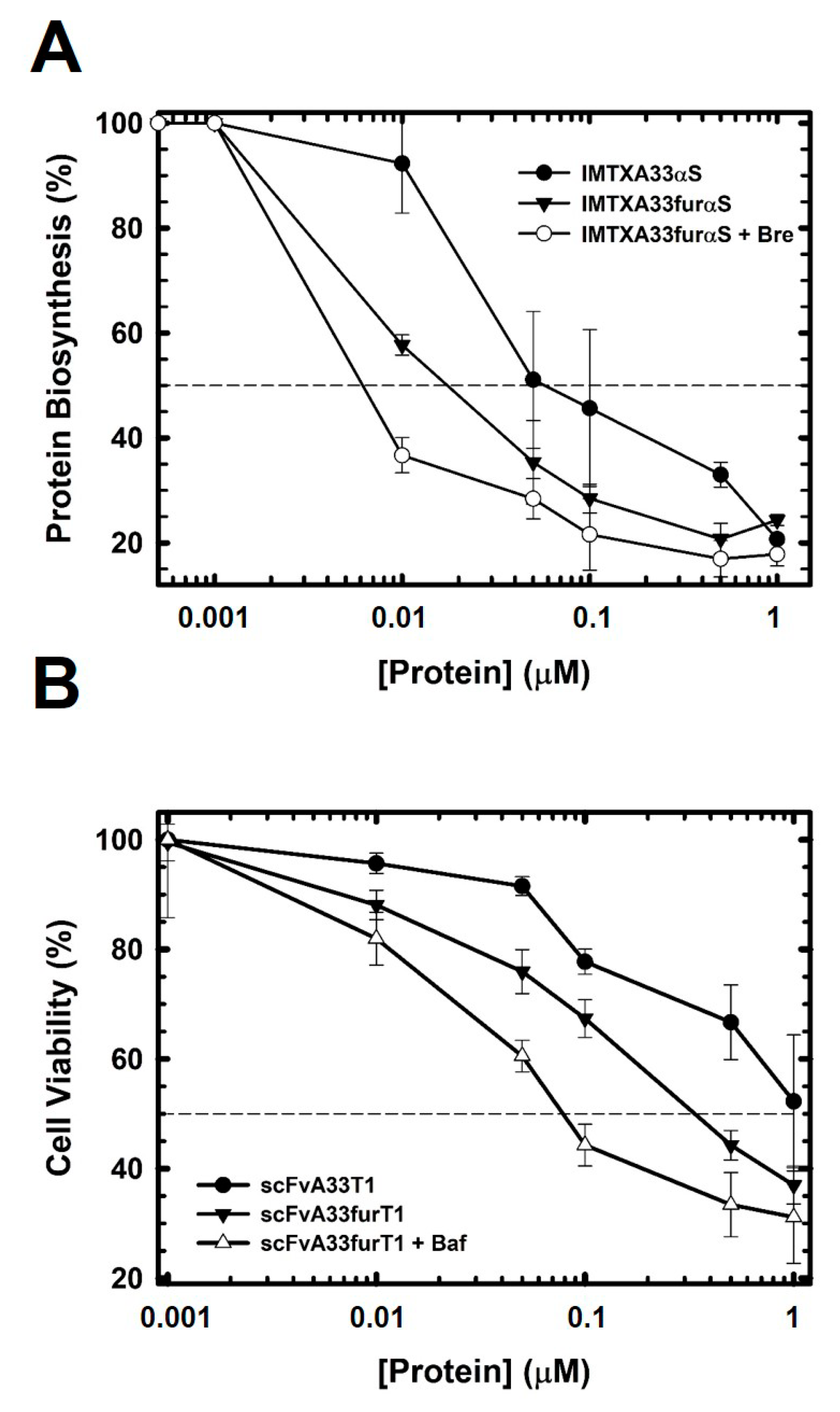
2.3. The Presence of the Furin Cleavage Site Significantly Increases the Cytotoxicity of Both Immunoconjugates
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Plasmid Design
5.2. Protein Production and Purification
5.3. Biophysical Characterization
5.4. Ribonucleolytic Activity Assays
5.5. Cell Lines Culture
5.6. Flow Cytometry Studies
5.7. Fluorescence Microscopy
5.8. MTT Viability Assay
5.9. Protein Biosynthesis Inhibition
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alewine, C.; Hassan, R.; Pastan, I. Advances in anticancer immunotoxin therapy. Oncologist 2015, 20, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Kavousipour, S.; Khademi, F.; Zamani, M.; Vakili, B.; Mokarram, P. Novel biotechnology approaches in colorectal cancer diagnosis and therapy. Biotechnol. Lett. 2017, 39, 785–803. [Google Scholar] [CrossRef] [PubMed]
- Nasiri, H.; Valedkarimi, Z.; Aghebati-Maleki, L.; Majidi, J. Antibody-drug conjugates: Promising and efficient tools for targeted cancer therapy. J. Cell. Physiol. 2018, 233, 6441–6457. [Google Scholar] [CrossRef]
- Frankel, A.E.; Woo, J.H.; Neville, D.M. Immunotoxins. In Principles of Cancer Biotherapy; Oldham, R.K., Dillman, R.O., Eds.; Springer: Dordrecht, The Netherlands, 2009; pp. 407–449. [Google Scholar]
- Madhumathi, J.; Verma, R.S. Therapeutic targets and recent advances in protein immunotoxins. Curr. Opin. Microbiol. 2012, 15, 300–309. [Google Scholar] [CrossRef]
- Pastan, I.; Hassan, R.; FitzGerald, D.J.; Kreitman, R.J. Immunotoxin treatment of cancer. Annu. Rev. Med. 2007, 58, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Kreitman, R.J. Recombinant immunotoxins containing truncated bacterial toxins for the treatment of hematologic malignancies. BioDrugs 2009, 23, 1–13. [Google Scholar] [CrossRef] [PubMed]
- King, E.M.; Mazor, R.; Cuburu, N.; Pastan, I. Low-dose methotrexate prevents primary and secondary humoral immune responses and induces immune tolerance to a recombinant immunotoxin. J. Immunol. 2018, 200, 2038–2045. [Google Scholar] [CrossRef] [PubMed]
- Becker, N.; Benhar, I. Antibody-Based Immunotoxins for the Treatment of Cancer. Antibodies 2012, 1, 39–69. [Google Scholar] [CrossRef]
- Weldon, J.E.; Xiang, L.; Zhang, J.; Beers, R.; Walker, D.A.; Onda, M.; Hassan, R.; Pastan, I. A recombinant immunotoxin against the tumor-associated antigen mesothelin reengineered for high activity, low off-target toxicity, and reduced antigenicity. Mol. Cancer Ther. 2013, 12, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Muyldermans, S. Nanobodies: Natural Single-Domain Antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [PubMed]
- Ecker, D.M.; Jones, S.D.; Levine, H.L. The therapeutic monoclonal antibody market. mAbs 2015, 1, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Cienfuegos, A.; Nuñez-Prado, N.; Compte, M.; Cuesta, A.M.; Blanco-Toribio, A.; Harwood, S.L.; Villate, M.; Merino, N.; Bonet, J.; Navarro, R.; et al. Intramolecular trimerization, a novel strategy for making multispecific antibodies with controlled orientation of the antigen binding domains. Sci. Rep. 2016, 6, 28643. [Google Scholar] [CrossRef] [PubMed]
- Kimiz-Gebologlu, I.; Gulce-Iz, S.; Biray-Avci, C. Monoclonal antibodies in cancer immunotherapy. Mol. Biol. Rep. 2018, 45, 2935–2940. [Google Scholar] [CrossRef] [PubMed]
- Appelbaum, F.R.; Bernstein, I.D. Gemtuzumab ozogamicin for acute myeloid leukemia. Blood 2017, 130, 2373–2376. [Google Scholar] [CrossRef] [PubMed]
- Kreitman, R.J.; Dearden, C.; Zinzani, P.L.; Delgado, J.; Karlin, L.; Robak, T.; Gladstone, D.E.; le Coutre, P.; Dietrich, S.; Gotic, M.; et al. Moxetumomab pasudotox in relapsed/refractory hairy cell leukemia. Leukemia 2018, 32, 1768–1777. [Google Scholar] [CrossRef]
- Avila, A.D.; de Acosta, C.M.; Lage, A. A new immunotoxin built by linking a hemolytic toxin to a monoclonal antibody specific for immature T lymphocytes. Int. J. Cancer 1988, 42, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Foss, F.M.; Saleh, M.N.; Krueger, J.G.; Nichols, J.C.; Murphy, J.R. Diphtheria toxin fusion proteins. Curr. Top. Microbiol. Immunol. 1988, 234, 663–681. [Google Scholar]
- LeMaistre, C.F.; Saleh, M.N.; Kuzel, T.M.; Foss, F.; Platanias, L.C.; Schwartz, G.; Ratain, M.; Rook, A.; Freytes, C.O.; Craig, F.; et al. Phase I Trial of a Ligand Fusion-Protein (DAB389IL-2) in Lymphomas Expressing the Receptor for Interkeukin-2. Blood 1998, 91, 399–405. [Google Scholar]
- O’Toole, J.E.; Esseltine, D.; Lynch, T.J.; Lambert, J.M.; Grossbard, M.L. Clinical trials with blocked ricin immunotoxins. Curr. Top. Microbiol. Immunol. 1998, 234, 35–56. [Google Scholar]
- Schnell, R.; Vitetta, E.; Schindler, J.; Barth, S.; Winkler, U.; Borchmann, P.; Hansmann, M.L.; Diehl, V.; Ghetie, V.; Engert, A. Clinical trials with an anti-CD25 ricin A-chain experimental and immunotoxin (RFT5-SMPT-dgA) in Hodgkin’s lymphomaLeuk. Lymphoma 1998, 30, 525–537. [Google Scholar] [CrossRef]
- García-Ortega, L.; Alegre-Cebollada, J.; García-Linares, S.; Bruix, M.; Martínez-Del-Pozo, A.; Gavilanes, J.G. The behavior of sea anemone actinoporins at the water-membrane interface. Biochim. Biophys. Acta 2011, 1808, 2275–2288. [Google Scholar] [CrossRef] [PubMed]
- Pirie, C.M.; Hackel, B.J.; Rosenblum, M.G.; Wittrup, K.D. Convergent potency of internalized gelonin immunotoxins across varied cell lines, antigens, and targeting moieties. J. Biol. Chem. 2011, 286, 4165–4172. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Duran-Struuck, R.; Crepeau, R.; Matar, A.; Hanekamp, I.; Srinivasan, S.; Neville, D.M., Jr.; Sachs, D.H.; Huang, C.A. Development of a diphtheria toxin based antiporcine CD3 recombinant immunotoxin. Bioconjug. Chem. 2011, 22, 2014–2020. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, X.F.; FitzGerald, D.J.; Pastan, I. The insulin receptor negatively regulates the action of Pseudomonas toxin-based immunotoxins and native Pseudomonas toxin. Cancer Res. 2013, 73, 2281–2288. [Google Scholar] [CrossRef] [PubMed]
- Rivera-de-Torre, E.; Palacios-Ortega, J.; Gavilanes, J.G.; Martínez-del-Pozo, Á.; García-Linares, S. Pore-Forming Proteins from Cnidarians and Arachnids as Potential Biotechnological Tools. Toxins (Basel) 2019, 11, 370. [Google Scholar] [CrossRef] [PubMed]
- De Lorenzo, C.; D’Alessio, G. From ImmunoToxins to ImmunoRNases. Curr. Pharm. Biotechnol. 2008, 9, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Balandin, T.G.; Edelweiss, E.; Andronova, N.V.; Treshalina, E.M.; Sapozhnikov, A.M.; Deyev, S.M. Antitumor activity and toxicity of anti-HER2 immunoRNase scFv 4D5-dibarnase in mice bearing human breast cancer xenografts. Investig. New Drugs 2011, 29, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Borriello, M.; Lacceti, P.; Terrazzano, G.; D’Alessio, G.; De Lorenzo, C. A novel fully human antitumor immunoRNase targeting ErbB2-positive tumors. Br. J. Cancer 2011, 104, 1716–1723. [Google Scholar] [CrossRef][Green Version]
- Tomé-Amat, J.; Menéndez-Méndez, A.; García-Ortega, L.; Batt, C.A.; Oñaderra, M.; Martínez-del-Pozo, A.; Gavilanes, J.G.; Lacadena, J. Production and characterization of scFvA33T1, an immunoRNase targeting colon cancer cells. FEBS J. 2012, 279, 3022–3032. [Google Scholar] [CrossRef]
- Shirmann, T.; Frenzel, A.; Linden, L.; Stelte-Ludwig, B.; Willuda, J.; Harrenga, A.; Dübel, S.; Müller-Tiemann, B.; Trautwein, M. Evaluation of human pancreatic RNase as effector molecule in a therapeutic antibody platform. mAbs 2014, 6, 367–380. [Google Scholar] [CrossRef]
- Sun, M.; Sun, L.; Sun, D.; Zhang, C.C.; Li, M. Targeted delivery of immuno-RNase may improve cancer therapy. Cancer Cell Int. 2018, 18, 58. [Google Scholar] [CrossRef] [PubMed]
- Wawrzynczak, E.J.; Henry, R.V.; Cumber, A.J.; Parnell, G.D.; Derbyshire, E.J.; Ulbrich, N. Biochemical, cytotoxic and pharmacokinetic properties of an immunotoxin composed of a mouse monoclonal antibody Fib75 and the ribosome-inactivating protein α-sarcin from Aspergillus giganteus. Eur. J. Biochem. 1991, 196, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Rathore, D.; Batra, J.K. Construction, expression and characterization of chimaeric toxins containing the ribonucleolytic toxin restrictocin: Intracellular mechanism of action. Biochem. J. 1997, 324, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Rathore, D.; Nayak, S.K.; Batra, J.K. Overproduction of fungal ribotoxin α-sarcin in Escherichia coli: Generation of an active immunotoxin. Gene 1997, 190, 31–35. [Google Scholar] [CrossRef]
- Carreras-Sangrà, N.; Tomé-Amat, J.; García-Ortega, L.; Batt, C.A.; Oñaderra, M.; Martínez-del-Pozo, A.; Gavilanes, J.G.; Lacadena, J. Production and characterization of a colon cancer-specific immunotoxin based on the fungal ribotoxin α-sarcin. Protein Eng. Des. Sel. 2012, 25, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Tomé-Amat, J.; Herrero-Galán, E.; Oñaderra, M.; Martínez-del-Pozo, A.; Gavilanes, J.G.; Lacadena, J. Preparation of an engineered safer immunotoxin against colon carcinoma based on the ribotoxin hirsutellin A. FEBS J. 2015, 282, 2131–2141. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.D.; Heam, A.R.; Holgate, R.G.; Kozub, D.; Fogg, M.H.; Carr, F.J.; Baker, M.P.; Lacadena, J.; Gehlsen, K.R. A deimmunised form of the ribotoxin, α-sarcin, lacking CD4+ T cell epitopes and its use as an immunotoxin warhead. Protein Eng. Des. Sel. 2016, 29, 531–540. [Google Scholar] [CrossRef]
- Tomé-Amat, J.; Ruiz-de-la-Herrán, J.; Martínez-del-Pozo, Á.; Gavilanes, J.G.; Lacadena, J. α-sarcin and RNase T1 based immunoconjugates: The role of intracellular trafficking in cytotoxic efficiency. FEBS J. 2015, 282, 673–684. [Google Scholar] [CrossRef]
- Tome-Amat, J.; Olombrada, M.; Ruiz-de-la-Herrán, J.; Pérez-Gómez, E.; Andradas, C.; Sánchez, C.; Martínez, L.; Martínez-Del-Pozo, Á.; Gavilanes, J.G.; Lacadena, J. Efficient in vivo antitumor effect of an immunotoxin based on ribotoxin α-sarcin in nude mice bearing human colorectal cancer xenografts. Springerplus 2015, 4, 168. [Google Scholar] [CrossRef]
- Olmo, N.; Turnay, J.; González de Buitrago, G.; de Silanes, I.L.; Gavilanes, J.G.; Lizarbe, M.A. Cytotoxic mechanism of the ribotoxin α-sarcin. Induction of cell death via apoptosis. Eur. J. Biochem. 2001, 268, 2113–2123. [Google Scholar] [CrossRef]
- Lacadena, J.; Alvarez-García, E.; Carreras-Sangrà, N.; Herrero-Galán, E.; Alegre-Cebollada, J.; García-Ortega, L.; Oñaderra, M.; Gavilanes, J.G.; Martínez-del-Pozo, A. Fungal ribotoxins: Molecular dissection of a family of natural killers. FEMS Microbiol. Rev. 2007, 1, 212–237. [Google Scholar] [CrossRef] [PubMed]
- Olombrada, M.; Lázaro-Gorines, R.; López-Rodríguez, J.C.; Martínez-del-Pozo, A.; Oñaderra, M.; Maestro-López, M.; Lacadena, J.; Gavilanes, J.G.; García-Ortega, L. Fungal ribotoxins: A Review of Potential Biotechnological Applications. Toxins 2017, 9, 71. [Google Scholar] [CrossRef] [PubMed]
- Weldon, J.E.; Pastan, I. A guide to taming a toxin-recombinant immunotoxins constructed from Pseudomonas exotoxin A for the treatment of cancer. FEBS J. 2011, 278, 4683–4700. [Google Scholar] [CrossRef] [PubMed]
- Weng, A.; Thakur, M.; von Mallinckradt, B.; Beceren-Braun, F.; Gilabert-Oriol, R.; Wiesner, B.; Eickhart, J.; Böttger, S.; Melzig, M.F.; Fuchs, H. Saponins modulate the intracellular trafficking of protein toxins. J. Control. Release 2012, 164, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Pasetto, M.; Antignani, A.; Ormanoglu, E.; Buehler, E.; Guha, R.; Pastan, I.; Martin, S.E.; FitzGerald, D.J. Whole-genome RNAi screen highlights components of the endoplasmic reticulum/Golgi as a source of resistance to immunotoxin-mediated cytotoxicity. Proc. Natl. Acad. Sci. USA 2015, 112, E1135–E1142. [Google Scholar] [CrossRef] [PubMed]
- Johannes, L.; Decaudin, D. Protein toxins: Intracellular trafficking for targeted therapy. Gene Ther. 2005, 12, 1360–1368. [Google Scholar] [CrossRef] [PubMed]
- Ritter, G.; Cohen, L.S.; Nice, E.C.; Catimel, B.; Burgess, A.W.; Moritz, R.L.; Ji, H.; Heath, J.K.; White, S.J.; Welt, S.; et al. Characterization of posttranslational modifications of human A33 antigen, a novel palmitoylated surface glycoprotein of human gastrointestinal epithelium. Biochem. Biophys. Res. Commun. 1997, 236, 682–686. [Google Scholar] [CrossRef] [PubMed]
- Pereira-Fantini, P.M.; Judd, L.M.; Kalantzis, A.; Peterson, A.; Ernst, M.; Heath, J.K.; Giraud, A.S. A33 antigen-deficient mice have defective colonic mucosal repair. Inflamm. Bowel Dis. 2010, 16, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H. The ribonuclease T1 family. Methods Enzymol. 2001, 341, 28–41. [Google Scholar]
- Goyal, A.; Batra, J.K. Inclusion of a furin-sensitive spacer enhances the cytotoxicity of ribotoxin restrictocin containing recombinant single-chain immunotoxins. Biochem. J. 2000, 345, 247–254. [Google Scholar] [CrossRef]
- Tortorella, L.L.; Pipalia, N.H.; Mukherjee, S.; Pastan, I.; Fitzgerald, D.; Maxfiled, F.R. Efficiency of Immunotoxin Cytotoxicity Is Modulated by the Intracellular Itinerary. PLoS ONE 2012, 7, e47320. [Google Scholar] [CrossRef] [PubMed]
- Weldon, J.E.; Scarzynski, M.; Therres, J.A.; Ostovitz, J.R.; Zhou, H.; Kreitman, R.J.; Pastan, I. Designing the furin-cleavable linker in recombinant immunotoxins based on Pseudomonas exotoxin A. Bioconjug. Chem. 2015, 26, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Mutter, N.L.; Soskine, M.; Huang, G.; Alburquerque, I.S.; Bernardes, G.J.L.; Maglia, G. Modular Pore-Forming Immunotoxins with Caged Cytotoxicity Tailored by Directed Evolution. ACS Chem. Biol. 2018, 13, 3153–3160. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, G.; Lee, F.; Onda, M.; Kolyvas, E.; Bhardwaj, G.; Baker, D.; Pastan, I. Protection of the Furin Cleavage Site in Low-Toxicity Immunotoxins Based on Pseudomonas Exotoxin A. Toxins 2016, 8, 217. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, G.; Mazor, R.; Lee, F.; Jang, Y.; Leshem, Y.; Pastan, I. Improving the In Vivo Efficacy of an Anti-Tac (CD25) Immunotoxin by Pseudomonas Exotoxin A Domain II Engineering. Large Mol. Ther. 2018. [Google Scholar] [CrossRef] [PubMed]
- Meng, P.; Dong, Q.C.; Tan, G.G.; Wen, W.H.; Wang, H.; Zhang, G.; Wang, Y.Z.; Jing, Y.M.; Wang, C.; Qin, W.J.; et al. Anti-tumor effects of a recombinant antiprostate specific membrane antigen immunotoxin against prostate cancer cells. BMC Urol. 2017, 17, 14. [Google Scholar] [CrossRef] [PubMed]
- Schapiro, F.B.; Soe, T.T.; Mallet, W.G.; Maxfield, F.R. Role of Cytoplasmic Domain Serines in Intracellular Trafficking of Furin. Mol. Biol. Cell 2014, 15, 2884–2894. [Google Scholar] [CrossRef]
- Wise, R.J.; Barr, P.J.; Wong, P.A.; Kiefer, M.C.; Brake, A.J.; Kaufman, R.J. Expression of a human proprotein processing enzyme: Correct cleavage of the von Willebrand factor precursor at a paired basic amino acid site. Proc. Natl. Acad. Sci. USA 1990, 87, 9378–9382. [Google Scholar] [CrossRef]
- Van de Ven, W.J.; Creemers, J.W.; Roebroek, A.J. Furin: The prototype mammalian subtilisin-like proprotein-processing enzyme. Endoproteolytic cleavage at paired basic residues of proproteins of the eukaryotic secretory pathway. Enzyme 1991, 45, 257–270. [Google Scholar] [CrossRef]
- Mattia, A.; Merker, R. Regulation of probiotic substances as ingredients in foods: Premarket approval or “generally recognized as safe” notification. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2008, 46, S115–S118. [Google Scholar] [CrossRef]
- Bader, O.; Krauke, Y.; Hube, B. Processing of predicted substrates of fungal Kex2 proteinases from Candida albicans, C. glabrata, Saccharomyces cerevisiae and Pichia pastoris. BMC Microbiol. 2008, 8, 116. [Google Scholar] [CrossRef] [PubMed]
- Martínez-del-Pozo, A.; Gasset, M.; Oñaderra, M.; Gavilanes, J.G. Conformational study of the antitumor protein α-sarcin. Biochim. Biophys. Acta 1988, 953, 280–288. [Google Scholar] [CrossRef]
- Pace, C.N.; Heinemann, U.; Hahn, U.; Saenger, W. Ribonuclease T1: Structure, function and stability. Angew. Chem. Int. Ed. Engl. 1991, 30, 343–454. [Google Scholar] [CrossRef]
- Pérez-Cañadillas, J.M.; Santoro, J.; Campos-Olivas, R.; Lacadena, J.; del Pozo, A.M.; Gavilanes, J.G.; Rico, M.; Bruix, M. The highly refined solution structure of the cytotoxic ribonuclease alpha-sarcin reveals the structural requirements for substrate recognition and ribonucleolytic activity. J. Mol. Biol. 2000, 299, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, J.A.; Power, B.E.; Garrett, T.P.J.; Yazaki, P.J.; Shively, J.E.; Raubischek, A.A.; Wu, A.M.; Hudson, P.J. The crystal structure of an anti-CEA scFv diabody assembled from T84.66 scFvs in V(L)-to-V(H) orientation: Implications for diabody flexibility. J. Mol. Biol. 2003, 326, 341–351. [Google Scholar] [CrossRef]
- Wilkinson, I.C.; Hall, C.J.; Veverka, V.; Shi, J.Y.; Muskett, F.W.; Stephens, P.E.; Taylor, R.J.; Henry, A.J.; Carr, M.D. High resolution NMR-based model for the structure of a scFv-IL-1beta complex: Potential for NMR as a key tool in therapeutic antibody design and development. J. Biol. Chem. 2009, 284, 31928–31935. [Google Scholar] [CrossRef] [PubMed]
- Oñaderra, M.; Mancheño, J.M.; Gasset, M.; Lacadena, J.; Schiavo, G.; del Pozo, A.M.; Gavilanes, J.G. Translocation of α-sarcin across the lipid bilayer of asolectin vesicle. Biochem. J. 1993, 295, 221–225. [Google Scholar] [CrossRef]
- Gruenberg, J. Lipids in endocytic membrane transport and sorting. Curr. Opin. Cell Biol. 2003, 15, 382–388. [Google Scholar] [CrossRef]
- Bissing, C.; Gruenberg, J. Lipid sorting and multivesicular endosome biogenesis. Cold Spring Harb. Perspect. Biol. 2013, 5, a016816. [Google Scholar]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Wherethey are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Zhao, P.; Liu, F.; Zhang, B.; Liu, X.; Wang, B.; Gong, J.; Yu, G.; Ma, M.; Lu, Y.; Sun, J.; et al. MAIGO2 is involved in abscisic acid-mediated response to abiotic stresses and Golgi-to-ER retrograde transport. Physiol. Plant. 2013, 148, 246–260. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Ruiz, A.; García-Ortega, L.; Kao, R.; Lacadena, J.; Oñaderra, M.; Mancheño, J.M.; Davies, J.; del Pozo, A.M.; Gavilanes, J.G. RNAse U2 and α-sarcin: A study of relationships. Methods Enzimol. 2001, 341, 335–351. [Google Scholar]
- Blanco-Toribio, A.; Lacadena, J.; Nuñez-Prado, N.; Álvarez-Ciénfuegos, A.; Villate, M.; Compte, M.; Sanz, L.; Blanco, F.J.; Álvarez-Vallina, L. Efficient production of single-chain fragment variable-based N-terminal trimerbodies in Pichia pastoris. Microb. Cell Factories 2014, 13, 116–124. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lázaro-Gorines, R.; Ruiz-de-la-Herrán, J.; Navarro, R.; Sanz, L.; Álvarez-Vallina, L.; Martínez-del-Pozo, A.; Gavilanes, J.G.; Lacadena, J. A novel Carcinoembryonic Antigen (CEA)-Targeted Trimeric Immunotoxin shows significantly enhanced Antitumor Activity in Human Colorectal Cancer Xenografts. Sci. Rep. 2019, 9, 11680. [Google Scholar] [CrossRef] [PubMed]
- Lacadena, J.; Martínez del Pozo, A.; Martínez-Ruiz, A.; Pérez-Cañadillas, J.M.; Bruix, M.; Mancheño, J.M.; Oñaderra, M.; Gavilanes, J.G. Role of histidine-50, glutamic acid-96, and histidine-137 in the ribonucleolytic mechanism of the ribotoxin alpha-sarcin. Proteins 1999, 37, 474–484. [Google Scholar] [CrossRef]
- Kao, R.; Martínez-Ruiz, A.; Martínez del Pozo, A.; Crameri, R.; Davies, J. Mitogillin and related fungal ribotoxins. Methods Enzymol. 2001, 341, 324–335. [Google Scholar] [PubMed]
- García-Ortega, L.; Masip, M.; Mancheño, J.M.; Oñaderra, M.; Lizarbe, M.A.; García-Mayoral, M.F.; Bruix, M.; Martínez-del-Pozo, A.; Gavilanes, J.G. Deletion of the NH2-terminal beta-hairpin of the ribotoxin alpha-sarcin produces a nontoxic but active ribonuclease. J. Biol. Chem. 2002, 277, 18632–18639. [Google Scholar] [CrossRef]
- Steyaert, J. A decade of protein engineering on ribonuclease T1-atomic dissection of the enzyme-substrate interactions. Eur. J. Biochem. 1997, 247, 1–11. [Google Scholar] [CrossRef]
- Yoshimori, T.; Yamamoto, A.; Moriyama, Y.; Futai, M.; Tashiro, Y. Bafilomycin A1, a Specific Inhibitor of Vacuolar-type H+-ATPase, Inhibits Acidification and Protein Degradation in Lysosomes of Cultured Cell. J. Biol. Chem. 1991, 266, 17707–17712. [Google Scholar]
- Yeung, T.M.; Shaan, C.G.; Wilding, J.L.; Muschel, R.; Bodmer, W.F. Cancer stem cells from colorectal cancer-derived cell lines. Proc. Natl. Acad. Sci. USA 2009, 107, 3722–3727. [Google Scholar] [CrossRef]
- Nazarewicz, R.R.; Salazar, G.; Patrushev, N.; San Martin, A.; Hilenski, L.; Xiong, S.; Alexander, R.W. Early endosomal antigen 1 (EEA1) is an obligate scaffold for angiotensin II-induced, PKC-α-dependent Akt activation in endosomes. J. Biol. Chem. 2011, 286, 2886–2895. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.G.; Orci, L. A view of acidic intracellular compartments. J. Cell Biol. 1988, 106, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Chen, T.L.; Vertel, B.M.; Colley, K.J. The CMP-syalic acid transporter is localized in the medial-trans Golgi and possesses two specific endoplasmic reticulum export motifs in its carboxyl-terminal cytoplasmic tail. J. Biol. Chem. 2006, 281, 31106–31118. [Google Scholar] [CrossRef] [PubMed]
- Zinchuk, V.; Zinchuk, O.; Okada, T. Quantitative Colocalization Analysis of Multicolor Confocal Immunofluorescence Microscopy Images: Pushing Pixels to Explore Biological Phenomena. Acta Histochem. Cytochem. 2007, 40, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Oda, K.; Yokota, S.; Takatsuki, A.; Ikehara, Y. Brefeldin A Causes Disassembly of the Golgi Complex and Accumulation of Secretory Proteins in the Endoplasmic Reticulum. J. Biol. Chem. 1988, 263, 18545–18552. [Google Scholar] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IMTXA33furαS | IMTXA33αS * | |
| Golgi colocalizationOverlap coefficient at 16 h (%) | 18.2 | 46.3 |
| IC50 (nM) | 15 | 30–40 |
| IC50 (nM) + Bafilomycin | 15–20 | 30 |
| IC50 (nM) + Brefefeldin A | 5 | >500 |
| IMTXA33furT1 | IMTXA33T1 * | |
| Lysosomes colocalizationOverlap coefficient at 16 h (%) | 23.4 | 58.1 |
| IC50 (nM) | 300 | >1 mM |
| IC50 (nM) + Bafilomycin | 70–90 | 70–90 |
| IC50 (nM) + Brefefeldin A | 300 | >1 mM |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruiz-de-la-Herrán, J.; Tomé-Amat, J.; Lázaro-Gorines, R.; Gavilanes, J.G.; Lacadena, J. Inclusion of a Furin Cleavage Site Enhances Antitumor Efficacy against Colorectal Cancer Cells of Ribotoxin α-Sarcin- or RNase T1-Based Immunotoxins. Toxins 2019, 11, 593. https://doi.org/10.3390/toxins11100593
Ruiz-de-la-Herrán J, Tomé-Amat J, Lázaro-Gorines R, Gavilanes JG, Lacadena J. Inclusion of a Furin Cleavage Site Enhances Antitumor Efficacy against Colorectal Cancer Cells of Ribotoxin α-Sarcin- or RNase T1-Based Immunotoxins. Toxins. 2019; 11(10):593. https://doi.org/10.3390/toxins11100593
Chicago/Turabian StyleRuiz-de-la-Herrán, Javier, Jaime Tomé-Amat, Rodrigo Lázaro-Gorines, José G. Gavilanes, and Javier Lacadena. 2019. "Inclusion of a Furin Cleavage Site Enhances Antitumor Efficacy against Colorectal Cancer Cells of Ribotoxin α-Sarcin- or RNase T1-Based Immunotoxins" Toxins 11, no. 10: 593. https://doi.org/10.3390/toxins11100593
APA StyleRuiz-de-la-Herrán, J., Tomé-Amat, J., Lázaro-Gorines, R., Gavilanes, J. G., & Lacadena, J. (2019). Inclusion of a Furin Cleavage Site Enhances Antitumor Efficacy against Colorectal Cancer Cells of Ribotoxin α-Sarcin- or RNase T1-Based Immunotoxins. Toxins, 11(10), 593. https://doi.org/10.3390/toxins11100593