Toxin Neutralization Using Alternative Binding Proteins

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
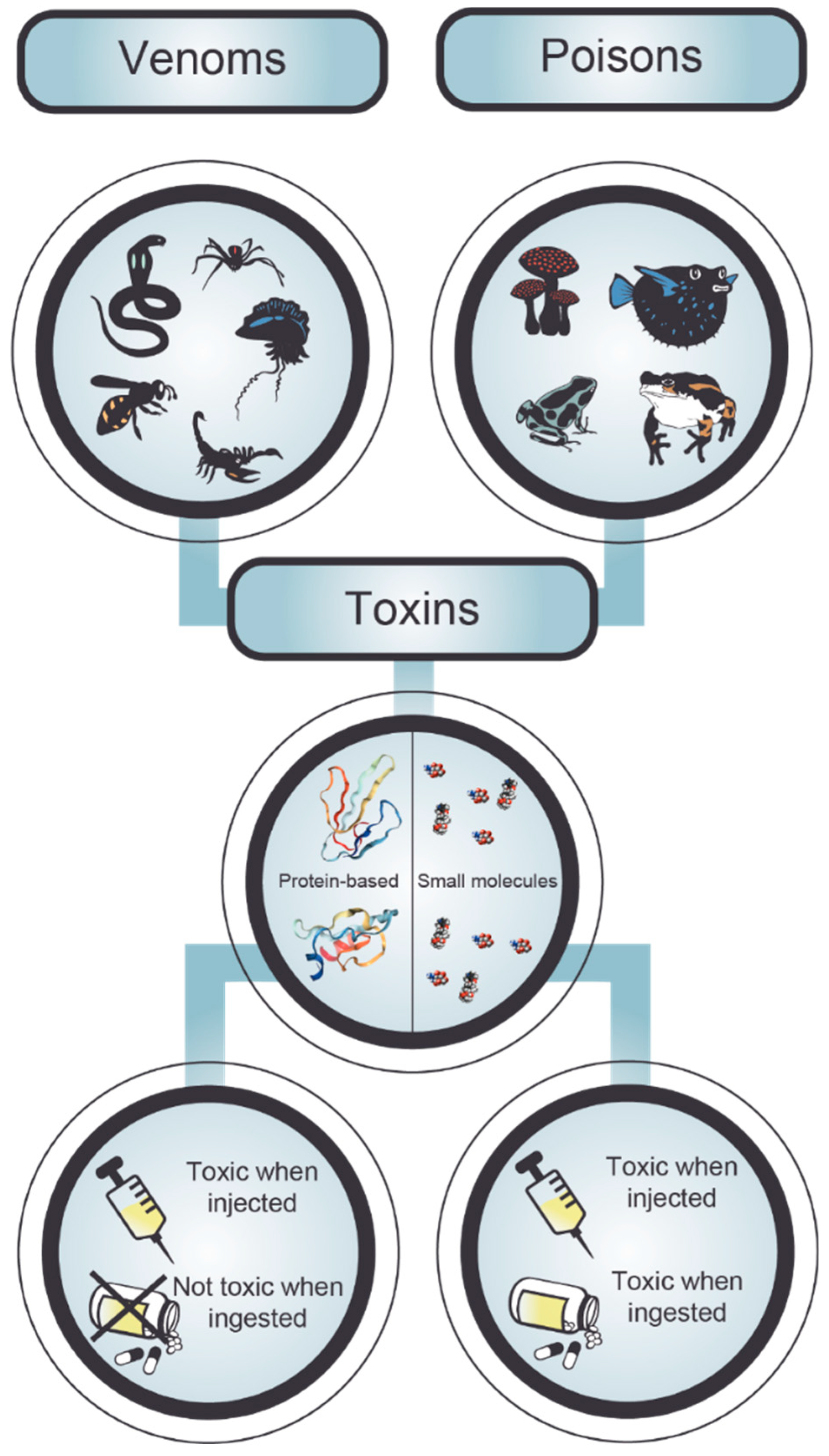
2. Poisonings and Envenomings
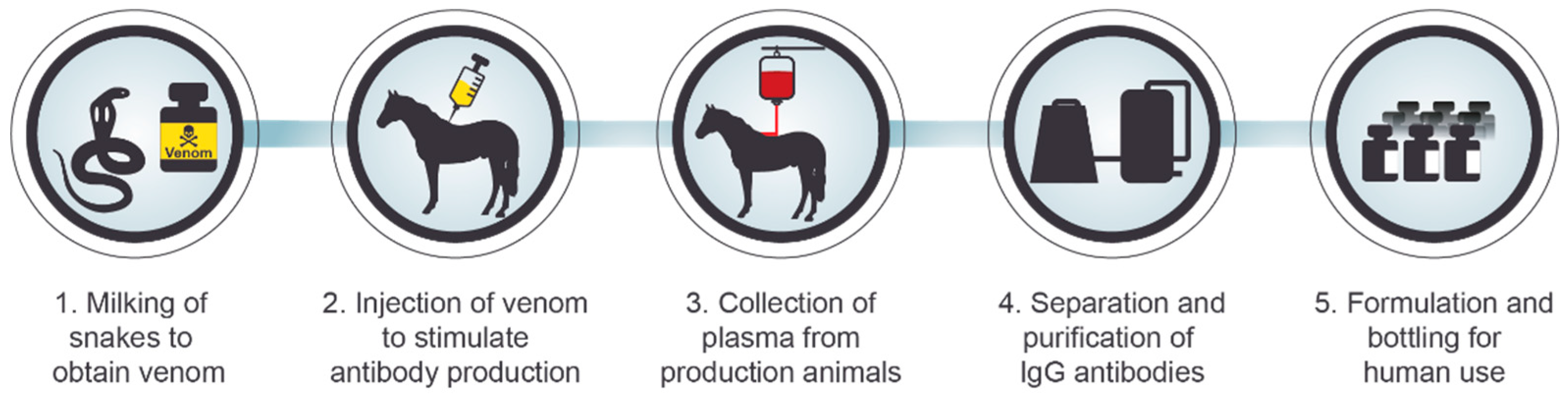
3. Serotherapy against Intoxication
4. Human Monoclonal Antibodies
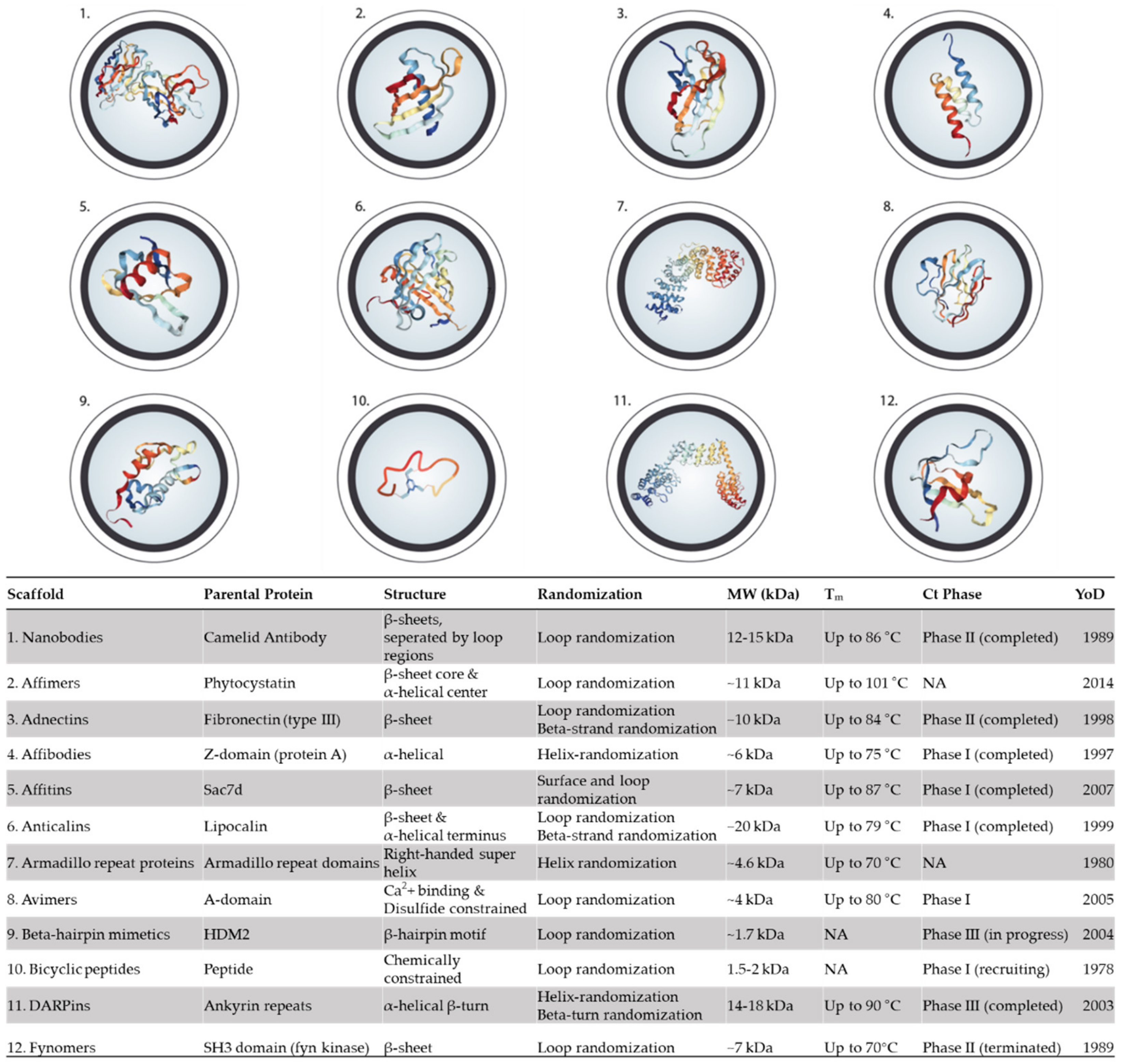
5. Alternative Binding Scaffolds
5.1. Nanobodies
5.2. Affimers
5.3. Adnectins (Monobodies)
5.4. Affibodies
5.5. Affitin (Nanofitins)
5.6. Anticalins
5.7. Armadillo Repeat Proteins
5.8. Avimers
5.9. β-Hairpin Mimetics
5.10. Bicyclic peptides
5.11. DARPins
5.12. Fynomers
6. Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Sunagar, K.; Casewell, N.R.; Varma, S.; Kolla, R.; Antunes, A.; Moran, Y. Deadly Innovations: Unraveling the Molecular Evolution of Animal Venoms. In Venom Genomics and Proteomics; Springer: Dordrecht, The Netherlands, 2014; pp. 1–23. [Google Scholar]
- Gutiérrez, J.M.; Calvete, J.J.; Habib, A.G.; Harrison, R.A.; Williams, D.J.; Warrell, D.A. Snakebite envenoming. Nat. Rev. Dis. Primer 2017, 3, 17063. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.A.; Hargreaves, A.; Wagstaff, S.C.; Faragher, B.; Lalloo, D.G. Snake Envenoming: A Disease of Poverty. PLoS Negl. Trop. Dis. 2009, 3, e569. [Google Scholar] [CrossRef]
- Chippaux, J.-P. Snakebite envenomation turns again into a neglected tropical disease! J. Venom. Anim. Toxins Incl. Trop. Dis. 2017, 23, 38. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Sustaining the Drive to Overcome the Global Impact of Neglected Tropical Diseases: Second WHO Report on Neglected Tropical Diseases; World Health Organization: Geneva, Switzerland, 2013; ISBN 978-92-4-156454-0. [Google Scholar]
- Calmette, A. L’immunisation artificielle des animaux contre le venin des serpents, et la thérapeutic expérimentale des morsures venimeuses. C. R. Soc. Biol. 1894, 46, 120–124. [Google Scholar]
- Phisalix, C.A.; Bertrand, G. Sur la propriété antitoxique du sang des animaux vaccinés contre le venin de vipère. C. R. Soc. Biol. 1894, 46, 111–113. [Google Scholar]
- Habib, A.G.; Brown, N.I. The snakebite problem and antivenom crisis from a health-economic perspective. Toxicon 2018, 150, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Laustsen, A.H.; Engmark, M.; Milbo, C.; Johannesen, J.; Lomonte, B.; Gutiérrez, J.M.; Lohse, B. From Fangs to Pharmacology: The Future of Snakebite Envenoming Therapy. Curr. Pharm. Des. 2016, 22, 5270–5293. [Google Scholar] [CrossRef] [PubMed]
- Laustsen, A.H.; Solà, M.; Jappe, E.C.; Oscoz, S.; Lauridsen, L.P.; Engmark, M. Biotechnological Trends in Spider and Scorpion Antivenom Development. Toxins 2016, 8, 226. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, C.; Laustsen, A.H. Recent Advances in Next Generation Snakebite Antivenoms. Trop. Med. Infect. Dis. 2018, 3, 42. [Google Scholar] [CrossRef] [PubMed]
- Laustsen, A.H.; María Gutiérrez, J.; Knudsen, C.; Johansen, K.H.; Bermúdez-Méndez, E.; Cerni, F.A.; Jürgensen, J.A.; Ledsgaard, L.; Martos-Esteban, A.; Øhlenschlæger, M.; et al. Pros and cons of different therapeutic antibody formats for recombinant antivenom development. Toxicon 2018, 146, 151–175. [Google Scholar] [CrossRef] [PubMed]
- Laustsen, A.H. Guiding recombinant antivenom development by omics technologies. New Biotechnol. 2018, 45, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Tkaczyk, C.; Hua, L.; Varkey, R.; Shi, Y.; Dettinger, L.; Woods, R.; Barnes, A.; MacGill, R.S.; Wilson, S.; Chowdhury, P.; et al. Identification of anti-alpha toxin mAbs that reduce severity of Staphylococcus aureus dermonecrosis and exhibit a correlation between affinity and potency. Clin. Vaccine Immunol. 2012, 19, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Leav, B.A.; Blair, B.; Leney, M.; Knauber, M.; Reilly, C.; Lowy, I.; Gerding, D.N.; Kelly, C.P.; Katchar, K.; Baxter, R.; et al. Serum anti-toxin B antibody correlates with protection from recurrent Clostridium difficile infection (CDI). Vaccine 2010, 28, 965–969. [Google Scholar] [CrossRef] [PubMed]
- Stanker, L.H.; Merrill, P.; Scotcher, M.C.; Cheng, L.W. Development and partial characterization of high-affinity monoclonal antibodies for botulinum toxin type A and their use in analysis of milk by sandwich ELISA. J. Immunol. Methods 2008, 336, 1–8. [Google Scholar] [CrossRef]
- Laustsen, A.H.; Karatt-Vellatt, A.; Masters, E.W.; Arias, A.S.; Pus, U.; Knudsen, C.; Oscoz, S.; Slavny, P.; Griffiths, D.T.; Luther, A.M.; et al. In vivo neutralization of dendrotoxin-mediated neurotoxicity of black mamba venom by oligoclonal human IgG antibodies. Nat. Commun. 2018, 9, 3928. [Google Scholar] [CrossRef]
- Presta, L.G. Molecular engineering and design of therapeutic antibodies. Curr. Opin. Immunol. 2008, 20, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.L.; Dhimolea, E.; Reichert, J.M. Development trends for human monoclonal antibody therapeutics. Nat. Rev. Drug Discov. 2010, 9, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; English, A.M.; Bai, D.L.; Ugrin, S.A.; Shabanowitz, J.; Ross, M.M.; Hunt, D.F.; Wang, W.-H. Analysis of Monoclonal Antibody Sequence and Post-translational Modifications by Time-controlled Proteolysis and Tandem Mass Spectrometry. Mol. Cell. Proteom. 2016, 15, 1479–1488. [Google Scholar] [CrossRef] [PubMed]
- Rouet, R.; Lowe, D.; Christ, D. Stability engineering of the human antibody repertoire. FEBS Lett. 2014, 588, 269–277. [Google Scholar] [CrossRef]
- Birch, J.R.; Racher, A.J. Antibody production. Adv. Drug Deliv. Rev. 2006, 58, 671–685. [Google Scholar] [CrossRef] [PubMed]
- Rouet, R.; Lowe, D.; Dudgeon, K.; Roome, B.; Schofield, P.; Langley, D.; Andrews, J.; Whitfeld, P.; Jermutus, L.; Christ, D. Expression of high-affinity human antibody fragments in bacteria. Nat. Protoc. 2012, 7, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Lombardi, R.; Phan, T.G.; Zimmermann, C.; Lowe, D.; Jermutus, L.; Christ, D. Challenges and opportunities for non-antibody scaffold drugs. Drug Discov. Today 2015, 20, 1271–1283. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.K. Poisons of animal origin. In Fundamentals of Toxicology; Gupta, P.K., Ed.; Academic Press: Cambridge, MA, USA, 2016; pp. 311–325. ISBN 978-0-12-805426-0. [Google Scholar]
- Casewell, N.R.; Wüster, W.; Vonk, F.J.; Harrison, R.A.; Fry, B.G. Complex cocktails: The evolutionary novelty of venoms. Trends Ecol. Evol. 2013, 28, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Church, J.E.; Hodgson, W.C. The pharmacological activity of fish venoms. Toxicon 2002, 40, 1083–1093. [Google Scholar] [CrossRef]
- De Graaf, D.C.; Aerts, M.; Danneels, E.; Devreese, B. Bee, wasp and ant venomics pave the way for a component-resolved diagnosis of sting allergy. J. Proteom. 2009, 72, 145–154. [Google Scholar] [CrossRef]
- Peiren, N.; Vanrobaeys, F.; de Graaf, D.C.; Devreese, B.; Van Beeumen, J.; Jacobs, F.J. The protein composition of honeybee venom reconsidered by a proteomic approach. Biochim. Biophys. Acta BBA Proteins Proteom. 2005, 1752, 1–5. [Google Scholar] [CrossRef]
- Mackessy, S.P. Evolutionary trends in venom composition in the Western Rattlesnakes (Crotalus viridis sensu lato): Toxicity vs. tenderizers. Toxicon 2010, 55, 1463–1474. [Google Scholar] [CrossRef]
- Glenn, J.L.; Straight, R. Mojave rattlesnake Crotalus scutulatus scutulatus venom: Variation in toxicity with geographical origin. Toxicon 1978, 16, 81–84. [Google Scholar] [CrossRef]
- Calvete, J.J.; Sanz, L.; Cid, P.; de la Torre, P.; Flores-Díaz, M.; Dos Santos, M.C.; Borges, A.; Bremo, A.; Angulo, Y.; Lomonte, B.; et al. Snake Venomics of the Central American Rattlesnake Crotalus simus and the South American Crotalus durissus Complex Points to Neurotoxicity as an Adaptive Paedomorphic Trend along Crotalus Dispersal in South America. J. Proteome Res. 2010, 9, 528–544. [Google Scholar] [CrossRef]
- Menezes, M.C.; Furtado, M.F.; Travaglia-Cardoso, S.R.; Camargo, A.C.M.; Serrano, S.M.T. Sex-based individual variation of snake venom proteome among eighteen Bothrops jararaca siblings. Toxicon 2006, 47, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Andrade, D.V.; Abe, A.S. Relationship of Venom Ontogeny and Diet in Bothrops. Herpetologica 1999, 55, 200–204. [Google Scholar]
- World Health Organization. Guidelines for the Production, Control and Regulation of Snake Antivenom Immunoglobulins; WHO Technical Report Series; WHO: Geneva, Switzerland, 2018; p. 964. [Google Scholar]
- Williams, D.; Gutiérrez, J.M.; Harrison, R.; Warrell, D.A.; White, J.; Winkel, K.D.; Gopalakrishnakone, P. The Global Snake Bite Initiative: An antidote for snake bite. Lancet 2010, 375, 89–91. [Google Scholar] [CrossRef]
- Jenner, E. An Inquiry into the Causes and Effects of the Variolae Vaccinae, a Disease Discovered in Some of the Western Counties of England, Particularly Gloucestershire, and Known by the Name of the Cow Pox; Sampson Low: London, UK, 1798. [Google Scholar]
- León, G.; Vargas, M.; Segura, Á.; Herrera, M.; Villalta, M.; Sánchez, A.; Solano, G.; Gómez, A.; Sánchez, M.; Estrada, R.; et al. Current technology for the industrial manufacture of snake antivenoms. Toxicon 2018, 151, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.; Landon, J. Antivenom: The most cost-effective treatment in the world? Toxicon 2010, 55, 1405–1407. [Google Scholar] [CrossRef]
- Williams, D.J.; Gutiérrez, J.-M.; Calvete, J.J.; Wüster, W.; Ratanabanangkoon, K.; Paiva, O.; Brown, N.I.; Casewell, N.R.; Harrison, R.A.; Rowley, P.D.; et al. Ending the drought: New strategies for improving the flow of affordable, effective antivenoms in Asia and Africa. J. Proteom. 2011, 74, 1735–1767. [Google Scholar] [CrossRef]
- Segura, A.; Herrera, M.; Villalta, M.; Vargas, M.; Gutiérrez, J.M.; León, G. Assessment of snake antivenom purity by comparing physicochemical and immunochemical methods. Biol. J. Int. Assoc. Biol. Stand. 2013, 41, 93–97. [Google Scholar] [CrossRef]
- Herrera, M.; Paiva, O.K.; Pagotto, A.H.; Segura, A.; Serrano, S.M.T.; Vargas, M.; Villalta, M.; Jensen, S.D.; León, G.; Williams, D.J.; et al. Antivenomic characterization of two antivenoms against the venom of the taipan, Oxyuranus scutellatus, from Papua New Guinea and Australia. Am. J. Trop. Med. Hyg. 2014, 91, 887–894. [Google Scholar] [CrossRef]
- Rawat, S.; Laing, G.; Smith, D.C.; Theakston, D.; Landon, J. A new antivenom to treat eastern coral snake (Micrurus fulvius fulvius) envenoming. Toxicon 1994, 32, 185–190. [Google Scholar] [CrossRef]
- Pucca, M.B.; Carlos, J.; Roncolato, E.C.; Bertolini, T.B.; Fossa, C.M.; Varanda, W.A.; Arantes, E.C.; Barbosa, J.E. Monoclonal antibody (Scfv) against the venom of the scorpion Tityus serrulatus, produced by phage display technic, is capable to recognize and inhibit the action of the ts1 toxin. Epeq/Fafibe 2011, 1, 18–23. [Google Scholar]
- Laustsen, A.H.; Engmark, M.; Clouser, C.; Timberlake, S.; Vigneault, F.; Gutiérrez, J.M.; Lomonte, B. Exploration of immunoglobulin transcriptomes from mice immunized with three-finger toxins and phospholipases A2 from the Central American coral snake, Micrurus nigrocinctus. PeerJ 2017, 5, e2924. [Google Scholar] [CrossRef] [PubMed]
- Leong, P.K.; Fung, S.Y.; Tan, C.H.; Sim, S.M.; Tan, N.H. Immunological cross-reactivity and neutralization of the principal toxins of Naja sumatrana and related cobra venoms by a Thai polyvalent antivenom (Neuro Polyvalent Snake Antivenom). Acta Trop. 2015, 149, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.H.; Tan, K.Y.; Lim, S.E.; Tan, N.H. Venomics of the beaked sea snake, Hydrophis schistosus: A minimalist toxin arsenal and its cross-neutralization by heterologous antivenoms. J. Proteom. 2015, 126, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.Y.; Tan, C.H.; Fung, S.Y.; Tan, N.H. Neutralization of the Principal Toxins from the Venoms of Thai Naja kaouthia and Malaysian Hydrophis schistosus: Insights into Toxin-Specific Neutralization by Two Different Antivenoms. Toxins 2016, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, E.R.R.; Umbarila, L.R.; Possani, L.D.; Becerril, B. Recombinant Neutralizing Antibodies, A New Generation of Antivenoms. In Scorpion Venoms; Springer: Dordrecht, The Netherlands, 2015; pp. 139–159. [Google Scholar]
- Chippaux, J.-P. Emerging options for the management of scorpion stings. Drug Des. Dev. Ther. 2012, 6, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Bermúdez-Méndez, E.; Fuglsang-Madsen, A.; Føns, S.; Lomonte, B.; Gutiérrez, J.M.; Laustsen, A.H. Innovative Immunization Strategies for Antivenom Development. Toxins 2018, 10, 452. [Google Scholar] [CrossRef] [PubMed]
- Chow, S.-K.; Casadevall, A. Monoclonal Antibodies and Toxins—A Perspective on Function and Isotype. Toxins 2012, 4, 430–454. [Google Scholar] [CrossRef]
- Saylor, C.; Dadachova, E.; Casadevall, A. Monoclonal antibody-based therapies for microbial diseases. Vaccine 2009, 27, G38–G46. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.B.; Cryz, S.J.; Schürch, U.; Ganss, M.T.; Bruderer, U. Immunotherapy with human monoclonal antibodies. Fragment A specificity of polyclonal and monoclonal antibodies is crucial for full protection against tetanus toxin. J. Immunol. 1993, 151, 466–472. [Google Scholar]
- Ho, M.; Silamut, K.; White, N.J.; Karbwang, J.; Looareesuwan, S.; Phillips, R.E.; Warrell, D.A. Pharmacokinetics of three commercial antivenoms in patients envenomed by the Malayan pit viper, Calloselasma rhodostoma, in Thailand. Am. J. Trop. Med. Hyg. 1990, 42, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Ismail, M.; Abd-Elsalam, M.A.; Al-Ahaidib, M.S. Pharmacokinetics of 125I-labelled Walterinnesia aegyptia venom and its distribution of the venom and its toxin versus slow absorption and distribution of IGG, F(AB’)2 and F(AB) of the antivenin. Toxicon 1998, 36, 93–114. [Google Scholar] [CrossRef]
- Keizer, R.J.; Huitema, A.D.R.; Schellens, J.H.M.; Beijnen, J.H. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin. Pharmacokinet. 2010, 49, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, M.; Bonagura, V.R.; Morrison, S.L.; Bjorkman, P.J. Analysis of the pH Dependence of the Neonatal Fc Receptor/Immunoglobulin G Interaction Using Antibody and Receptor Variants. Biochemistry 1995, 34, 14649–14657. [Google Scholar] [CrossRef]
- Tabrizi, M.A.; Tseng, C.-M.L.; Roskos, L.K. Elimination mechanisms of therapeutic monoclonal antibodies. Drug Discov. Today 2006, 11, 81–88. [Google Scholar] [CrossRef]
- Wang, W.; Wang, E.Q.; Balthasar, J.P. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin. Pharmacol. Ther. 2008, 84, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.; Jin, F.; Prabhu, S.; Iyer, S. Monoclonal antibodies: What are the pharmacokinetic and pharmacodynamic considerations for drug development? Expert Opin. Drug Metab. Toxicol. 2012, 8, 141–160. [Google Scholar] [CrossRef]
- Berry, J.D.; Gaudet, R.G. Antibodies in infectious diseases: Polyclonals, monoclonals and niche biotechnology. New Biotechnol. 2011, 28, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Harmsen, M.M.; De Haard, H.J. Properties, production, and applications of camelid single-domain antibody fragments. Appl. Microbiol. Biotechnol. 2007, 77, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.M.; Chen, W.; Raubitschek, A.; Williams, L.E.; Neumaier, M.; Fischer, R.; Hu, S.; Odom-Maryon, T.; Wong, J.Y.C.; Shively, J.E. Tumor localization of anti-CEA single-chain Fvs: Improved targeting by non-covalent dimers. Immunotechnology 1996, 2, 21–36. [Google Scholar] [CrossRef]
- Wu, A.M. Anti-carcinoembryonic antigen (CEA) diabody for rapid tumor targeting and imaging. Tumor Target. 1999, 4, 47–58. [Google Scholar]
- Hansel, T.T.; Kropshofer, H.; Singer, T.; Mitchell, J.A.; George, A.J.T. The safety and side effects of monoclonal antibodies. Nat. Rev. Drug Discov. 2010, 9, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Morine, N.; Matsuda, S.; Terada, K.; Eto, A.; Ishida, I.; Oku, H. Neutralization of hemorrhagic snake venom metalloproteinase HR1a from Protobothrops flavoviridis by human monoclonal antibody. Toxicon 2008, 51, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Krautz-Peterson, G.; Chapman-Bonofiglio, S.; Boisvert, K.; Feng, H.; Herman, I.M.; Tzipori, S.; Sheoran, A.S. Intracellular Neutralization of Shiga Toxin 2 by an A Subunit-Specific Human Monoclonal Antibody. Infect. Immun. 2008, 76, 1931–1939. [Google Scholar] [CrossRef] [PubMed]
- Koon, H.W.; Shih, D.Q.; Hing, T.C.; Yoo, J.H.; Ho, S.; Chen, X.; Kelly, C.P.; Targan, S.R.; Pothoulakis, C. Human monoclonal antibodies against Clostridium difficile toxins A and B inhibit inflammatory and histologic responses to the toxins in human colon and peripheral blood monocytes. Antimicrob. Agents Chemother. 2013, 57, 3214–3223. [Google Scholar] [CrossRef] [PubMed]
- Drozdowski, B.; Zhou, Y.; Kline, B.; Spidel, J.; Chan, Y.Y.; Albone, E.; Turchin, H.; Chao, Q.; Henry, M.; Balogach, J.; et al. Generation and characterization of high affinity human monoclonal antibodies that neutralize staphylococcal enterotoxin B. J. Immune Based Ther. Vaccines 2010, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Slyke, G.V.; Sully, E.K.; Bohorova, N.; Bohorov, O.; Kim, D.; Pauly, M.H.; Whaley, K.J.; Zeitlin, L.; Mantis, N.J. Humanized Monoclonal Antibody That Passively Protects Mice against Systemic and Intranasal Ricin Toxin Challenge. Clin. Vaccine Immunol. 2016, 23, 795–799. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, M.T.; Li, H.; Williamson, E.D.; LeButt, C.S.; Flick-Smith, H.C.; Quinn, C.P.; Westra, H.; Galloway, D.; Mateczun, A.; Goldman, S.; et al. Human Monoclonal Antibodies against Anthrax Lethal Factor and Protective Antigen Act Independently To Protect against Bacillus anthracis Infection and Enhance Endogenous Immunity to Anthrax. Infect. Immun. 2007, 75, 5425–5433. [Google Scholar] [CrossRef]
- Adekar, S.P.; Takahashi, T.; Jones, R.M.; Al-Saleem, F.H.; Ancharski, D.M.; Root, M.J.; Kapadnis, B.P.; Simpson, L.L.; Dessain, S.K. Neutralization of Botulinum Neurotoxin by a Human Monoclonal Antibody Specific for the Catalytic Light Chain. PLoS ONE 2008, 3, e3023. [Google Scholar] [CrossRef]
- Laustsen, A.H.; Johansen, K.H.; Engmark, M.; Andersen, M.R. Recombinant snakebite antivenoms: A cost-competitive solution to a neglected tropical disease? PLoS Negl. Trop. Dis. 2017, 11, e0005361. [Google Scholar] [CrossRef]
- Walsh, G. Biopharmaceutical benchmarks 2014. Nat. Biotechnol. 2014, 32, 992–1000. [Google Scholar] [CrossRef]
- Grilo, A.L.; Mantalaris, A. The Increasingly Human and Profitable Monoclonal Antibody Market. Trends Biotechnol. 2019, 37, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Chadd, H.E.; Chamow, S.M. Therapeutic antibody expression technology. Curr. Opin. Biotechnol. 2001, 12, 188–194. [Google Scholar] [CrossRef]
- Frenzel, A.; Hust, M.; Schirrmann, T. Expression of Recombinant Antibodies. Front. Immunol. 2013, 4, 217. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, S.K.; Settanni, G.; Kenig, M.; Binz, H.K.; Plückthun, A. Folding and unfolding mechanism of highly stable full-consensus ankyrin repeat proteins. J. Mol. Biol. 2008, 376, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Steinwand, M.; Droste, P.; Frenzel, A.; Hust, M.; Dübel, S.; Schirrmann, T. The influence of antibody fragment format on phage display based affinity maturation of IgG. mAbs 2014, 6, 204–218. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Douthwaite, J.A.; Chen, Y.; Kemp, B.; Kidd, S.; Percival-Alwyn, J.; Smith, A.; Goode, K.; Swerdlow, B.; Lowe, D.; et al. A high-throughput platform for population reformatting and mammalian expression of phage display libraries to enable functional screening as full-length IgG. mAbs 2017, 9, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Mazor, Y.; Van Blarcom, T.; Carroll, S.; Georgiou, G. Selection of full-length IgGs by tandem display on filamentous phage particles and Escherichia coli fluorescence-activated cell sorting screening: Selection of IgG by tandem phage panning-FACS. FEBS J. 2010, 277, 2291–2303. [Google Scholar] [CrossRef]
- Feldwisch, J.; Tolmachev, V.; Lendel, C.; Herne, N.; Sjöberg, A.; Larsson, B.; Rosik, D.; Lindqvist, E.; Fant, G.; Höidén-Guthenberg, I.; et al. Design of an Optimized Scaffold for Affibody Molecules. J. Mol. Biol. 2010, 398, 232–247. [Google Scholar] [CrossRef]
- Weinstock, M.T.; Francis, J.N.; Redman, J.S.; Kay, M.S. Protease-resistant peptide design-empowering nature’s fragile warriors against HIV. Biopolymers 2012, 98, 431–442. [Google Scholar] [CrossRef]
- Binz, H.K.; Bakker, T.R.; Phillips, D.J.; Cornelius, A.; Zitt, C.; Göttler, T.; Sigrist, G.; Fiedler, U.; Ekawardhani, S.; Dolado, I.; et al. Design and characterization of MP0250, a tri-specific anti-HGF/anti-VEGF DARPin® drug candidate. mAbs 2017, 9, 1262–1269. [Google Scholar] [CrossRef]
- Zahnd, C.; Wyler, E.; Schwenk, J.M.; Steiner, D.; Lawrence, M.C.; McKern, N.M.; Pecorari, F.; Ward, C.W.; Joos, T.O.; Plückthun, A. A Designed Ankyrin Repeat Protein Evolved to Picomolar Affinity to Her2. J. Mol. Biol. 2007, 369, 1015–1028. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Krippendorff, B.-F.; Sharma, S.; Walz, A.C.; Lavé, T.; Shah, D.K. Influence of molecular size on tissue distribution of antibody fragments. mAbs 2016, 8, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Hamano, Y.; Grunkemeyer, J.A.; Sudhakar, A.; Zeisberg, M.; Cosgrove, D.; Morello, R.; Lee, B.; Sugimoto, H.; Kalluri, R. Determinants of Vascular Permeability in the Kidney Glomerulus. J. Biol. Chem. 2002, 277, 31154–31162. [Google Scholar] [CrossRef] [PubMed]
- Holt, L.J.; Herring, C.; Jespers, L.S.; Woolven, B.P.; Tomlinson, I.M. Domain antibodies: Proteins for therapy. Trends Biotechnol. 2003, 21, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, T.; Chao, G.; Sitkoff, D.; Lo, F.; Monshizadegan, H.; Meyers, D.; Low, S.; Russo, K.; DiBella, R.; Denhez, F.; et al. Pharmacologic profile of the Adnectin BMS-962476, a small protein biologic alternative to PCSK9 antibodies for low-density lipoprotein lowering. J. Pharmacol. Exp. Ther. 2014, 350, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Gille, H.; Hülsmeyer, M.; Trentmann, S.; Matschiner, G.; Christian, H.J.; Meyer, T.; Amirkhosravi, A.; Audoly, L.P.; Hohlbaum, A.M.; Skerra, A. Functional characterization of a VEGF-A-targeting Anticalin, prototype of a novel therapeutic human protein class. Angiogenesis 2016, 19, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Weidle, U.H.; Auer, J.; Brinkmann, U.; Georges, G.; Tiefenthaler, G. The Emerging Role of New Protein Scaffold-based Agents for Treatment of Cancer. Cancer Genom. Proteom. 2013, 10, 155–168. [Google Scholar]
- Plückthun, A. Designed ankyrin repeat proteins (DARPins): Binding proteins for research, diagnostics, and therapy. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 489–511. [Google Scholar] [CrossRef]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef]
- Lauwereys, M. Potent enzyme inhibitors derived from dromedary heavy-chain antibodies. EMBO J. 1998, 17, 3512–3520. [Google Scholar] [CrossRef]
- Pravda, L.; Berka, K.; Svobodová Vařeková, R.; Sehnal, D.; Banáš, P.; Laskowski, R.A.; Koča, J.; Otyepka, M. Anatomy of enzyme channels. BMC Bioinform. 2014, 15, 379. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Cai, J.; Xu, Q.; Chen, Z.W. Atomic force bio-analytics of polymerization and aggregation of phycoerythrin-conjugated immunoglobulin G molecules. Mol. Immunol. 2004, 41, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Lewin, M.; Samuel, S.; Merkel, J.; Bickler, P. Varespladib (LY315920) Appears to Be a Potent, Broad-Spectrum, Inhibitor of Snake Venom Phospholipase A2 and a Possible Pre-Referral Treatment for Envenomation. Toxins 2016, 8, 248. [Google Scholar] [CrossRef] [PubMed]
- Schilling, J.; Schöppe, J.; Plückthun, A. From DARPins to LoopDARPins: Novel LoopDARPin Design Allows the Selection of Low Picomolar Binders in a Single Round of Ribosome Display. J. Mol. Biol. 2014, 426, 691–721. [Google Scholar] [CrossRef] [PubMed]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hammers, C.; Songa, E.B.; Bendahman, N.; Hammers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Els Conrath, K.; Lauwereys, M.; Wyns, L.; Muyldermans, S. Camel single-domain antibodies as modular building units in bispecific and bivalent antibody constructs. J. Biol. Chem. 2001, 276, 7346–7350. [Google Scholar] [CrossRef] [PubMed]
- Kolkman, J.A.; Law, D.A. Nanobodies—From llamas to therapeutic proteins. Drug Discov. Today Technol. 2010, 7, e139–e146. [Google Scholar] [CrossRef] [PubMed]
- Arbabi-Ghahroudi, M. Camelid Single-Domain Antibodies: Historical Perspective and Future Outlook. Front. Immunol. 2017, 8, 1589. [Google Scholar] [CrossRef]
- Muyldermans, S. Nanobodies: Natural Single-Domain Antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef]
- Steeland, S.; Vandenbroucke, R.E.; Libert, C. Nanobodies as therapeutics: Big opportunities for small antibodies. Drug Discov. Today 2016, 21, 1076–1113. [Google Scholar] [CrossRef]
- Stanfield, R.L.; Dooley, H.; Flajnik, M.F.; Wilson, I.A. Crystal structure of a shark single-domain antibody V region in complex with lysozyme. Science 2004, 305, 1770–1773. [Google Scholar] [CrossRef] [PubMed]
- Dolk, E.; van der Vaart, M.; Lutje Hulsik, D.; Vriend, G.; de Haard, H.; Spinelli, S.; Cambillau, C.; Frenken, L.; Verrips, T. Isolation of Llama Antibody Fragments for Prevention of Dandruff by Phage Display in Shampoo. Appl. Environ. Microbiol. 2005, 71, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Omidfar, K.; Rasaee, M.J.; Kashanian, S.; Paknejad, M.; Bathaie, Z. Studies of thermostability in Camelus bactrianus (Bactrian camel) single-domain antibody specific for the mutant epidermal-growth-factor receptor expressed by Pichia. Biotechnol. Appl. Biochem. 2007, 46, 41–49. [Google Scholar] [PubMed]
- Conrath, K.; Vincke, C.; Stijlemans, B.; Schymkowitz, J.; Decanniere, K.; Wyns, L.; Muyldermans, S.; Loris, R. Antigen Binding and Solubility Effects upon the Veneering of a Camel VHH in Framework-2 to Mimic a VH. J. Mol. Biol. 2005, 350, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Van Audenhove, I.; Gettemans, J. Nanobodies as Versatile Tools to Understand, Diagnose, Visualize and Treat Cancer. EBioMedicine 2016, 8, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Staus, D.P.; Wingler, L.M.; Strachan, R.T.; Rasmussen, S.G.F.; Pardon, E.; Ahn, S.; Steyaert, J.; Kobilka, B.K.; Lefkowitz, R.J. Regulation of β2-adrenergic receptor function by conformationally selective single-domain intrabodies. Mol. Pharmacol. 2014, 85, 472–481. [Google Scholar] [CrossRef]
- Manglik, A.; Kobilka, B.K.; Steyaert, J. Nanobodies to Study G Protein-Coupled Receptor Structure and Function. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 19–37. [Google Scholar] [CrossRef]
- De Meyer, T.; Muyldermans, S.; Depicker, A. Nanobody-based products as research and diagnostic tools. Trends Biotechnol. 2014, 32, 263–270. [Google Scholar] [CrossRef]
- Hwang, C.W.; Flach, F.E. Recurrent Coagulopathy after Rattlesnake Bite Requiring Continuous Intravenous Dosing of Antivenom. Case Rep. Emerg. Med. 2015, 2015, 719302. [Google Scholar] [CrossRef]
- Anderson, G.P.; Liu, J.H.; Zabetakis, D.; Liu, J.L.; Goldman, E.R. Thermal stabilization of anti-α-cobratoxin single domain antibodies. Toxicon 2017, 129, 68–73. [Google Scholar] [CrossRef]
- Richard, G.; Meyers, A.J.; McLean, M.D.; Arbabi-Ghahroudi, M.; MacKenzie, R.; Hall, J.C. In Vivo Neutralization of α-Cobratoxin with High-Affinity Llama Single-Domain Antibodies (VHHs) and a VHH-Fc Antibody. PLoS ONE 2013, 8, e69495. [Google Scholar] [CrossRef] [PubMed]
- Hmila, I.; Cosyns, B.; Tounsi, H.; Roosens, B.; Caveliers, V.; Abderrazek, R.B.; Boubaker, S.; Muyldermans, S.; El Ayeb, M.; Bouhaouala-Zahar, B.; et al. Pre-clinical studies of toxin-specific Nanobodies: Evidence of in vivo efficacy to prevent fatal disturbances provoked by scorpion envenoming. Toxicol. Appl. Pharmacol. 2012, 264, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Hmila, I.; Saerens, D.; Abderrazek, R.B.; Vincke, C.; Abidi, N.; Benlasfar, Z.; Govaert, J.; Ayeb, M.E.; Bouhaouala-Zahar, B.; Muyldermans, S. A bispecific nanobody to provide full protection against lethal scorpion envenoming. FASEB J. 2010, 24, 3479–3489. [Google Scholar] [CrossRef] [PubMed]
- Hmila, I.; Abdallah R, B.A.; Saerens, D.; Benlasfar, Z.; Conrath, K.; Ayeb, M.E.; Muyldermans, S.; Bouhaouala-Zahar, B. VHH, bivalent domains and chimeric Heavy chain-only antibodies with high neutralizing efficacy for scorpion toxin AahI′. Mol. Immunol. 2008, 45, 3847–3856. [Google Scholar] [CrossRef]
- Tiede, C.; Tang, A.A.S.; Deacon, S.E.; Mandal, U.; Nettleship, J.E.; Owen, R.L.; George, S.E.; Harrison, D.J.; Owens, R.J.; Tomlinson, D.C.; et al. Adhiron: A stable and versatile peptide display scaffold for molecular recognition applications. Protein Eng. Des. Sel. 2014, 27, 145–155. [Google Scholar] [CrossRef]
- Stadler, L.K.J.; Hoffmann, T.; Tomlinson, D.C.; Song, Q.; Lee, T.; Busby, M.; Nyathi, Y.; Gendra, E.; Tiede, C.; Flanagan, K.; et al. Structure-function studies of an engineered scaffold protein derived from Stefin A. II: Development and applications of the SQT variant. Protein Eng. Des. Sel. 2011, 24, 751–763. [Google Scholar] [CrossRef] [PubMed]
- Tiede, C.; Bedford, R.; Heseltine, S.J.; Smith, G.; Wijetunga, I.; Ross, R.; AlQallaf, D.; Roberts, A.P.; Balls, A.; Curd, A.; et al. Affimer proteins are versatile and renewable affinity reagents. eLife 2017, 6, e24903. [Google Scholar] [CrossRef] [PubMed]
- Škrlec, K.; Štrukelj, B.; Berlec, A. Non-immunoglobulin scaffolds: A focus on their targets. Trends Biotechnol. 2015, 33, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Koutsoumpeli, E.; Tiede, C.; Murray, J.; Tang, A.; Bon, R.S.; Tomlinson, D.C.; Johnson, S. Antibody Mimetics for the Detection of Small Organic Compounds Using a Quartz Crystal Microbalance. Anal. Chem. 2017, 89, 3051–3058. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.I.; Baxter, E.W.; Owen, R.L.; Thomsen, M.; Tomlinson, D.C.; Waterhouse, M.P.; Win, S.J.; Nettleship, J.E.; Tiede, C.; Foster, R.J.; et al. Affimer proteins inhibit immune complex binding to FcγRIIIa with high specificity through competitive and allosteric modes of action. Proc. Natl. Acad. Sci. USA 2018, 115, E72–E81. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.J.; Tiede, C.; Penswick, N.; Tang, A.A.-S.; Trinh, C.H.; Mandal, U.; Zajac, K.Z.; Gaule, T.; Howell, G.; Edwards, T.A.; et al. Generation of specific inhibitors of SUMO-1- and SUMO-2/3-mediated protein-protein interactions using Affimer (Adhiron) technology. Sci. Signal. 2017, 10, eaaj2005. [Google Scholar] [CrossRef] [PubMed]
- Carter, P.J. Introduction to current and future protein therapeutics: A protein engineering perspective. Exp. Cell Res. 2011, 317, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Lipovšek, D. Adnectins: Engineered target-binding protein therapeutics. Protein Eng. Des. Sel. 2011, 24, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Ramamurthy, V.; Krystek, S.R.; Bush, A.; Wei, A.; Emanuel, S.L.; Das Gupta, R.; Janjua, A.; Cheng, L.; Murdock, M.; Abramczyk, B.; et al. Structures of adnectin/protein complexes reveal an expanded binding footprint. Structure 2012, 20, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Koide, A.; Bailey, C.W.; Huang, X.; Koide, S. The fibronectin type III domain as a scaffold for novel binding proteins. J. Mol. Biol. 1998, 284, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Aha, P.; Gu, K.; Kuimelis, R.G.; Kurz, M.; Lam, T.; Lim, A.C.; Liu, H.; Lohse, P.A.; Sun, L.; et al. Directed evolution of high-affinity antibody mimics using mRNA display. Chem. Biol. 2002, 9, 933–942. [Google Scholar] [CrossRef]
- Parker, M.H.; Chen, Y.; Danehy, F.; Dufu, K.; Ekstrom, J.; Getmanova, E.; Gokemeijer, J.; Xu, L.; Lipovsek, D. Antibody mimics based on human fibronectin type three domain engineered for thermostability and high-affinity binding to vascular endothelial growth factor receptor two. Protein Eng. Des. Sel. 2005, 18, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Plaxco, K.W.; Spitzfaden, C.; Campbell, I.D.; Dobson, C.M. A comparison of the folding kinetics and thermodynamics of two homologous fibronectin type III modules. J. Mol. Biol. 1997, 270, 763–770. [Google Scholar] [CrossRef]
- Cota, E.; Hamill, S.J.; Fowler, S.B.; Clarke, J. Two proteins with the same structure respond very differently to mutation: The role of plasticity in protein stability. J. Mol. Biol. 2000, 302, 713–725. [Google Scholar] [CrossRef]
- Batori, V.; Koide, A.; Koide, S. Exploring the potential of the monobody scaffold: Effects of loop elongation on the stability of a fibronectin type III domain. Protein Eng. Des. Sel. 2002, 15, 1015–1020. [Google Scholar] [CrossRef]
- Koide, A.; Abbatiello, S.; Rothgery, L.; Koide, S. Probing protein conformational changes in living cells by using designer binding proteins: Application to the estrogen receptor. Proc. Natl. Acad. Sci. USA 2002, 99, 1253–1258. [Google Scholar] [CrossRef] [PubMed]
- Bloom, L.; Calabro, V. FN3: A new protein scaffold reaches the clinic. Drug Discov. Today 2009, 14, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Hackel, B.J.; Wittrup, K.D. The full amino acid repertoire is superior to serine/tyrosine for selection of high affinity immunoglobulin G binders from the fibronectin scaffold. Protein Eng. Des. Sel. 2010, 23, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Hackel, B.J.; Kapila, A.; Wittrup, K.D. Picomolar affinity fibronectin domains engineered utilizing loop length diversity, recursive mutagenesis, and loop shuffling. J. Mol. Biol. 2008, 381, 1238–1252. [Google Scholar] [CrossRef] [PubMed]
- Hackel, B.J.; Ackerman, M.E.; Howland, S.W.; Wittrup, K.D. Stability and CDR composition biases enrich binder functionality landscapes. J. Mol. Biol. 2010, 401, 84–96. [Google Scholar] [CrossRef]
- Nord, K.; Gunneriusson, E.; Ringdahl, J.; Ståhl, S.; Uhlén, M.; Nygren, P.-Å. Binding proteins selected from combinatorial libraries of an α-helical bacterial receptor domain. Nat. Biotechnol. 1997, 15, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Wahlberg, E.; Lendel, C.; Helgstrand, M.; Allard, P.; Dincbas-Renqvist, V.; Hedqvist, A.; Berglund, H.; Nygren, P.-Å.; Härd, T. An affibody in complex with a target protein: Structure and coupled folding. Proc. Natl. Acad. Sci. USA 2003, 100, 3185–3190. [Google Scholar] [CrossRef]
- Nygren Per-Åke Alternative binding proteins: Affibody binding proteins developed from a small three-helix bundle scaffold. FEBS J. 2008, 275, 2668–2676. [CrossRef] [PubMed]
- Gunneriusson, E.; Nord, K.; Uhlén, M.; Nygren, P.-Å. Affinity maturation of a Taq DNA polymerase specific affibody by helix shuffling. Protein Eng. Des. Sel. 1999, 12, 873–878. [Google Scholar] [CrossRef]
- Westerlund, K.; Altai, M.; Mitran, B.; Konijnenberg, M.; Oroujeni, M.; Atterby, C.; de Jong, M.; Orlova, A.; Mattsson, J.; Micke, P.; et al. Radionuclide Therapy of HER2-Expressing Human Xenografts Using Affibody-Based Peptide Nucleic Acid-Mediated Pretargeting: In Vivo Proof of Principle. J. Nucl. Med. 2018, 59, 1092–1098. [Google Scholar] [CrossRef]
- Murzin, A.G. OB(oligonucleotide/oligosaccharide binding)-fold: Common structural and functional solution for non-homologous sequences. EMBO J. 1993, 12, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Mouratou, B.; Schaeffer, F.; Guilvout, I.; Tello-Manigne, D.; Pugsley, A.P.; Alzari, P.M.; Pecorari, F. Remodeling a DNA-binding protein as a specific in vivo inhibitor of bacterial secretin PulD. Proc. Natl. Acad. Sci. USA 2007, 104, 17983–17988. [Google Scholar] [CrossRef]
- Correa, A.; Pacheco, S.; Mechaly, A.E.; Obal, G.; Béhar, G.; Mouratou, B.; Oppezzo, P.; Alzari, P.M.; Pecorari, F. Potent and Specific Inhibition of Glycosidases by Small Artificial Binding Proteins (Affitins). PLoS ONE 2014, 9, e97438. [Google Scholar] [CrossRef] [PubMed]
- Béhar, G.; Bellinzoni, M.; Maillasson, M.; Paillard-Laurance, L.; Alzari, P.M.; He, X.; Mouratou, B.; Pecorari, F. Tolerance of the archaeal Sac7d scaffold protein to alternative library designs: Characterization of anti-immunoglobulin G Affitins. Protein Eng. Des. Sel. 2013, 26, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Béhar, G.; Renodon-Cornière, A.; Mouratou, B.; Pecorari, F. Affitins as robust tailored reagents for affinity chromatography purification of antibodies and non-immunoglobulin proteins. J. Chromatogr. A 2016, 1441, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Kalichuk, V.; Renodon-Cornière, A.; Béhar, G.; Carrión, F.; Obal, G.; Maillasson, M.; Mouratou, B.; Préat, V.; Pecorari, F. A novel, smaller scaffold for Affitins: Showcase with binders specific for EpCAM. Biotechnol. Bioeng. 2018, 115, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Beste, G.; Schmidt, F.S.; Stibora, T.; Skerra, A. Small antibody-like proteins with prescribed ligand specificities derived from the lipocalin fold. Proc. Natl. Acad. Sci. USA 1999, 96, 1898–1903. [Google Scholar] [CrossRef] [PubMed]
- Schiefner, A.; Skerra, A. The Menagerie of Human Lipocalins: A Natural Protein Scaffold for Molecular Recognition of Physiological Compounds. Acc. Chem. Res. 2015, 48, 976–985. [Google Scholar] [CrossRef] [PubMed]
- Skerra, A. Alternative binding proteins: Anticalins—Harnessing the structural plasticity of the lipocalin ligand pocket to engineer novel binding activities. FEBS J. 2008, 275, 2677–2683. [Google Scholar] [CrossRef] [PubMed]
- Pervaiz, S.; Brew, K. Homology of β-Lactoglobulin, Serum Retinol-Binding Protein, and Protein HC. Science 1985, 228, 335–337. [Google Scholar] [CrossRef]
- Rothe, C.; Skerra, A. Anticalin® Proteins as Therapeutic Agents in Human Diseases. Biodrugs 2018, 32, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Eggenstein, E.; Eichinger, A.; Kim, H.-J.; Skerra, A. Structure-guided engineering of Anticalins with improved binding behavior and biochemical characteristics for application in radio-immuno imaging and/or therapy. J. Struct. Biol. 2014, 185, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, M.; Skerra, A. Chapter seven—Anticalins: Small Engineered Binding Proteins Based on the Lipocalin Scaffold. In Methods in Enzymology; Wittrup, K.D., Verdine, G.L., Eds.; Academic Press: New York, NY, USA, 2012; Volume 503, pp. 157–188. [Google Scholar]
- Eyer, F.; Steimer, W.; Nitzsche, T.; Jung, N.; Neuberger, H.; Müller, C.; Schlapschy, M.; Zilker, T.; Skerra, A. Intravenous application of an anticalin dramatically lowers plasma digoxin levels and reduces its toxic effects in rats. Toxicol. Appl. Pharmacol. 2012, 263, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Nüsslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Riggleman, B.; Schedl, P.; Wieschaus, E. Spatial expression of the Drosophila segment polarity gene armadillo is posttranscriptionally regulated by wingless. Cell 1990, 63, 549–560. [Google Scholar] [CrossRef]
- Tewari, R.; Bailes, E.; Bunting, K.A.; Coates, J.C. Armadillo-repeat protein functions: Questions for little creatures. Trends Cell Biol. 2010, 20, 470–481. [Google Scholar] [CrossRef]
- Reichen, C.; Hansen, S.; Plückthun, A. Modular peptide binding: From a comparison of natural binders to designed armadillo repeat proteins. J. Struct. Biol. 2014, 185, 147–162. [Google Scholar] [CrossRef]
- Madhurantakam, C.; Varadamsetty, G.; Grütter, M.G.; Plückthun, A.; Mittl, P.R.E. Structure-based optimization of designed Armadillo-repeat proteins. Protein Sci. 2012, 21, 1015–1028. [Google Scholar] [CrossRef]
- Conti, E.; Uy, M.; Leighton, L.; Blobel, G.; Kuriyan, J. Crystallographic Analysis of the Recognition of a Nuclear Localization Signal by the Nuclear Import Factor Karyopherin α. Cell 1998, 94, 193–204. [Google Scholar] [CrossRef]
- Andrade, M.A.; Perez-Iratxeta, C.; Ponting, C.P. Protein Repeats: Structures, Functions, and Evolution. J. Struct. Biol. 2001, 134, 117–131. [Google Scholar] [CrossRef]
- Varadamsetty, G.; Tremmel, D.; Hansen, S.; Parmeggiani, F.; Plückthun, A. Designed Armadillo repeat proteins: Library generation, characterization and selection of peptide binders with high specificity. J. Mol. Biol. 2012, 424, 68–87. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.; Tremmel, D.; Madhurantakam, C.; Reichen, C.; Mittl, P.R.E.; Plückthun, A. Structure and Energetic Contributions of a Designed Modular Peptide-Binding Protein with Picomolar Affinity. J. Am. Chem. Soc. 2016, 138, 3526–3532. [Google Scholar] [CrossRef] [PubMed]
- Alfarano, P.; Varadamsetty, G.; Ewald, C.; Parmeggiani, F.; Pellarin, R.; Zerbe, O.; Plückthun, A.; Caflisch, A. Optimization of designed armadillo repeat proteins by molecular dynamics simulations and NMR spectroscopy. Protein Sci. 2012, 21, 1298–1314. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.; Ernst, P.; König, S.L.B.; Reichen, C.; Ewald, C.; Nettels, D.; Mittl, P.R.E.; Schuler, B.; Plückthun, A. Curvature of designed armadillo repeat proteins allows modular peptide binding. J. Struct. Biol. 2018, 201, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, N.; Vlachakis, D.; Memou, A.; Leandrou, E.; Valkimadi, P.-E.; Melachroinou, K.; Re, D.B.; Przedborski, S.; Dauer, W.T.; Stefanis, L.; et al. A motif within the armadillo repeat of Parkinson’s-linked LRRK2 interacts with FADD to hijack the extrinsic death pathway. Sci. Rep. 2018, 8, 3455. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Song, H.; Mei, H.; Fang, E.; Wang, X.; Yang, F.; Li, H.; Chen, Y.; Huang, K.; Zheng, L.; et al. Armadillo repeat containing 12 promotes neuroblastoma progression through interaction with retinoblastoma binding protein 4. Nat. Commun. 2018, 9, 2829. [Google Scholar] [CrossRef]
- Candida Barisson Villares Fragoso, M.; Pontes Cavalcante, I.; Meneses Ferreira, A.; Marinho de Paula Mariani, B.; Ferini Pacicco Lotfi, C. Genetics of primary macronodular adrenal hyperplasia. Presse Méd. 2018, 47, e139–e149. [Google Scholar] [CrossRef]
- Silverman, J.; Lu, Q.; Bakker, A.; To, W.; Duguay, A.; Alba, B.M.; Smith, R.; Rivas, A.; Li, P.; Le, H.; et al. Multivalent avimer proteins evolved by exon shuffling of a family of human receptor domains. Nat. Biotechnol. 2005, 23, 1556–1561. [Google Scholar] [CrossRef]
- North, C.L.; Blacklow, S.C. Structural Independence of Ligand-Binding Modules Five and Six of the LDL Receptor. Biochemistry 1999, 38, 3926–3935. [Google Scholar] [CrossRef]
- Hulme, J.T.; D’Souza, W.N.; McBride, H.J.; Yoon, B.-R.P.; Willee, A.M.; Duguay, A.; Thomas, M.; Fan, B.; Dayao, M.R.; Rottman, J.B.; et al. Novel protein therapeutic joint retention strategy based on collagen-binding Avimers. J. Orthop. Res. 2018, 36, 1238–1247. [Google Scholar] [CrossRef]
- Fasan, R.; Dias, R.L.A.; Moehle, K.; Zerbe, O.; Vrijbloed, J.W.; Obrecht, D.; Robinson, J.A. Using a β-Hairpin To Mimic an α-Helix: Cyclic Peptidomimetic Inhibitors of the p53–HDM2 Protein–Protein Interaction. Angew. Chem. Int. Ed. 2004, 43, 2109–2112. [Google Scholar] [CrossRef] [PubMed]
- Fasan, R.; Dias, R.L.A.; Moehle, K.; Zerbe, O.; Obrecht, D.; Mittl, P.R.E.; Grütter, M.G.; Robinson, J.A. Structure–Activity Studies in a Family of β-Hairpin Protein Epitope Mimetic Inhibitors of the p53–HDM2 Protein–Protein Interaction. ChemBioChem 2006, 7, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Favre, M.; Moehle, K.; Jiang, L.; Pfeiffer, B.; Robinson, J.A. Structural Mimicry of Canonical Conformations in Antibody Hypervariable Loops Using Cyclic Peptides Containing a Heterochiral Diproline Template. J. Am. Chem. Soc. 1999, 121, 2679–2685. [Google Scholar] [CrossRef]
- Karpova, D.; Dauber, K.; Spohn, G.; Chudziak, D.; Wiercinska, E.; Schulz, M.; Pettit, A.R.; Levesque, J.P.; Romagnoli, B.; Patel, K.; et al. The novel CXCR4 antagonist POL5551 mobilizes hematopoietic stem and progenitor cells with greater efficiency than Plerixafor. Leukemia 2013, 27, 2322–2331. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Moehle, K.; Dhanapal, B.; Obrecht, D.; Robinson, J.A. Combinatorial Biomimetic Chemistry: Parallel Synthesis of a Small Library ofβ-Hairpin Mimetics Based on Loop III from Human Platelet-Derived Growth Factor B. Helv. Chim. Acta 2000, 83, 3097–3112. [Google Scholar] [CrossRef]
- Rhodes, C.A.; Pei, D. Bicyclic Peptides as Next-Generation Therapeutics. Chem. Eur. J. 2017, 23, 12690–12703. [Google Scholar] [CrossRef]
- Heinis, C.; Rutherford, T.; Freund, S.; Winter, G. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat. Chem. Biol. 2009, 5, 502–507. [Google Scholar] [CrossRef]
- Baeriswyl, V.; Heinis, C. Polycyclic Peptide Therapeutics. ChemMedChem 2013, 8, 377–384. [Google Scholar] [CrossRef]
- Angelini, A.; Cendron, L.; Chen, S.; Touati, J.; Winter, G.; Zanotti, G.; Heinis, C. Bicyclic Peptide Inhibitor Reveals Large Contact Interface with a Protease Target. ACS Chem. Biol. 2012, 7, 817–821. [Google Scholar] [CrossRef]
- Hacker, D.E.; Hoinka, J.; Iqbal, E.S.; Przytycka, T.M.; Hartman, M.C.T. Highly-constrained bicyclic scaffolds for the discovery of protease-stable peptides via mRNA display. ACS Chem. Biol. 2017, 12, 795–804. [Google Scholar] [CrossRef]
- Bennett, G.; Harrison, H.; Campbell, S.; Teufel, D.; Langford, G.; Watt, A.; Bonny, C. Development of BT1718, a Bicycle Drug Conjugate® (BDC) targeting MT1-MMP for treatment of solid tumours. Eur. J. Cancer 2016, 69, S21. [Google Scholar] [CrossRef]
- Bork, P. Hundreds of ankyrin-like repeats in functionally diverse proteins: Mobile modules that cross phyla horizontally? Proteins 1993, 17, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Binz, H.K.; Stumpp, M.T.; Forrer, P.; Amstutz, P.; Plückthun, A. Designing Repeat Proteins: Well-expressed, Soluble and Stable Proteins from Combinatorial Libraries of Consensus Ankyrin Repeat Proteins. J. Mol. Biol. 2003, 332, 489–503. [Google Scholar] [CrossRef]
- Stumpp, M.T.; Binz, H.K.; Amstutz, P. DARPins: A new generation of protein therapeutics. Drug Discov. Today 2008, 13, 695–701. [Google Scholar] [CrossRef]
- Zahnd, C.; Kawe, M.; Stumpp, M.T.; de Pasquale, C.; Tamaskovic, R.; Nagy-Davidescu, G.; Dreier, B.; Schibli, R.; Binz, H.K.; Waibel, R.; et al. Efficient Tumor Targeting with High-Affinity Designed Ankyrin Repeat Proteins: Effects of Affinity and Molecular Size. Cancer Res. 2010, 70, 1595–1605. [Google Scholar] [CrossRef] [PubMed]
- Münch, R.C.; Janicki, H.; Völker, I.; Rasbach, A.; Hallek, M.; Büning, H.; Buchholz, C.J. Displaying High-affinity Ligands on Adeno-associated Viral Vectors Enables Tumor Cell-specific and Safe Gene Transfer. Mol. Ther. 2013, 21, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Souied, E.H.; Devin, F.; Mauget-Faÿsse, M.; Kolář, P.; Wolf-Schnurrbusch, U.; Framme, C.; Gaucher, D.; Querques, G.; Stumpp, M.T.; Wolf, S.; et al. Treatment of exudative age-related macular degeneration with a designed ankyrin repeat protein that binds vascular endothelial growth factor: A phase I/II study. Am. J. Ophthalmol. 2014, 158, 724–732.e2. [Google Scholar] [CrossRef]
- Rodon, J.; Omlin, A.; Herbschleb, K.H.; Garcia-Corbacho, J.; Steiner, J.; Dolado, I.; Zitt, C.; Feurstein, D.; Turner, D.; Dawson, K.M.; et al. Abstract B25: First-in-human Phase I study to evaluate MP0250, a DARPin blocking HGF and VEGF, in patients with advanced solid tumors. Mol. Cancer Ther. 2015, 14, B25. [Google Scholar] [CrossRef]
- Stahl, A.; Stumpp, M.T.; Schlegel, A.; Ekawardhani, S.; Lehrling, C.; Martin, G.; Gulotti-Georgieva, M.; Villemagne, D.; Forrer, P.; Agostini, H.T.; et al. Highly potent VEGF-A-antagonistic DARPins as anti-angiogenic agents for topical and intravitreal applications. Angiogenesis 2013, 16, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.P.; Perlmutter, R.M. Expression of a novel form of the fyn proto-oncogene in hematopoietic cells. New Biol. 1989, 1, 66–74. [Google Scholar] [PubMed]
- Grabulovski, D.; Kaspar, M.; Neri, D. A Novel, Non-immunogenic Fyn SH3-derived Binding Protein with Tumor Vascular Targeting Properties. J. Biol. Chem. 2007, 282, 3196–3204. [Google Scholar] [CrossRef] [PubMed]
- Silacci, M.; Baenziger-Tobler, N.; Lembke, W.; Zha, W.; Batey, S.; Bertschinger, J.; Grabulovski, D. Linker Length Matters, Fynomer-Fc Fusion with an Optimized Linker Displaying Picomolar IL-17A Inhibition Potency. J. Biol. Chem. 2014, 289, 14392–14398. [Google Scholar] [CrossRef] [PubMed]
- Filimonov, V.V.; Azuaga, A.I.; Viguera, A.R.; Serrano, L.; Mateo, P.L. A thermodynamic analysis of a family of small globular proteins: SH3 domains. Biophys. Chem. 1999, 77, 195–208. [Google Scholar] [CrossRef]
- Bertschinger, J.; Grabulovski, D.; Neri, D. Selection of single domain binding proteins by covalent DNA display. Protein Eng. Des. Sel. 2007, 20, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Simeon, R.; Chen, Z. In vitro-engineered non-antibody protein therapeutics. Protein Cell 2018, 9, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Wuellner, U.; Klupsch, K.; Buller, F.; Attinger-Toller, I.; Santimaria, R.; Zbinden, I.; Henne, P.; Grabulovski, D.; Bertschinger, J.; Brack, S. Bispecific CD3/HER2 Targeting FynomAb Induces Redirected T Cell-Mediated Cytolysis with High Potency and Enhanced Tumor Selectivity. Antibodies 2015, 4, 426–440. [Google Scholar] [CrossRef]
- Silacci, M.; Lembke, W.; Woods, R.; Attinger-Toller, I.; Baenziger-Tobler, N.; Batey, S.; Santimaria, R.; von der Bey, U.; Koenig-Friedrich, S.; Zha, W.; et al. Discovery and characterization of COVA322, a clinical-stage bispecific TNF/IL-17A inhibitor for the treatment of inflammatory diseases. MAbs 2016, 8, 141–149. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jenkins, T.P.; Fryer, T.; Dehli, R.I.; Jürgensen, J.A.; Fuglsang-Madsen, A.; Føns, S.; Laustsen, A.H. Toxin Neutralization Using Alternative Binding Proteins. Toxins 2019, 11, 53. https://doi.org/10.3390/toxins11010053
Jenkins TP, Fryer T, Dehli RI, Jürgensen JA, Fuglsang-Madsen A, Føns S, Laustsen AH. Toxin Neutralization Using Alternative Binding Proteins. Toxins. 2019; 11(1):53. https://doi.org/10.3390/toxins11010053
Chicago/Turabian StyleJenkins, Timothy Patrick, Thomas Fryer, Rasmus Ibsen Dehli, Jonas Arnold Jürgensen, Albert Fuglsang-Madsen, Sofie Føns, and Andreas Hougaard Laustsen. 2019. "Toxin Neutralization Using Alternative Binding Proteins" Toxins 11, no. 1: 53. https://doi.org/10.3390/toxins11010053
APA StyleJenkins, T. P., Fryer, T., Dehli, R. I., Jürgensen, J. A., Fuglsang-Madsen, A., Føns, S., & Laustsen, A. H. (2019). Toxin Neutralization Using Alternative Binding Proteins. Toxins, 11(1), 53. https://doi.org/10.3390/toxins11010053