Gut Microbiota Profiling of Aflatoxin B1-Induced Rats Treated with Lactobacillus casei Shirota


Abstract
1. Introduction
2. Results and Discussion
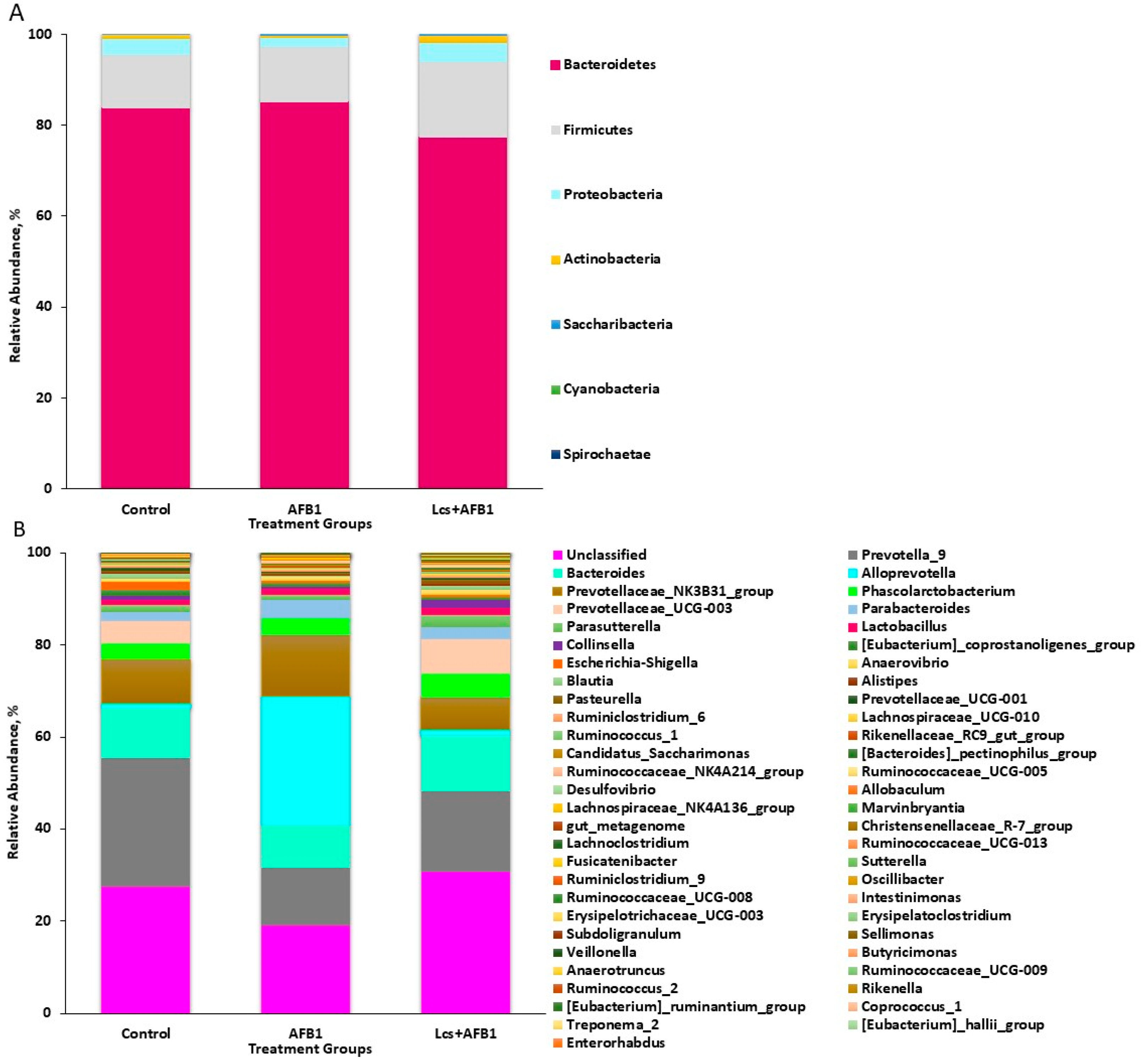
2.1. Sequencing and Bacterial Abundance
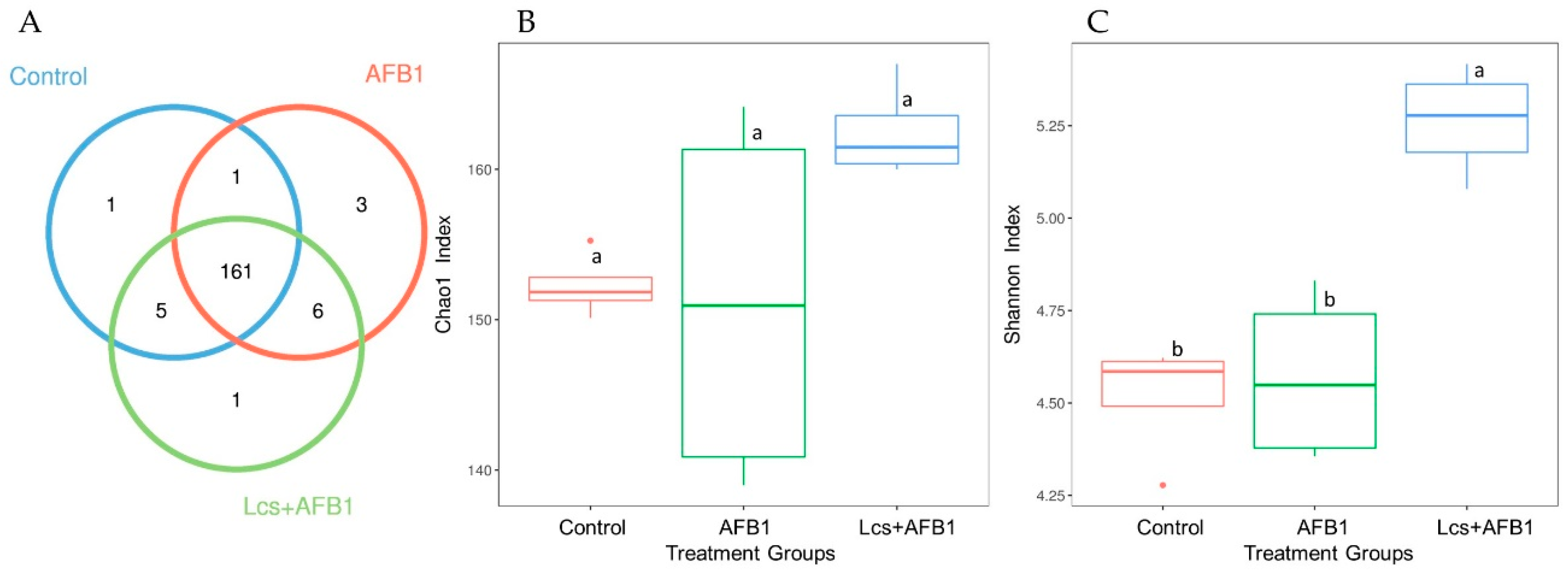
2.2. Alpha and Beta Diversity
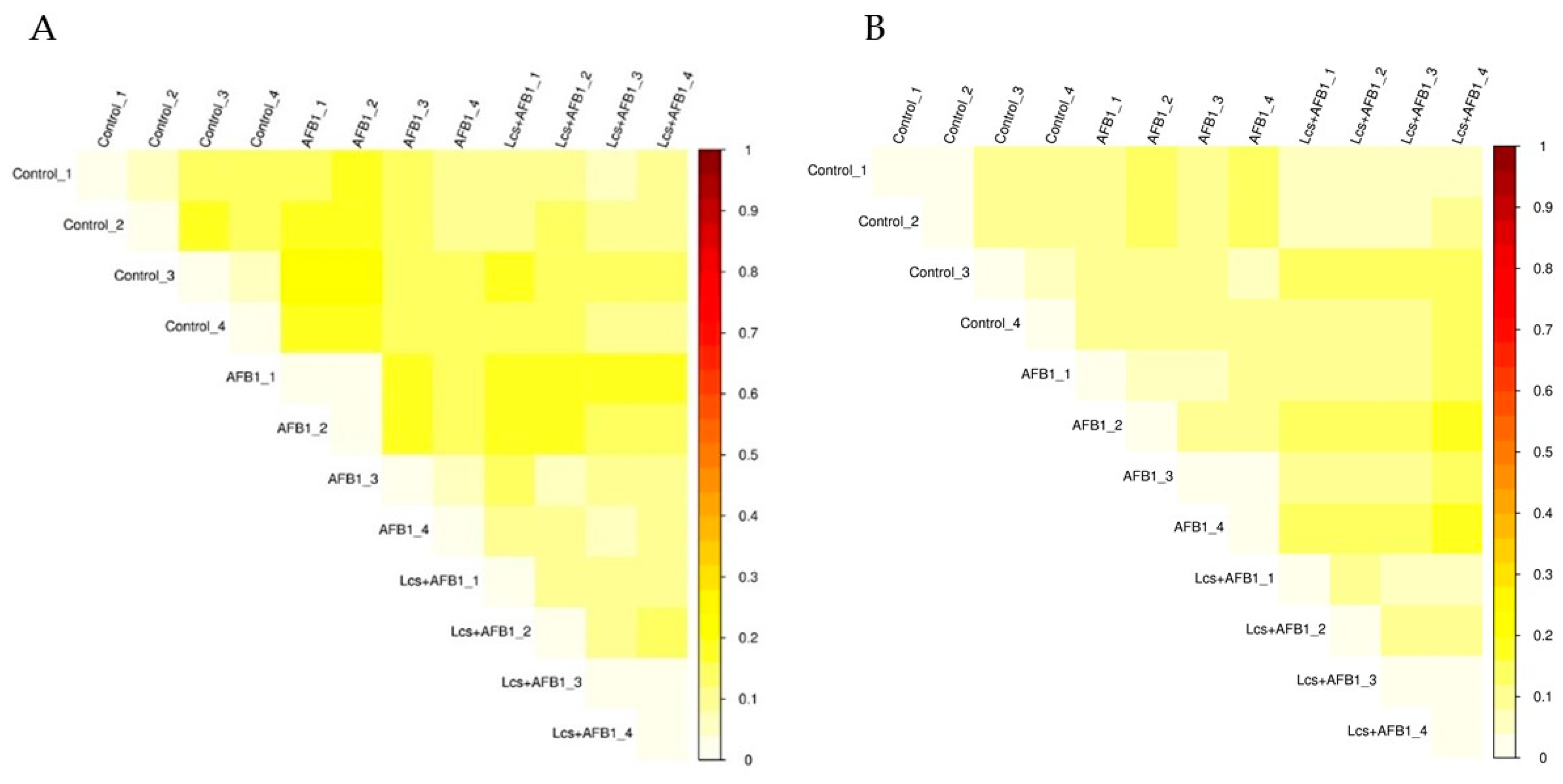
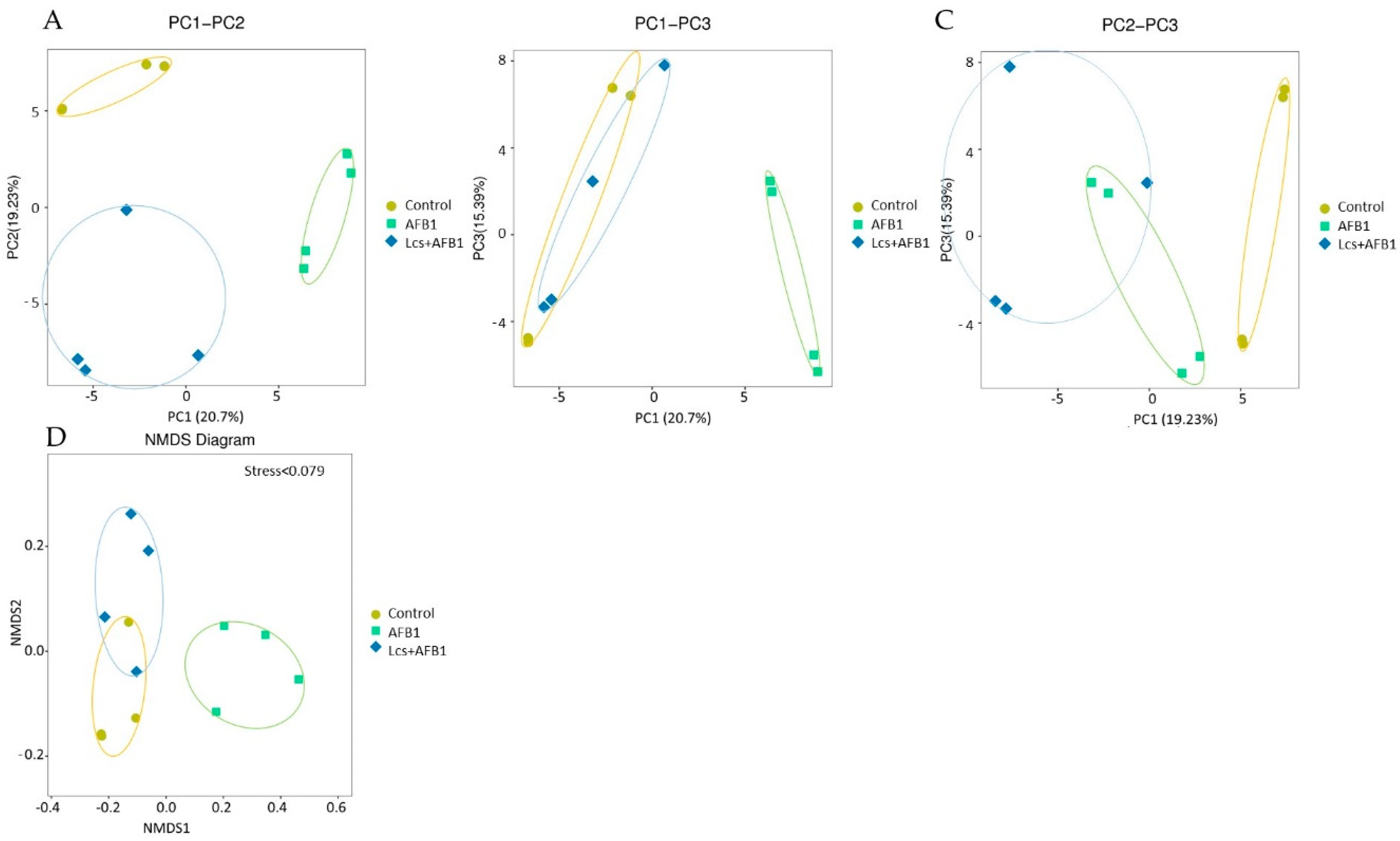
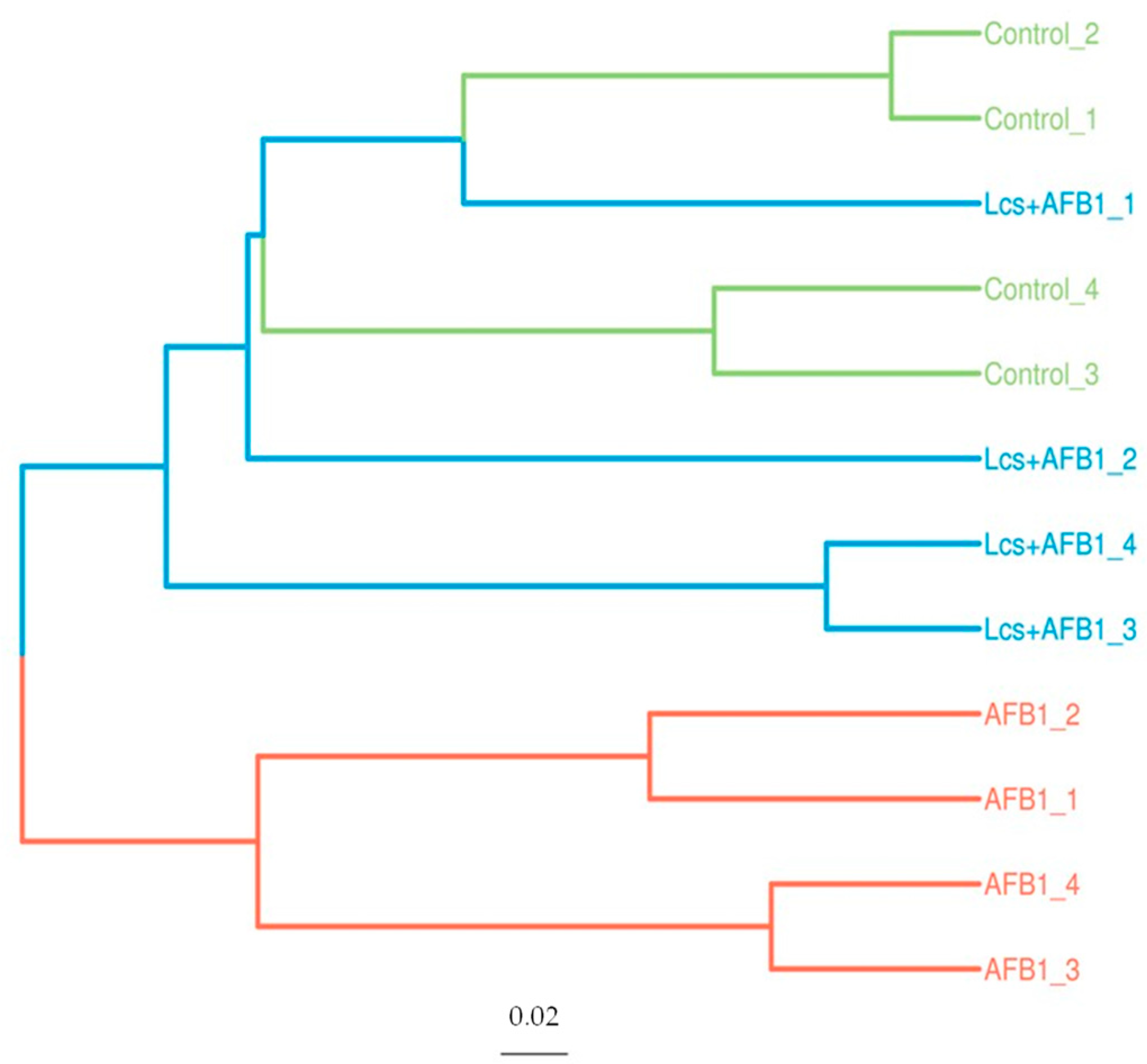
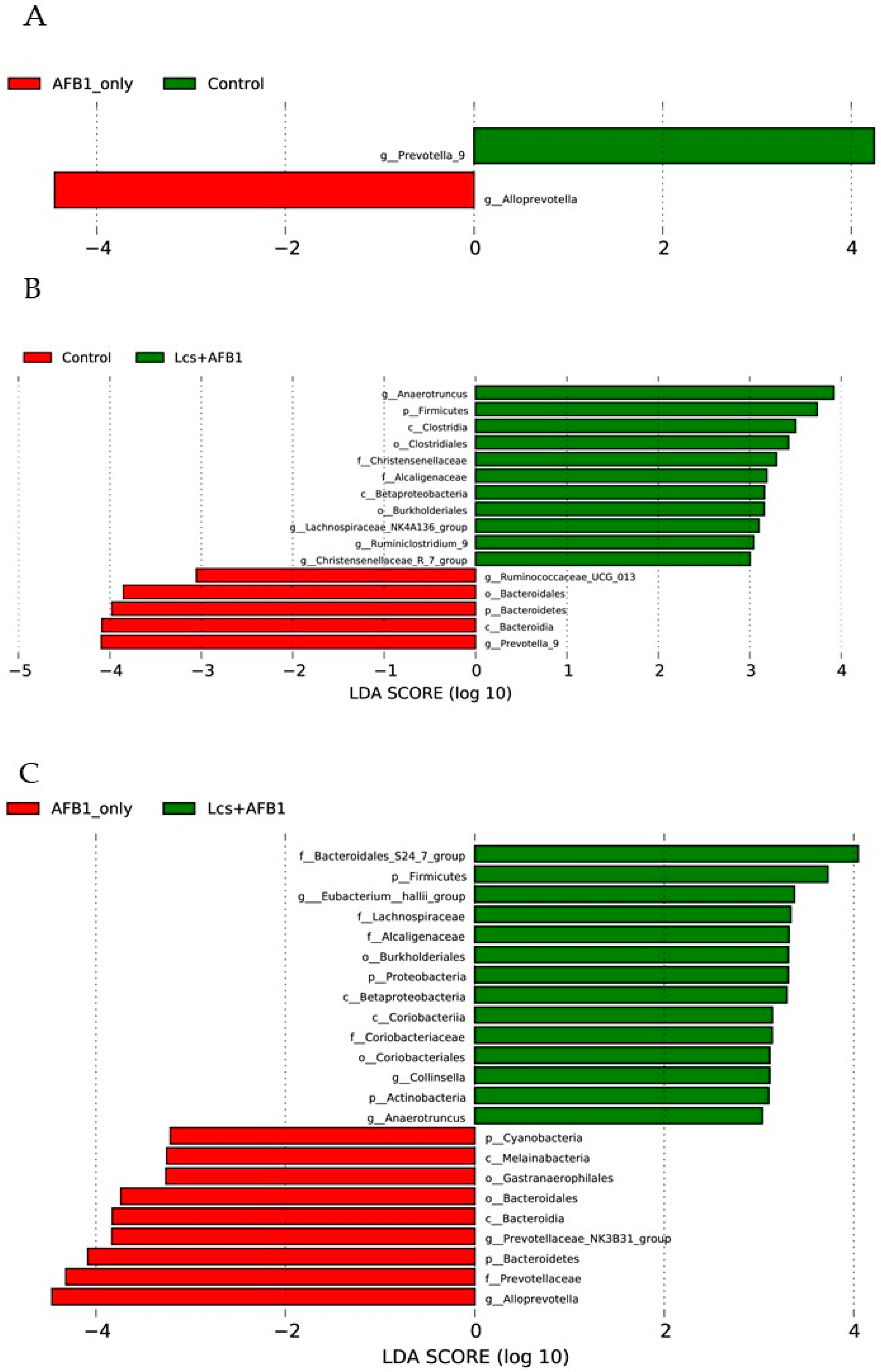
2.3. Variation Analysis Between Groups
3. Conclusions
4. Materials and Methods
4.1. Ethics Statement
4.2. Bacterial Culture
4.3. Experimental Animals
4.4. Experimental Protocol
4.5. Gut Microbiome Modulation via Administration of Lactobacillus casei Shirota on AFB1-Induced Rat
4.5.1. Fecal Sample Collection
4.5.2. Extraction of Fecal Sample DNA
4.5.3. Metagenomic Sequencing of Gut Microbiota
4.5.4. Data Analysis
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Neme, K.; Mohammed, A. Mycotoxin occurrence in grains and the role of postharvest management as a mitigation strategies. A review. Food Control 2017, 78, 412–425. [Google Scholar] [CrossRef]
- Dai, Y.; Huang, K.; Zhang, B.; Zhu, L.; Xu, W. Aflatoxin B1-induced epigenetic alterations: An overview. Food Chem. Toxicol. 2017, 109, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar-Mathur, P.; Sunkara, S.; Bhatnagar-Panwar, M.; Waliyar, F.; Sharma, K.K. Biotechnological advances for combating aspergillus flavus and aflatoxin contamination in crops. Plant Sci. 2015, 234, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Marchese, S.; Polo, A.; Ariano, A.; Velotto, S.; Costantini, S.; Severino, L. Aflatoxin B1 and M1: Biological properties and their involvement in cancer development. Toxins 2018, 10, 214. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.Y.; Watson, S.; Routledge, M.N. Aflatoxin exposure and associated human health effects, a review of epidemiological studies. Food Saf. 2016, 4, 14–27. [Google Scholar] [CrossRef]
- Prakash, B.; Kedia, A.; Mishra, P.K.; Dubey, N. Plant essential oils as food preservatives to control moulds, mycotoxin contamination and oxidative deterioration of agri-food commodities–potentials and challenges. Food Control 2015, 47, 381–391. [Google Scholar] [CrossRef]
- Robert, H.; Payros, D.; Pinton, P.; Théodorou, V.; Mercier-Bonin, M.; Oswald, I.P. Impact of mycotoxins on the intestine: Are mucus and microbiota new targets? J. Toxicol. Environ. Health Part B 2017, 20, 249–275. [Google Scholar] [CrossRef]
- Liew, W.-P.-P.; Mohd-Redzwan, S. Mycotoxin: Its impact on gut health and microbiota. Front. Cell. Infect. Microbiol. 2018, 8, 60. [Google Scholar] [CrossRef]
- Lynch, S.V.; Pedersen, O. The human intestinal microbiome in health and disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef]
- Palm, N.W.; de Zoete, M.R.; Flavell, R.A. Immune–microbiota interactions in health and disease. Clin. Immunol. 2015, 159, 122–127. [Google Scholar] [CrossRef]
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Reddy, D.N. Role of the normal gut microbiota. World J. Gastroenterol. WJG 2015, 21, 8787. [Google Scholar] [CrossRef] [PubMed]
- D’Argenio, V.; Salvatore, F. The role of the gut microbiome in the healthy adult status. Clin. Chim. Acta 2015, 451, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, B.O.; Bäckhed, F. Signals from the gut microbiota to distant organs in physiology and disease. Nat. Med. 2016, 22, 1079. [Google Scholar] [CrossRef]
- Ussar, S.; Griffin, N.W.; Bezy, O.; Fujisaka, S.; Vienberg, S.; Softic, S.; Deng, L.; Bry, L.; Gordon, J.I.; Kahn, C.R. Interactions between gut microbiota, host genetics and diet modulate the predisposition to obesity and metabolic syndrome. Cell Metab. 2015, 22, 516–530. [Google Scholar] [CrossRef] [PubMed]
- Sonnenburg, E.D.; Smits, S.A.; Tikhonov, M.; Higginbottom, S.K.; Wingreen, N.S.; Sonnenburg, J.L. Diet-induced extinctions in the gut microbiota compound over generations. Nature 2016, 529, 212. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, J.R.; Adams, D.H.; Fava, F.; Hermes, G.D.; Hirschfield, G.M.; Hold, G.; Quraishi, M.N.; Kinross, J.; Smidt, H.; Tuohy, K.M. The gut microbiota and host health: A new clinical frontier. Gut 2015, 65, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, B.; Delgado, S.; Blanco-Míguez, A.; Lourenço, A.; Gueimonde, M.; Margolles, A. Probiotics, gut microbiota, and their influence on host health and disease. Mol. Nutr. Food Res. 2017, 61, 1600240. [Google Scholar] [CrossRef]
- Bauer, E.; Williams, B.A.; Smidt, H.; Mosenthin, R.; Verstegen, M.W. Influence of dietary components on development of the microbiota in single-stomached species. Nutr. Res. Rev. 2006, 19, 63–78. [Google Scholar] [CrossRef]
- FAO/WHO. Joint FAO/WHO Expert Consultation on Evaluation of Health and Nutritional Properties of Probiotics in Food including Powder Milk with Live Lactic Acid Bacteria; FAO/WHO: Cordoba, Argentina, 2001. [Google Scholar]
- Martín, R.; Bermúdez-Humarán, L.G.; Langella, P. Searching for the bacterial effector: The example of the multi-skilled commensal bacterium faecalibacterium prausnitzii. Front. Microbiol. 2018, 9, 346. [Google Scholar] [CrossRef]
- Heeney, D.D.; Gareau, M.G.; Marco, M.L. Intestinal lactobacillus in health and disease, a driver or just along for the ride? Curr. Opin. Biotechnol. 2018, 49, 140–147. [Google Scholar] [CrossRef]
- Fochesato, A.; Cuello, D.; Poloni, V.; Galvagno, M.; Dogi, C.; Cavaglieri, L. Aflatoxin B1 adsorption/desorption dynamics in the presence of lactobacillus rhamnosus rc 007 in a gastrointestinal tract simulated model. J. Appl. Microbiol. 2018. [Google Scholar] [CrossRef]
- Apás, A.L.; González, S.N.; Arena, M.E. Potential of goat probiotic to bind mutagens. Anaerobe 2014, 28, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Damayanti, E.; Istiqomah, L.; Saragih, J.; Purwoko, T. Characterization of Lactic Acid Bacteria as Poultry Probiotic Candidates with Aflatoxin B1 Binding Activities. In IOP Conference Series: Earth and Environmental Science; IOP Publishing: Bristol, UK, 2017; p. 012030. [Google Scholar]
- Saladino, F.; Posarelli, E.; Luz, C.; Luciano, F.; Rodriguez-Estrada, M.; Mañes, J.; Meca, G. Influence of probiotic microorganisms on aflatoxins B1 and B2 bioaccessibility evaluated with a simulated gastrointestinal digestion. J. Food Compos. Anal. 2018, 68, 128–132. [Google Scholar] [CrossRef]
- Matsumoto, K.; Takada, T.; Shimizu, K.; Moriyama, K.; Kawakami, K.; Hirano, K.; Kajimoto, O.; Nomoto, K. Effects of a probiotic fermented milk beverage containing lactobacillus casei strain shirota on defecation frequency, intestinal microbiota, and the intestinal environment of healthy individuals with soft stools. J. Biosci. Bioeng. 2010, 110, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Winnie-Pui-Pui, L.; Adilah, Z.N.; Leslie, T.T.L.; Sabran, M.-R. The binding efficiency and interaction of lactobacillus casei shirota toward aflatoxin B1. Front. Microbiol. 2018, 9, 1503. [Google Scholar]
- Mohd Redzwan, S.; Mutalib, M.S.A.; Wang, J.-S.; Ahmad, Z.; Kang, M.-S.; Nasrabadi, E.N.; Jamaluddin, R. Effect of supplementation of fermented milk drink containing probiotic lactobacillus casei shirota on the concentrations of aflatoxin biomarkers among employees of universiti putra malaysia: A randomised, double-blind, cross-over, placebo-controlled study. Br. J. Nutr. 2016, 115, 39–54. [Google Scholar] [CrossRef]
- Wos-Oxley, M.L.; Bleich, A.; Oxley, A.P.; Kahl, S.; Janus, L.M.; Smoczek, A.; Nahrstedt, H.; Pils, M.C.; Taudien, S.; Platzer, M. Comparative evaluation of establishing a human gut microbial community within rodent models. Gut Microbes 2012, 3, 234–249. [Google Scholar] [CrossRef]
- Lun, H.; Yang, W.; Zhao, S.; Jiang, M.; Xu, M.; Liu, F.; Wang, Y. Altered gut microbiota and microbial biomarkers associated with chronic kidney disease. Microbiol. Open 2018. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Chen, H.; Mao, B.; Yang, Q.; Zhao, J.; Gu, Z.; Zhang, H.; Chen, Y.Q.; Chen, W. Microbial biogeography and core microbiota of the rat digestive tract. Sci. Rep. 2017, 7, 45840. [Google Scholar] [CrossRef]
- Nguyen, T.L.A.; Vieira-Silva, S.; Liston, A.; Raes, J. How informative is the mouse for human gut microbiota research? Dis. Models Mech. 2015, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Gwinn, D.C.; Allen, M.S.; Bonvechio, K.I.; Hoyer, M.V.; Beesley, L.S. Evaluating estimators of species richness: The importance of considering statistical error rates. Methods Ecol. Evol. 2016, 7, 294–302. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, M.; Ho, C.-T.; Guo, X.; Wu, Z.; Weng, P.; Yan, M.; Cao, J. Metagenomics analysis of gut microbiota modulatory effect of green tea polyphenols by high fat diet-induced obesity mice model. J. Funct. Foods 2018, 46, 268–277. [Google Scholar] [CrossRef]
- Gharechahi, J.; Zahiri, H.S.; Noghabi, K.A.; Salekdeh, G.H. In-depth diversity analysis of the bacterial community resident in the camel rumen. Syst. Appl. Microbiol. 2015, 38, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. Unifrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169. [Google Scholar] [CrossRef]
- Paliy, O.; Shankar, V. Application of multivariate statistical techniques in microbial ecology. Mol. Ecol. 2016, 25, 1032–1057. [Google Scholar] [CrossRef] [PubMed]
- Dexter, E.; Rollwagen-Bollens, G.; Bollens, S.M. The trouble with stress: A flexible method for the evaluation of nonmetric multidimensional scaling. Limnol. Oceanogr. Methods 2018, 16, 434–443. [Google Scholar] [CrossRef]
- Comunian, R.; Ferrocino, I.; Paba, A.; Daga, E.; Campus, M.; Di Salvo, R.; Cauli, E.; Piras, F.; Zurru, R.; Cocolin, L. Evolution of microbiota during spontaneous and inoculated tonda di cagliari table olives fermentation and impact on sensory characteristics. LWT-Food Sci. Technol. 2017, 84, 64–72. [Google Scholar] [CrossRef]
- Xia, Y.; Sun, J. Statistical models and analysis of microbiome data from mice and humans. In Mechanisms Underlying Host-Microbiome Interactions in Pathophysiology of Human Diseases; Springer: Boston, MA, USA, 2018; pp. 303–371. [Google Scholar]
- Bicalho, M.; Machado, V.; Higgins, C.; Lima, F.; Bicalho, R. Genetic and functional analysis of the bovine uterine microbiota. Part i: Metritis versus healthy cows. J. Dairy Sci. 2017, 100, 3850–3862. [Google Scholar] [CrossRef]
- Stearns, J.C.; Davidson, C.J.; McKeon, S.; Whelan, F.J.; Fontes, M.E.; Schryvers, A.B.; Bowdish, D.M.; Kellner, J.D.; Surette, M.G. Culture and molecular-based profiles show shifts in bacterial communities of the upper respiratory tract that occur with age. ISME J. 2015, 9, 1246. [Google Scholar] [CrossRef]
- Wang, J.; Tang, L.; Glenn, T.C.; Wang, J.-S. Aflatoxin B1 induced compositional changes in gut microbial communities of male f344 rats. Toxicol. Sci. 2015, 150, kfv259. [Google Scholar]
- Cheng, W.; Lu, J.; Li, B.; Lin, W.; Zhang, Z.; Wei, X.; Sun, C.; Chi, M.; Bi, W.; Yang, B. Effect of functional oligosaccharides and ordinary dietary fiber on intestinal microbiota diversity. Front. Microbiol. 2017, 8, 1750. [Google Scholar] [CrossRef] [PubMed]
- Longo, P.L.; Dabdoub, S.; Kumar, P.; Artese, H.P.C.; Dib, S.A.; Romito, G.A.; Mayer, M.P.A. Glycemic status affects the subgingival microbiome of diabetic patients. J. Clin. Periodontol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.H.; Oh, I.J.; Jung, C.; Lee, S.Y.; Lee, J. The effect of protectants and pH changes on the cellular growth and succinic acid yield of mannheimia succiniciproducens LPK7. J. Microbiol. Biotechnol. 2010, 20, 1677–1680. [Google Scholar] [PubMed]
- Zhao, T.; Mu, X.; You, Q. Succinate: An initiator in tumorigenesis and progression. Oncotarget 2017, 8, 53819. [Google Scholar] [CrossRef]
- Gupta, R.A.; Motiwala, M.N.; Dumore, N.G.; Danao, K.R.; Ganjare, A.B. Effect of piperine on inhibition of FFA induced TLR4 mediated inflammation and amelioration of acetic acid induced ulcerative colitis in mice. J. Ethnopharmacol. 2015, 164, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Jacques, P.; Elewaut, D. Joint expedition: Linking gut inflammation to arthritis. Mucosal Immunol. 2008, 1, 364. [Google Scholar] [CrossRef]
- Nurul Adilah, Z.; Liew, W.-P.-P.; Mohd Redzwan, S.; Amin, I. Effect of high protein diet and probiotic lactobacillus casei shirota supplementation in aflatoxin B1-induced rats. BioMed Res. Int. 2018, 2018, 9568351. [Google Scholar] [CrossRef]
- Knipstein, B.; Huang, J.; Barr, E.; Sossenheimer, P.; Dietzen, D.; Egner, P.A.; Groopman, J.D.; Rudnick, D.A. Dietary aflatoxin-induced stunting in a novel rat model: Evidence for toxin-induced liver injury and hepatic growth hormone resistance. Pediatr. Res. 2015, 78, 120–127. [Google Scholar] [CrossRef]
- Pitt, J.M.; Vétizou, M.; Waldschmitt, N.; Kroemer, G.; Chamaillard, M.; Boneca, I.G.; Zitvogel, L. Fine-tuning cancer immunotherapy: Optimizing the gut microbiome. Cancer Res. 2016, 76, 4602–4607. [Google Scholar] [CrossRef]
- Di Cerbo, A.; Palmieri, B.; Aponte, M.; Morales-Medina, J.C.; Iannitti, T. Mechanisms and therapeutic effectiveness of lactobacilli. J. Clin. Pathol. 2015, 69, 187–203. [Google Scholar] [CrossRef] [PubMed]
- Kato-Kataoka, A.; Nishida, K.; Takada, M.; Kawai, M.; Kikuchi-Hayakawa, H.; Suda, K.; Ishikawa, H.; Gondo, Y.; Shimizu, K.; Matsuki, T. Fermented milk containing lactobacillus casei strain shirota preserves the diversity of the gut microbiota and relieves abdominal dysfunction in healthy medical students exposed to academic stress. Appl. Environ. Microbiol. 2016, 82, 3649–3658. [Google Scholar] [CrossRef] [PubMed]
- Spanhaak, S.; Havenaar, R.; Schaafsma, G. The effect of consumption of milk fermented by lactobacillus casei strain shirota on the intestinal microflora and immune parameters in humans. Eur. J. Clin. Nutr. 1998, 52, 899. [Google Scholar] [CrossRef] [PubMed]
- Bonvalet, M.; Daillère, R.; Roberti, M.P.; Rauber, C.; Zitvogel, L. The impact of the intestinal microbiota in therapeutic responses against cancer. In Oncoimmunology; Springer: Cham, Swithland, 2018; pp. 447–462. [Google Scholar]
- Aguilar-Toalá, J.; Santiago-López, L.; Peres, C.; Peres, C.; Garcia, H.; Vallejo-Cordoba, B.; González-Córdova, A.; Hernández-Mendoza, A. Assessment of multifunctional activity of bioactive peptides derived from fermented milk by specific lactobacillus plantarum strains. J. Dairy Sci. 2017, 100, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Donkor, O.N.; Henriksson, A.; Vasiljevic, T.; Shah, N. A-galactosidase and proteolytic activities of selected probiotic and dairy cultures in fermented soymilk. Food Chem. 2007, 104, 10–20. [Google Scholar] [CrossRef]
- Potula, H.-H.; Richer, L.; Werts, C.; Gomes-Solecki, M. Pre-treatment with lactobacillus plantarum prevents severe pathogenesis in mice infected with leptospira interrogans and may be associated with recruitment of myeloid cells. PLoS Negl. Trop. Dis. 2017, 11, e0005870. [Google Scholar] [CrossRef]
- Nikbakht Nasrabadi, E.; Jamaluddin, R.; Mutalib, A.; Khaza’ai, H.; Khalesi, S.; Mohd Redzwan, S. Reduction of aflatoxin level in aflatoxin-induced rats by the activity of probiotic lactobacillus casei strain shirota. J. Appl. Microbiol. 2013, 114, 1507–1515. [Google Scholar] [CrossRef]
- Qian, G.; Tang, L.; Guo, X.; Wang, F.; Massey, M.E.; Su, J.; Guo, T.L.; Williams, J.H.; Phillips, T.D.; Wang, J.S. Aflatoxin B1 modulates the expression of phenotypic markers and cytokines by splenic lymphocytes of male f344 rats. J. Appl. Microbiol. 2014, 34, 241–249. [Google Scholar]
- Daniel, J.H.; Lewis, L.W.; Redwood, Y.A.; Kieszak, S.; Breiman, R.F.; Flanders, W.D.; Bell, C.; Mwihia, J.; Ogana, G.; Likimani, S. Comprehensive assessment of maize aflatoxin levels in eastern Kenya, 2005–2007. Environ. Health Perspect. 2011, 119, 1794. [Google Scholar] [CrossRef]
- Li, M.; Shu, X.; Xu, H.; Zhang, C.; Yang, L.; Zhang, L.; Ji, G. Integrative analysis of metabolome and gut microbiota in diet-induced hyperlipidemic rats treated with berberine compounds. J. Transl. Med. 2016, 14, 237. [Google Scholar] [CrossRef]
- Ferrand, J.; Patron, K.; Legrand-Frossi, C.; Frippiat, J.-P.; Merlin, C.; Alauzet, C.; Lozniewski, A. Comparison of seven methods for extraction of bacterial DNA from fecal and cecal samples of mice. J. Microbiol. Methods 2014, 105, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.R.; Borre, Y.; O’Brien, C.; Patterson, E.; El Aidy, S.; Deane, J.; Kennedy, P.J.; Beers, S.; Scott, K.; Moloney, G. Transferring the blues: Depression-associated gut microbiota induces neurobehavioural changes in the rat. J. Psychiatr. Res. 2016, 82, 109–118. [Google Scholar] [CrossRef]
- Chan, C.S.; Chan, K.-G.; Tay, Y.-L.; Chua, Y.-H.; Goh, K.M. Diversity of thermophiles in a malaysian hot spring determined using 16s rRNA and shotgun metagenome sequencing. Front. Microbiol. 2015, 6, 177. [Google Scholar] [CrossRef]
- Jesser, K.J.; Noble, R.T. Characterizing the ecology of Vibrio in the Neuse River Estuary, North Carolina using heat shock protein 60 (hsp60) next-generation amplicon sequencing. Appl. Environ. Microbiol. 2018, 84, e00333-18. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.R.; Wang, Q.; Cardenas, E.; Fish, J.; Chai, B.; Farris, R.J.; Kulam-Syed-Mohideen, A.S.; McGarrell, D.M.; Marsh, T.; Garrity, G.M. The ribosomal database project: Improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009, 37, 141–145. [Google Scholar] [CrossRef]
- Kraemer, J.G.; Ramette, A.; Aebi, S.; Oppliger, A.; Hilty, M. Influence of pig farming on the human’s nasal microbiota: The key role of the airborne microbial communities. Appl. Environ. Microbiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Astudillo-García, C.; Bell, J.J.; Webster, N.S.; Glasl, B.; Jompa, J.; Montoya, J.M.; Taylor, M.W. Evaluating the core microbiota in complex communities: A systematic investigation. Environ. Microbiol. 2017, 19, 1450–1462. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; Van, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factor | R-Value | p-Value |
|---|---|---|
| Control vs AFB1 | 1 | 0.025 |
| Control vs Lcs + AFB1 | 0.292 | 0.094 |
| AFB1 vs Lcs + AFB1 | 0.87 | 0.025 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liew, W.-P.-P.; Mohd-Redzwan, S.; Than, L.T.L. Gut Microbiota Profiling of Aflatoxin B1-Induced Rats Treated with Lactobacillus casei Shirota. Toxins 2019, 11, 49. https://doi.org/10.3390/toxins11010049
Liew W-P-P, Mohd-Redzwan S, Than LTL. Gut Microbiota Profiling of Aflatoxin B1-Induced Rats Treated with Lactobacillus casei Shirota. Toxins. 2019; 11(1):49. https://doi.org/10.3390/toxins11010049
Chicago/Turabian StyleLiew, Winnie-Pui-Pui, Sabran Mohd-Redzwan, and Leslie Thian Lung Than. 2019. "Gut Microbiota Profiling of Aflatoxin B1-Induced Rats Treated with Lactobacillus casei Shirota" Toxins 11, no. 1: 49. https://doi.org/10.3390/toxins11010049
APA StyleLiew, W.-P.-P., Mohd-Redzwan, S., & Than, L. T. L. (2019). Gut Microbiota Profiling of Aflatoxin B1-Induced Rats Treated with Lactobacillus casei Shirota. Toxins, 11(1), 49. https://doi.org/10.3390/toxins11010049